

ABSTRACT
KRAS inhibitors perform inferior to other targeted drugs in vitro and fail clinical trials. To investigate a possible reason for this, we treated human and murine tumor cells with KRAS inhibitors deltarasin (targeting phosphodiesterase-δ), cysmethynil (targeting isoprenylcysteine carboxylmethyltransferase), and AA12 (targeting KRASG12C), and silenced/overexpressed mutant KRAS using custom-designed vectors. We show that KRAS-mutant tumor cells exclusively respond to KRAS blockade in vivo, because the oncogene co-opts host myeloid cells via a C-C-motif chemokine ligand 2/interleukin-1β-mediated signaling loop for sustained tumorigenicity. Indeed, KRAS-mutant tumors did not respond to deltarasin in Ccr2 and Il1b gene-deficient mice, but were deltarasin-sensitive in wild-type and Ccr2-deficient mice adoptively transplanted with wild-type murine bone marrow. A KRAS-dependent pro-inflammatory transcriptome was prominent in human cancers with high KRAS mutation prevalence and predicted poor survival. Our findings support that in vitro cellular systems are suboptimal for anti-KRAS drug screens since the latter drugs function to suppress interleukin-1 receptor 1 expression and myeloid IL-1β-delivered pro-growth effects in vivo. Moreover the findings support that interleukin-1β blockade might be suitable for the therapy of KRAS-mutant cancers.
INTRODUCTION
Since its discovery, the KRAS proto-oncogene GTPase (encoded by the human KRAS and the murine Kras genes) has become the holy grail of anticancer therapy [1,2]. The KRAS oncoprotein possesses a unique molecular structure that potentiates it as a driver of multiple cancer cell hallmarks (including proliferation, migration, metastasis, angiogenesis, inflammation, and apoptosis evasion), but also renders it non-actionable due to the absence of a druggable deep pocket [2,3]. KRAS point mutations that constitutively activate GTPase function occur most frequently in codons 12, 13, and 61 and are particularly frequent in pancreatic (70%), colorectal (35%), and lung (20%) adenocarcinomas [3,4]. However, full KRAS GTPase activity and downstream signaling additionally prerequisites its integration into the cell membrane, which is facilitated by post-translational lipidation and membrane transport of KRAS by various enzymes such as farnesyltransferase (FT), geranylgeranytransferase (GGT), isoprenylcysteine carboxyl methyltransferase (ICMT), phosphodiesterase-δ (PDEδ), and others [3,5]. To this end, therapeutic attempts to inhibit KRAS lipidation by targeting FT/GGT/ICMT were recently coupled by the development of PDEδ blockers and of allosteric and covalent inhibitors of mutated KRASG12C [6–9].
Despite coordinated efforts [1], anti-KRAS drug discovery is lagging behind other oncogene targets [3]. In addition to molecular structural considerations [5], the mode of KRAS oncogenic functions could be a reason for this. To this end, Janes and collaborators recently reported a discordance between the in vitro and the in vivo effects of a newly developed covalent KRASG12C inhibitor [9]. This observation is relevant to other reports describing how KRAS-dependence is linked to signatures of intravital-restricted processes like inflammation and epithelial-to-mesenchymal transition [10–12] and how pro-inflammatory properties of KRAS mutations potentiate malignant pleural effusions in mice [13,14].
Here we hypothesized that KRAS effects and druggability are preferentially at play in vivo. We tested the efficacy of three different KRAS inhibitors with divergent modes of action in vitro and in vivo using a battery of 30 natural and transduced human and murine cancer cell lines and four different methods to integrally assess tumor cell growth. We consistently show that KRAS inhibitors exert ubiquitous in vitro effects irrespective of cellular KRAS mutation status, but are specifically effective against KRAS-mutant tumors in vivo. Using transcriptome analyses of cell lines expressing endogenous or exogenous wild-type or mutant Kras alleles, Ccr2 and Il1b gene-deficient mice, as well as adoptive bone marrow transfer, we show that mutant Kras establishes a proinflammatory C-C motif chemokine ligand 2 (CCL2)/interleukin-1β (IL-1β)-mediated signaling loop to host myeloid cells in vivo, which is required for KRAS-mediated tumorigenicity, but also for specific KRAS inhibitor efficacy. The KRAS/CCL2/IL1B transcript signature is further shown to be enriched in human tumors with higher KRAS mutation frequencies and to portend poor survival. Our data show that intact inflammatory tumor-to-host interactions are required for full KRAS inhibitor efficacy and imply that in vitro drug screens might not be optimal for KRAS inhibitor discovery.
MATERIALS AND METHODS
Cell culture
NCI-H358, NCI-H358M, NCI-H460, NCI-H520, NCI-H1299, NCI-H1944, NCI-H3122 (referred to hereafter omitting NCI), EKVX, A549, LLC, B16F10, and PANO2 cell lines were from the National Cancer Institute (Frederick, MD); MC38 cells were a gift from Dr. Timothy S. Blackwell (Vanderbilt University, Nashville, TN) and AE17 cells from Dr. YC Gary Lee (University of Western Australia, Perth, Australia) [13–15]. FULA1 (FVB urethane-induced lung adenocarcinoma 1) and CULA (C57BL/6 urethane-induced lung adenocarcinoma) cell lines were isolated from the lungs of FVB and C57BL/6 mice, respectively, harboring primary lung adenocarcinomas induced by urethane [13,16,17]. Human and murine cell lines were cultured, respectively, in Roswell Park Memorial Institute (RPMI)-1640 and Dulbecco’s Modified Eagle Medium (DMEM), both supplemented with 10% FBS and 100 IU/mL penicillin/streptomycin, and were maintained in a humidified incubator at 37 °C with 95% air–5% CO2. Cell lines were authenticated annually using the short tandem repeat method and were tested negative for Mycoplasma Spp. biannually by MycoAlert Mycoplasma Detection Kit (LONZA; Verviers, Belgium).
Drugs
Deltarasin (CAS #1440898-82-7; Tocris Bio-Techne #5424; Wiesbaden-Nordenstadt, Germany), KRASG12C inhibitor 12 (AA12; CAS #1469337-95-8; Selleckchem #S7331; Houston, TX), and cysmethynil (CAS #851636-83-4; Cayman Chemicals #14745; Ann Arbor, MI) were dissolved in dimethyl sulfoxide (DMSO) to 10 mM stock concentration and stored at −80 °C. For in vitro and in vivo experiments, drugs were further diluted in normal saline and equimolar DMSO solutions were used as control.
Cellular Assays
In vitro cell proliferation was determined using water soluble tetrazolium-1 [2-(4-iodophenyl)-3-(4-nitrophenyl)-5-(2,4-disulphophenyl)-2H-teterazolium; WST-1] assay (Bimake; Munich, Germany). For this, 3000 cells/well were plated in triplicates in 96-well plates in 5% FBS-containing media and allowed to adhere overnight, followed by treatment with different drug concentrations. WST-1 reagent was added 72 hours later according to the manufacturer’s protocol and absorbance at 450 nm was measured 1-4 hours later on a TECAN Sunrise microplate reader (Männedorf, Switzerland). For colony formation assay, 300 cells were plated in triplicates in 6-well plates in 5% FBS-containing media, were treated 24 hours later with 1-2 µM deltarasin, media were replaced with drug-free media 72 hours later, and cells were incubated until ≤ 50 colonies formed. Colonies were fixed with 80% ethanol, stained with 0.5% crystal violet, counted and photographed. All cellular experiments were independently repeated at least thice.
Western Immunoblotting
Cellular protein lysates were prepared using radioimmunoprecipitation assay (RIPA) buffer containing phosphatase/protease inhibitor cocktail (Thermo Fisher, Waltham, MA), separated by SDS-PAGE, and transferred to nitrocellulose membranes according to standard protocols. Anti-total (t)-extracellular-signal regulated kinase (ERK), anti-phospho (p)-ERK, and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibodies were from Santa Cruz Biotechnology (Houston, TX)(Supplementary Table S1).
Constructs
Short-hairpin (sh) RNA-mediated Kras-silenced (shKras) LLC, AE17, and MC38 cells, as well as B16F10 and PANO2 cells overexpressing custom-made plasmid encoding KrasG12C (pKrasG12C; Addgene #64372; GFP-KrasG12C_2B_puro) were produced as described elsewhere [13]. NCI-H3122 and EKVX cells were stably transfected with pKrasG12C or its homologous GFP backbone plasmid without KrasG12C (pC; Addgene #64336; Bicistronic_GFP_ires_puro) using previously established methods [13]. All plasmids were made in-house, deposited, validated, and re-purchased from Addgene (Watertown, MA). Lentiviral shRNA targeting murine Ccl2 was purchased from Santa Cruz Biotechnology (random control shRNA sc-108080-V, GFP control shRNA sc-108084-V, murine shCcl2 sc-43914-SH). For stable shRNA and plasmid transfection, 105 tumor cells in 6-well culture vessels were transfected with 5 μg DNA using XFect (Takara, Kusatsu, Japan) and clones were selected by puromycin (2-10 μg/mL).
Mice
FVB/NJ (FVB; #001800), C57BL/6J (C57BL/6; #000664), B6.129P2-Cxcr1tm1Dgen/J (Cxcr1-/-; #005820) [15], B6.129S4-Ccr2tm1Ifc/J(Ccr2-/-; #004999) [16], B6.129S2(C)-Cxcr2tm1Mwm/J (Cxcr2+/-; #006848) [15], and B6(Cg)-Rag2tm1.1Cgn/J (Rag2-/-; #008449) mice were from the Jackson Laboratory (Bar Harbor, ME) and Il1btm1Yiw mice (Il1b-/-; MGI #2157396) [18] were a kind gift from Dr. Yoichiro Iwakura (Tokyo University of Sciences, Japan). All mice were bred at the Center for Animal Models of Disease of the University of Patras. Ccr2-/- mice were back-crossed to the FVB strain for > F12. Experimental mice were weight-(20-30 g), sex-, and age-(6-12 weeks) matched; both female and male mice were used. In these studies, 284 mice were enrolled in total. In more detail, 25 FVB (21 for tumor experiments and 4 as bone marrow donors), 151 C57BL/6 (all for tumor experiments), 15 Cxcr1-/-(all on the C57BL/6 background for tumor experiments), 34 Ccr2-/-(12 on the C57BL/6 and 18 on the FVB backgrounds for tumor experiments and 4 on the FVB background as bone marrow donors), 12 Cxcr2+/-(all on the C57BL/6 background for tumor experiments), 32 Rag2-/-(all on the C57BL/6 background for tumor experiments), and 15 Il1b-/-(all on the C57BL/6 background for tumor experiments) mice were used.
In vivo tumor models and drug treatments
For in vivo injections, 106 cells suspended in 50 µL PBS were implanted subcutaneously (sc) in the rear flank. Tumor dimensions (length, L; width, W; depth, D) were monitored serially using calipers and tumor volume (V) was calculated as V = π ∗ L ∗ W ∗ D ⁄ 6. Drug treatments were initiated when tumors reached 100 mm3 volume and consisted of daily intraperitoneal (ip) deltarasin (15 mg/Kg in 100 μL saline 1% DMSO) or 100 μL saline 1% DMSO. Animals were monitored daily for sickness and were euthanized using CO2 when in distress or when tumors reached 2-3 cm3 volume, whichever came first.
Microarrays, PCR, GSEA, and Kaplan-Meier analyses
Isogenic cell line doublets stably expressing shC or shKras (LLC, MC38, and AE17 cells) and pC or pKrasG12C (PANO2 and B16F10 cells) were generated as described elsewhere [13]. Benign samples including whole murine lungs, tracheal epithelial cells (TEC; cultured out from murine tracheas), and bone marrow-derived macrophages (BMDM; cultured from murine bone marrow by weekly incubation with 20 ng/mL M-CSF) and mast cells (BMMC; cultured from murine bone marrow by monthly incubation with 100 ng/mL IL-3 plus KITL) were prepared as described elsewhere [14, 17, 18]. Cellular RNA was isolated using Trizol (Thermo Fisher) followed by RNAeasy column purification and genomic DNA removal (Qiagen, Hilden, Germany). One μg RNA was reverse-transcribed using oligo(dT)18 and iScript Advanced cDNA synthesis kit for RT-qPCR (Bio-Rad Laboratories; Hercules, CA). Il1r1/IL1R1 and Gapdh/GAPDH qPCR was performed using specific primers (Supplementary Table S2) and Lightcycler 480 Sybr Green I Master (Roche; Mannheim, Germany) in a Lightcycler 480 II (Roche Diagnostics). Ct values from triplicate reactions were analyzed with the 2-ΔCT method as detailed elsewhere [15]. mRNA abundance was determined relative to Gapdh/GAPDH and is given as 2-ΔCT = 2 -(Ct of Il1r1/IL1R1)-(Ct of Gapdh/GAPDH). Mouse microarrays were done as described elsewhere [13–15]. Briefly, triplicate cultures of 106 cells were subjected to RNA extraction as above, 5 μg of pooled total RNA were tested for RNA quality on an ABI2000 Bioanalyzer (Agilent; Santa Clara, CA), labelled, and hybridized to GeneChip Mouse Gene 2.0 ST arrays (Affymetrix; Santa Clara, CA). Analyses using Affymetrix Expression/Transcriptome Analysis Consoles consisted of normalization of all arrays together using Lowess multi-array algorithm, intensity-dependent estimation of noise for statistical analysis of differential expression, and unsupervised hierarchical clustering of microarray data and WikiPathway analysis. Murine microarray data are publicly available at the Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/; Accession ID: GSE58190). Gene set enrichment analyses (GSEA) was done using publicly available Human Gene 1.0 ST microarray data obtained from GEO. The following datasets were used: GSE31852 with gene expression profiles of 121 biopsies from patients with lung adenocarcinoma (LADC) with EGFR (n = 17), KRAS (n = 21), or none of the two (n = 83) mutations [Biomarker-integrated Approaches of Targeted Therapy for Lung Cancer Elimination (BATTLE) trial]; GSE43458 with gene expression profiles of LADC from smokers and never-smokers (n = 40 each), as well as normal lung tissue from never-smokers (n = 30) also from the BATTLE trial; and GSE103512 with gene expression profiles of breast (n = 65), colorectal (n = 55), and non-small-cell lung (n = 60) cancer patients from a Roche dataset. Kaplan-Meier analyses were done using KM-plotter (http://www.kmplot.com) [19]. All patients were included and overall survival and all stages/grades were set as parameters.
ELISA
Murine and human CCL2 levels of cell culture supernatants were detected using appropriate ELISA kits (Peprotech; London, UK). For sample preparation, cells were incubated with IC60 deltarasin for 72 hours before collecting cell-free supernatants for CCL2 measurements and whole cellular lysates for normalization of CCL2 levels to total cellular protein.
Immunofluorescence
Paraffin-embedded mouse tissue blocks were cut into 3 µ m-thick sections, deparaffinized by ethanol gradient, rehydrated, and boiled in sodium citrate pH 6.0 for 10 minutes for antigen retrieval. After post-fixation and permeabilization, tissue sections were co-stained with either AlexaFluor488-conjugated mouse monoclonal anti-IL-1β antibody and rabbit polyclonal anti-CCR2 antibody or AlexaFluor488-conjugated mouse monoclonal anti-IL1R1 antibody and rabbit polyclonal anti-CCL2 antibody (Supplementary Table S2). After counterstaining with 300 nM 4’,6-diamidino-2-phenylindole (DAPI), slides were evaluated on an AxioImager.M2 (Zeiss; Jena, Germany) and digital images were processed with Fiji academic software (https://fiji.sc/). Control stains were carried out with isotype controls for normal mouse IgG1/ IgG2a (Alexa Fluor® 488 conjugated; sc-3891/ sc-3890) and secondary antibody only.
Bone marrow replacement
For adoptive bone marrow transplants (BMT), bone marrow cells were flushed from both femurs and tibias of wild-type (WT) or Ccr2-/- mice (all back-crossed >F12 to the FVB background) using fully supplemented DMEM. Ccr2-/- mice (all FVB) received ten million intravenous (iv) bone marrow cells from WT or Ccr2-/- mice 12 hours after total-body irradiation (900 Rad), as described elsewhere [14,15,18]. One mouse in each experiment was not engrafted and was observed till moribund on days 5-15 post-irradiation. One month was allowed for full bone marrow reconstitution of chimeras prior to tumor cell injections.
Statistics
Sample size was calculated using G*power (http://www.gpower.hhu.de/) [20]. In specific, we set out to determine biologically (> 50%) and statistically (α = 0.05; β = 0.20) significant differences between two unmatched independent groups with SD ∼ 30% of mean using two-tailed t-tests, yielding n = 7/group. Hence experiments with n = 5 mice/group were contemplated in batches, till the achievement of probability (P) < 0.05 with α < 0.05 or P > 0.05 with β < 0.20, whichever came first. Two-way ANOVA was employed to achieve further reduction. Results are given as mean ± SD. Sample size (n) refers to biological replicates. Differences between means were assessed using one-way or two-way ANOVA with Bonferroni post-tests. Fifty and 60% inhibitory concentrations (IC50/60) were calculated using nonlinear regression, a logarithmic inhibitor-response model, unweighted least squares regression without outlier elimination and constraints, and extra sum-of-squares F-test comparisons. P < 0.05 was considered significant. Statistics and plots were done on Prism versions 5.0, 6.0, and 8.0 (GraphPad; San Diego, CA).
Study approval
Experiments were approved by the Veterinary Administration of the Prefecture of Western Greece (approval # 366456/1461) and by the Government of Upper Bavaria (approval # 55.2-1-54-2532-194-2016) and were conducted according to Directive 2010/63/EU (http://eurlex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32010L0063).
RESULTS
Mutation-independent effects of KRAS inhibitors in vitro
We initially investigated the cellular responses of a battery of human and murine cell lines with known KRAS/Kras mutation status [4,13–15] (Supplementary Figures S1A, S1B) to three preclinical KRAS inhibitors: deltarasin targeting PDEδ [7], AA12 allosterically targeting KRASG12C [8], and cysmethynil targeting ICMT [6] (Figure 1A). For this, widely used assays were employed based on literature searches (Supplementary Figure S1C). Initially, IC50 values were calculated from WST-1 assays done after 72 hours of treatment with half-log-incremental drug concentrations. Unexpectedly, all three KRAS inhibitors showed comparable efficacy across all cell lines tested, independent of their KRAS/Kras mutation status (Figures 1B-1D; Supplementary Figure S2). Importantly, overall in vitro efficacy of all three drugs was modest, with IC50 values between 1-50 µM (Supplementary Tables S3–S5). A literature search revealed that this was generally true for developmental KRAS inhibitors compared with tyrosine kinase inhibitors (Supplementary Figure S1D). To extend these results, we analyzed the response of eight selected murine and human cell lines to IC60 concentrations of deltarasin in an in vitro colony formation assay. Again, deltarasin efficacy was independent of KRAS/Kras mutation status (Figures 1E, 1F; Supplementary Figure S3). Since KRAS activates the mitogen-activated protein kinase cascade inducing phosphorylation of ERK, we quantified t-and p-ERK relative to GAPDH in 12 murine and human cell lines treated with saline or IC60 deltarasin. In line with the above, deltarasin inhibited p-ERK independent from cellular KRAS/Kras mutation status (Figures 1G, 1H; Supplementary Figure S4). Thus, pharmacologic KRAS inhibition does not reveal KRAS-dependence in vitro.
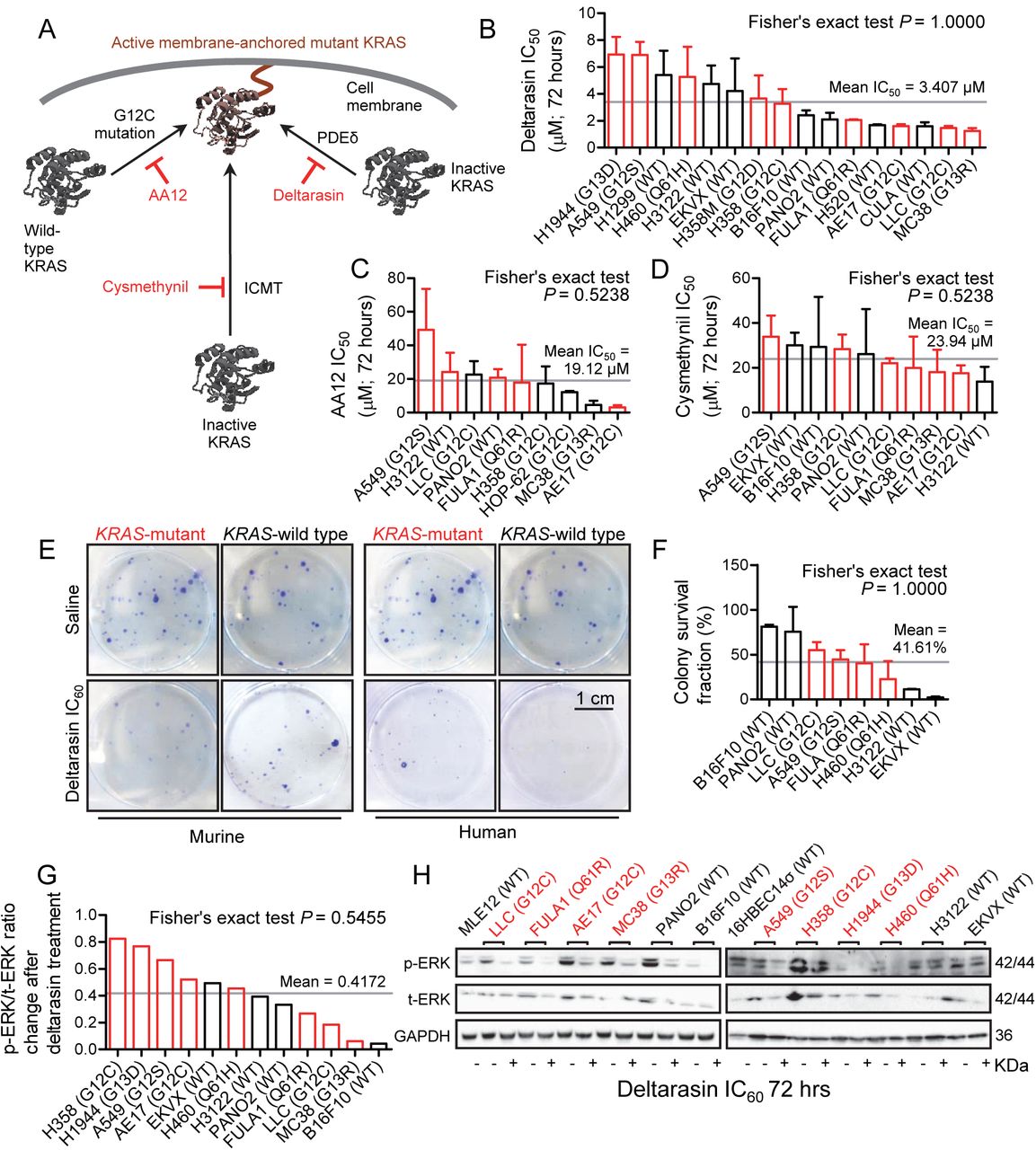
Different mouse and human tumor cell lines with (red) and without (black) Kras/KRAS mutations (codon changes are given in parentheses) were assessed for cell viability by colorimetric WST-1-assay, for colony formation by crystal violet-stained colony counts, and for ERK phosphorylation by phospho (p)- and total (t)-ERK immunoblots after 72-hour treatments with three different KRAS inhibitors (n = 3/data-point).
(A) Graphical abstract showing molecular targets of preclinical KRAS inhibitors AA12, cysmethynil, and deltarasin.
(B) Fifty percent inhibitory concentrations (IC50) of deltarasin (B), AA12 (C), and cysmethynil (D) by WST-1 assay.
(E, F) Representative images of colonies after saline or IC60 deltarasin treatment (E) and colony survival fraction (F) after IC60 deltarasin normalized to saline treatment.
(G, H) Quantification of normalized p-ERK/t-ERK signal change after IC60 deltarasin normalized to saline treatment (G) and representative immunoblots (H).
(B-D, F, G) Data presented as mean ± SD. Grey lines represent the mean of all cell lines tested, which was used to dichotomize cell lines into sensitive and resistant. P, probability by Fisher’s exact test for cross-tabulation of Kras/KRAS mutation status to drug sensitivity/resistance. KRAS, KRAS proto-oncogene GTPase; WT, wild-type; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
Specific in vivo effects of deltarasin against KRAS-mutant tumors
To replicate these results in vivo, we induced sc tumors in C57BL/6, FVB, and Rag2-/- mice using six different cancer cell lines and initiated daily ip saline or deltarasin (15 mg/Kg in saline) treatments after tumor establishment (tumor volume ≥ 100 mm3; latency ≥ 14 days post-sc injection). Interestingly, deltarasin selectively inhibited the sc growth of murine and human KRAS-mutant tumors, but had no effect on KRAS-WT tumors (Figures 2A, 2B). Moreover, forced overexpression of pKrasG12C in KRAS-WT mouse and human cancer cells accelerated tumor growth and restored the response to the drug (Figure 2C). Taken together, these data show that deltarasin-mediated KRAS inhibition selectively halts the growth of KRAS-mutant cancer cells in vivo.
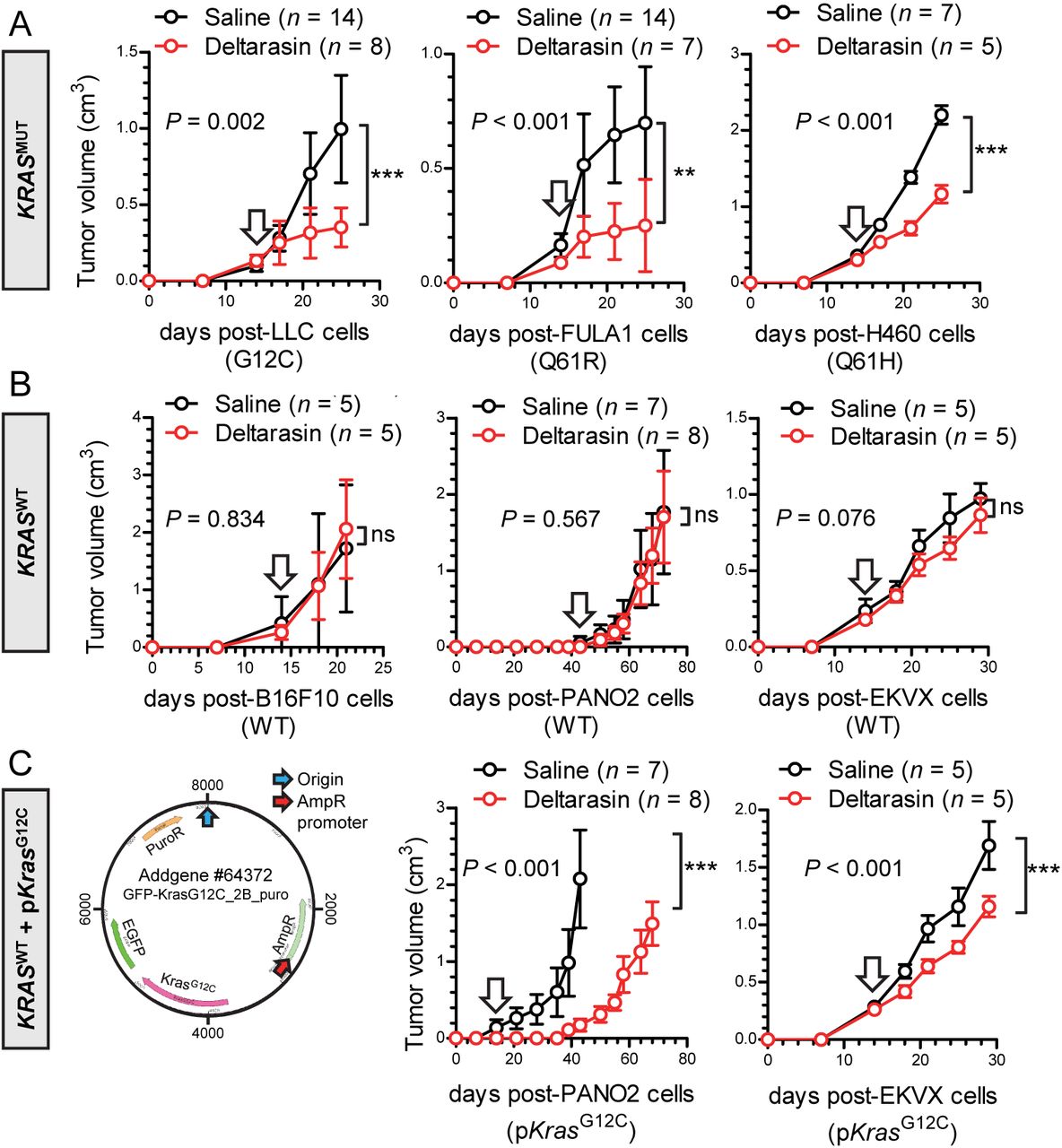
Different mouse and human tumor cell lines with (A; KRASMUT) and without (B; KRASWT) endogenous Kras/KRAS mutations (codon changes are given in parentheses), as well as KRASWT cell lines forcedly expressing a plasmid encoding mutant murine KrasG12C (C; pKrasG12C), were injected into the rear flank (106 tumor cells sc) of C57BL/6 (LLC, B16F10, and PANO2 cells), FVB (FULA1 cells), or Rag2-/- (H460 and EKVX cells) mice. After tumor establishment (tumor volume > 100 mm3; arrows), mice were randomly allocated to daily ip treatments with 100 μL saline (black) or 15 mg/ Kg deltarasin in 100 μL saline (red). Tumor growth was assessed by measuring three vertical tumor dimensions.
Data presented as mean ± SD. n, sample size; P, overall probability, 2-way ANOVA; ns, **, and ***: P > 0.05, P < 0.01, and P < 0.001, respectively, Bonferroni post-test.
Genetic KRAS manipulation reveals contrasting KRAS-dependencies in vitro and in vivo
To further validate the observed in vivo-restricted specificity of deltarasin, we overexpressed anti-Kras-specific shRNA (shKras) in Kras-mutant parental cell lines or pKrasG12C in Kras-WT parental cell lines [13]. In accord with pharmacologic KRAS inhibition, genetic Kras modulation did not impact the in vitro response of cancer cell lines to deltarasin, as determined by WST-1 IC50 values and ERK activation levels (Figures 3A-3E, Supplementary Figures S5, S6). In contrast to the lack of Kras-dependence in vitro, mutant Kras was required and sufficient for sustained tumor growth in vivo (Figure 3F): murine cell lines expressing shKras displayed statistically (P < 0.001) and biologically (50-90% inhibition) significantly decreased tumor growth compared with parental cell lines expressing shC. Vice versa, pKrasG12C overexpression accelerated tumor growth compared with overexpression of pC. Collectively, these results support that, similar to drug-based KRAS inhibition, genetic Kras modulation selectively impacts tumor growth in vivo.

(A) Different murine parental (black/grey: stably expressing random shRNA, shC, or control plasmid, pC) or Kras-modified (red: stably expressing shKras; green: stably expressing mutant KrasG12C plasmid, pKrasG12C) tumor cell lines were assessed for cell viability (IC50 by WST-1-assay; n = 2-4/data-point) after 72 hours of deltarasin treatment.
(B) Summary of averaged deltarasin IC50 values from all cell lines from (A) (n = 2-3 cell lines/group).
(C) Human parental (black/grey: stably expressing control plasmid pC) or KRAS-modified (green: stably expressing pKrasG12C) tumor cell lines were assessed for cell viability by WST-1 assay (n = 2-5/data-point) after 72 hours of deltarasin treatment.
(D) Immunoblots of cell lines from (A) for p-ERK, t-ERK and GAPDH.
(E) Quantification of normalized p-ERK/t-ERK signal from (D). Dara were summarized by mutation status and origin.
(F) The five cell line doublets from (A) were injected into the rear flank (106 tumor cells sc) of C57BL/6 mice for induction of flank tumors by genetically modified cells (red, shKras; green, pKrasG12C) or control cells (black, shC or pC).
P, overall probability by one-way (A-C) and two-way (E, F) ANOVA. ns, **, and ***: P > 0.05, P < 0.01, and P < 0.001, respectively, for the indicated comparisons by Bonferroni post-tests. Data are presented as mean ± SD.
A mutant Kras transcriptome signature contains Ccl2 and Il1b
In an effort to identify mutant-Kras-driven genes responsible for in vivo restricted KRAS-dependence, we analyzed the global transcriptomes of the parental and Kras-modulated murine cell lines described above and of benign samples [whole lungs, tracheal epithelial cells (TEC), and bone marrow-derived macrophages (BMDM) and mast cells (BMMC)]. Unsupervised hierarchical clustering showed an absolute segregation of benign, Kras-WT, and Kras-mutant samples by 1408 differentially expressed genes (ΔGE) using an ANOVA P < 0.05 threshold (Figure 4A). Paired analyses of five isogenic cancer cell line doublets with modulated Kras (LLC, MC38, and AE17 cells expressing shC versus shKras and PANO2 and B16F10 cells expressing pC versus pKrasG12C) identified another 3432 Kras-responsive transcripts. Out of the 170 transcripts that were present in both gene sets, 42 were both differentially represented in benign, Kras-WT, and Kras-mutant samples and responsive (ΔGE > 1.40) to Kras modulation, including Kras per se (Figure 4B, Supplementary Table S6). Interestingly, Il1r1, Ccl7, and Ccl2 were among those genes and clustered tightly together (Figure 4C) and chemokine signaling was the pathway most significantly perturbed by Kras modulation on WikiPathway analysis (Figure 4D) [21]. We next translated our 42-gene murine mutant Kras signature to their 37 human orthologues using Orthoretriever (http://lighthouse.ucsf.edu/orthoretriever/) and ran GSEA (http://software.broadinstitute.org/gsea/index.jsp) [22]. Interestingly, our humanized mutant KRAS signature was enriched in only two out of the Broad Institute’s 50 hallmark signatures: positively in the signature “inflammatory response” and negatively in the signature “G2M-checkpoint” (Figure 4E). Moreover, this mutant KRAS signature was significantly positively enriched in KRAS- versus EGFR-mutant LADC (GSE43458) from the BATTLE trial [23, 24] (Figure 4F). In this connection, we recently reported that mutant KRAS drives CCL2 and Il1R1 expression to establish inflammatory feedback loops with IL-1β-secreting myeloid cells in malignant pleural effusions [13, 14, 18]. Collectively, the data further supported that in vivo-restricted mutant KRAS-dependence is mediated by proinflammatory signals to CCR2+ IL-1β-secreting host cells.

(A) Unsupervised hierarchical clustering of gene expression of Kras-mutant and Kras-WT cancer cell lines, as well as benign cells and tissues.
(B) Venn diagram of analytical strategy of transcriptome analysis.
(C) Unsupervised hierarchical clustering of gene expression of Kras-modified cancer cell line doublets reveals co-clustering of Il1r1 and Ccl2.
(D) WikiPathway analysis showing pathways significantly overrepresented in the KRAS signature.
(E) GSEA of 37 human orthologues of the murine KRAS signature against the Broad Institute’s 50 hallmark signatures showing positive enrichment in the “inflammatory response” and negative enrichment in the “G2M checkpoint” signatures. NES, normalized enrichment score; P, family-wise error rate probability.
(F) GSEA of 37 human orthologues of the murine KRAS signature against KRAS-(n = 21) versus EGFR-(n = 17)-mutant lung adenocarcinomas (LADC) from BATTLE reveals positive enrichment of our KRAS signature in human KRAS-mutant LADC. NES, normalized enrichment score; P, family-wise error rate probability.
CCR2+ IL-1β-secreting myeloid cells potentiate in vivo KRAS-dependence
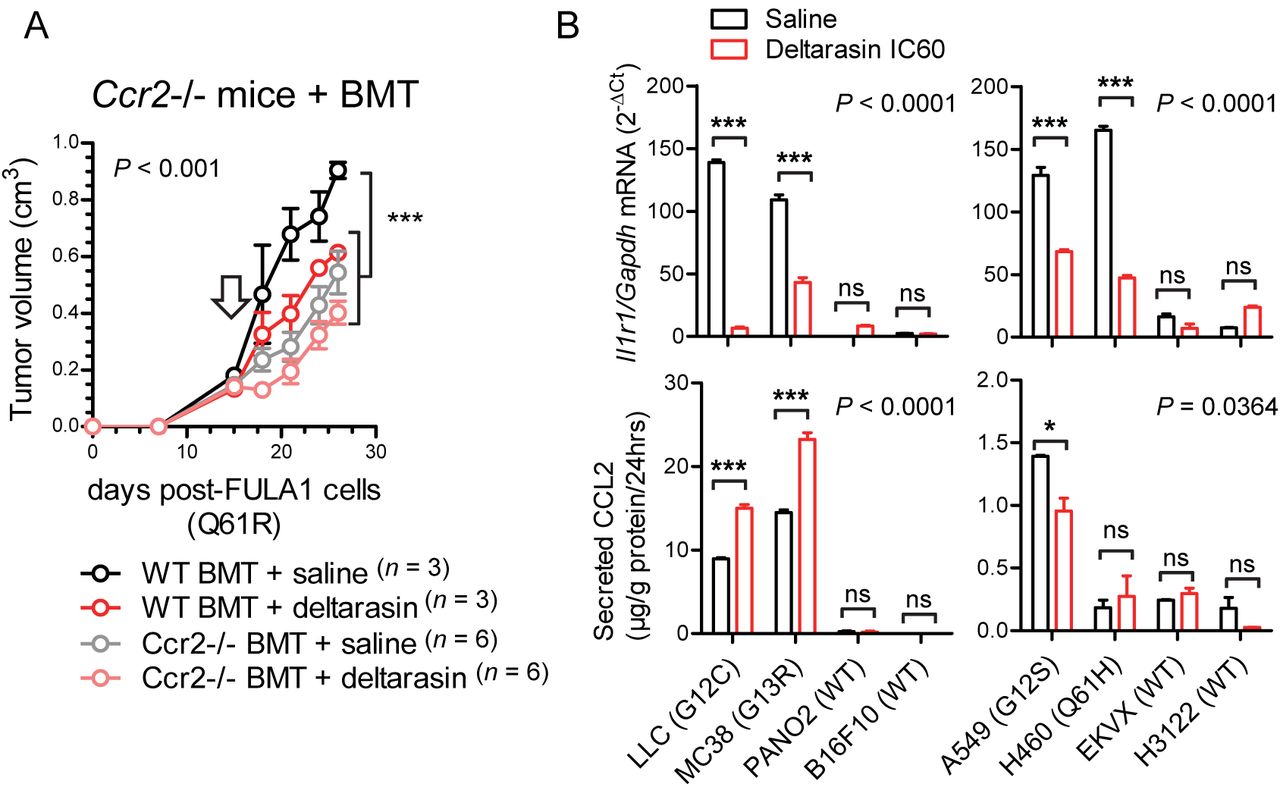
These results led us to the hypothesis that CCR2+ IL-1β-secreting myeloid cells are required for in vivo KRAS-dependence (Figure 5A). Indeed, numerous such cells co-expressing CCR2 and IL-1β were identified in the stromata of our experimental KRAS-mutant tumors by immunohistochemistry (Figure 5B). To definitively test our hypothesis, we induced flank tumors by injecting one million LLC cells (KrasG12C) sc into syngeneic C57BL/6 mice competent (WT) or deficient (Il1b-/-, Ccr2-/-) [16, 18] in the Il1b and Ccr2 genes. Mice haplo/diplo-insufficient in the Cxcr1 and Cxcr2 chemokine receptor genes (Cxcr1-/-, Cxcr2+/-) [15] were also employed as additional controls for Ccr2-/- mice and daily ip saline or 15 mg/Kg deltarasin treatments were initiated when tumors reached 100 mm3 volumes. Expectedly, deltarasin treatment statistically and biologically significantly inhibited LLC tumor growth in WT, Cxcr1-/-, and Cxcr2+/- mice. However, deltarasin effects were diminished in Il1b-/- and completely abrogated in Ccr2-/- mice (Figure 5C). To exclude the possibility of developmental effects of knockout mice, we total-body irradiated (900 Rad) Ccr2-/- mice and performed adoptive BMT from WT or Ccr2-/- donors, as described previously [14, 18]. For this experiment, WT and Ccr2-/- mice back-crossed > F12 to the FVB strain were used together with syngeneic FULA1 cells (KrasQ61R) to obtain results with another cell line harboring a different Kras mutation and a broad mutation spectrum relevant to human KRAS-mutant LADC [17]. Again, daily ip saline or deltarasin treatments were started when tumors > 100 mm3 were established. Expectedly, Ccr2-/- chimeras receiving Ccr2-/- BMT did not respond to deltarasin, but Ccr2-/- chimeras receiving WT BMT displayed markedly increased tumor growth as well as a statistically and biologically significant inhibition by deltarasin treatment (Figure 6A). Collectively, these results indicate that myeloid CCR2 and IL-1β are required for deltarasin efficacy against Kras-mutant tumors in vivo.

(A) Graphical abstract of the proposed mechanism of in vivo restricted KRAS dependence.
(B) Representative image of CCR2/IL-1β-co-staining of a KRAS-mutant tumor from a Rag2-/- mouse showing co-localization of the two proteins in the tumor stroma. Image was taken using an AxioImager.M2 (Zeiss; Jena, Germany) and a 60x objective.
(C) Syngeneic C57BL/6 mice competent (WT) or deficient (Il1b-/-, Ccr2-/-) [16, 18] in the Il1b and Ccr2 genes or haplo/diplo-insufficient in the Cxcr1 and Cxcr2 chemokine receptor genes (Cxcr1-/-, Cxcr2+/-) received 106 LLC cells (KrasG12C) sc followed by daily ip saline (black) or 15 mg/Kg deltarasin (red) treatments initiated when tumors reached 100 mm3 volumes (arrows). Data are presented as mean ± SD. P, overall probabilities by 2-way ANOVA; ns, *, and ***: P > 0.05, P < 0.05, and P < 0.001 for the indicated comparisons by Bonferroni post-tests. Table shows animal numbers used and percentile tumor inhibition by deltarasin compared with saline.
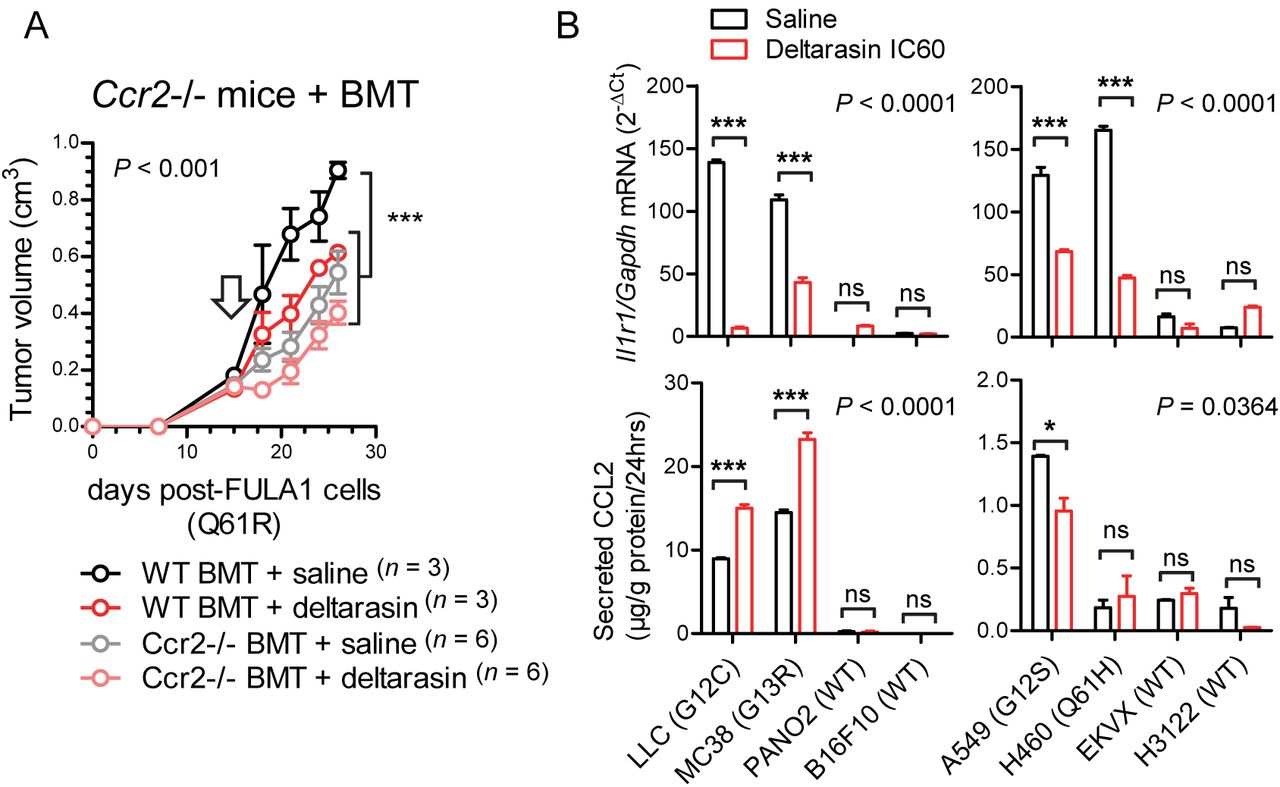
(A) Total-body irradiated (900 Rad) Ccr2-/- mice received adoptive BMT from WT or Ccr2-/- donors (all back-crossed > F12 to the FVB strain). After one month allowed for chimeric bone marrow reconstitution, chimeras received 106 syngeneic FULA1 cells (KrasQ61R) sc [17]. Daily ip saline or deltarasin (15 mg/Kg in saline) treatments were started when tumors > 100 mm3 were established (arrow). Data are presented as mean ± SD. P, overall probabilities by 2-way ANOVA; ***: P < 0.001 for the indicated comparisons by Bonferroni post-tests.
(B) Il1r1/IL1R1 mRNA expression by qPCR (top) and CCL2 protein secretion by ELISA (bottom) of mouse (left) and human (right) cancer cell lines treated with saline or deltarasin IC60 for 72 hours. Data are presented as mean ± SD. P, overall probabilities by 2-way ANOVA; ns, *, and ***: P > 0.05, P < 0.05 and P < 0.001, respectively, for the indicated comparisons by Bonferroni post-tests.
Deltarasin limits IL-1β sensing by KRAS-mutant tumor cells
We next interrogated the mechanism of in vivo-restricted deltarasin dependence. Based on the microarray-derived mutant Kras signature that encompassed Ccl2 and Il1r1 (Figure 4) and our previous reports of mutant KRAS-mediated transcriptional regulation of CCL2 and IL1R1 [13, 14], we tested whether deltarasin blocks expression of these two genes. Indeed, KRAS-mutant mouse and human cancer cell lines displayed markedly increased baseline Il1r1/IL1R1 mRNA expression compared with WT cell lines, and significantly downregulated Il1r1/IL1R1 transcript levels after deltarasin treatment. On the contrary, only some KRAS-mutant cell lines displayed increased baseline CCL2 protein secretion compared with WT cell lines, and CCL2 elaboration was not consistently blocked by deltarasin treatment, suggesting that deltarasin-mediated downregulation of Il1r1/IL1R1 expression delivers the bulk of the drug’s in vivo effects.
An inflammatory CCL2/IL1B signature in KRAS-mutant human cancers
To investigate the relevance of our findings to KRAS-mutant human cancers, we analyzed the average expression of KRAS, CCL2, and IL1B genes in public data (GSE43458) from the BATTLE trial [23, 24]. Interestingly, mean KRAS/CCL2/IL1B expression was statistically significantly increased in smokers’ LADC (n = 40) compared with never-smokers’ LADC (n =40) and normal lung tissue samples (n = 30) (Figure 7A). Since KRAS mutations are more frequent in LADC of smokers [25], this finding suggested that our inflammatory signature was overrepresented in tumors with higher KRAS mutation frequencies. This was also true in another dataset from patients with breast, colorectal, and lung cancer (GSE103512) [26], where mean KRAS/CCL2/IL1B expression was significantly higher in lung and colorectal cancer, which have higher KRAS mutation rates [4], compared with breast cancer (Figure 7B). Finally, online Kaplan-Meier analyses (http://www.kmplot.com) [19] using lung cancer patient data were done (Figure 7C). These revealed that in patients with LADC (a tumor with high KRAS mutation frequency) high KRAS/CCL2/IL1B expression levels portended 93% increased odds of death regardless of smoking status. On the contrary, KRAS/CCL2/IL1B expression did not impact the survival of patients with squamous cell lung carcinoma (a tumor with low KRAS mutation frequency). When exclusively smokers were examined (thereby enriching the sample for KRAS-mutant patients), high KRAS/CCL2/IL1B expression levels portended 128% increased odds of death in LADC and continued to have no impact on the survival of patients with squamous cell lung carcinoma. Taken together, these data suggest that KRAS/CCL2/IL1B transcripts are overexpressed in human KRAS-mutant cancers and detrimentally affect survival. Moreover, that the proposed KRAS-driven inflammatory loop may be clinically relevant.

(A) Average KRAS/CCL2/IL1B expression normalized to ACTB in lung adenocarcinomas (LADC) from smokers and never-smokers and normal lung tissue from never-smokers from the BATTLE study (GSE43458) [23,24].
(B) KRAS/CCL2/IL1B expression normalized to ACTB in breast, non-small cell lung, and colorectal cancer (ROCHE study GSE103512). KRAS mutation frequencies of these tumor types are from COSMIC [4].
(C) Kaplan-Meier analyses of lung cancer patients stratified by average KRAS/CCL2/IL1B expression done on http://www.kmplot.com [19]. KRAS mutation frequencies are from the Campbell cohort [25]. Top: all patients; Bottom: ever-smokers only.
(A, B) Data are presented as violin plots. P, overall probability by one-way ANOVA. ns, *, **, and ***: P > 0.05, P < 0.05, P < 0.01, and P < 0.001, respectively, for the indicated comparisons by Bonferroni post-tests.
DISCUSSION
We hypothesized that mutant KRAS dependence occurs non-cell-autonomously and that KRAS inhibitor effects are delivered in vivo. We used 30 cancer cell lines with different KRAS mutations and multiple in vitro assays to show that both pharmacologic and genetic KRAS inhibition is selectively effective against KRAS-mutant murine and human tumors in vivo. Using isogenic cell lines with intact or compromised mutant KRAS signaling, we identify a novel KRAS-mutation-specific transcriptome signature that is surprisingly predominated by inflammatory response genes including CCL2 and IL1B. We further employ several transgenic mouse strains and adoptive bone marrow transfer experiments to show that effective pharmacologic KRAS blockade in vivo is dependent on the presence of CCR2+ IL-1β-secreting myeloid cells in the tumor microenvironment. Finally, we show that the KRAS blocker deltarasin functions to downregulate IL1R1 expression in KRAS-mutant tumor cells and that the proposed KRAS/CCL2/IL1B signature is enriched in human cancers with high KRAS mutation frequencies in which it portends a dismal prognosis. Our results imply that conventional cell-based screens for the discovery and development of novel KRAS blockers might be suboptimal, and that IL-1β inhibition may be specifically effective against KRAS-mutant cancers.
A long line of evidence supports that homotypic two-dimensional cancer cell cultures are not optimal for the study of KRAS-dependence. Singh et al. established a “RAS-dependency index” in a large panel of human lung and pancreatic cancer cell lines systematically addressing the variable in vitro efficacy of KRAS inhibition [10]. Project DRIVE, a comprehensive synthetic lethality screen applying > 150000 shRNAs on 7,837 genes and 398 cancer cell lines (https://oncologynibr.shinyapps.io/drive/), identified no lethal interaction partners for KRAS in vitro, a finding that urged the authors to state: “… the data here raise the likelihood that no single synthetic lethal gene will be found across all KRAS mutant tumors … commonly used KRAS mutant models are not KRAS dependent, when interrogated as monolayer cell cultures … ablating KRAS dependence will need to carefully consider these findings…” [12]. Recently, Janes et al. developed ARS-1620, a new covalent G12C-specific KRAS inhibitor that is highly effective in vivo, but not in vitro [9]. The authors developed three-dimensional co-culture systems and state: “We use ARS-1620 to dissect oncogenic KRAS dependency and demonstrate that monolayer culture formats significantly underestimate KRAS dependency in vivo”. Despite the tremendous progress contributed by the above-referenced work, the mechanism(s) of the observed in vivo-restricted KRAS-dependence remained obscure prior to this report.
To this end, multiple lines of work support the notion that the paracrine effects of KRAS and other RAS oncogenes overshadow their cell-autonomous impact. A pioneering report identified how RAS oncogenes utilize paracrine IL-8 signaling to induce angiogenesis in vivo [11, 27]. We determined how KRAS-mutant cancer cells depend on paracrine CCL2 signaling to myeloid cells including mononuclear and mast cells to induce vascular permeability and angiogenesis during malignant pleural effusion development [13, 18]. In turn, myeloid-derived IL-1β was found to selectively trigger non-canonical nuclear factor (NF)-κB activation in KRAS-mutant cancer cells via IL1R1 and inhibitor of NF-κB kinase α (IΚΚα), with the latter presenting a marked therapeutic target in mouse models of pre-metastatic and advanced lung cancer [14, 28]. Here we show how deltarasin functions to abrogate a mutant KRAS-initiated in vivo inflammatory loop of tumor-derived CCL2 and myeloid-secreted IL-1β by downregulating IL1R1 expression of KRAS-mutant tumor cells and thereby abolishing their receptivity to myeloid IL-1β signals. We identify CCR2+ myeloid cells that provide IL-1β to the microenvironment of KRAS-mutant tumors and show that they are required for mutant KRAS dependence in vivo. Data from syngeneic mouse models of global host Ccr2 and Il1b gene deficiency and of focal myeloid Ccr2 reconstitution are further supported by human cancer xenograft experiments in Rag2-/- mice, which lack B- and T-cell function but feature intact myeloid cells [29], to collectively identify the proposed inflammatory loop that potentiates KRAS blockade.
In addition to Kras, Ccl2, and Il1b, a battery of other transcripts emanated within the signature of KRAS-mutant cancers derived from the transcriptomes of our cell lines, providing synthetic lethality candidates for in vivo KRAS dependency for future research. This signature includes signal transducers Ranbp3l, Gpr149, and Rassf8, inflammatory messengers Ccl7, Cxcl1, and Casp3, cell surface receptors Pdgfra and Ttk, cell cycle genes and tumor suppressors Cdca5, Hist2h3c2, Plag1, Fanca, and Gmnn, among others. The importance of some of these candidates is worth mentioning: Cxcl1 was recently found to mediate the effects of KRAS-IKKα addiction during malignant pleural effusion development [14]; Casp3 is a central effector of compensatory tumor proliferation and radiotherapy resistance [30]; and Gmnn was recently found to function as a tumor suppressor in lung and colon cancer [31]. Surprisingly, Kras mutation status imprinted the transcriptomes of our cell lines more profoundly than their tissues of origin, making them cluster together in an unsupervised fashion. Furthermore, our KRAS-mutation signature was enriched in human KRAS-mutant cancers and predicted poor survival, a fact that is further validating this gene set. Most importantly, the mutant KRAS signature was dominated by the inflammatory response pathway on both WikiPathways analysis and GSEA, highlighting the notion that the oncogene functions in a proinflammatory fashion.
In addition to fostering the battle to drug KRAS, the present work bears significant clinical implications by pinning CCL2 and IL-1β as key inflammatory addiction partners of mutant KRAS. Although targeting CCL2 with neutralizing antibodies yielded promising preclinical results [13, 18, 32–35], clinical trials of the anti-human CCL2 antibody carlumab were hampered by limited drug efficacy and tolerability [36–38]. In contrast, targeting IL-1β with canakinumab has raised enthusiasm and holds great premise in cancer therapy. In this regard, the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS), a randomised trial of the role of IL-1β inhibition in atherosclerosis, secondarily aimed at establishing whether low (50 mg), medium (150 mg), or high (300 mg)-dose canakinumab given sc every three months might alter cancer incidence [39,40]. The results astonished, with total cancer mortality decreasing by 51% in the high-dose group, incident lung cancer decreasing by39% in the medium-dose and by 67% in the high-dose groups, and with lung cancer mortality decreasing by 77% in the high-dose canakinumab group. Although our results of diminished deltarasin efficacy with Il1b-/- mice were less impressive compared with the complete abrogation of deltarasin effects in Ccl2-/- mice, we believe that this is attributable to redundant IL-1α signaling in the former and that targeting IL-1β might be specifically effective against KRAS-mutant cancers [41–45]. This is plausible from CANTOS results, since canakinumab effects in decreasing lung cancer incidence and mortality were double in current than in past smokers overall and quadruple when the high-dose group was examined alone, with current smokers having higher KRAS mutation rates than never-smokers [4,23–25]. Our results suggest that canakinumab might be selectively effective against KRAS-mutant cancers and warrant a posteriori analysis of CANTOS results by KRAS mutation status.
In summary, we show that KRAS-mutant cancer cells express CCL2 and IL1R1 to initiate an inflammatory signaling loop with CCR2/IL-1β-expressing myeloid cells. Our work indicates that this crosstalk is required for KRAS-dependence and blockade, which targets IL1R1 expression. The data set a rational framework for the future development of effective KRAS inhibitors and design of clinical trials aimed at targeting IL-1β in cancer.
Financial support
European Research Council 2010 Starting Independent Investigator and 2015 Proof of Concept Grants (260524 and 679345, respectively, to GTS).
Conflict of interest statement
The authors declare no potential conflicts of interest.
AUTHORS’ CONTRIBUTIONS
KAMA performed in vitro experiments, transcriptome analyses, histology and microscopy, analyzed the data, and wrote the manuscript draft; GN, VA, and DK performed in vivo experiments; CH, LVK, and ASL performed in vitro experiments; GAG performed GSEA; MAAP performed pathway analysis and deposited microarray data at GEO; RAH and SK provided critical intellectual input; GTS designed, funded, and guided the study, analyzed the data, is the guarantor of the study’s integrity, wrote the final version of the manuscript, and designed the final version of the figures. All authors reviewed and edited the paper and approved the final version before submission.
ACKNOWLEDGEMENTS
This work was supported by European Research Council 2010 Starting Independent Investigator and 2015 Proof of Concept Grants (260524 and 679345, to GTS), and by a European Respiratory Society 2013 Romain Pauwels Research Award (to GTS). The authors thank the University of Patras Center for Animal Models of Disease for experimental support. The authors have no financial conflict of interest.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}