

Abstract
Background The urine from healthy human is traditionally considered to be sterile. And researches on urine microbiome were mainly focused on infection and urinary incontinence. There were few studies linking female urine and reproductive tract microbial communities.
Results Here we sampled the urinary microbiota in 147 Chinese women, and explored the nature of colonization by 16S rRNA gene amplicon sequencing, real-time qPCR and live bacteria culture. The results reflect distinct bacterial communities in urine, indicative of a non-sterile environment. Lactobacillus, Streptococcus and Pseudomonas were the most three dominated genera in urine. We also examined the relationship between urine and six sites of the reproductive tract, and found microbiota in the urine were ranged between upper and lower reproductive tract. The urinary microbiota was also closely linked to the natural and diseased physiological conditions of subjects by associating with their lifestyle and gynecology clinical factors.
Conclusion Our research showed the connectivity of the microbiota in the female urogenital system and provided insight into the exploration of urethra and genital tract diseases.
Introduction
The role of microbiota played in vaginal environment has received a lot of attention over the past decade, while the female upper reproductive tract is traditionally believed to be sterile and mostly studied in the context of infections or incontinence [1]. Recently, Chen et al. has elaborated that the female upper reproductive tract is colonised a microbiota and associated with the life style, pregnant history and gynaecological diseases. Like the female upper reproductive tract[2], the sterile hypothesis of urine also been overturned by emerging evidences that indicate the existence of microorganisms in urinary tract by cultural or sequencing methods [3,4], and it is significantly associated with the infection of bladder and urinary tract [5]. A recent culture-dependent study also has proposed a similarity between bacterial isolates from the bladder and from the vagina from the same individuals [6], however the microbial biomass in urine is generally much lower [7].
Here we report the urinary microbiota for a relatively large cohort of 147 women of reproductive age. Together with our recently published peritoneal fluid, uterine and vaginal samples from the same individuals, we show that although with greater amounts of Lactobacillus and Streptococcus, the urinary microbiota is more similar to the microbiota in cervix and uterine cavity, in accordance with the anatomical opening of the bladder. Together with a wealth of metadata, we also explore the potential of the urinary microbiota for diagnosis.
Results
Microbiota composition of the urine
Morning midstream urine (UR) was self-collected on the day of surgery from an exploratory study cohort of 137 Chinese women recruited at Peking University Shenzhen Hospital, China. As with our previous vagino-uterine microbiota study [2], all the volunteers had conditions that were not known to involve infections (Supplementary Table 1). 95 women in the cohort also collected samples from six locations within the female reproductive tract, including lower third of vagina (CL), posterior fornix(CU), cervical mucus (CV), endometrium (ET), left and right fallopian tubes (FLL and FRL), and peritoneal fluid (PF).Their vagino-uterine microbiome information have been published previously [2]. After 16S rRNA gene amplicon sequencing, the sequencing reads were pre-processed for quality control and filtering, then clustered into operational taxonomic units (OTUs) (Supplementary Table 2, 3).
The urinary bacterial communities in genus level were clustered into three subtypes: two were dominated by Lactobacillus or Streptococcus, whereas the third had diverse organisms (Fig. 1). Pseudomonas, Staphylococcus, Acinetobacter and Vagococcus also constituted a significant part of the urinary bacterial communities. Although urethral orifice is next to vaginal orifice, the median relative abundance of Lactobacillus, Pseudomonas and Acinetobacter in the UR samples were more similar to the ET samples (Supplementary Fig. 1). At the phylum level, urine was dominated by Firmicutes and Proteobacteria (Supplementary Fig. 1).

The relative abundance of genus detected in each individual was shown in the bar chart. Samples derive from the initial cohort of 137 Chinese reproductive-age women (Supplementary Table 1)
Cultivation of live bacteria from Transurethral catheterized urine
The question whether bacterial DNA signals are made up of living bacteria or fragments in the urine samples has aroused heated discussion. Here we performed a validation study by the combination of 16S rRNA gene amplicon sequencing and live bacteria culture of urine samples from an additional cohort of 10 women.
According to the sequencing results, 8 out of 9 negative controls had lower number of reads than the respective urine samples (Supplementary Fig. 2), confirming that few biomass was detected in the non-bio control samples, which eliminated the deviation introduced by sample contamination. Then we tried to culture and isolate bacterial colony from newly collected urine samples. Urine samples were serial diluted and spread on three different kinds of agar plates and incubated under both aerobically and anaerobically conditions. Six different positive isolates belonging to 5 genera, including Lactobacillus, Staphylococcus, Clostridium, Enterococcus and Propionibacterium were obtained from 3 out of 10 subjects (Supplementary Table 4). The 5 genera were also found as dominate in our 16S rRNA gene amplicon sequencing data, and consistent with previous cultivation results of published papers. [8–11] (Supplementary Table 4). Reassuringly, no isolates were detected from the negative controls. Therefore, we verified the existence of live bacteria in the urine by obtaining isolates using conventional culturing methods.
A considerable amount of bacterial biomass revealed by qPCR
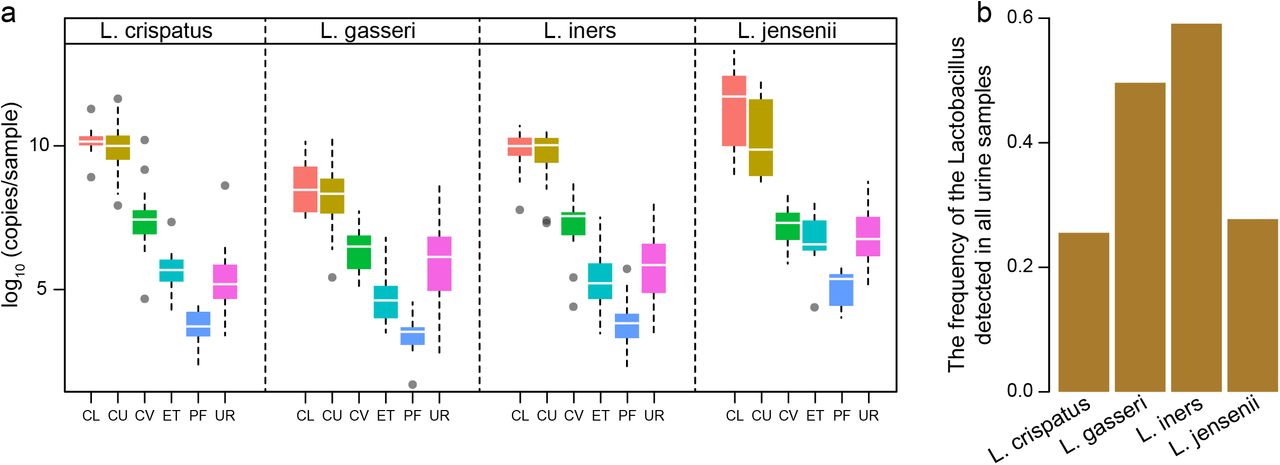
To provide some additional compelling evidences of the bacterial communities in the urine, a species-specific real-time qPCR method was utilized to focus on the four dominant vaginal Lactobacillus species, i.e. L. crispatus, L. iners, L. jensenii and L. gasseri. The Lactobacillus species we examined presented the similar distribution and abundance along the reproductive tract, and the corresponding urinary Lactobacillus ranged between upper and lower reproductive tract (Fig. 2a). Among them, L. iners occurred most frequently in the urine samples (Fig. 2b). The total bacterial biomass is expressed by the ratio of the copy number from the result of qPCR to relative abundance according to the result of 16S rRNA gene sequencing in same sample. The result gave an estimation of 107, placing the urinary bacterial biomass between the vaginal-cervical sites (1010-1011) and the endometrium (ET) and the peritoneal fluid (PF) samples [2] (Fig. 2), all of which were orders of magnitude above potential background noise (Supplementary Fig. 2) [12]. These results were interestingly consistent with a weakly acidic pH of the urine, in contrast to pH < 4.5 in the vagina or pH ~ 8 in the peritoneal fluid [13].

(a) The abundance of Lactobacillus iners, Lactobacillus jensenii, Lactobacillus crispatus and Lactobacillus gasseri calculated by qPCR results in different samples. Boxes denote the interquartile range (IQR) between the first and third quartiles (25th and 75th percentiles, respectively), and the line inside the boxes denote the median. The whiskers denote the lowest and highest values within 1.5 times the IQR from the first and third quartiles, respectively. Samples derive from the initial cohort of 137 Chinese reproductive-age women (Supplementary Table 1). (b) The frequency of the related Lactobacillus detected in all urine samples.
Microbiota in the urine is similar to that in the reproductive tract
To further assess the microbiota relationship between urine and the 6 positions of the reproductive tract, the intra-individual distances between samples of different sit from each individual were calculated. Weighted and unWeighted intra-individual UniFrac distance relative to urine samples increased from CL to ET and PF gradually. The result showed that weighted and unWeighted intra-individual UniFrac distance relative to urine samples increased from CL to ET and PF gradually (Supplementary Fig. 3). Although the urine and reproductive tract samples also clustered together when subjected to principal coordinate analyses (PCoA), the similar distribution of microbiota was presented between urine and ET/CV (Fig 3).

Samples were taken from UR, CL, CU and CV before operation, and from ET and PF during operation. Samples derive from the initial cohort of 137 Chinese reproductive-age women. Each dot represents one sample (n = 94 CL, 95 CU, 95 CV, 80 ET, 93 PF, 9 FLL, 10 FRL and 137 UR).
(a) The scatter plot with arrows is specific to the coinertia analysis, and represents the individuals. The beginning of the arrow is the position of the individual described by the OTU set in urine samples; the end of the arrow is the position of the individual described by the urinary metabolites. (b) The relationship between the metabolites and the OTUs provided by the coinertia analysis. A higher coinertia value indicates a higher explanation degree. Samples derive from the initial cohort of 137 Chinese reproductive-age women.
Lifestyle and clinical factors influencing the urinary microbiota
With our comprehensive collection of demographic and baseline clinical characteristics from women of reproductive age (Supplementary Table 1), we explored whether lifestyle and various clinical factors were related to changes in urinary bacterial communities. Urinary microbial composition was significantly associated with these factors, such as age, surgical history, abortion, vaginal deliveries, experience of given birth (multipara vs. nullipara), infertility due to endometriosis and hysteromyoma (PERMANOVA, P < 0.05, q < 0.05, Table 1).Although the urinary microbiota also correlated with some other factors, such as menstrual phase, contraception, endometriosis, pelvic adhesiolysis and anemia, the statistical significance was not achieved after controlling for multiple testing (PERMANOVA, P < 0.05 but q > 0.05, Table 1). Therefore, urinary microbiota closely linked to the natural and diseased physiological conditions of reproductive-aged women inferred the signal or critical indicator to the female health.
PERMANOVA for the influence of phenotypes on the urinary microbiota
Discussion
Using the self-collected and catheterized urine from women without infection and other complications, here we demonstrate the existence of urinary microbiome. Similar to the female reproductive tract, the urinary tract and bladder are inhabited by the commensal microorganisms, such as Lactobacillus, Streptococcus, Pseudomonas and Acinetobacter. And its bacterial biomass is between the lower (vagina and cervix) and the upper (endometrium and the peritoneal fluid) genital tract samples. The female urinary tract is proximity to the vagina with rich microbial environments, which may be related to the composition of urine microbes.
Previous related studies about the female urogenital system indicated the alterations in the urinary microbiota were associated with cesarean delivery [14] and bacterial vaginosis [15]. The urinary microbial diversity has also been reported to be increased after trauma [14][16]. We also explored the association between the composition of urinary microbiota and that of reproductive tract microbiota and uterine-related factors. It is valuable to note the diseases, such as hysteromyoma, endometriosis, and infertility due to endometriosis, were significantly associated with urinary microbiota. The variance in microbial composition of endometrium and the peritoneal fluid samples also can be significant explained by the urinary metabolites.
Therefore, we need to be aware of the changes in urinary microbiota associated with uterine-related diseases, which requires detailed exploration. To our knowledge, this is the first comprehensive study to investigate the relationship between urinary microbiota and various uterine-related factors which provide the reference for clinical. The discoveries inspire a novel scientific explanation of the uterine-related diseases by longitudinal studies on the microbiome of urinary and reproductive tract.
Materials and Methods
Ethical approval and study population
The study was approved by the Institutional Review Board of BGI-Shenzhen and Peking University Shenzhen Hospital. All participants gave written informed consent prior to their recruitment into the study.
In this study, a total of 147 reproductive age women (age 22-48) were recruited by Peking University Shenzhen Hospital (Supplementary Table 1). All participants were reproductive age women who underwent hysteroscopy and/or laparoscopy for conditions without infections, such as hysteromyoma, adenomyosis, endometriosis, salpingemphraxis, etc. Subjects with other related diseases, such as vaginal inflammation, severe pelvic adhesion, endocrine or autoimmune disorders were removed. Pregnant women, breastfeeding women and Menstrual women of sampling were also excluded. None of the subjects received any hormones, antibiotics and vaginal medications within a month. In addition, no cervical treatment was performed in 7 days, no vaginal douching was performed in 5 days, and no sexual activity was performed in 2 days.
Sample collection
137 self-sampling morning mid-stream urine samples were collected between December 2013 and July 2014 prior to the surgery (Supplementary Table 1), and stored at −80°C until transported on dry ice to BGI-Shenzhen for sequencing. The samples from additional 14 women for validation were collected by the doctor during the surgery in July 2017. For each operation, urine catheter was inserted into the urethra to collect mid-stream urine, and same amount of sterile saline which collected through urine catheter was set as control samples. The samples were then placed at 4°C, transported to BGI-Shenzhen, and processed within 6 hours. A portion of each sample was used for culturing live bacteria and the other part was used for sequencing.
16S rRNA amplicon sequencing
Genomic DNA was extracted as previously described [2]. The primers 515F and 907R were appiled for PCR amplification of the hypervariable regions V4-V5 of the bacterial 16S rRNA gene. The 907R primer include a unique barcoded fusion. The primers were as follow: 515F: 5’-GTGCCAGCMGCCGCGGTAA-3’ and 907R: 5’-CCGTCAATTCMTTTRAGT −3’, where M denotes A or C and R denotes purine.
The condition of PCR amplification was as follows: 3 min of denaturation at 94°C, followed by 25 cycles of 45 s at 94 °C (denaturing), 60 s at 50 °C (annealing)r, 90 s at 72 °C (elongation), with a final elongation for 10 min at 72 °C. The amplification products were purified by the AxyPrepTM Mag PCR Clean-Up Kit (Axygen, USA). The amplicon libraries generated by emulsion PCR were sequenced by Ion Torrent Personal Genome Machine (PGM) platform on 318 chips using the 400 bp sequencing kit.
Processing of sequencing reads
The raw sequencing reads were put to Mothur firstly (V1.33.3) [17] for filtering the low-quality reads followed by 3 criteria: 1) reads with less than 200bp; 2) reads not matching the degenerated PCR primers for up to two errors; 3) reads with an average quality score less than 25. A total of 8,812,607 reads, with an average of 57,225 reads per sample (a minimum of 1,113 reads and a maximum of 194,564 reads) were obtained. Subsequently, The sequences with identification threshold greater than 97% were clustered into Operational Taxonomic Units (OTUs) using QIIME uclust programme [18], where each cluster was thought of as representing a species. The seed sequences of each OTU were aligned against the Greengene reference sequences (gg_13_8_otus) to annotate using the UCLUST taxonomy assigner.
We also calculated the Unifrac distance using QIIME based on taxonomic abundance profiles at the OTU level.
PERMANOVA on the influence of phenotypes
Permutational multivariate analysis of variance (PERMANOVA) was used to assess the effect of different covariate based on the relative abundances of OTUs of the samples [19,20] by using Bray-Curtis and UniFrac distance and 9999 permutations from vegan package in R [20,21].
Real-time quantitative PCR
We quantified the four Lactobacillus species, including L.iners, L jensenii, L. crispatus and L. gasseri using the modified real-time quantitative PCR [22]. SYBR Premix Ex Taq GC (TAKARA) was applied to run on StepOnePlus Real-time PCR System (Life Technologies). PCR reaction mixture contained 10 μl of 2×SYBR Premix Ex Taq GC, 0.2 μM forward primer, 0.2 μM reverse primer, 1.6 μl of DNA sample and 8.2 μl ultrapure water to make up the final reaction volume of 20 μl. Each run included a standard curve and all samples were amplified in triplicate. The ultrapure water was also included as the blank control template.
To construct the standard curves, the sequencing-confirmed plasmids of four species were used after quantification with Qubit Fluorometer and serial 10-fold dilutions. The amplification efficiency was (100±10)% and linearity values were all ⩾0.99.
Bacterial culturing
The urine samples and controls from additional 10 subjects were cultivated in the laboratory by spreading 100 μl of sample on the different agars with 5% horse blood, such as PYG agar (DSMZ 104 medium), BHI agar and EG agar. The plates were incubated in both aerobic and anaerobic conditions at 37 °C for 72 hours. To keep the medium anaerobically during culture, resazurin and cysteine-HCl were added as reducing agents. The genomic DNA of the isolates was extracted by the Bacterial DNA Kit (OMEGA), and then performed 16S rRNA gene amplification by the universal primers 27F/1492R [23]. The amplicons were purified and sent for Sanger sequencing. The generated sequences were blast on the EzBioCloud (https://www.ezbiocloud.net/) for identification.
Coinertia analysis
The PCoA were performed based on the canberra distance using two data tables. The two tables are linked by the same individuals. The Coinertia analysis were performed with the function “coinertia” (ade4 package, R version 3.5.0).
Notes
Data availability
The sequence reads used in this paper have been deposited in the European Nucleotide Archive with the accession number PRJEB29341. The sequence reads have been also deposited in the CNSA (https://db.cngb.org/cnsa/) of CNGB database with accession code CNP0000166.
Author contributions
H.J. and R.W. organized this study. W.W., J.D., H.D., L.Z., H.T., T.W and R.W. performed the sample collection, and phenotypic information collection. F.L., L.S., C.C. and J.L. performed the molecular biology experiments. F.L., C.C. and L.H. performed the bioinformatic analyses. F.L., X.Z., C.C. and H.J., wrote the manuscript.
Competing interests
There were no competing financial interests.
Acknowledgments
The study was supported by the Shenzhen Municipal Government of China (JCYJ20160229172757249 and JCYJ20170817145523036) and Shenzhen Peacock Plan (No. KQTD20150330171505310). The authors really appreciate colleagues at BGI-Shenzhen for DNA extraction, library construction, and sequencing.





{kind=link}
{kind=link}
{kind=link}