

Summary
Oncogenic transformation alters the metabolism of cellular nutrients to sustain tumor growth. We here define a mechanism by which modifications in cholesterol metabolism control the formation of pancreatic ductal adenocarcinoma (PDAC). Disruption of distal cholesterol biosynthesis by means of conditional inactivation of Nsdhl in mice bearing a tumor-inducing Kras mutation (KrasG12D) prevented PDAC formation in the context of a heterozygous Trp53f/+genotype without impairing normal pancreatic development. In mice with pancreatic Nsdhl ablation and homozygous loss of Trp53, the emerging tumors presented with the aggressive basal (mesenchymal) phenotype as opposed to the classic (glandular) PDAC. This paralleled significantly reduced expression of cholesterol metabolic pathway genes in human basal PDAC subtype. Mechanistically, we demonstrate that genetic or metabolic cholesterol deprivation stabilizes the transforming growth factor beta (TGFβ) receptor to activate pro-mesenchymal effectors in human and murine PDAC, providing a direct mechanism by which cholesterol metabolism can condition tumor differentiation.
Introduction
The incidence of pancreatic ductal adenocarcinoma (PDAC) has been rising for the past decades, and by 2030, it is poised to become the second leading cause of cancer deaths in the USA (Chari et al., 2015; Rahib et al., 2014). The nearly identical numbers of PDAC diagnoses and PDAC deaths have been associated with high metastatic propensity of this cancer. Epithelial-to-mesenchymal transition (EMT) has been proposed as one of the key mechanisms of drug resistance (Sabnis and Bivona, 2019; Zheng et al., 2015), cancer metastatic dissemination and invasive growth (Aiello et al., 2017).
Gene expression analyses have established at least two molecular subtypes of PDAC, the classic (also known as glandular), and the basal (i.e., mesenchymal (Aung et al., 2018; Moffitt et al., 2015)), each of which is associated with distinct prognoses and sensitivity to chemotherapy, so that the median survival of basal PDAC is nearly half of that in classic subtype (Aung et al., 2018). The defining features of the basal subset of PDAC include increased signaling by transforming growth factor beta (TGFβ), and expression of TGFβ-induced genes associated with a mesenchymal phenotype, such as transcription factors ZEB1, ZEB2, TWIST, SNAI1, and SNAI2 (Scheel et al., 2011), as well as reduced expression of epithelial adhesion molecule E-cadherin (CDH1) and epithelial lineage transcription factors GATA6, SOX17, and HNF4A (Bailey et al., 2016; Collisson et al., 2011; Moffitt et al., 2015; TCGA., 2017). Although TGFβ signaling plays a central role in EMT of established tumors, it has a potent tumor suppressive role in untransformed cells, as reflected by frequent inactivation of components of the TGFβ pathway (TGFBR1, TGFBR2, SMAD4) in about a quarter of PDAC cases (David et al., 2016; Stankic et al., 2013). At present, little is known of the epigenetic or metabolic factors that act upstream of TGFβ in EMT, and influence PDAC evolution into the basal versus classic subtypes.
In epidemiological studies, the rising incidence of PDAC has been linked to elevated serum cholesterol (dyslipidemia) and obesity (Genkinger et al., 2015), which are attributable in part to high fat and calorie Western diets. Oncogene-transformed cancer cells accelerate uptake and endogenous biosynthesis of cholesterol and phospholipids (Pitroda et al., 2009; Silvente-Poirot and Poirot, 2014). In mouse models of PDAC, driver mutations in the oncogene Kras coupled with loss of the tumor suppressor Trp53 reprogram cellular metabolism in numerous ways affecting utilization of energy sources, accelerating cholesterol biosynthesis and uptake (Freed-Pastor et al., 2012; Ying et al., 2012). Although cholesterol has been proposed to drive PDAC carcinogenesis, this idea has been challenged by the relatively modest clinical activity of cholesterol-lowering medications such as statins aimed to reduce PDAC risk (Bang et al., 2018). Instead, the lack of correlation between PDAC mortality and serum cholesterol levels (Huang et al., 2017) raises the possibility of a tumor cell-centric mechanism by which cholesterol can modulate oncogenic functions and modify disease course at earlier stages of PDAC formation. Supporting this idea, data implicating cholesterol and its metabolites in cancer initiation and progression are beginning to emerge (Gabitova et al., 2015; Nelson et al., 2013). In one notable example, liver X-receptor pathway activation, an important nodule of the cholesterol metabolism, in a mouse model of breast cancer in conjunction with genetically induced accumulation of 27-hydroxycholesterol, fostered the development of aggressive basal adenocarcinoma with mesenchymal differentiation. These findings suggested that the network of genes and metabolites regulating cholesterol homeostasis can directly condition cancer differentiation (Nelson et al., 2013). Despite these advances, the mechanism for this dramatic effect of cholesterol metabolism on cancer epigenetic program remains to be defined.
In this work, we employed a genetic mouse model to explore cholesterol dependency in PDAC. The enzymatic activity of NSDHL (NAD(P)-dependent steroid dehydrogenase-like) mediates an irreversible step in the oxidative decarboxylation of two methyl moieties at the C4 position of a cholesterol precursor, which is essential for endogenous cholesterol biosynthesis (Cunningham et al., 2015; Gabitova et al., 2015). Conditional inactivation of Nsdhl in pancreatic epithelial cells via Pdx1-Cre transgene enabled us to study the role of endogenous cholesterol biosynthesis in the context of pancreatic cancer model combining Cre-activated oncogenic KrasG12D allele and conditional knockout of “floxed” Trp53 (aka, KPC mice, (Bardeesy et al., 2006)). Remarkably, genetically induced cholesterol auxotrophy in the pancreatic epithelium blocked malignant progression of pancreatic precursor lesions and protected Trp53+/−mice from PDAC development. In unexpected contrast, in the context of bi-allelic rather than heterozygous inactivation of Trp53, loss of Nsdhl promoted basal phenotype of KrasG12D-induced PDAC. This metabolically-determined dichotomy of PDAC differentiation was mediated by cholesterol-sensitive regulation of TGFβ signaling. Importantly, the results that we obtained from animal model paralleled in our analyses the human PDAC transcriptome data in which cholesterol pathway mRNA signatures are strikingly different and independently prognostic when comparing between classic and basal subtypes. Finally, we define a mechanism by which cholesterol abundance controls TGFβ signaling. Contingent on complete p53 inactivation and under cancer cell cholesterol auxotrophy, cellular cholesterol regulated TGFβ ligand abundance and dynamics of receptor activation via canonical SMAD2/3 effector cascade, promoted the EMT transcriptional program to attain an aggressive and poorly differentiated, basal, PDAC phenotype.
Results
Disruption of epithelial cholesterol biosynthesis prevents PDAC development
To test dependency of pancreatic carcinogenesis on tumor-intrinsic cholesterol biosynthesis, we used mice in which conditional inactivation of Nsdhl via a Pdx1-Cre transgene is expected to take place in the developing pancreatic bud at embryonic day 8.5 (Gabitova et al., 2015; Offield et al., 1996). The efficiency of Nsdhl inactivation in pancreatic epithelium was evident from complete Cre-mediated Nsdhlf/fgenomic rearrangement and loss of detectable NSDHL expression in pancreatic tissue (NsdhlΔPanc mice, Fig. S1A, B). These NsdhlΔPanc mice produced normal-sized litters at term with an approximate 1:1 male to female ratio; given the location of Nsdhl on the X chromosome, this implied no embryonic lethality. Comparison of pancreatic tissues of NsdhlΔPanc and wild type mice at 6-8 weeks of age demonstrated normal pancreas size (Fig. S1C), normal pancreatic ductal and acinar anatomy (Fig. S1D), and proliferation rate (Fig. S1E). To probe the regenerative capacity of the Nsdhl-deficient pancreas, we treated adult NsdhlΔPanc and wild type animals with a two-day course of caerulein, an established inducer of acute pancreatitis (Renner et al., 1983). We saw no significant morphological differences during acinar regeneration following acute caerulein-induced injury, and no differences in proliferation (Ki67, Fig. S1F, G), suggesting normal regenerative capacity of NsdhlΔPanc epithelial cells.
We next evaluated the effects of conditional Nsdhl knockout in an established model of pancreatic carcinogenesis induced by oncogenic KrasG12D (KC mice) in which mice develop pre-malignant lesions by 6 months of age (Hingorani et al., 2005; Jackson et al., 2001; Tuveson et al., 2004). KC and KCN animals (denoting the LSL-KrasG12D;Pdx1-Cre genotype with or without Nsdhlf/f allele, respectively) developed pre-malignant epithelial lesions represented by acinar-to-ductal metaplasia (ADM) and low grade pancreatic intraepithelial neoplasia (PanIN) at a similar low preponderance per pancreas (Fig. S2A). These results indicate that arresting endogenous epithelial cholesterol biosynthesis, at the level of NSDHL, is insufficient to counter the increased proliferative effect of KrasG12Dseen in pancreatic epithelial cells.
Progression of PanIN to PDAC, in the pancreas of both mice and humans, requires inactivation of the P53 tumor suppressor in conjunction with constitutive KRAS activity (Hingorani et al., 2005). Control animals with monoallelic loss of Trp53 (denoted as Pdx1-Cre;LSL-KrasG12D;Trp53f/+, or KPC mice) developed pancreatic adenocarcinoma starting from 2 months of age with a medium overall survival of about 5 months (Fig. 1A). The majority of evaluable PDAC in these KPC animals were represented by grade 1 and 2 adenocarcinoma with glandular differentiation features (Table S1). In contrast, the KPCN mice developed PDAC at much later term and only in 3 out of 37 animals (Fig. 1A). This translated to a markedly superior PDAC-free survival at 6 months of age in KPCN compared to KPC (92% vs. 30%, respectively, log rank test p<0.0001), despite a similar preponderance of proliferating Ki67+ cells (Fig. S2B).

Effects of NSDHL deficiency on pancreatic adenocarcinoma development. (A) Kaplan-Meier PDAC-free survival of KPC (n=34) and KPCN (n=37) mice. p<0.0001, Logrank test. (B) Frequency of pancreatic epithelial lesion by grade per section in KPC and KPCN mice at 5-6 months of age; p=0.035 for PDAC, Fisher’s exact test; p=0.02 for PanIN2/3; p=0.0006 for ADM, Wilcoxon test. (C, D) Expression of cleaved caspase 3 (C) and phosphorylated ERK1/2 (D) in pancreatic ADM and PanIN lesions. (E) Kaplan-Meier survival of KPPC (n=64) and KPPCN (n=76) mice. p<0.0001, Logrank test. (F) Delayed progression of pancreatic epithelial lesion in KPPCN mice compared to age-matched KPPC at 4 and 7 weeks shown as relative lesion area per section; p<0.02 for comparisons of PDAC and normal areas, Wilcoxon test. (G) Hematoxylin and eosin stained pancreas sections at 7 weeks of age of KPPC and KPPCN mice, respectively. Small foci of grade 4 PDAC (arrow) are seen on the background of nearly normal KPPCN pancreatic tissue with ADM (A) and early PanIN (P) lesions. Scale bars, as shown. (H) Histological grading of advanced PDAC tumors; p<0.01 for grades 1, 3 and 4. (I) Comparison of grade 4 PDAC areas per section; (1) and (2), p<0.0001. (J) Comparisons of the highest PDAC grades per animal; (3) p=0.0009. In all figures, data are represented as mean±SEM, p-values by Wilcoxon test. See also Figure S2.
Histological evaluation of pancreatic tissues of age-matched mice at 5-6 months demonstrated a correspondingly low burden of advanced PanIN and PDAC lesions in KPCN mice (Fig. 1B) compared to KPC. Further, KPCN pancreatic tissues exhibited a robust expression of cleaved caspase 3 within ADM and PanIN lesions, compared to KPC (Fig. 1C, S2C), which were not observed in normally appearing acinar cells (Fig. 1C). We further investigated possible effects of NSDHL inactivation on the signaling downstream of KRAS and found slightly increased expression of phosphorylated form of MAPK1/2 in KPCN pancreatic premalignant lesions (Fig. 1D, S2D) indicating no inhibitory effects of NSDHL deficiency on proliferation-associated MAPK signaling. The phosphorylated ribosomal protein S6, an mTOR kinase target and a correlate of metabolic activity (Peterson and Sabatini, 2005), tended to be diminished in KPCN PanIN lesions (Fig. S2E).
Deficiency of Nsdhl has been reported to cause accumulation of C4-methylsterols, a group of metabolites arising from cholesterol pathway inhibition that activate the transcription factor LXRα (Gabitova et al., 2015). Confirming significant inhibition of cholesterol biosynthesis in the KPCN pancreas, we found significantly elevated expression of the LXRα–dependent transcript ATP-binding cassette transporter member 1 (ABCA1) in ADM and PanIN lesions, in contrast to KPC (Fig. S2F). Notably, detailed examination demonstrated that the small number of PDAC arising in KPCN mice were of poorly differentiated histology (Table S1).
Cholesterol restriction promotes loss of differentiation and epithelial-mesenchymal transition (EMT) in Kras-induced PDAC in the context of complete Trp53 loss
In the KPPC model (Pdx1-Cre;LSL-KrasG12D;Trp53f/f), complete loss of Trp53 induces rapid development of PDAC (Bardeesy et al., 2006). We next compared mice with bi-allelic inactivation of Trp53, with or without conditional knockout of Nsdhlf/f (designated KPPC or KPPCN) (Fig. 1E). Inactivation of NSDHL in the absence of P53 only marginally improved the median survival of KPPCN compared to approximately 2 months attained in KPPC despite the markedly delayed progression of premalignant lesions to PDAC in KPCN mice (Fig. 1A, B). Most KPPCN animals commenced significant weight loss around day 70, resulting in a relatively small although statistically significant difference in median survival compared to KPPC mice (Fig. 1E, median survival, 76 vs. 64 days, respectively; p<0.0001, log rank test).
To investigate the pathological features of KPPCN carcinomas, we evaluated pancreatic tissues at 4 and 7 weeks of age which surprisingly showed a markedly reduced burden of advanced PanIN pancreatic lesions and PDAC in the KPPCN compared to KPPC group (Fig. 1F). At 4 weeks, PDAC was detectable in 4/7 KPPC pancreatic tissues, while no evidence of tumors was apparent in the age-matched KPPCN animals. At 7 weeks, 8 of 9 KPPC mice had developed multifocal large pancreatic tumors comprising more than 50% of the organ area. At the same age, only 3 of 8 KPPCN mice had developed microscopic PDAC foci, comprising <5% of the pancreas (Fig. 1F). Importantly, these microscopic foci were comprised of poorly differentiated adenocarcinoma (indicated by arrow, Fig. 1G) and appeared on the background of low grade PanIN and ADM lesions. Micro-magnetic resonance imaging (mMRI) of larger cohorts demonstrated advanced large PDAC tumors and a typically enlarged pancreas in 7 week old KPPC mice, while KPPCN mice typically had a normally sized pancreas (Fig. S3A), which was also reflected in a marked difference in pancreas weight between the two genotypes (Fig. S3B). We next compared pathological features of advanced PDAC tumors present in KPPC and KPPCN mice at the time of euthanasia. Whereas in KPPC mice PDAC were typically grade 1-2, tumors in KPPCN mice demonstrated nearly uniform development of grade 4 PDAC (Fig. 1H-J, Fig. S3C for grading, and Table S1), echoing the decreased differentiation of KPCN versus KPC tumors (Table S1).
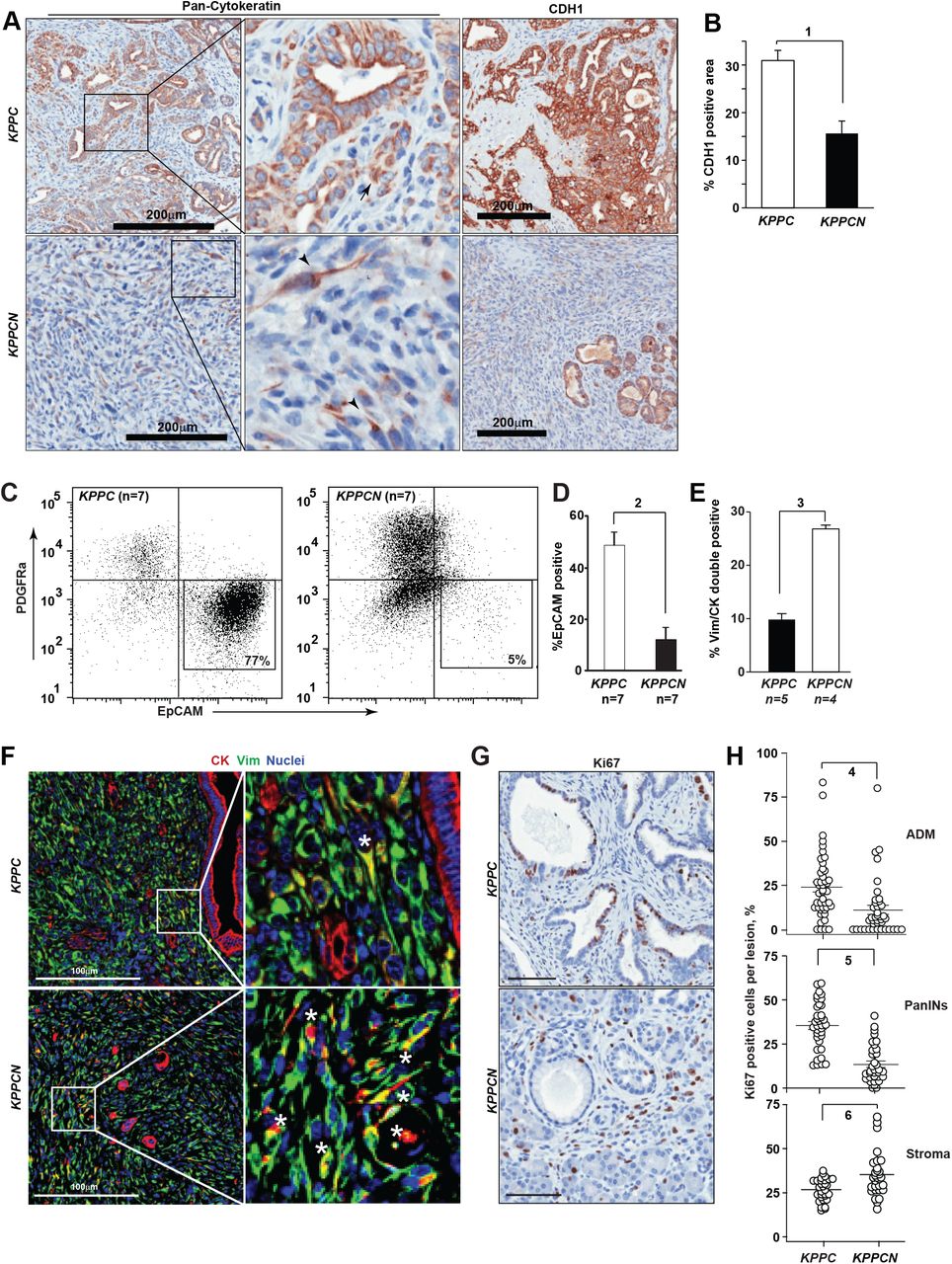
Analyzing tumor differentiation in greater depth, KPPC tumors were characterized by cytokeratin-positive pseudoglandular structures with strong expression of E-cadherin (CDH1 in Fig 2A), as was previously established for this model of PDAC (Bardeesy et al., 2006). In contrast, the majority of KPPCN carcinomas were comprised of single cells or small clusters of spindle-shaped tumor cells with weak expression of cytokeratin and absence of CDH1 expression (Fig 2B). The KPPCN carcinomas also had a lower density of CD31+ blood vessels (Fig. S3D, E), ruling out an Nsdhl-dependent angiogenesis switch previously implicated in PDAC progression (Rhim et al., 2014). Since loss of CDH1 expression is a feature of EMT (Scheel et al., 2011)), we further examined the differentiation markers of KPPCN carcinomas. Direct labeling of cells isolated from tumor tissues with EPCAM and PDGFRα antibodies allowed us to discern the populations of epithelial carcinoma cells and cancer-associated fibroblasts (CAFs) (Fig. S4A). Fluorescence-activated cell sorting (FACS) of cells isolated from the spontaneous pancreatic tumors showed significant reduction in EPCAM-positive cells in KPPCN as compared to KPPC (10% vs. 50%, respectively; Fig. 2C, D). This reduction could either reflect the possibility that KPPCN tumors were disproportionally composed of CAFs, or alternatively could indicate that a significant number of KPPCN cancer cells had lost epithelial lineage markers. The cultures of FACS-isolated cellular populations from KPPCN tumors (predominantly PDGFRα-/EPCAM-double-negative, or PDGFRα+) carried Cre-mediated rearrangements of Kras and Nsdhl indicating epithelial origin of these cells (Fig. S4B-E).

Development of basal pancreatic adenocarcinoma with mesenchymal differentiation in Nsdhl knockouts. (A) Representative pan-cytokeratin (CK) and E-cadherin (CDH1) immunohistochemical staining of pancreatic tumor tissues. Scale bars, 200 µm. (B) Quantification of CDH1 positive areas; (1) p=0.001. (C) Fluorescent activated cell sorting (FACS) of primary pancreatic tumor tissue shows markedly lower percentage of EPCAM+ cells in KPPCN compared to KPPC. CD45+ myeloid cells were excluded by gating. (D) Percentage of EPCAM-positive epithelial cells assessed by FACS in primary KPPC and KPPCN tumors; (2) p=0.0012, Wilcoxon test. (E) Quantification of CK+/VIM+ double positive cells by multicolor immunofluorescence; (2) p=0.016, Wilcoxon test. (F) Representative images of multiplex immunofluorescence characterization of KPPC and KPPCN carcinoma, depicting cytokeratin (CK) positive cells in red, vimentin (Vim) positive cells in green, and nuclei in blue. Asterisks, denote double-positive (CK+, VIM+) cells (G) Quantification of Ki67+ cells in pancreatic ADM, PanIN lesions and surrounding stroma in KPPC (n=7) and KPPCN (n=9) 4-week old mice. Data are represented as percent of Ki67+ in individual lesions; (4) p=0.001; (5) p<0.0001; (6) p=0.0015, Student t-test. (H) Representative Ki67 expression in stroma and epithelial lesions. In all figures, data are represented as mean±SEM. See also Figures S2-S4.
In keeping with the established pathology for the murine pancreatic adenocarcinoma induced by oncogenic KrasG12Dand Trp53 inactivation (Aiello et al., 2018; Bardeesy et al., 2006; Hingorani et al., 2005), the KPPC tumors exhibited glandular CK+ and CDH1+ epithelial structures (Fig. 2A, F) clearly separated from the cancer-associated fibroblasts expressing vimentin (VIM). As an alternative approach to assessing the interaction of the epithelial and the stromal compartmentalization of KPPC and KPPCN tumors, we used immunofluorescent labeling of cytokeratin (CK) and VIM markers, respectively. Only a small fraction (<10%, Fig. 2E) of cells in KPPC tumor co-expressed VIM and CK; these were typically single cells or small clusters of carcinoma cells localized outside of larger glandular structures (Fig. 2F). In contrast, in KPPCN tumors the overall frequency of double-positive VIM+/CK+ cells was significantly elevated (Fig. 2F), with these cells comprising the bulk of the tumor. We conclude that the higher prevalence of VIM+/CK+ double positive cells and loss of CDH1 expression in KPPCN tumors reflects their mesenchymal differentiation, consistent with the reported basal subtype of PDAC(Moffitt et al., 2015).
Independent of the KPPCN tumors adapting a more mesenchymal morphology, these tumors might differentially condition the essential tumor-supporting activity of pancreatic CAFs. The heterogeneous CAF population in pancreatic cancer is comprised of secretory fibroblasts and α-smooth muscle actin-positive (αSMA+) CAFs (also known as myofibroblasts) (Ohlund et al., 2017). The latter are responsible for enhanced deposition of collagen I, a characteristic of fibrotic desmoplasia in classic PDAC, and have been suggested to restrain PDAC progression in some models(Ozdemir et al., 2014). Assessment of the stroma of KPPCN tumors showed markedly reduced collagen deposition and fibrosis, based on use of type I collagen-directed antibodies and by Masson’s trichrome staining, respectively (Fig. S5A, B), and paucity of αSMA+ myofibroblasts (Fig. S5C, D), altogether indicating that basal PDAC induced an expansion of CAF populations expressing high level of GLI1 (Fig. S5E, F) and phosphorylated focal adhesion kinase (pY397-FAK, Fig. S5G, H) compared to classic (i.e. glandular) PDAC(Ohlund et al., 2017). The activated state of the tumor-associated stroma was also apparent at the premalignant stage, as assessed by Ki-67 positivity in pancreatic stromal elements surrounding KPPCN ADM and PanIN lesions compared to KPPC (Fig. 2G, H). These results together suggest that development of basal PDAC is distinctly different from the canonical ADM-PanIN-PDAC linear progression in classic glandular subtype of pancreatic cancer, and is associated with hyperactivated stroma.
KPPCN PDAC cell-intrinsic activation of TGFβ and other pro-EMT signaling
To understand cell-intrinsic differences in EMT-related signaling mechanisms distinguishing KPPC and KPPCN PDAC, we generated an extended panel of cell lines from KPPC (n=10) and KPPCN (n=10) tumors using differential trypsinization followed by FACS sorting enrichment of PDAC cells, which were analyzed at early passages (passage 3-5). The epithelial identity of these cell lines was validated by expression of CK, albeit sometimes low, and by completeness of Kras and Trp53 gene rearrangements (Fig. S6A, B). In vitro, we observed about half of KPPCN cell lines grew in a pattern of scattered single cells suggesting an EMT phenotype (Scheel et al., 2011) (Fig. S4C). Importantly, the observed in vitro EMT phenotypes of KPPCN cells were stable and correlated with development of diffuse undifferentiated carcinomas following syngeneic orthotopic implantation and resulted in shorter survival of animals compared to similar implantations of KPPC PDAC cells (Fig. S6C, D).
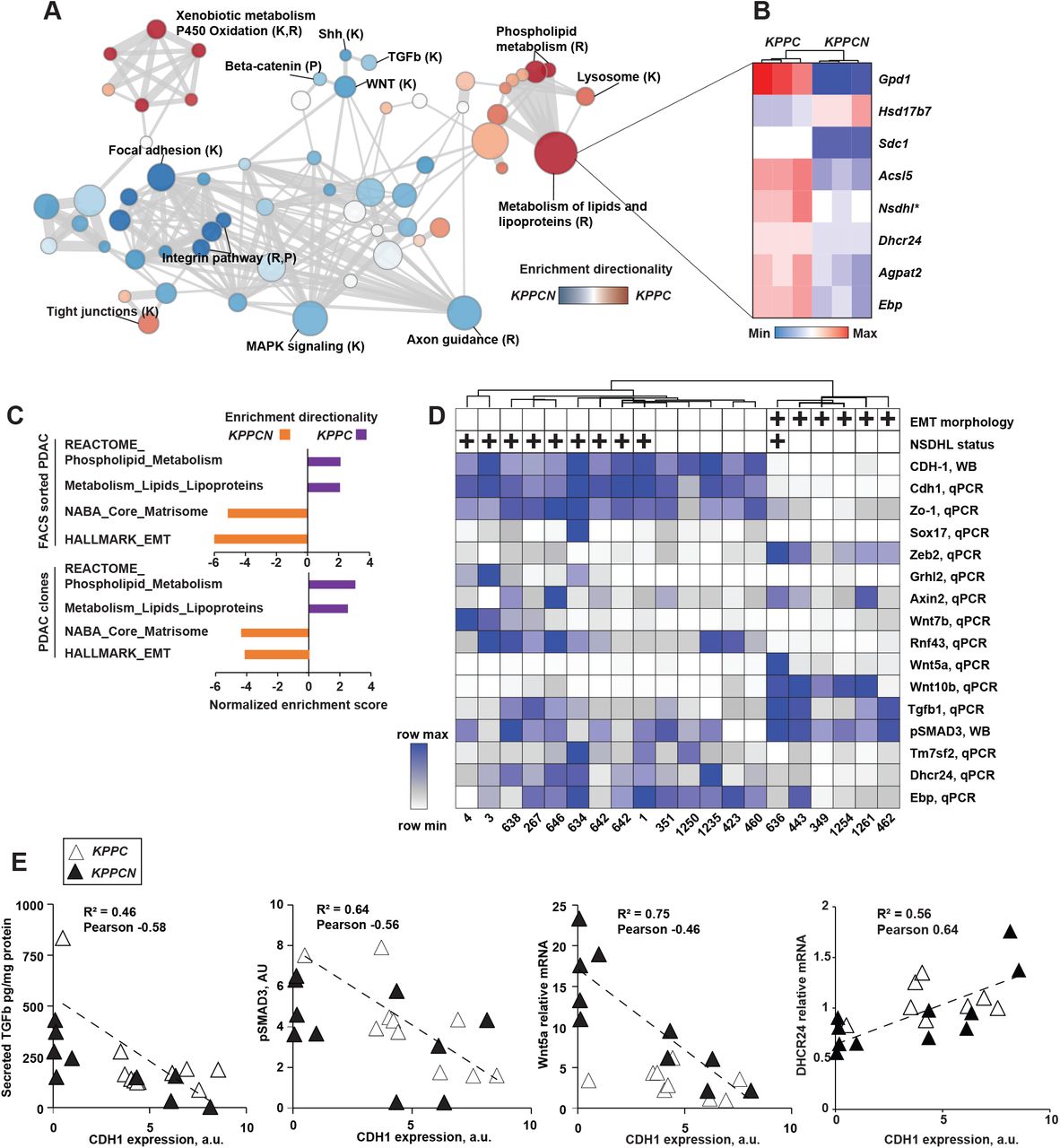
Five of 10 KPCN and 1/10 KPC cell lines were classified as mesenchymal due to loss of CDH1 on the cell surface, as determined by flow cytometry (Fig. S6E). To identify the mechanistic basis of the enhanced EMT of KPPCN cancer cells, we analyzed mRNA from 3 independent PDAC clones derived from the KPPC and KPPCN tumors using next generation sequencing, focusing initially on lines with the typical phenotypes: epithelial for KPPC and mesenchymal for KPPCN (Fig. 3A and Table S2). Confirming a sustained cell-intrinsic effect of Nsdhl deficiency of lipid metabolism, KPPCN carcinoma cells showed globally suppressed expression of multiple genes involved in biosynthesis of phospholipids and cholesterol (Fig. 3A, B). In addition to genetically induced Nshdl deficiency, expression of the distal cholesterol biosynthesis pathway gene Ebp and Dhcr24, along with other canonical SREBP targets were markedly reduced (Fig. 3B).

Conditional knockout of Nsdhl promotes epithelial-to-mesenchymal transition (EMT) switch in mouse pancreatic adenocarcinoma. (A) Gene Set Enrichment Analysis (GSEA) of differentially expressed genes as determined by mRNA sequencing of KPPC and KPPCN carcinoma clones, n=3 per genotype. Nodes sizes in the graph are proportionate to the number of genes in a given gene set, thickness of edges is proportionate to the number of shared genes between the gene sets. Sources of signatures: K, KEGG (Kyoto Encyclopedia of Genes and Genome); R, www.Reactome.org. (B) Heat map of a selected list of differentially expressed cholesterol and lipid metabolism genes. (C) Normalized enrichment scores for each genotype of FACS sorted primary PDAC cells and PDAC clones (shown, family-wise error rate <5%). (D) Unsupervised hierarchical clustering of quantified expression of indicated genes. qPCR, quantitative reverse transcription PCR; WB, Western blot. Mesenchymal morphology in vitro and NSDHL status is indicated above the heatmap by (+) positive. (E) Correlation of CDH1 expression and secreted TGFβ1, phosphorylated SMAD2/3, WNT5A and DHCR24 mRNA in KPPC and KPPCN clones. See also Figure S5 and Table S2.
Reciprocally, the KPPCN transcriptome was significantly enriched in gene targets associated with the EMT-promoting signaling pathways including TGFβ (Scheel et al., 2011), as well as PDGFR, integrin, and WNT, whereas expression of components of tight junctions (KEGG), a hallmark of epithelial differentiation, was enriched in the KPPC transcriptome (Fig. 3A). These differentially expressed mRNA signatures were similarly reproduced in RNA sequencing analyses of KPPC and KPPCN PDAC cells freshly isolated by FACS sorting from primary tumors (Fig. 3C); we observed highly significant overrepresentation of phospholipid and lipid metabolism gene transcripts in KPPC cells, and overrepresentation of signatures for EMT and extracellular matrix genes in KPPCN cells (Fig. 3C and Table S2).
We then used qRT-PCR to explore patterns of gene expression for key components of these pathways across the entire cell line panel (Fig. 3D). Among the KPPCN cell lines, those characterized as epithelial based on retention of CDH1 clustered with 9/10 KPPC cell lines, implying loss of Nsdhl predominantly acted by increasing the likelihood of mesenchymal transition. Mesenchymal cell lines also expressed high levels of the EMT transcription factor Zeb2, together with reduced expression of the epithelial Zo1, Sox17, and Ghrl2 genes (Fig. 3D). Mesenchymal cell lines showed increased motility in a wound closure assay in vitro compared to CDH1-positive cell lines, independent of tumor genotype (Fig. S6F). Several genes of canonical and non-canonical WNT signaling were upregulated in mesenchymal PDAC cells including Wnt5a, Wnt10b, while a negative regulator of WNT signaling, Rnf43 (Wu et al., 2011), was nearly undetectable (Fig. 3D). As anticipated, the expression of CDH1, a hallmark feature of epithelial lineage, negatively correlated with expression of Zeb2, Wnt5a, Wnt10a and secreted Tgfb1 to the media, whereas epithelial biomarkers Zo1, Sox17, cholesterol pathway Tm7sf2, Dhcr24 and Ebp, and a negative regulator of WNT signaling Rnf43 correlated with CDH1 expression (Fig. 3E, S6G). In addition to mRNA expression differences, we specifically probed the signaling activity of the TGFβ pathway reflected by levels of phosphorylated SMAD3, a canonical TGFβ pathway effector (Kretzschmar et al., 1999), and found a strong negative correlation with CDH1 and cholesterol pathway genes (Fig. 3E, S6G).
Notably, TGFβ1 is a major regulator of stromal activation in PDAC (Franco-Barraza et al., 2017), and increased secretion of this protein could explain the increased proliferation of stromal CAFs observed in KPPCN premalignant lesions (Fig 2G, H). An additional common regulator of PDAC stromal activation is Sonic hedgehog (Shh) (Rhim et al., 2014). Comparison of Shh secretion in cell culture supernatants produced by PDAC cells derived from KPPC and KPPCN pancreatic tumors (Fig. S7A) indicated no difference in Shh secretion. This suggested NSDHL deficiency alone, in the absence of stromal cells or TGFβ1, was not inducing tumoral Shh secretion in vitro.
Cholesterol deprivation activates TGFβ signaling
To establish a functional linkage between Nsdhl deficiency, decrease in the expression of cholesterol biosynthesis pathway enzymes, and EMT, we first established that total cellular cholesterol of KPPCN cell lines was approximately 25% lower than in KPPC cells when grown in cell culture medium containing lipids (with fetal bovine serum, FBS) indicating that cellular uptake of exogenous lipids and cholesterol could not fully compensate for the reduced endogenous cholesterol biosynthesis. These differences were further exaggerated in cultures supplemented with lipid-depleted serum (LDS, Fig. 4A). We proceeded next to investigate a possible causal link between cellular cholesterol levels and activation of the TGFβ pathway in PDAC cells. To induce a decrease in total cellular cholesterol comparable to KPPCN cell lines, we cultured KPPC PDAC cell lines (KPC3 and KPC634) for 48 hours in LDS medium with non-toxic concentrations of the 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor, compactin(Brown et al., 1978) (Fig. 4B). Strikingly, KPPC cell lines grown in LDS with compactin in the absence of added TGFβ1 showed levels of phosphorylated SMAD2/3 comparable to a 30-minutes pulse with TGFβ1 in cholesterol-replete cells (Fig. 4C, D). The increased levels of pSMAD2/3 induced by LDS+compactin were eliminated by SB431542, a selective inhibitor of the type I TGFβ receptor tyrosine kinase 1 (TGFBR1) and the homologous activin receptors ACVR1B and ACVR1C (Fig. 4C). Silencing of TGFBR1, but not of ACVR1B and ACVR1C, with siRNA demonstrated abrogation of pSMAD2/3 in cholesterol-depleted KPC3, indicating TGFβ family receptors predominated in the activation of SMAD2/3 in these cells (Fig. 4E, S6H).
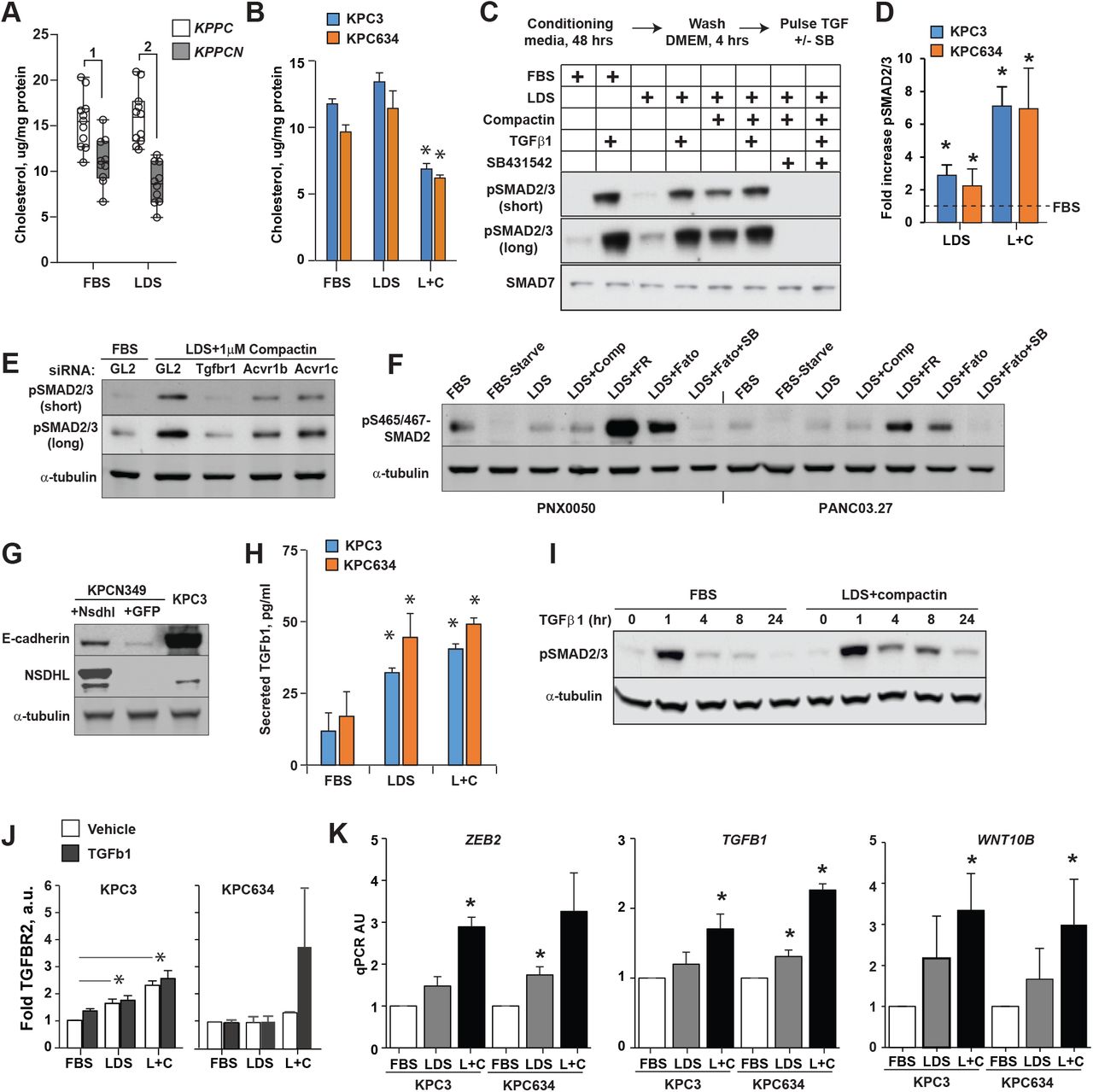
Cholesterol deprivation activates TGFβ pathway signaling in PDAC cells. (A) Cholesterol level in KPPC (n=10) and KPPCN (n=10) clones grown for 48 hours in FBS or LDS media; (1) p=0.004; (2) p=0.0001, Wilcoxon test. (B) Cholesterol levels in PDAC cells conditioned for 48 hours as indicated; L+C, 5%LDS with 1 µM Compactin. *, p<0.05, two-way Student t-test. Cholesterol depletion increased SMAD2/3 phosphorylation. (C) Representative Western blot of phosphorylated pSMAD2 (Ser465/467) and pSmad3 (Ser423/425) in KPC3 cells cultured for 48 hours in fetal bovine serum or lipid depleted serum (LDS) in DMEM, followed by incubation in serum-free DMEM for 4 hours. Indicated samples were treated with TGFβ1 at 10 ng/ml for 30 minutes, or SB431542 at 25 µM for 1 hour. (D) Summary results of cholesterol depletion on levels of phosphorylated pSMAD2(Ser465/467) and pSmad3 (Ser423/425). Shown are results from 3 independent experiments normalized to α-tubulin. (E) Silencing of TGFBR1, ACVR1B and ACVR1C with siRNA abrogated SMAD2/3 phosphorylation in KPC3 cells preconditioned for 48 hours in LDS with compactin. (F) Effect of cholesterol depletion on expression of pSMAD2/3 in human PDAC cell lines. Cells were conditioned for 48 hours in the presence of 1 µM Compactin (Comp), 10 µM FR171456 (NSDHL inhibitor, FR), 20 µM Fatostatin (SREBP1/2 inhibitor, Fato), or 25 µM SB431542 (TGFBR1 inhibitor, SB). (G) Expression of E-cadherin in KPCN349 cells modified to express full length Nsdhl. Cells modified with lentiviral vector to express GFP served as control. (H) Increased TGFβ1 in supernatants of KPC3 cells conditioned as in (A). (I) Persistence in SMAD2/3 phosphorylation upon TGFβ1 treatment. KPC3 cells were conditioned in FBS or LDS+compactin with for 48 hours, pulsed with 2.5 ng/ml of TGFβ1 for 60 min, washed and lysed at the times indicated. Levels of pSMAD2 and tubulin as a loading control were assayed by Western blotting. (J) Quantified expression of TGFBR2 in KPC3 and KPC634 cells conditioned in FBS, LDS, or LDS+compactin 1 µM (L+C). (K) Expression of Zeb2, Tgfb1 and Wnt10b mRNA as assessed by qRT-PCR in KPC3 and KPC634 PDAC cells cultured for 48 hours in media supplemented with FBS, LDS or LDS+compactin (1 µM); *, p<0.05, two-way Student t-test.
The induction of pSMAD2/3 by an agent targeting the cholesterol biosynthesis pathway was not specific to compactin or to murine PDAC models, as inhibiting endogenous cholesterol biosynthesis with FR171456, a selective NSDHL inhibitor (Helliwell et al., 2015), or fatostatin, an SREBP inhibitor (Chen et al., 2018; Talebi et al., 2018), similarly upregulated pSMAD2/3 in two human PDAC cell lines (Fig. 4F). In addition to increasing SMAD2/3 phosphorylation, cholesterol starvation induced formation of actin stress fibers (Fig. S6G), which represent an EMT-associated, SMAD-independent response to TGFβ pathway activation, mediated by the CDC42 and RhoA GTPases (Edlund et al., 2002). Importantly, restoration of cholesterol biosynthetic competency in mesenchymal/basal-like KPCN349 PDAC cells, via lentiviral re-expression of Nsdhl cDNA, partially restored CDH1 expression, indicating a direct effect on epithelial differentiation (Fig. 4G).
The fact that inhibitors of TGFβ receptors blocked induction of pSMAD2/3 by cholesterol depletion suggested a mechanism by which cholesterol reduction activates this signaling cascade. We had noted that mesenchymal PDAC cell lines expressed higher levels of Tgfb1 mRNA (Fig. 3D). Similarly, KPC3 and KPC634 cells preconditioned for 48 hours in LDS, with or without compactin, secreted higher levels TGFβ1 in culture media (Fig. 4H). Several reports have implicated membrane cholesterol as influencing endocytic trafficking of TGFβ receptors, thus affecting TGFβ pathway signaling activity (Di Guglielmo et al., 2003; Miller et al., 2018). We conducted TGFβ1 pulse-chase experiments in which PDAC cells were pre-conditioned in cholesterol-replete or cholesterol-poor cultures and pulsed using low dose, 2.5 ng/ml, TGFβ1. Activation of SMAD2/3 was then followed in the absence of added TGFβ1. These experiments demonstrated markedly prolonged phosphorylation of SMAD2/3 in cholesterol depleted PDAC cells (Fig. 4I). Signaling activity of TGFβ receptors is restricted by their endocytic trafficking to late endosomes and lysosomes (Miller et al., 2018), while protracted SMAD2/3 phosphorylation is associated with increased constitutive levels of TGFβ receptor as we determined in cholesterol depleted cells (Fig. 4J). We further determined that cholesterol starvation of epithelial PDAC cells increased the expression level of the mesenchymal biomarkers, such as Tgfβ1, Zeb2 and Wnt10b (Fig. 4K) defined by transriptome analyses of PDAC cell lines (Fig 3D), implying the switch between subclasses is dynamic and reversible.
To determine whether in vitro observations of cholesterol-dependent modulation of TGFβ signaling activity correlated with the effects on PDAC development observed in the KPC mouse model, we evaluated activation of this pathway in murine tumor tissues. In agreement, NSDHL-deficient premalignant pancreatic lesions demonstrated more activated pSMAD2, than did lesions in KPC and KPPC mice (Fig. S7D).
Distinct cholesterol metabolic programs define transcriptional subsets of human pancreatic cancer
To determine whether the relationship between the cholesterol biosynthesis and EMT signaling observed in mouse models is also reflected in human PDAC, we compared transcriptional profiles between classic and basal subsets of PDAC using data available for 76 high purity pancreatic adenocarcinoma samples by The Cancer Genome Atlas Research Network (TCGA., 2017). Notably, basal PDAC had significantly increased expression of the gene signatures of TGFβ and EMT signaling, among other hallmark pathways of tumor aggressiveness such as the PI3K-mTOR pathway, mitosis, and hypoxia (Fig. 5A). In contrast, the fatty acid and cholesterol metabolism gene signatures (Molecular Signature Database, (Liberzon et al., 2015)) were significantly higher in classic PDAC compared to basal (Fig. 5B). In confirmatory analysis, we found that the cholesterol biosynthesis gene signature (Reactome, (Fabregat et al., 2017)) was significantly lower in basal PDAC, compared to classic, from independent cohorts curated by the International Cancer Genome Consortium (ICGC, Fig. 5C).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cholesterol homeostasis regulates classic versus basal differentiation of human PDAC. (A) Comparison of mRNA transcriptional signatures between classic and basal PDAC among 76 TCGA cases with estimated tumor cell fraction greater than 30% and p-values <0.01 (shown as –log10, orange line). (B-D) Comparison of cholesterol homeostasis genes expression in human basal and classic PDAC. In B, expression of “hallmark_cholesterol_homeostasis” signature (Molecular Signature Database(Liberzon et al., 2015) in 76 TCGA PDAC cases; in C, shown are “reactome_cholesterol_biosynthesis” mRNA signatures in 102 PDAC cases from ICGC (GSE50827); and D represents “hallmark_cholesterol_homeostasis” mRNA signature expression in 85 patient-derived xenograft samples stratified by human GATA6 mRNA Z-score above or below zero. y-axes illustrate enrichment score comparing basal and classic subtypes of PDAC. Positive and negative scores indicate positive and negative enrichment, respectively. Boxplots represent median (black bar) and range (25-75th percentile) of enrichment scores for individual cases shown as red dots for each sample in that subtype. (E) Heat map of normalized expression of representative genes in the “hallmark_cholesterol_homeostasis” signature for TCGA and human PDAC PDX cases. Z-scores calculated for each gene are plotted on a red (higher expression) and blue (low expression) scale. Top color bar, subtype of PDAC. Mutations, shown as black lines if present. (F, G) Kaplan-Meier survival of Moffitt classic and basal PDAC in the TCGA (F) and ICGC (G) cohorts stratified by positive or negative enrichment for “hallmark_cholesterol_homeostasis” signature. See also Tables S3-4.
Separately, we classified a panel of 85 human PDAC patient-derived xenografts (PDXs) by a validated clinical biomarker(Aung et al., 2018), i.e. high or low GATA6 mRNA expression, as classic or basal PDAC, respectively. In accord with the TCGA and the ICGC datasets, mRNA analysis of GATA6low PDX showed significantly lower expression of hallmark cholesterol homeostasis genes (Fig. 5D). The cholesterol homeostasis genes underrepresented in basal PDAC (Fig. 5E) included 16 canonical genes of cholesterol biosynthesis, LDLR (the principal receptor for exogenous cholesterol uptake (Guillaumond et al., 2015)), and SREBF2, a master transcription factor of cholesterol biosynthesis pathway (Hua et al., 1995). These data strongly suggested functional cholesterol metabolic auxotrophy in basal subset of human PDAC. Importantly, the cholesterol homeostasis gene signature independently impacted the overall survival of patients. For these survival analyses, we compared PDAC cases from TCGA and ICGC datasets stratified by their positive or negative cholesterol gene signature enrichment scores (Fig. 5B, C). The cases with positive enrichment for cholesterol homeostasis genes had a similar, longer survival, irrespective of the Moffitt basal or classic subclass. Contrastingly, low expression of cholesterol homeostasis genes was associated with dismally shorter survival of the basal PDAC patients compared to the classic variant (Fig. 5F, G) thus indicating poorer prognosis in patients with low cholesterol pathway genes expression.
Discussion
Pancreatic cancer remains one of the most lethal human cancers. EMT and uncontrolled metastases are the primary cause of mortality in PDAC, and particularly associated with the basal subtype (Aiello et al., 2018; Rhim et al., 2012). This study provides evidence for a direct role of cholesterol metabolism in induction of EMT and basal PDAC development. In the mouse model, disruption of cholesterol biosynthesis consistently decreased tumor differentiation in Kras-dependent pancreatic tumorigenesis, in a phenotype we defined as cell-intrinsic. Mechanistically, this arose from upregulated expression of TGFβ1, which acted in an autocrine manner through SMAD2/3 to promote EMT in PDAC cancer cells, and in a paracrine manner to activate CAFs surrounding the cancer cells, which are known for their increased TGFβ production(Kojima et al., 2010). We also showed that starvation for cholesterol in cells with intact NSDHL separately resulted in similar induction of EMT, based on activation of TGFβ1 signaling. Finally, in human PDAC, we established a relationship between a signature indicative of reduced cholesterol homeostasis and poor survival, specifically in the basal subset of PDAC tumors, which constitute ∼25% of PDAC overall. Given the known linkages between diet, obesity, and the risk and prognosis of aggressive cancers (Golemis et al., 2018), there has long been interest in the relationship between cholesterol and PDAC pathogenesis (Huang et al., 2017). The complex signaling mechanism revealed in this study offers one explanation as to why such relationships have been difficult to establish, and for the poor clinical outcomes in trials of statins for PDAC (Hong et al., 2014). Experimental models and clinical trials of statins aimed to define the role of cholesterol metabolism in disease risk and treatment have yielded complicated and sometimes conflicting results (Chen et al., 2019; Huang et al., 2017). Importantly, our data provides a mechanistic framework for interpretation of contradictions arising from some of these experiments. For instance, our results raise the possibility that potential beneficial effects cholesterol restriction could be confined to cancers with functional P53: a relatively small cohort. In the broader population of PDAC patients, cholesterol lowering interventions may have inadvertently favored the selection of poorly differentiated cancer cells that have undergone EMT and contributed to resistance and worse outcomes. Further, based on our transcriptome analyses (Fig. 5), the effects of statins might only be relevant to the classic rather than the basal subset of PDACs. In the latter, lowering cholesterol may be detrimental by further promoting TGFβ signaling. Our retrospective analyses may be of interest for further prospective validation in the clinic and for the applicability of cholesterol suppression as a therapeutic modality in PDAC. As an example, our results may explain why statin use has been associated with modestly improved overall survival in patients with existing pancreatic tumors, but where the reductions in level of serum cholesterol did not correlate with survival in individual patients (Huang et al., 2017).
Genetic ablation of Nsdhl provides a useful model for dissection of cholesterol signaling, as loss of its activity in catalyzing an irreversible step of oxidative C4-demethylation is more selective to cholesterol instead of mevalonate pathway inhibitors having pleiotropic effects on farnesylation and geranylation-based signaling (Deng et al., 2018). While inactivating mutations in NSDHL or other cholesterol pathway genes are extremely rare in pancreatic cancer (cbioportal.org), low NSDHL mRNA expression was evident in the majority of “basal” PDAC (Fig. 5). Conversely, a signature of higher expression of multiple additional cholesterol pathway genes is a feature of classic PDAC. An intriguing feature of our study is the fact that non-equivalent results were obtained when Nsdhl loss was accompanied by heterozygous versus homozygous loss of Trp53. Nsdhl inactivation in the context of the heterozygous loss of Trp53 dramatically delayed malignant progression of the precursor lesions and nearly completely abrogated PDAC development (Fig. 1A). These results indicate that in the context of partial retention of Trp53 tumor suppressor activity, cholesterol pathway proficiency is critical for the continuous progression from ADM to PanINs, and ultimately to the classic (glandular/classic) variant of PDAC associated with an activating Kras mutation (Fig. 1B). Conversely, the linear ADM-PanIN-classic PDAC progression was impaired in KPPCN mice with complete deletion of Trp53, and hence with reduced restriction on proliferation (Fig. 1C, (Morton et al., 2010)). Despite this reduced ability to induce classic PDAC, all KPPCN mice died of fulminant basal PDAC originating from rare microscopic foci, expressing elevated TGFβ1 and surrounded by highly activated yet non-canonical CAFs.
The mechanisms by which cholesterol deprivation stimulates TGFβ signaling and EMT require further analysis. Among candidate mechanisms, depletion of the cellular membrane cholesterol pools has been shown to increase membrane fluidity promoting EMT (Zhao et al., 2016). In line with our findings, genetic studies in breast cancer mouse model have demonstrated induction of the EMT by increased levels of 27-hydroxycholestrol, an established LXR agonist (Nelson et al., 2013). Based on these data, reduced biosynthesis of cholesterol could be nominated as an EMT-promoting metabolic switch by reinforcing TGFβ1 pathway signaling in the absence of P53. In human PDAC, we hypothesize that a nutrient- and oxygen-restricted tumor microenvironment may lower the levels of cholesterol in early stage PanINs, activating TGFβ signaling and supporting emergence of basal PDAC. It is possible that basal PDAC are ultimately selected to bypass a strong anti-proliferative and pro-apoptotic effects of TGFβ signaling elicited through SMADs and P53 interactions (Cordenonsi et al., 2003) induced in cholesterol-poor tumors. Therefore, P53 is nearly universally mutates in basal PDAC (Fig. 5E) where it exerts a dominant negative effect to overcome the KRAS-induced senescence and promote PDAC metastases(Hingorani et al., 2005; Morton et al., 2010). Our use of conditional Trp53f/f mice instead of P53 mutant allowed us to functionally probe the complex interactions between P53, TGFβ pathway and cholesterol metabolism during KRAS-induced pancreatic carcinogenesis. Some data suggests that a role of cholesterol in regulating TGFβ pathway signaling may also be relevant to other human pathologies in which cholesterol depletion or accumulation takes place. One interesting example is in formation of atherosclerotic plaques, where progressive cholesterol accumulation abolishes TGFβ signaling and SMAD3 phosphorylation in smooth muscle cells (Chen et al., 2016).
Author contributions
L.G.C., S.P., and I.A. designed research; L.G.C., S.P., D.R., A.K., T.R.H, R.F., J.F.-B., N.S., E.N., E.H., K.Q.C., I.S. and R.A.M. performed research; R.A.M., R.C. and A.M.O. contributed new reagents/analytical tools; L.G.C., S.P., E.H., E.C. and I.A. analyzed data; L.G.C., E.A.G, E.C. and I.A. wrote the paper.
METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse strains
Cell lines
METHOD DETAILS
PCR genotyping
Induction of acute pancreatitis in mice with caerulein
Micro-magnetic resonance imaging (mMRI)
Orthotopic reimplantations
Immunohistochemistry
Histology images analysis
Lesion and tumor grading criteria
Simultaneous multi-channel immunofluorescent (SMI) labeling of formalin-fixed paraffin-embedded mouse pancreatic tumors
mNSDHL expression in KPCN cells
WB analysis of protein expression levels in cell lines and tissues
Quantitative RT-PCR
Clustering analysis
RNAseq and its bioinformatical analysis
Wound healing assay
TGFβ ELISA
Confocal image acquisition
Cholesterol measurement
siRNA transfection
Shh ELISA
Phalloidin staining of stress fibers
STATISTICAL ANALYSIS
DATA AND SOFTWARE AVAILABILITY
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Igor Astsaturov (Igor.Astsaturov{at}fccc.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
Mice carrying the conditional knockout allele of Nsdhl (Cunningham et al., 2015) were kindly provided by Dr. Gail Herman (The Research Institute at Nationwide Children’s Hospital, Columbus, OH). These mice (official name: Nsdhltm1.1Hrm, MGI:5581334, to be designated as Nsdhlf/fhere) are congenic on a C57BL/6J background. KPC (LSL-KrasG12D;Trp53f/f; Pdx1-Cre) and KC (LSL-KrasG12D; Pdx1-Cre) mice were kindly provided by Dr. Kerry Campbell (Fox Chase Cancer Center, Philadelphia, PA) and those mice are congenic on a C57BL/6J background as well. Mice were bred to obtain the desired genotype with Nsdhl depletion in normal pancreas (Nsdhlf/f; Pdx1-Cre, to be designated as NsdhlΔPanc here) and Nsdhl depletion in pancreas of KPC mice with different status of p53 expression (KPC mice carrying Trp53f/f or Trp53f/+, or KC mice). All mice were bred and kept under defined-flora pathogen-free conditions at the AAALAC-approved Animal Facility of the Fox Chase Cancer Center, Philadelphia, PA. Mice of both genders, equally distributed, were used for experiments. Tumor bearing mice were observed once weekly until signs of sickness appeared or animals showed distress or weight loss of more than 10%, per the local Institutional Animal Care and Use Committee (IACUC) guidelines.
Cell Lines
Mouse pancreatic cancer cell lines (KPC and KPCN) were derived from mouse pancreatic tumors by tumor dissociation and subsequent fluorescence-activating cell sorting (FACS). Tumor dissociation was performed with the use of the Gentle MACS Tumor Dissociation Kit (Miltenyi Biotec, Order No. 130-096-730) according to the manufacturer’s instructions. Briefly, each tumor was isolated from the animal in a sterile environment and washed in PBS. A 1mm3 piece was taken for genotyping and a larger piece was taken for histopathology analysis. The rest of the tumor tissue was placed in the dissociation enzyme mix and minced quickly to get pieces ∼2mm3 in size. Then the enzyme-tissue mixture was transferred into the gentleMACS C tube and incubated at 37°C with constant rotation for 40 minutes. After that the tissue was further mechanically processed by the gentleMACS Dissociator. The single-cell suspension was obtained by passing the tissue mixture through the 70µm cell strainer. Dead cells were subsequently removed by the Dead Cell Removal Kit (Miltenyi Biotec, Order No. 130-090-101).
In order to separate cells of different origin we further stained the cell suspension with antibodies against CD45 (Biolegend #103107; 1:200), FAP (Abcam #ab28244; 1:50), EPDACAM (Biolegend #118212; 1:200) and PDGFRa (CD140a) (Biolegend #135905; 1:80). To prevent antibodies from binding to Fc-receptors we treated the cell suspension with Fc-block (Biolegend #101301; 1:50) prior using other antibodies. To detect cells labeled with FAP we used BV421 anti-rabbit secondary antibody (BD Biosciences #565014; 1:50). Live cells were selected based on Propidium Iodide staining (Biolegend #421301). Fluorescence detection and sorting were performed with BD FACS Aria II flow cytometer. Sorted cells were propagated for first two passages in enriched media (RPMI-1640 supplemented with 15% v/v FBS, 2mM L-glutamine, 100µg/ml Penicillin/Streptomycin, 20ng/ml EGF, 25µg/ml Insulin, NEAA 1x, 1mM Na-Pyruvate and 2µg/ml Hydrocortizone) and for subsequent passages in DMEM supplemented with 10% v/v FBS and 2mM L-glutamine with 100µg/ml Penicillin/Streptomycin. Cells were used at passages 3-5.
Human pancreatic cancer cell line PANC03.27 (#CRL-2549, ATCC) was kindly provided by Dr. Paul Campbell (Fox Chase Cancer Center, Philadelphia, PA).
Human pancreatic cancer cell line PNX0050 was derived as described earlier (Beglyarova et al., 2016).
METHOD DETAILS
PCR genotyping
Small (1-2mm) pieces from mouse tails or mouse pancreata were used for genotyping. For cell line genotyping we isolated genomic DNA with QIAamp® DNA Micro Kit (#56304, Qiagen). Primers for genotyping are listed in Supplementary Table S5. The PCR reaction was performed using Terra PCR Direct Red Dye Premix (#639286, TaKaRa) for LSL-KRas construct detection and GoTaq Green Master Mix (#M7122, Promega) for all other constructs.
Induction of acute pancreatitis in mice with caerulein
Acute pancreatitis in mice was induced by caerulein (#C9026, Sigma-Aldrich) treatment as previously described (Morris et al., 2010). Briefly, at day “-1”, mice were injected with 50µg/kg of caerulein i/p every hour for 6 hours (6 injections total). In 24 hours (day “0”) the course of injections was repeated. Control mice were injected with saline. At days “2”, “5”, “7” pancreatic tissues were collected and immediately fixed in formalin. Fixed tissues were embedded in paraffin and stained with H&E and Ki-67 for further histological analysis.
Micro-magnetic resonance imaging (mMRI)
mMRI was performed on 7-8 week old KPPC and KPPCN mice following the standard procedure of Small Animal Imaging Facility (Dr. Harvey Hensley, Fox Chase Cancer Center, Philadelphia, PA). Briefly, animals were imaged in a 7 Tesla vertical wide-bore magnet, using a Bruker DRX 300 spectrometer (Billerica, MA) with a micro-imaging accessory, which included a micro 2.5 gradient set, a 30 cm radiofrequency coil, and Paravision software (Bruker, Billerica, MA). In order to increase the contrast of the imaging, Magnevist (#50419-188-82, Bayer Healthcare Pharmaceuticals Inc.) was diluted in sterile PBS to achieve 46.9mg/ml concentration and injected subcutaneously into the shoulder region, 200µl/mouse, immediately preceding the scan. Prior to Magnevist injection, and throughout the whole imaging process, mice were anesthetized with a mixture of oxygen and isoflurane (2-3%) according to the local IACUC guidelines. Scout scans were performed in the axial and sagittal orientations, permitting us to accurately prescribe an oblique data set in coronal and sagittal orientations that included the organs of interest. A two dimensional spin echo pulse sequence was employed with echo time 15msec, repetition time 630msec, field of view=2.56cm, acquisition matrix=256×256, slice thickness=0.75mm, 2 averages, scan time=5 minutes. Fat suppression (standard on Bruker DRX systems) was used for all scans.
Orthotopic reimplantations
Orthotopic reimplantations of KPC and KPCN tumor cells in mouse pancreas were performed as described by Kim et al. (Kim et al., 2009). Total 106 cells per 50µl of 30%/70% Matrigel/PBS mixture were injected into the pancreatic head region of syngeneic C57BL/6J mice. Anesthetics and analgesics were used according to the local IACUC guidelines. Tumor bearing mice were observed twice weekly until signs of sickness appeared or animals showed distress or weight loss of more than 10%, per the local IACUC guidelines.
Immunohistochemistry
For immunohistochemistry, formalin-fixed pancreatic tissue was embedded in paraffin and stained with indicated antibodies diluted per the manufacturer’s instructions (listed in Key Resources Table). Antibody binding was visualized using the Liquid DAB+ Substrate Chromogen System (Dako). Samples were counterstained for 1 minute with hematoxylin. Collagen deposition and organization were directly visualized by the standard Masson’s Trichrome Stain.
Histology images analysis
Slides were scanned by an Aperio ScanScope CS scanner (Aperio) and selected regions of interest were outlined manually. The surface area taken by different lesions, the expression levels of ABCA1, pS-S6, cleaved Caspase-3, Ki-67, pErk, CDH1, CD31, pSMAD2 and trichrome positive area were measured using the ImageScope software (Leica Biosystems Imaging, Inc.).
Lesion and tumor grading criteria
Normal tissue, acinar-to-ductal metaplasia (ADM), pancreatic intraepithelial neoplasia (PanINs) and pancreatic ductal adenocarcinoma (PDAC) were determined according to the standard classification (Hruban et al., 2006). For histopathological scoring, tumors were classified using the standard pathological grading scheme into either well differentiated (grade 1), moderately differentiated (grade 2), poorly differentiated (grade 3) or undifferentiated (which includes sarcomatoid) (grade 4).
Multi-channel immunofluorescent labeling of mouse pancreatic tumors
For vimentin, cyto-keratin, Gli1 and pFAK immunofluorescent co-staining of formalin-fixed paraffin-embedded mouse pancreatic tumors, we utilized the simultaneous multi-channel immunofluorescent (SMI) labeling approach described earlier (Franco-Barraza et al., 2017). Briefly, Gli1 and pFAK primary antibodies were conjugated with Q-dot probes by using antibody Q-dot labeling kits (#S10469 and #S10450, ThermoFisher Scientific). Slides were deparaffinized and stained with Q-dot pre-labeled antibodies (Franco-Barraza et al., 2017). To detect epithelial/tumoral locations and mesenchymal (stromal) components slides were further stained with the mouse monoclonal “cocktail” of anti-pan-cytokeratin (clones AE1/AE3, DAKO) and anti-vimentin (EPR3776, Abcam) antibodies with subsequent staining with secondary donkey anti-mouse Cy2 and donkey anti-rabbit Cy3 antibodies. Nuclei were stained using DR (1:105, #DR50050, Biostatus) (Franco-Barraza et al., 2017). Images were collected using Caliper’s multispectral imaging system (Perkin Elmer) and analyzed with SMIA-CUKIE (SCR_014795) software, as described earlier (Franco-Barraza et al., 2017).
WB analysis of protein expression levels in cell lines and tissues
For Western blot analysis, the piece of tissue sample or cultured cells were homogenized in RIPA buffer (#24928, Santa Cruz) with phosphatase and protease inhibitors (#1862495, #1861278, Thermo Scientific) on ice and cleared then by centrifugation. The protein concentration was measured with Pierce BCA Protein Assay Kit (#23225, Thermo Scientific). Proteins were separated on the 4-12% Bis-Tris Protein gels (Invitrogen) and then horizontally transferred to the Immobilon-FL PVDF membrane (#IPFL00010, Millipore). Primary and secondary antibodies were used in concentrations indicated in the Key Resources Table according to manufacturer’s instructions. The density of obtained bands was quantified with Image Studio software (LI-COR).
Quantitative RT-PCR
For evaluation of target gene expression, total RNA was extracted using the RNeasy Mini Kit (#74104, Qiagen). RNA was reverse transcribed (RT) using Moloney murine leukemia virus (MMLV) reverse transcriptase (#28025013, Ambion) and a mixture of anchored oligo-dT and random decamers (IDT). Two reverse-transcription reactions were performed for each sample using either 100 or 25ng of input RNA in a final volume of 50µl. Taqman or SYBR Green assays were used (see Supplementary Table S5) in combination with Life Technologies Universal Master mixes and run on a 7900 HT sequence detection system (Life Technologies). Cycling conditions were 95°C, 15 minutes, followed by 40 (two-step) cycles (95°C, 15s; 60°C, 60s). Ct (cycle threshold) values were converted to quantities (in arbitrary units) using a standard curve (five points, four fold dilutions) established with a calibrator sample.
Clustering analysis
Hierarchical clustering analysis of KPC and KPCN cell lines was performed based on data obtained from qPCR and WB results. The expression level of each gene (for qPCR) or the level of each protein in total cell lysates (for WB) for all analyzed cell lines was first normalized by converting them to a percentage, wherein the highest level of expression for each gene was set to 100%. The obtained results were further clustered using Morpheus on-line software (Morpheus, https://software.broadinstitute.org/morpheus) with using average linkage method and one minus Pearson correlation metric.
RNA sequencing of mouse pancreatic cancer cells
Murine PDAC cells were freshly isolated from dissociated pancreatic tumor tissues followed by FACS sorting (see Cell Lines). Total RNA was isolated using TRIzol (#15596-026, Life technologies) according to the manufacturer’s protocol. RNA from established cell lines was extracted with the use of an RNeasy Mini Kit (#74104, Qiagen). RNA samples were further submitted for RNAseq analysis. Total RNA libraries were prepared by using Pico Input SMARTer Stranded Total RNA-Seq Kit (Takara). In short, 250pg-10ng total RNA from each sample was reverse-transcribed via random priming and reverse transcriptase. Full-length cDNA was obtained with SMART (Switching Mechanism At 5’ end of RNA Template) technology. The template-switching reaction kept the strand orientation of the RNA. The ribosomal cDNA was hybridized to mammalian-specific R-Probes and then cleaved by ZapR. Libraries containing Illumina adapter with TruSeq HT indexes were subsequently pooled and loaded to the Hiseq 2500. Single end reads at 75bp were generated for gene expression analyses.
Sequencing reads were analyzed for quality issues using FastQC (S.Andrews, http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Reads were aligned to mouse genome (mm10) using TopHat2 (PMID: 23618408) and absolute gene counts were quantified using HTSeq (PMID: 25260700). The resulting gene counts were used as input for differential expression analysis between KPC and KPCN clones and primary cells using DESeq2 (PMID: 25516281). Genes that are differentially expressed were selected for subsequent downstream analysis for identification of biological pathways and ontologies using Gene Set Enrichment Analysis (GSEA) (p-value < 0.001) (PMID: 16199517) using MsigDB datasets (PMID: 26771021). To graphically represent the significantly enriched datasets (False Discovery Rate < 25%) Enrichment Map was used (PMID: 21085593).
Analysis of PDAC patient tumor tissues
The data of mRNA expression and patient outcomes data for human pancreatic ductal adenocarcinoma were extracted from publicly available data portals. RNA-Seq data for the TCGA PDAC tumor tissues were downloaded from Braod GDAC Firehose poratal (https://gdac.broadinstitute.org/) Subsequent analyses were limited to 76 cases with inferred PDAC fraction >30% by ESTIMATE method (Yoshihara et al., 2013) as described in (TCGA., 2017). The mRNA gene expression array data from 103 primary pancreatic ductal adenocarcinoma samples were obtained from GSE50827 and matched to survival information from the International Cancer Genome Consortium (ICGC) data portal (http://dcc/icgc.org). One case was excluded from the ICGC series due to early death. The PDAC cases in both, TCGA and ICGC series were classified as classic or basal according to a weighted gene expression algorithm (Moffitt et al., 2015). The RNA sequencing data from 85 human PDAC patient-derived xenografts was obtained from Champions Oncology, Inc., Hackensack, NJ, via TumorGraft® database (https://database.championsoncology.com/). The raw gene expression data used for analyzing subtype-specific pathway alterations in PDAC were normalized using Robust Multi-array Average (RMA) procedure (PMID: 12925520). Gene set variation analysis (GSVA, (Hanzelmann et al., 2013)) on the RMA normalized expression data was applied to identify pathways that are enriched in a single sample (method arguments: function=’gsva’; mx. Diff=TRUE; verbose=FALSE). Pathways that differ significantly between basal and classical cases were identified using Wilcoxon-Rank sum test using enrichment scores resulting from GSVA. Analyses to compare the overall-survival among the basal and classical subtypes were performed by comparing survival curves with log-rank tests. These were calculated using the R ‘survival’ package Therneau, T. M. & Grambsch, P. M. Modeling Survival Data: Extending the Cox Model (Springer-Verlag, 2010).
Wound healing assay
For the wound healing assay, cells were seeded in 12-well plates in concentration 2*105 cells/ml/well in DMEM supplemented with 10% v/v FBS and 2mM L-glutamine with 100µg/ml Penicillin/Streptomycin and cultured to 95% confluence. The wound was created by scratching the cell monolayer with 200µl sterile pipette tip. To remove the debris and smooth the edge of the scratch, cells were washed once with the growth medium. Images of cells migrating into the wound were captured by time lapse microscopy (Nikon TE300 epi-fluorescent inverted microscope, 20x lens NA 0.45) every 15 minutes for duration of 20 hours at 37°C. Results were analyzed with ImageJ software.
TGFβ ELISA
The amount of TGFβ secreted by mouse pancreatic tumor cells was quantified by using the Mouse TGFβ 1 DuoSet ELISA (#DY1679-05, R&D systems), complemented with Sample Activation Kit 1 (#DY010, R&D systems) and DuoSet ELISA Ancillary Reagent Kit 1 (#DY007, R&D systems) according to the manufacturer’s instructions. Conditioned media for TGFβ secretion quantification was collected from KPC and KPCN tumor cells growing in DMEM supplemented with 1% v/v FBS and 2mM L-glutamine with 100µg/ml Penicillin/Streptomycin for 48 hours. Protein concentration of lysates, prepared from remaining cell pellets, was used for data normalization.
Confocal image acquisition
Confocal images were acquired with Leica TCS SP8 laser scanning confocal microscope with a 63x oil lens, NA 1.4. Images within each particular experiment were acquired under identical exposure conditions per channel.
Shh ELISA
The amount of Shh secreted by mouse pancreatic tumor cells was quantified by using the Mouse Shh-N ELISA Kit (#RAB0431, Millipore), according to the manufacturer’s instructions. KPC and KPCN cells were plated in DMEM supplemented with 10% v/v FBS and 2mM L-glutamine with 100µg/ml Penicillin/Streptomycin. The next day, when cell confluence achieved ∼90%, cells were washed twice with Hank’s Balanced Salt Solution (HBSS) and starved in HBSS overnight. Then fresh HBSS with/without Cholesterol Lipid Concentrate (#12531018, Gibco) was added and collected in 8 hours for analysis of the amount of secreted Shh. Protein concentration of lysates, prepared from remaining cell pellets, was used for data normalization.
mNSDHL expression in KPCN cells
In order to create KPCN cells expressing mouse NSDHL protein we used lentiviral construct pLEX-HA-MYC (#OHS4492, Thermo Scientific Open Biosystems). Lentivirus production was performed by co-transfecting HEK293T packaging cells with mNSDHL coding transfer vector and packaging plasmid mix included in Trans-Lentiviral ORF packaging kit with Calcium Phosphate (#TLP5916, Thermo Scientific Open Biosystems) according to the manufacturer’s protocol. KPCN cell lines were further infected with obtained lentiviruses and desired cells were selected by Puromycin (#P8833, Sigma) treatment (20µg/ml).
Cholesterol measurement
Cholesterol level in KPC and KPCN cells as well as in FBS and LDS containing media was measured by Amplex Red Kit (#A12216, Life Technologies), according to the manufacturer’s instructions.
siRNA transfection
siRNAs targeting Tgfbr1, Acvr1b, Acvr1c genes and control (Gl2) were obtained from Qiagen (#SI02735194, #SI01447040, #SI01447033, #SI01447019, #SI00888916, #SI00888909, #SI00888902, #SI00888895, #SI00888944, #SI00888937, #SI00888930, #SI00888923, #SI03650353, Qiagen). For control (Gl2) one siRNA was used and for targets of our interest (Tgfbr1, Acvr1b, Acvr1c) mix of four siRNAs per one gene was used. Cells were transfected with siRNA at 30 nM (total for four siRNAs) concentrations mixed with HiPerfect Transfection Reagent (#301704, Qiagen) according to the manufacturer’s reverse transfection protocol. In 24 hours after plating, cells were treated with media containing LDS or LDS with 1uM Compactin (#sc-200853, Santa Cruz). RNA and protein lysates were collected in 48 hours after treatment.
Phalloidin staining of stress fibers
To detect activation of stress fibers we performed phalloidin staining. KPC3 cells were plated on coverslips in DMEM supplemented with 10% v/v FBS and 2mM L-glutamine with 100µg/ml Penicillin/Streptomycin. The next day cells were washed with serum free medium and placed in DMEM supplemented with 2mM L-glutamine with 100µg/ml Penicillin/Streptomycin and 5% v/v FBS or 5% v/v LDS and 1uM compactin (#sc-200853, Santa Cruz). After 48 hours of incubation cells were starved for 4 hours in serum free medium. Then coverslips were fixed in 4%PFA and cells were permeabilized with 0.25% Triton X-100 (#BP151-100, Fisher Scientific), blocked in 3%BSA with 0.01% Triton X-100 and stained with Alexa Fluor 488 Phalloidin (#A12379, Invitrogen).
STATISTICAL ANALYSIS
For analysis of continuous data we used Wilcoxon tests, and binary outcomes were compared using Fisher’s exact test. Repeated measures (i.e. multiple measures within mouse) were analyzed using generalized linear regression models with Generalized Estimating Equations (Liang and Zeger, 1986). Growth curves were modeled using linear regression with interactions between treatment and time, again using GEE to account for within-sample correlation. Survival time outcomes were assessed using Kaplan-Meier curves with log-rank tests.
DATA AND SOFTWARE AVAILABILITY
RNAseq data reported in Figure 3 has been deposited into the Sequence Read Archive (SRA) database, deposition PRJNA530747 (https://www.ncbi.nlm.nih.gov/sra).
Acknowledgments
We are grateful to Catherine Reiner and the FCCC Immune Monitoring Facility for technical assistance with histological analyses and immunohistochemistry experiments. Grant Support: This work was supported by NIH core grant CA-06927, by the Pew Charitable Fund, and by a generous gift from Mrs. Concetta Greenberg to the M&C Greenberg Pancreatic Cancer Institute at Fox Chase Cancer Center. Some of the authors were supported by NIH R01 CA188430, K22 CA160725, R21 CA164205, R21 CA231252 (I.A., E.C.), a career development award from Genentech; by Tobacco Settlement funding from the State of Pennsylvania (I.A., E.C.), and by a grant from the Bucks County Board of Associates (L.G.C., I.A.) as well as by NIH R01 CA63366 (E.A.G.); R01CA113451 (E.C.); WX81XWH-15-1-0170 (E.C., R.F., J.F.-B.), R01 CA232256 (E.C., J.F.-B.), T32CA009035 (R.F., L.G.C.), R01 HD065800 (A.M.O.) and by the Program of Competitive Growth of Kazan Federal University (L.G.C.); cloning experiments were supported by the grant from Russian Scientific Foundation (project 15-15-20032) to I.A.
References
SUPPLEMENTARY REFERENCES




