

Abstract
Early surrogates for long-lived immunity after inactivated influenza vaccination (IIV) are lacking. Antigen-specific memory B cells (Bmem) after IIV have been recently identified. We show that the antigen-specific Bmem compartment after IIV is heterogenous and comprises a clonotypically and transcriptionally distinct T-bethi subset that persists in circulation over time after vaccination and exclusively correlates with the long-lived antibody response. We demonstrate that this subset has an effector memory transcriptome and is epigenetically remodeled to facilitate intracellular immunoglobulin production. Finally, via clonal sharing, we show an enriched in vivo ontologic relationship between the secondary plasmablast response that develops after vaccine boost and the T-bethi fraction of the flu-specific Bmem response that forms after initial prime. Collectively, our data identify a novel biomarker of durable humoral immunity after influenza vaccination.
Introduction
After inactivated influenza vaccination (IIV), durable humoral immunity is mediated by long-lived antibody (Ab) secreting cells (ASCs) and memory B cells (Bmem).1 In the influenza experienced adult, inactivated influenza vaccination (IIV) mobilizes pre-existing hemagglutinin (HA) antigen-specific memory B cell (Bmem) subsets.2 These responding Bmem may assume different fates. They may proliferate and affinity mature in the germinal center (GC) response to produce daughter cohorts of HA-specific Bmem; they may die immediately upon activation; they may directly differentiate into ASCs; or they may persist as long-term memory. The early cues that direct vaccine-elicited antigen-specific Bmem toward any of these various fates and the relationship of these Bmem fate decisions to the development of durable humoral immunity are not completely clear.
Traditionally defined according to expression of the canonical memory marker, CD27,3–6 human Bmem are now appreciated to be a heterogeneous subset by transcriptional programming, differentiation capacity and responsiveness to antigen. For instance, a subset of atypical Bmem that express the lineage defining transcription factor (TF), T-bet, have been identified in the context of infection, vaccination, aging and autoimmunity.7 We recently reported in a murine model of influenza infection that T-bet expression in flu-specific B cells as necessary for the development of long-lived ASCs to primary influenza infection as well as for the development of the secondary ASC (plasmablast or PB) in response to challenge infection.8 This finding led us to query the relationship between vaccine-elicited durable immunity and the early vaccine-specific Bmem response after IIV of healthy human subjects.
Here we study the clonotype, transcriptome and epigenome of circulating human Bmem after IIV strictly classified according to antigen reactivity and T-bet expression. We find that the BCR inhibitory signaling molecule, FcRL5,9 accurately identifies HA-specific Bmem by T-bet expression. We also find HApos T-bethi Bmem are transcriptionally remodeled as effector memory with increased accessibility at an Xbp1 enhancer locus that aligns with an established T-bet binding site.10 We show that the early HApos T-bethi subset predicts the development of long-lived Ab response to the vaccine. Finally, we demonstrate that HApos T-bethi Bmem clones are preferentially recalled into the early antigen-specific PB repertoire after re-vaccination.
Results
I. T-bet parses phenotypically distinct HA-specific humoral immune readouts after IIV
After inactivated influenza vaccination (IIV), there is an early expansion of the circulating follicular helper T cell (cTfh, CD4+CXCR5+PD1+ICOS+ T cell)11, 12 and plasmablast fractions (CD27posCD38hi, PB) within 5-10 days of vaccine receipt. 13, 14 The plasmablast fraction has been extensively studied as a measure of vaccine induced antigen-specific humoral immunity.15–18 However, plasmablasts are rapidly cycling, short-lived cells and therefore do not reflect longitudinal immune protection after vaccination. Using fluorochrome labeled tetramer or monomer reagents, hemaggulutinin (HA)-specific Bmems have been identified within one month after IIV. 19–21 This antigen-specific Bmem compartment after IIV has been described as heterogeneous, comprising typical and atypical memory B cells.19–21 We wanted to assess the phenotype and kinetics of the HA-specific Bmem compartment after IIV and the relationship of this compartment to the plasmablast and long-lived antibody (Ab) response. We hypothesized that the HA-specific Bmem and plasmablast humoral immune responses are distinct vaccine outputs. In order to test this hypothesis, we administered the 2015 IIV to 19 healthy subjects and drew blood at sequential proximal time points (day 0, 7, 14, 21, 28) as well as distant time points from vaccine receipt (day 120). Detailed vaccination and infectious histories from subjects were not recorded. Gating strategies are shown in Supplemental Fig 1 A-C. Specificity of our HA tetramer has been described22 and, unless specifically indicated, HA tetramer matches relevant vaccine antigen strain (H1 or H3). Our 2015 cohort responded to the vaccine as evidenced by a significant increase in the circulating cTfh and PB fraction from day 0 to day 7 of IIV (Supplemental Figure 1 B,1C). Also within 7 days of IIV, two phenotypically unique IgDnegCD38med/lo HA-specific (H1) Bmem populations were identified in circulation. These two populations are shown in representative FACS plots (Figure 1A) and are phenotypically distinct from CD38hi B cells and from each other according to HA (H1)-tetramer, Ki-67, T-bet expression and SSC-A parameters. These HA-specific Bmem subsets were further phenotyped as shown in Supplemental Figure 1D. Both subsets were enriched in the expression of the canonical memory B cell marker, CD27 (Supplemental Figure 1D). Consistent with prior description of atypical T-bethi B cells,8, 19, 20, 23–29 we found day 7 HApos T-bet hi Bmem to have increased expression of the integrin CD11c and the inhibitory B cell receptor (BCR) signaling molecule FcRL5 as well as decreased expression of the chemokine receptor CXCR5 and the complement receptor CD21. Underscoring these phenotypic differences, we interrogated the morphology of H1pos T-bethi Bmem using ImageStream and found them to be larger and more granular than H1pos T-betlo Bmem (Supplemental Fig 1E).

(A-C) Gating strategy that defines the IgDneg, non-plasma cell (NPC) CD38 hi and naïve B cell (A), plasmablast, PB (B), and circulating Tfh (cTfh) (C) populations. Among 19 subjects who received the 2015 IIV, the magnitude of the PB response expressed as percent live CD19+ B cells between day 0 to day 7 after IIV (B) and the cTfh response expressed as percent live CD4 T cells between day 0 and day 7 (C) is also depicted.
(D) FACS plots depict phenotyping of H1pos T-bet hi (red), H1pos T-bet lo (blue), non-H1pos T-bet lo (gray), and non H1pos T-bet hi (pink) populations according to CD27, CD11c, Fcrl5, CXCR5 and CD21 expression.
(E) ImageStream analysis of B cells within 7 days of IIV. Representative Brightfield images of 15 CD38 hi, H1pos T-bet hi Bmem, and H1 pos T-bet lo Bmem are depicted.
(F-S) These data refer to correlations between various subsets among 19 healthy subjects who received the 2015 IIV that included the California H1 (Ca-H1) and Switzerland (Sw-H3) antigens.
(F-I) Correlations between the fold change in cTfh from day 0 to day 7 after IIV with percent day 7 antigen specific (Ca-H1 or Sw-H3) T-bet hi and T-bet lo Bmem out of the total NPC gate.
(J-K) Correlation between fold change in H1 and H3 IgG titer from day 0 to day 120 after IIV and the fold change in the day 7 PB and cTfh response expressed as percent from live CD19+ B and CD4+ T cells respectively.
(L) Correlation between fold change in H3 IgG titer from day 0 to day 120 after IIV and the percent day 7 Ca-H1pos T-bethi Bmem subset from the parent NPC gate.
(M) Correlation between the fold change in H1 IgG titer from day 0 to day 120 after IIV and the percent day 7 Sw-H3 T-bet hi Bmem subset from the parent NPC gate.
(N) Correlation between the fold change in H1 titer from day 0 to day 120 after IIV and the percent day 14 Ca-H1 pos T-bet hi Bmem subset from the parent NPC gate.
(O) Correlation between the fold change in H3 titer from day 0 to day 120 after IIV and the percent day 14 Ca-H1pos T-bethi Bmem subset from the parent NPC gate.
(P) Correlation between the fold change in H1 titer from day 0 to day 120 after IIV and the percent D14 Ca-H1 T-bet lo Bmem subset from the parent NPC gate.
(Q) Correlation between the fold change in H3 IgG from day 0 to day 120 and the percent day 14 Sw-H3 T-bet hi Bmem subset from the parent NPC gate.
(R) Correlation between the fold change in H1 IgG from day 0 to day 120 and the percent day 14 Sw-H3 T-bet hi Bmem subset from the parent NPC gate.
(S) Correlation between the fold change in H3 IgG from day 0 to day 120 and the day 14 Sw-H3 T-bet lo Bmem subset from the parent NPC gate.
Statistical analysis was done with Student’s t-test (B, C) and Spearman correlation (F-S). *p< 0.05, **, p<0.01, *** p<0.001, **** p <0.0001 ns= non-significant

(A) Representative flow panel depicting CD19pos IgDneg B cells from the peripheral mononuclear blood cells (PBMCs) of one subject 7 days after IIV classified into 4 subsets according to expression of H1-tetramer and Ki-67. Accompanying histograms demonstrate differences in Ki-67, CD38, T-bet, H1-tetramer expression as well as side scatter (SSC-A) parameters in these 4 subsets.
(B) Geometric mean fluorescence intensity (gMFI) of Ki-67 stain in HA-specific T-bethi (red) and HA-specific T-betlo Bmem (blue) in 8 subjects at weekly time points after IIV
(C-J) Nineteen healthy subjects received the 2015 IIV that included the California-H1 (Ca-H1) and Switzerland-H3 (Sw-H3) vaccine antigens.
(C-F) Frequencies of circulating T-bethi (red) and T-betlo (blue) Ca-H1 or Sw-H3 Bmem subsets were assessed at weekly time points one month after vaccine. Populations are represented as percent of parent non-plasma cell gate (CD19pos IgDneg CD38med/lo, NPCs). Gating strategy to identify NPCs is depicted in Supplemental Fig 1.
(G-H) Correlation between fold change in plasmablasts (CD27pos CD38hi or PB) between days 0 and 7 and the frequency of circulating T-bethi (red) and T-betlo (blue) California H1 (Ca-H1) or Switzerland H3 (Sw-H3) Bmem expressed as a percent of the parent non-plasma cell (NPC) compartment. Gating strategy to identify PBs is shown in Supplemental Figure 1.
(I-J) Correlation between the circulating day 7 Ca-H1 and Sw-H3-specific T-bethi and T-betlo Bmem response from 19 subjects given the 2015 IIV. HA-specific Bmem response is expressed as a percent of the parent NPC gate. Statistical analyses were performed with one way ANOVA testing (B), Wilcoxon rank sum testing (C-F), and Spearman correlation coefficient (G-J), paired Student’s t-test. *p< 0.05, **, p<0.01, *** p<0.001, **** p <0.0001 ns= non-significant
Ki67 is not a binary marker of proliferation and its expression at any individual timepoint does not necessarily reflect ongoing or future proliferative potential but may rather reflective prior replicative history.30, 31 Thus we evaluated for phenotypic changes in Ki-67 expression in the HA-specific Bmem compartment over time after IIV. We found that Ki-67 expression was not sustained over time in the circulating HApos T-bethi Bmem compartment nor did the circulating HApos T-betlo compartment develop significant Ki-67 expression over time after vaccination (Figure 1B). In our 2015 IIV cohort, the HApos (H1 and H3) T-bethi Bmem expanded in the circulation within one month of IIV (Figure 1C, 1E). However, we did not consistently observe a parallel expansion in the HApos T-betlo Bmem fraction in this cohort (Figure 1D, 1F). We found an exclusive correlation between the day 7 cTfh and the day 7 HApos T-bethi Bmem responses to the 2015 IIV (Supplemental Figure 1 F-I). Finally in our 2015 IIV cohort, we found no correlations between the magnitudes of the day 7 HApos T-bet hi and HApos T-bet lo Bmem to either the PB response (Figure 1 G-H) or to one another (Figure 1 I-J). Collectively, the data suggest that the day 7 HApos T-bethi and T-betlo Bmem are unique humoral immune outputs to the IIV.
II. HApos T-bethi Bmem transcriptionally and epigenetically resemble effector memory
T-bet is a lineage defining master transcriptional regulator of terminally differentiated effector cell subsets32, 33 and its role in B cell differentiation has been studied in various pathologic in vivo and in vitro contexts.8, 19, 20, 23–29 Murine studies of influenza infection in wild-type versus B-Tbx21-/- chimeric mice demonstrate that T-bet is necessary for long-lived IgG2c antibody production after primary infection and dispensable for the development and maintenance of the flu-specific memory pool after primary infection.8 However, flu-specific memory B-TBX21-/- cells cannot mount a recall antibody-secreting cell (ASC) response upon antigen re-challenge. These data suggest that T-bet expression in antigen-specific B cells may either directly mediate the initial transcriptional commitment toward terminal ASC differentiation as “pre-ASCs,” or may mediate transcriptional commitment toward effector memory.34 Existing transcriptional data sets of circulating human Bmem after IIV have compared the aggregate Bmem compartment according to expression of atypical cell surface markers, CD7119 and CD21.20 The latter study concluded that CD21lo Bmem after IIV had enhanced T-bet expression and had coincident up-regulation of key ASC transcription factors (TFs), like PRDM1.20, 35–38 We hypothesized that the transcriptional evaluation of antigen-specific Bmem after IIV strictly defined by T-bet expression would similarly show T-bet mediated transcriptional commitment toward terminal ASC differentiation.
To test our hypothesis and characterize the transcriptome of HA-specific Bmem by T-bet expression we used the cell surface marker FcRL5 to sort-purify Ca-H1pos T-bethi Bmem, Ca-H1pos T-bet lo Bmem, naïve B cells and ASCs for RNA-seq from 6 donors at day 7 after 2017 IIV The 2017 IIV included the Michigan 2015 (Mi) H1 antigen which only differs from the sort Ca-H1 tetramer by a single mutation and glycosylation residue.39, 40 Figure 2A shows the principal component analysis (PCA) of this transcriptional data. Utilizing FcRL5 as a cell surface surrogate for T-bet expression accurately classified HApos Bmem according to T-bet and FcRL5 expression status (Figure 2B, Supplemental Fig 2A). For purposes of brevity, hereafter we refer to the HA-Tetint Ki-67int T-bethi SSC-Ahi FcrRL5hi Bmem response to IIV as “HApos T-bet hi“ and the HA-Tethi Ki-67loT-betlo SSC-Alo FcRL5lo response as “HApos T-bet lo.”

(A) Comparison of DEGs (defined as log 2FC, q <0.05) between published CD21lo over CD21hi Bmem after IIV20 and day HA pos T-bet hi (Fcrl5hi) over day 7 HA pos T-bet lo Fcrl5lo Bmem studied here. Significant DEGs that are shared between gene lists and different between gene lists are distingusihed by color. Target genes of interest are annotated.
(B) Gene set enrichment analysis (GSEA) comparing the transcriptome profile of day 7 H1pos T-bethi over day 7 H1pos T-betlo Bmem to additional published gene sets of effector memory T cells over central memory T cells.48–50 Data is reported as enrichment score (ES) plotted against ranked gene list (n = 10992 genes) of day 7 H1pos T-bethi over day 7 H1pos T-betlo Bmem with the dotted line indicating the leading edge of genes and the purple triangle demarcating the change in gene expression polarity. Normalized enrichment score (NES) and p value are also reported.
(C) Circulating plasmablasts (PB, green), naïve B cells (brown, CD19pos CD27neg IgDpos, gating strategy in Supplemental Figure 1), and H1pos Bmem classified by FcrL5 expression (T-bet/Fcrl5hi, red; T-bet/Fcrl5lo, blue) were sort-purified from (4) subjects at day 14 after 2017 IIV for comparative transcriptional analysis via RNA-seq. Principal component analysis of these data is shown.
(D) Circulating H1 pos T-bet hi (N=4) Bmem, H1 pos T-bet lo Bmem (N=4), and day 7 PB (N=5) were sort-purified for ATAC-seq using Fcrl5 as a cell surface surrogate for T-bet expression. Principal component analysis of these data is show here.
(E) Defined as Reads per Peak per Million (RPPM), accessibility at genes known to function as target genes of IRF4 in human plasma cells51 was assessed in each population sorted for ATAC-seq as shown in Supplemental Figure 2D.
(F-H) Live CD19+ B cells were purified in one subject within 7 days of IIV for ImageStream resolution of the H1pos Bmem compartment by T-bet expression. FLOCK analysis of Imagestream data is shown as heat map with cluster size indicated by an accompanying bar plot. Five target clusters of interest were identified and colorized (green, pink, magenta, red, blue). Expression of intracellular and extracellular tetramer stain of these 5 target clusters shown by dot plot (G-H).
(I) Genome plot of chromatin accessibility for the XBP1 locus is shown in D7 PB, D14 H1pos T-bet hi Bmem, D14 H1pos T-bet lo Bmem aligned with previously published T-bet binding sites as assessed by ChiP-seq.10 Data is reported as RPPM.

(A-B) Circulating plasmablasts (PB, green), naïve B cells (brown, CD19pos CD27neg IgDpos, gating strategy in Supplemental Figure 1), and Ca-H1pos Bmem classified by FcrL5 expression (T-bet/Fcrl5hi, red; T-bet/Fcrl5lo, blue) were sort-purified from (6) subjects at day 7 after 2017 IIV for comparative transcriptional analysis via RNA-seq. Principal component analysis of these populations is shown in (A).
Reads per Kilobase per Million (RPKM) for target genes of interest by sorted population is shown in (B) with significance of gene expression differences between day 7 Ca-H1pos T-bethi over day 7 Ca-H1pos T-betlo Bmem indicated. Significance of target gene expression differences between other depicted populations is provided in the Supplemental File 2.
(C) Heat maps of gene expression in various subsets (I: naïve, II: PB, III: D7 Ca-H1pos T-bethi Bmem, IV: D7 Ca-H1pos T-betlo Bmem) curated by function as related to cell cycle and apoptosis (Qiagen RT2 profiler). Differentially expressed genes (DEGs) between populations are represented as dots with colors of dots indicating the identity of comparator subsets.
(D) Gene set enrichment analysis (GSEA) comparing the transcriptome profile of day 7 Ca-H1pos T-bethi over day 7 Ca-H1pos T-betlo Bmem to published gene sets of effector memory T cells over central memory T cells.48 Data is reported as enrichment score (ES) plotted against ranked gene list (n = 10992 genes) of day 7 Ca-H1pos T-bethi over day 7 Ca-H1pos T-betlo Bmem with the dotted line indicating the leading edge of genes and the purple triangle demarcating the change in gene expression polarity. Normalized enrichment score (NES) and p value are also reported. (E-F) Circulating PB (green, n=5), D7 H1pos T-bethi Bmem (red, n=2), D7 H1pos T-betlo Bmem (blue, n=5), and D7 H1neg T-bethi Bmem (pink, n=3) were sort-purified from healthy subjects after 2018 IIV for analysis of chromatin accessibility by ATAC-seq. Fcrl5 was used as a cell surface surrogate for T-bet expression (see Supplemental Fig 2). Principal component analysis of ATAC-seq data with relevant populations indicated by color and label is shown in (E). (F) Defined as Reads per Peak per Million (RPPM), accessibility at genes known to function as target genes of IRF4 in human plasma cells51 was assessed in each population sorted for ATAC-seq as shown in Figure 2E.
(G) Transcription factors (TFs) that regulate the D7 H1pos T-bethi Bmem network over D7 H1pos T-betlo Bmem network were identified using PageRank (PR) analysis. PR log fold change (FC) versus differential gene expression (DEG) by RNA-seq analysis of these TFs is depicted. Statistical analysis was performed using two-way ANOVA (B) and multiple t-test testing (F). *p< 0.05, **, p<0.01, *** p<0.001, **** p <0.0001 ns= non-significant
Expectedly, naïve, Bmem and PB subsets group separately on the PC1 axis. There were 762 differentially expressed genes (DEGs) identified between H1pos T-bethi Bmem and H1pos T-betlo Bmem. There were near equivalent DEGs identified between PBs and H1pos T-bethi (3154) versus PBs and H1pos T-betlo Bmem (3571) and H1pos T-bethi Bmem did not group more closely to PBs on either the PC1 or PC2 axis relative to H1pos T-betlo Bmem. Consistent with this observation, we saw no differences in the gene expression (reads per kilobase per million, RPKM) between HApos T-bet lo Bmem and HApos T-bet hi Bmem at key ASC defining TFs like PRDM1,36–38 XBP1,41, 42 IRF443, 44 (Figure 2B). Instead we saw differential expression between HApos T-bethi Bmem and HApos T-betlo Bmem at key effector cell TFs, like Zeb2,45, 46 chemokine receptor, CXCR3 that is a direct downstream target of TBX21,33 and a chemokine that distinguishes effector from central memory, CCR7.47 We compared our transcriptional analysis to curated gene lists of cell cycle genes and apoptosis genes (Qiagen RT2 profiler) (Figure 2C). Expectedly we found that the vaccine elicited PBs, which are known to proliferate and are short-lived, had differential expression at a number of cell cycle and apoptosis genes. Consistent with transient expression of Ki-67 over time in the HApos T-bethi Bmem, we did not find significant differences in cell cycle genes between day 7 H1pos T-bethi and day 7 H1pos T-betlo Bmem. Consistent with terminal effector cell remodeling, we found that day 7 H1pos T-bethi Bmem upregulated pro-apoptotic genes, like Fas, and down-regulated anti-apoptotic genes, like Bcl2, in comparison to day 7 H1pos T-bet lo Bmem. Finally we performed gene set enrichment analysis (GSEA) against curated gene sets with effector T cell genes (Figure 2D, Supplemental 2B)48–50 and found significant enrichment between the DEGs of day 7 HApos T-bethi Bmem over day 7 HApos T-bet lo Bmem and these gene lists.
Next we asked whether HApos T-bethi Bmem were epigenetically more similar to PBs than HApos T-bet lo Bmem. To test this we used the cell surface marker FcRL5 to sort purify day 7 H1pos T-bethi, H1 pos T-bet lo Bmem, PBs and H1neg T-bet hi Bmem to assess chromatin accessibility in these subsets using ATAC-seq. We found 1923 significant differentially accessible regions (DARs) between HApos T-bet hi and HApos T-bet lo populations. We also identified 10,464 and 11,362 DARs between ASCs and HApos T-betlo and T-bethi Bmem respectively. PCA representation of this ATAC-seq data is shown Figure 2E with PBs grouping distinctly from both HApos Bmem subsets. We assessed the accessibility of genes known to be targets of the ASC TF IRF4 in plasma cells51 across these various populations and we found no difference in accessibility at these genes among HApos T-bethi and HApos T-betlo Bmem (Figure 2F). Finally, we analyzed our accessibility and transcriptional data concordantly using PAGERANK (PR) analysis52 to discern TF motifs that result in changes in gene expression of target genes. We identified 706 TFs that were predicted by PR to contribute to the transcription gene networks of D7 HApos T-bethi over D7 HApos T-betlo Bmem (Figure 2G). Notably no ASC defining TF network was identified by the PR algorithm with a log2 fold change of >1. However, PR predicted the significant up-regulation of TF networks TBX21, BATF,53 BHLHE40,54 and the downregulation of TF network TCF7,55 consistent with effector immune cell programming. Collectively, these data do not support that HApos T-bethi Bmem at day 7 after vaccination are transcriptionally or epigenetically committed to terminal differentiation as ASCs. Rather these data support that day 7 HApos T-bethi Bmem are distinguished by an effector memory cell program. Further analysis comparing the transcriptome and chromatin accessibility of day 14 HAposT-bethi over day 14 HApos T-betlo Bmem replicated the transcriptional and epigenetic findings described above at the day 7 time point. (Supplemental Fig 2C, 2D, 2E)
III. HApos T-bethi Bmem are transcriptionally remodeled to facilitate intracellular HA-specific immunoglobulin production
B cell differentiation into ASCs is known to be epigenetically regulated in a division dependent manner56, 57 and cell cycle arrest facilitates terminal immune cell differentiation.58 Top up-regulated TFs in the PR analysis of the day 7 antigen-specific T-bet hi over T-bet lo Bmem network, include SOX5 and E2F7 (Figure 2L), and these TFs inhibit cell proliferation and/or effect cell cycle arrest.59–61 Concordantly, the PAX5 target molecule, Bach2,62, 63 is downregulated in the PR analysis of the day 7 HApos T-bet hi over T-bet lo network, and this downregulation facilitates ASC differentiation in a division dependent manner.64, 65 PR identified CDKN2C or p18INK4 as a significant DEG target in the predicted E2F7 regulatory network of day 7 HApos T-bethi over HApos T-betlo Bmem (Figure 2E). Interestingly, CDKN2C has previously been demonstrated as critical to the development of the immunoglobulin production and secretory function of ASCs by inhibiting G1/S cell cycle progression.66–68 From these transcriptional and epigenetic network analyses we hypothesized that day 7 HApos T-bethi Bmem may be distinguished by intracellular HA-specific immunoglobulin (Ig) production. To test this hypothesis, we used ImageStream to visualize, enumerate and compare the external (extracellular, EC) and internalized (internalized, intracellular IC) H1-tetramer intensity score (IDEAS software) among circulating PBs, T-bet hi and T-bet lo Bmem within 7 days of IIV. Representative images of day 7 T-bet lo, T-bet hi and CD38 hi B cells are shown in Figure 3A. HA intensity score (Figures 3B, 3C) among aggregate cells demonstrate, expectedly, that CD38hi cells have the highest intensity score for H1-tetramer internal stain among these three subsets. Interestingly, the intracellular intensity score of H1-tetramer stain in the T-bethi Bmem compartment significantly exceeds that of T-betlo Bmem. We also assessed our ImageStream data using an unsupervised analytic platform, FLOCK, to identify clusters of cells with shared expression patterns of fluorochrome labeled targets. FLOCK independently identified a cluster of cells with high CD38 and intracellular H1-tetramer expression (green) as well as a cluster of cells with intermediate expression of intracellular H1-tetramer, low CD38 expression and high expression of T-bet (red) (Supplemental 2F-H). These data provide functional evidence that day 7 HApos T-bethi Bmem produce more intracellular flu-Ig than HApos T-bet lo Bmem.
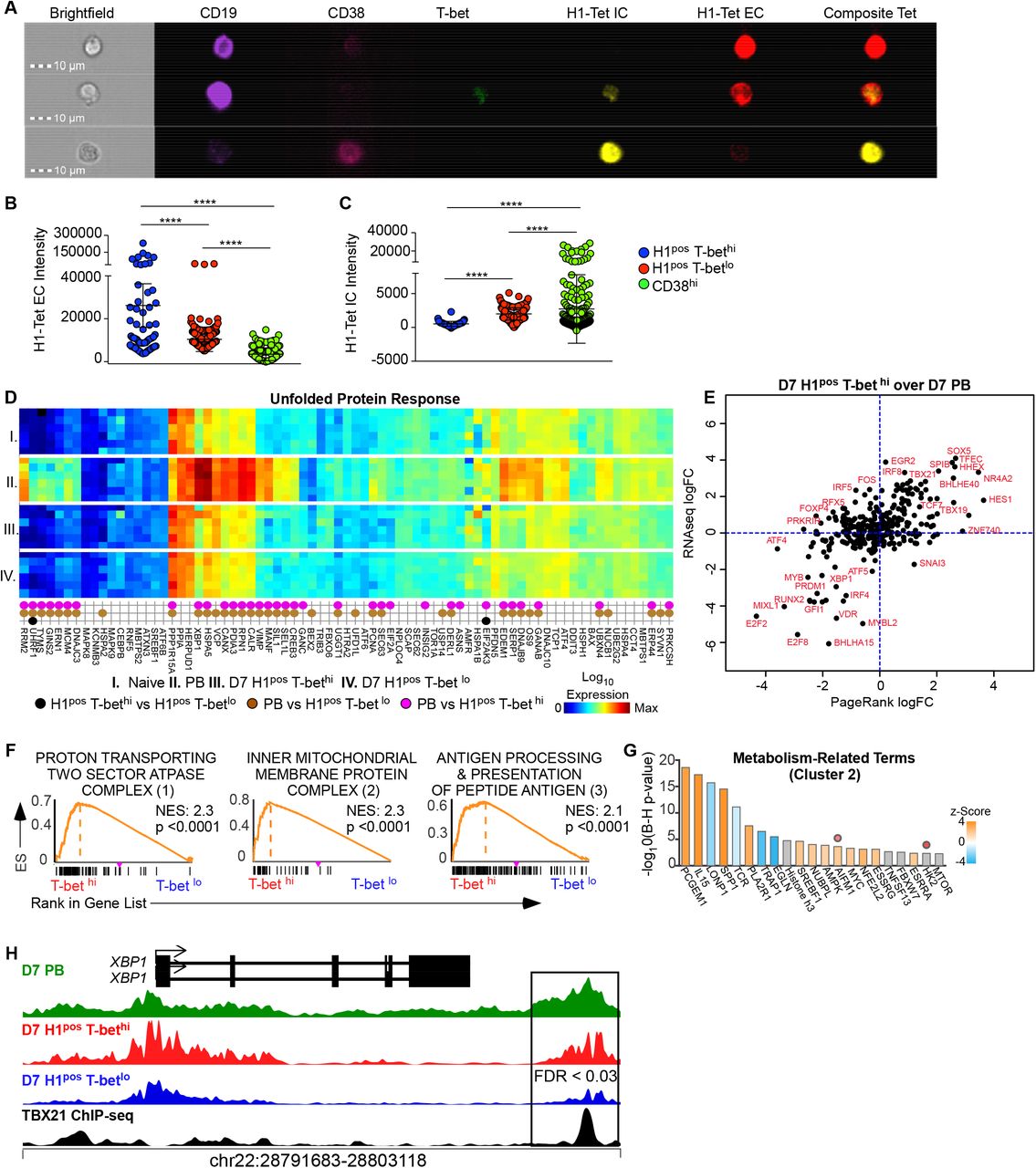
(A-C) A healthy individual received the 2018 IIV and had PBMCs harvested at day 7 after vaccination for staining of B cells with H1 tetramer extracellularly and intracellularly. B cells were then visualized using ImageStream. Images of three candidate cells are shown with relevant fluorochrome labeled targets depicted (A). Intensity of H1 tetramer extracellular (B) and intracellular (C) stain as calculated by IDEAS software for individual T-bet hi, T-bet lo or CD38 hi B cells.
(D) Heat map of gene expression in various subsets (I: naïve, II: PB, III: D7 H1pos T-bet hi Bmem, IV: D7 H1pos T-bet lo Bmem) curated by function as related to the unfold protein response (Qiagen RT2 profiler). Differentially expressed genes (DEGs) between populations are represented as dots with colors of dots indicating the identity of comparator subsets.
(E) Using PageRank (PR) analysis, transcription factors (TFs) that regulate the D7 H1pos T-bet hi Bmem network over D7 PB network were identified and shown as PR log fold change plotted against DEGs from RNA-seq data set (see Figure 2).
(F) GSEA comparing the transcriptome profile of day 7 Ca-H1pos T-bet hi over day 7 Ca-H1pos T-bet lo Bmem against Gene Ontology terms was performed. Network analysis on the output identified 3 clusters of gene ontology terms grouped by shared leading edge genes. Representative GSEA plots from each of these clusters, labeled 1-3, is shown.
(G) Ingenuity pathway analysis (IPA) was performed on the genes that comprised clusters 2. Predicted upstream regulators of cluster 2 are shown in a bar plot with bar color indicating z-score. Regulators that are predicted with bias are demarcated with a dot above that regulator.
(H) Genome plot of chromatin accessibility for the XBP1 locus is shown in D7 PB, D7 H1pos T-bet hi Bmem, D7 H1pos T-bet lo Bmem aligned with previously published T-bet binding sites as assessed by CHiP-seq.10 Data is reported as RPPM.
Statistical analysis was performed using one-way ANOVA testing (B, C). *p< 0.05, **, p<0.01, *** p<0.001, **** p <0.0001 ns= non-significant
T-bet expression has been associated with altered metabolic programming of effector T cells.69, 70 Our finding that HApos T-bethi Bmem have intermediate production of intracellular immunoglobulin prompted us to query the relationship between T-bet expression in HApos Bmem and the unfolded protein response (UPR). First we compared the DEGs between day 7 naïve B cells, PBs, HApos T-bethi Bmem and HApos T-betlo Bmem to a curated list of genes associated with UPR (Qiagen RT2 Profiler) (Figure 3D). We found that PBs almost exclusively upregulated the genes known to be associated with UPR. Next we used PR analysis to identify TFs that regulate the gene network of day 7 HApos T-bethi Bmem over day 7 PBs. Not surprisingly, we found that PR predicted down-regulation of master ASC TFs, Xbp1, PRDM1 and IRF4, as well as master regulators of UPR, ATF4, in day 7 HApos T-bethi Bmem (Figure 3E).71 Thus, our transcriptional and epigenetic data do not demonstrate that T-bet expression in HA-specific Bmem is associated with the development of UPR.
We interrogated our transcriptional gene set further to understand the role of T-bet expression in B cell metabolism by using GSEA to compare the DEGs between day 7 H1posT-bethi Bmem over H1pos T-betlo Bmem to Gene Ontology (GO) gene sets. We found enrichment against GO terms that clustered into 3 groups according to shared leading edge gene lists. Representative GSEA plots of these 3 clusters are shown in Figure 3F and clusters 1 and 2 pertain to cellular metabolism and mitochondrial respiration. We used Ingenuity Pathway Analysis IPA) on cluster 2 leading edge genes to understand predicted upstream regulators of that gene set (Figure 3G). This cluster was chosen because there were sufficient leading edge genes to permit this type of analysis. IPA predicted regulators of the metabolism cluster include modulators of metabolic transitions to aerobic glycolysis (PCGEM1,72 LONP173 and TRAP174), as well as established mediators of effector cell sustained respiratory capacity (SRC), Il-15.75 Finally we examined the genome plot of our ATAC-seq data at the Xbp1 locus to assess for DARs at this gene. Of note, Xbp-1 is not a target gene in the Staudt et al Nature 2008 gene list and accessibility at this locus is not shown in Figure 2F. We identified a DAR (FDR = 0.03) at an Xbp-1 enhancer site between day 7 HApos T-bethi over HApos T-betlo Bmem that persists at day 14 after vaccination (FDR <0.0004) (Figure 3H, Supplemental 2I) with predicted RUNX, Ets and PU.IRF binding motifs. Although there are no predicted TBX21 binding motifs at this site, this DAR directly aligns to peaks identified by published Tbx21 ChIP-seq data of GM12878 cells.10 Collectively, these data suggest that T-bet expression in HApos Bmem is associated with altered mitochondrial respiration and cellular metabolism parameters and may facilitate intracellular immunoglobulin production/UPR by associating with TF complexes at an Xbp1 enhancer locus to effect increased Xbp1 chromatin accessibility.
IV. Clonotypes from HApos T-bet hi Bmem Exclusively Persist in the Circulation
Our data show that HApos T-bethi Bmem have distinct kinetics from HApos T-betlo Bmem after vaccination suggesting that this subset may also be clonotypically distinct from HApos T-betlo Bmem. To test this possibility we used FcRL5 as a cell surface marker of T-bet expression [Supplmental Fig 1D] to sort purify HApos Bmem by T-bet expression from the peripheral blood of three subjects (donor 1, 2, 3) at various timepoints after 2016-7 IIV for BCR sequencing. Unique clones across subsets were identified as having the same VH- and JH-gene annotations, identical length at the CDR3-H and at least 85% sequence similarity between the CDR3.
Figure 4A shows representative clonality plots of H3pos T-bethi and H3pos T-betlo Bmem with the number of sequences retrieved from each subset and the number of lineages resolved from these sequences also represented. BCR repertoire diversity within each subset was assessed through a variety of metrics including the number of clonotypes that composed the top 20% or 50% of all sequences (D20, D50). Circulating day 7 H3pos T-betlo Bmem were clonotypically more diverse than circulating day 7 H3pos T-bethi Bmem (Figure 4A) in both donor 1 and 2 (Donor 1: D20, T-bet lo 29 vs. T-bethi 14, D50, T-betlo 151 vs T-bethi 82; Donor 2: D20, T-bet lo 3 vs. T-bet hi 1, D50 T-bet lo 11 vs T-bet hi 3).

Three individuals (Donors 1-3) were given the 2016 IIV and had PBMCs collected at serial time points within one month of vaccination for assessment of the heavy chain repertoire (Vh families 1-7) of flu-specific subsets using next generation sequencing. Data from Donors 1 and 2 are shown here. Data from donor 3 is shown in Supplemental Figure 3. The cumulative percentage of sequences (Y-axis) versus lineage (clonal) size (X-axis) ranked by lineage size is shown in (A).
(B) Relationship between the day 7 H3pos T-bethi Bmem and the day 7 H3pos T-betlo Bmem Vh repertoire is shown as an alluvial plot for donor 1 and 2. Cumulative percentage of sequences are ordered into lineages and ribbons connect lineages that have 85% CDR3 similarity between the two populations.
(C-F) Alluvial plots and accumulation curves depict connectivity of clonotypes in donor 1 over time after IIV. Corresponding summary table depicting percent of shared lineages between Bmem over time is shown in the Supplementary File 2. Connectivity between D7 H3pos T-bet lo Bmem and D7 H3pos T-bet hi clonotypes and D14 H3pos T-bet lo Bmem clonotypes (C) or D14 H3pos T-bet hi Bmem clonotypes (D) is shown. Connectivity is defined according to 85% CDR3 sequence similarity. Red-yellow ribbons correspond to clones shared across 3 populations. Green-blue ribbons correspond to clones shared across 2 populations. Only those lineages that have connectivity with day 14 clones are depicted here. (E-F) Lineages of day 14 H3pos Bmem populations are rank ordered by descending size and the percent of day 7 H3pos Bmem lineages that are shared by rank ordered day 14 clonotype is plotted. Day 7 H3pos Bmem lineages are divided as total T-bet hi clones (solid red line), T-bet hi exclusive clones (dotted red line), total T-bet lo clones (solid blue line), T-bet lo exclusive clones (dotted blue line).
(G) Three donors (donor 1, donor 3, donor 4) received 2017 IIV with single cell sort-purification of Ca-H1pos T-bet hi Bmem and Ca-H1pos T-bet lo Bmem within 7-14 days of vaccine for recombinant monoclonal antibody (rMAb) generation. Luminex diagram shows reactivity of these rMAbs to various H1 antigens, Michigan 15 (Mi15), California07 (Ca7), Puerto Rico/8 (PR8). Clones with high PR8 reactivity (gMFI >4000) are colorized by T-bet expression status and shown on CA7 and MI15 Luminex diagrams.
Statistical analysis was done with Student’s t-test and post-testing for cumulative distribution. (G). *p< 0.05, **, p<0.01, *** p<0.001, **** p <0.0001 ns= non-significant

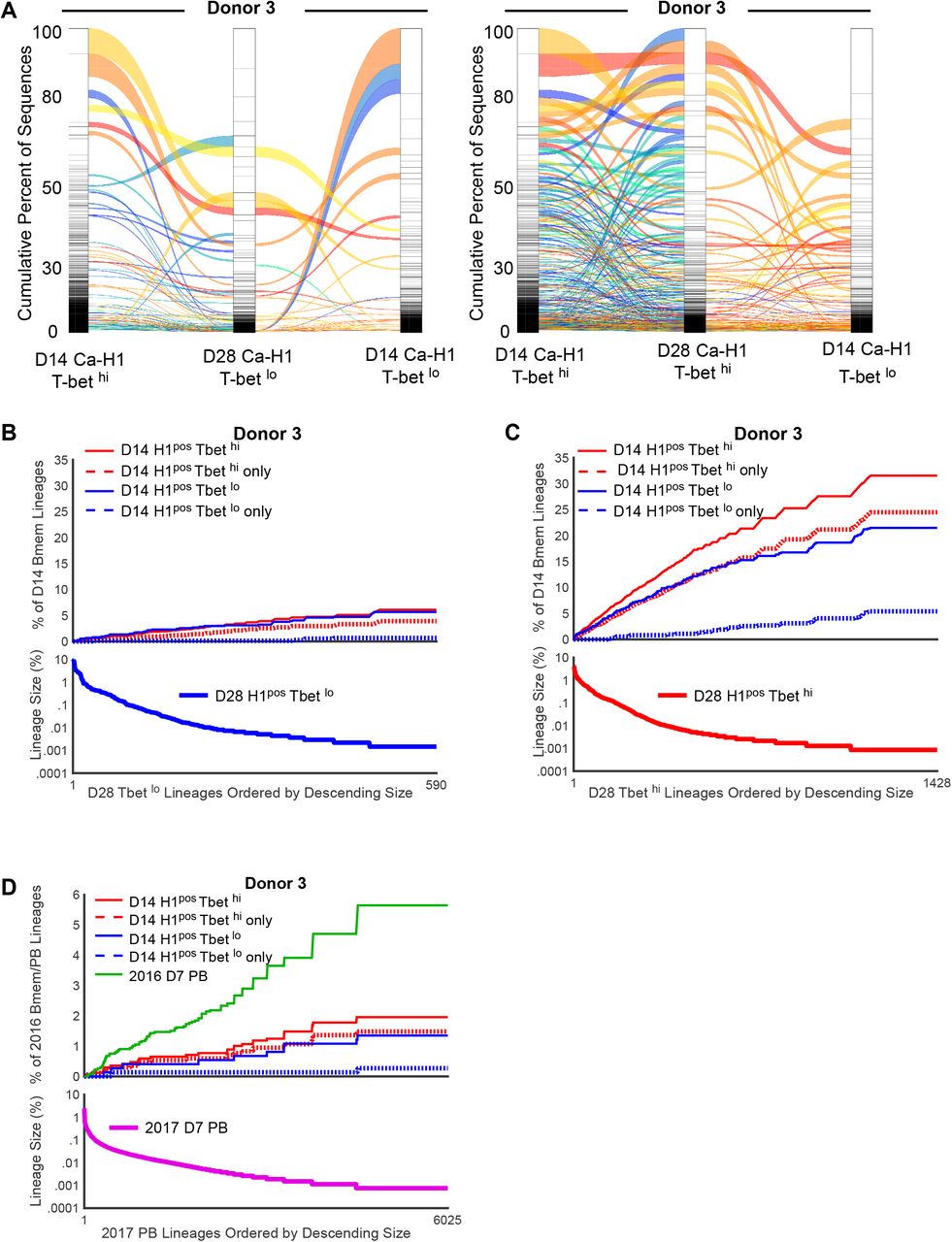
(A) Alluvial plots depict connectivity of clonotypes in donor 3 over time after IIV. Connectivity between D14 Ca-H1 T-betlo Bmem and D7 Ca-H1 Fcrl5hi clonotypes and D14 HK-H3pos T-betlo Bmem clonotypes or D14 HK-H3 Fcrl5hi Bmem clonotypes is shown. Connectivity is defined according to 85% CDR3 sequence similarity. Red-yellow ribbons correspond to clones shared across 3 populations. Green-blue ribbons correspond to clones shared across 2 populations. Only those lineages that have connectivity with day 14 clones are depicted here. (B-C) Lineages of day 28 H1pos Bmem populations were rank ordered by descending size and the percent of day 14 H1pos T-bet hi (B) or day 14 H1 pos T-bet lo (C) Bmem lineages that are shared by rank ordered D28 clonotype is plotted. Day 14 H3pos Bmem lineages are divided as total T-bet hi clones (solid red line), T-bet hi exclusive clones (dotted red line), total T-bet lo clones (solid blue line), T-bet lo exclusive clones (dotted blue line). Corresponding summary table depicting percent of shared lineages between Bmem over time is shown in the Supplementary File 2.
(D) Lineages of D7 2017 PBs were rank ordered by descending size and the percent of D14 2016 Bmem and 2016 PB lineages that are shared with these rank ordered D7 2017 PB clonotypes are plotted. Day 14 H3pos Bmem lineages are divided as total T-bet hi clones (solid red line), T-bet hi exclusive clones (dotted red line), total T-bet lo clones (solid blue line), T-bet lo exclusive clones (dotted blue line). Corresponding summary table depicting percent of shared lineages between Bmem and PB subsets over time is shown in the Supplementary File 2.
Next we examined the frequency of shared clones between the H3pos T-bethi or H3pos T-betlo Bmem populations in donors 1 and 2 (Figure 4B). We found that 6.1% and 5.1% of H3pos T-bethi Bmem clones from donors 1 and 2 respectively were shared with clones from H3pos T-betlo Bmem. Conversely, we found that 4.2% and 1.9% of H3pos T-bet lo Bmem clones from donors 1 and 2 respectively were shared with clones from the corresponding H3pos T-bethi Bmem subset. These data indicate that HApos T-bet hi Bmem are largely clonotypically distinct from HApos T-bet lo Bmem at a single time point. However, we also wanted to understand if the relationships between these populations over time. Therefore we amplified the BCR sequences at day 14 after IIV in donor 1 according to H3 and T-bet expression for comparison to day 7 antigen-specific clones. We found that a greater proportion of large day 14 H3pos T-bet hi Bmem clones were enriched in unique clones from day 7 H3pos T-bethi Bmem population as compared to day 7 H3pos T-betlo Bmem population (Figure 4 D, Figure 4 F). In contrast, we found that clones from day 14 H3pos T-bet lo Bmem were related to very few unique clones from day 7 H3pos T-bet lo Bmem subset (Figure 4 C, Figure 4 E). These observations remained true even at 100% CDR3 similarity (Supplemental File 2). We evaluated clonotypic relationships between HApos T-bet hi and HApos T-bet lo Bmem at additional time points after IIV in a separate donor and again detected that large clones of day 28 H1pos T-bet hi Bmem were enriched for unique clonotypes from day 14 H1pos T-bethi Bmem over day 14 H1pos T-bet lo Bmem (Supplemental 3A-C).
V. HAposT-bethi Bmem Exclusively Correlate with the Long-lived Ab Response after IIV
In order to understand the relationship of circulating HApos T-bet hi and HApos T-bet lo Bmem to each other and to any vaccine elicited germinal center response, we assessed the heavy chain mutation rates between total and related antigen-specific T-bethi and T-bet lo Bmem clones at matched time points (donor 1: day 7, day 14; donor 2: day 7, donor 3: day 14, day 28) We also assessed the heavy chain mutation rates among shared antigen-specific T-bethi clones across time in donor 1 (day 7 to day 14) and donor 3 (day 14 to day 28). We were unable to resolve any consistent directionality (increased or decreased) in total, silent or non-silent mutation rate differences among total or shared clonotypes by T-bet expression status.
The influenza antigen-specific Bmem compartment is significantly mutated as a result of repeated infectious exposure and/or inoculations. Thus, the incremental differences in heavy chain mutation across antigen-specific Bmem subsets after vaccination may be subtle and this may not be the appropriate read-out of the germinal center response to vaccination.20 Therefore we cloned single Ca-H1pos Bmem by T-bet/FcRL5 expression for recombinant monoclonal antibody (rMAb) generation from three donors after 2017 IIV (donor 1, day 7; donor 3, day 14; donor 4 day 14) to assess for binding to a panel of H1 antigens (California-H1, Ca7, Michigan-15, Mi15, and Puerto Rico/8, PR8) in a multiplex bead assay as has been described previously.76, 77 We did not find any difference in relative binding reactivity to either the vaccine antigen, Mi15, or the closely related sort antigen, Ca7 (Figure 4G). However, we observed an enrichment in PR8 reactive clones among rMAbs generated from individual Ca-H1pos T-bet hi Bmem (Figure 4G). Regardless of T-bet expression status, those clones with highest PR8 reactivity had relatively lower relative binding avidity for the vaccine H1 antigen, Mi15 or the closely related Ca7 antigen than non-PR8 reactive clones. PR8 is antigenically more similar to pre-2009 circulating and inoculating H1N1 strains that diverged from swine early in the 20th century78 and is less similar to more recently circulating and inoculating H1N1 strains like Mi15 and Ca7 that are directly adapted from contemporaneous swine H1 strains.79 Thus, enrichment of PR8 reactive clones in the T-bethi repertoire suggests that the repertoire of this fraction may preferentially contain clones from temporally distant exposures to influenza viral variants. Collectively, these BCR repertoire data suggest that HApos T-bethi Bmem are clonally dissimilar from the HApos T-betlo Bmem repertoire.
Although we were unable to resolve differences in the mutation rates or relative binding avidity to vaccine antigen of HApos Bmem by T-bet expression status, the unique persistence of HApos T-bet hi clonotypes in the circulation across one month after IIV suggests that these may mark the presence and or represent direct outputs of an ongoing germinal center response. We further hypothesized that the long-lived antibody response would therefore correlate with the magnitude of the HApos T-bethi Bmem response as the long-lived antibody responses to antigen is impaired in the absence of germinal center responses.80
As a measure of durable humoral immunity induced by vaccination, we calculated the fold change in the HA IgG Ab titer between days 0 and 120 after IIV. Expectedly, we found no correlation between the magnitude of the day 7 PB response and long-lived antibody after 2015 IIV (Supplemental Figure 1J-K). However, the H1 and H3 long-lived Ab response correlated with the peak day 7 H1 and H3 T-bet hi Bmem response (Figure 5 A-D). Notably, this correlation was antigen-specific as the day 7 H1posT-bet hi Bmem did not correlate with the long-lived H3 Ab response and vice versa (Supplemental Fig 1L-M). We found similar antigen-specific correlations exclusively between the D14 HA-specific T-bethi Bmem and the long-lived Ab response (Supplemental Figure 1 N-S). Therefore, these data suggest that HA-specific T-bethi Bmem may serve as a putative early biomarker of the long-lived antibody response after IIV.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
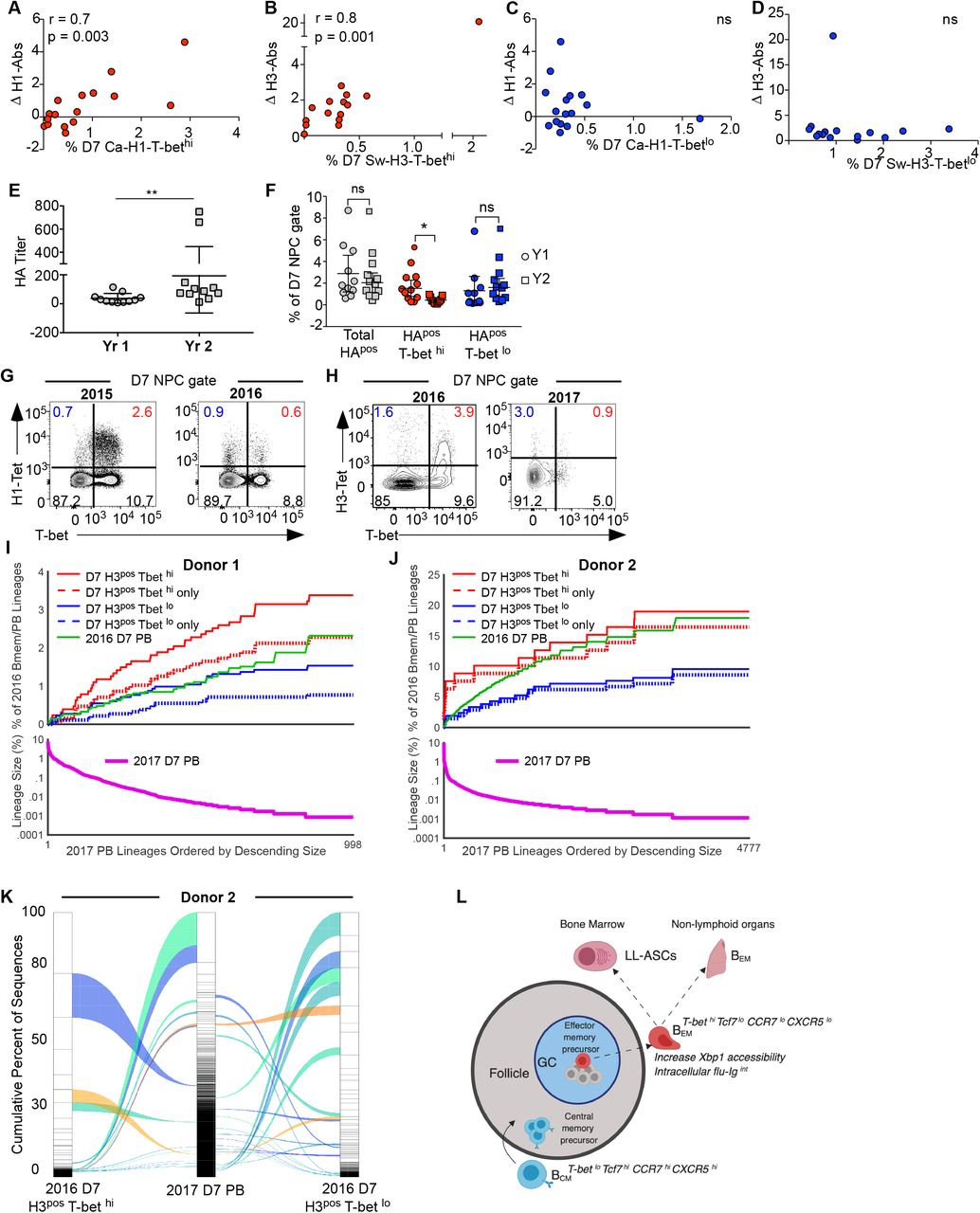
(A-D) Correlation between fold change in HA-IgG (day 0 to day 120) and the magnitude of the day 7 HApos T-bethi and day 7 HApos T-betlo Bmem response (expressed as percent of total NPCs) in 19 healthy individuals who received the 2015 IIV. The 2015 IIV included the California H1 (Ca-H1) and Switzerland H3 (Sw-H3) antigens. Correlations between HA antigen-specific populations and corresponding HA titer is shown. Other relevant correlations are shown in Supplemental Figure 1.
(E-K) Ten individuals under study received serial IIV and had the HA-specific B cell subset assessed by flow cytometry. Three of these individuals (donor 1, 2, and 3) were examined using next-generation sequencing for repertoire analysis in both vaccine years. Data from donor 3 is shown in Supplemental Figure 3.
(E) Pre-vaccine HA-IgG titer in subjects under study who received the IIV in sequential vaccine seasons.
(F) Dot plot shows the magnitude by year of the total, T-bet hi and T-bet lo D7 HA-specific Bmem population expressed as a percent of the NPC gate in 10 individuals given sequential IIV.
(G) Representative FACS plot from a single individual vaccinated in 2015 and 2016 showing the frequency of the H1 population by T-bet expression within 7 days of IIV. The H1 vaccine antigen was conserved between the 2015 and 2016 IIV.
(H) Representative FACS plot from a single individual vaccinated in 2016 and 2017 showing the frequency of the H3 population by T-bet expression within 7 days of IIV. The H3 vaccine antigen was conserved between the 2016 and 2017 IIV.
(I-J) Lineages of D7 2017 PBs (thick green line) were rank ordered by descending size and the percent of day 7 2016 Bmem and 2016 PB lineages (thin green line) that are shared with these rank ordered day 7 2017 PB clonotypes is plotted. 2016 D7 H3pos Bmem lineages are divided as total T-bet hi clones (solid red line), T-bethi exclusive clones (dotted red line), total T-bet lo clones (solid blue line), T-bet lo exclusive clones (dotted blue line). Corresponding summary table depicting percent of shared lineages between Bmem and PB subsets over time is shown in the Supplementary File 2.
(K) Alluvial plot depicts the clonal relatedness of 2017 D7 PB clones to those from the 2016 D7 H3 T-bet hi and H3 T-bet lo Bmem subsets from Donor 2. Brown ribbons correspond to clones shared across 3 populations. Green-blue ribbons correspond to clones shared across 2 populations. Only those lineages that have connectivity with 2017 D7 PB clones are depicted here.
(L) Candidate model. Abbreviations for long-lived antibody-secreting cells (LL-ASCs), effector Bmem (BEM) and central Bmem (BCM) are used.
Statistical analyses were performed with Spearman correlations (A-D), Student’s t-test (E) and one-way ANOVA (Friedman test) (F). *p< 0.05, **, p<0.01, *** p<0.001, **** p <0.0001 ns= non-significant
VI. T-bethi Bmem clonotypes are preferentially recalled into the PB repertoire
Effector memory B cells have been described as resident in the murine lung tissue and directly differentiate into ASCs after antigen re-challenge.22 Our transcriptional data suggests that HApos T-bet hi Bmem are effector memory B cells and therefore we hypothesized that this subset will directly form ASCs after antigen re-challenge. To test this hypothesis we re-vaccinated 2 cohorts of subjects across sequential vaccine years (2015 and 2016; 2016 and 2017) and assessed the magnitude of the HApos T-bethi and HApos T-betlo Bmem response within 7 days of vaccine receipt in each year. We found a significant increase in HA-titer after vaccine (Figure 5E), demonstrating that all 10 subjects responded to the prime and had relatively high titers of HA-Ab prior to vaccine boost. We found that re-vaccination contracted the HApos T-bet hi Bmem fraction but did not significantly alter the magnitude of the total day 7 HApos Bmem or the day 7 HApos T-bet lo Bmem response (Figure 5F-H) across vaccine seasons. Next we compared the 2016 HApos T-bethi and HApos T-bet lo Bmem clonotypes to those of the 2016 and 2017 PB subset in 3 subjects who received IIV in 2016 and 2017. Expanded clones in the 2017 PB repertoire were preferentially related to clones from the 2016 HApos T-bethi Bmem subset under both 85% and 100% CDR3 similarity constraints (Figure 5 I-K, Supplemental Figure 3 D, Supplemental File 2). These data suggest that clonotypes HApos T-bethi Bmem directly seed the secondary ASC compartment to provide immune protection after challenge infection.
Discussion
Here we define two HA-specific Bmem subsets that circulate within seven days of IIV and have differential expression of the lineage defining TF T-bet. We show that T-bet expression divides the HA-specific Bmem compartment into effector memory (TCF7lo, CCR7lo) and central memory compartments (TCF7hi, CCR7hi). Consistent with an effector memory profile, we show that HApos T-bethi Bmem exclusively persist in the blood compartment within one month of vaccination and that clonotypes of this subset are recalled into the circulating expanded PB lineages upon antigen re-challenge.
T-bet expressing B cells have been described in the context of aging, autoimmunity, infection, and vaccination as atypical memory and this subset is now regarded as heterogeneous.7 For instance, CD27neg T-bethi Bmem (double negative 2, DN2) have been shown in lupus subjects as an activated extrafollicular B cell subset that arise directly from naïve B cells and respond to IL-21 and TLR7 signaling alone to differentiate into ASCs.25, 81 However, this same population has been characterized in patients with chronic infection from HIV, HCV and malaria as exhausted and unresponsive to BCR signaling.82–85 In contrast to these studies, our study reports DEGs between antigen-specific Bmem that are enriched in canonical CD27 expression by T-bet expression (Supplemental Figure 1D) While there are multiple shared targets in our gene list and gene lists that compare DN2 to canonical memory (e.g. TBX21, CXCR5, Tcf7, Zeb2, Bach2, ITGAX) there are also important differences. Notably DN2 exclusively upregulate IL21R expression relatively to canonical memory, suggesting the DN2 subset and the HApos T-bethi Bmems described here have different ontogeny, cytokine responsiveness and consequent functions in vivo.
In the context of vaccination against influenza, atypical memory B cells have been identified in the HA-specific Bmem compartment with T-bet expression.19, 20 Our study is distinguished from these previous reports because we did not discern a pre-ASC transcriptional signature in the atypical Bmem compartment after IIV. Rather we found that the antigen-specific T-bethi Bmem subset to retain transcriptional and epigenetic programming as effector memory. This discrepancy in findings may pertain to differences in study design with respect to the inclusion of antigen-specific versus aggregate vaccine-elicited atypical Bmem in transcriptional analysis, the transcriptional timepoint under study after vaccination (day 7 vs day 14), and the use of different cell surface surrogates to parse Bmem into subsets with consequent heterogeneity in the resolution of the T-bet dependent programming signature (Supplemental Fig 1D, Supplemental Figure 2A).
Effector immune cells are distinguished from central immune cells according to cellular metabolism, respiration and mitochondrial dynamics.75, 86, 87 Although it is currently unclear if metabolic re-programming alone directly shapes cellular fate decisions,88 TFs that orchestrate terminal cellular fate commitments concordantly orchestrate cellular metabolic program changes.70 ASCs are a terminally differentiated effector cell of the humoral immune arm. This differentiation is mediated by PRDM1 beyond an early timepoint.36 Production of immunoglobulin by ASCs is a nutrient intensive process that requires cellular adaptation termed the unfolded protein response (UPR). ASCs exhibit a transcriptionally unique UPR from other cell types that is felt to occur early in ASC differentiation and prior to high Ig secretion.89–91 Xbp1/IRE-1 is a key regulator of the ASC UPR41, 42 and its expression in B cells is directly modulated by PRDM1 as part of terminal ASC differentiation.91, 92 Here we show a significant DAR at an Xbp1 enhancer locus between the HApos T-bethi over HApos T-bet lo Bmem subsets that is located at a published TBX21 ChIP-seq binding site.10 Although Xbp1 has been shown to be dispensable for plasma cell terminal differentiation, absence of Xbp1 attenuates Ig secretory function.93 In fact, PRDM1 has been demonstrated as dispensable for the initiation of Ab secretion.36 In keeping with this finding, the change in Xbp1 locus accessibility that we observed in HApos T-bethi Bmem is unlikely to be regulated by PRDM1 as there are no predicted PRDM1 binding motifs at this enhancer site. Thus, while our data do not suggest that HApos T-bethi Bmem have made a transcriptional or epigenetic commitment to PRDM1-mediated terminal ASC differentiation, our data do suggest that T-bet manipulates the metabolic programming of HApos Bmem via DNA binding effects at the Xbp1 locus to facilitate chromatin accessibility at that site with consequent intracellular immunoglobulin production. It is also possible that there are DNA binding independent effects of T-bet on HApos B cell metabolism through functional Bcl-6 antagonism,69 as has been described in the context of CD4 T cell effector function, but these are not clearly elucidated in the transcriptional network analysis presented here.
Bone marrow resident long-lived ASCs mediated durable immunity and are distinguished from other ASC subsets, including PBs, by metabolic programs like autophagy as well as morphologic and phenotypic characteristics like the expression of surface Ig.94, 95 Current early immune readouts after IIV, include early antigen-specific PBs and cTfh and are not known to predict durable immunity after vaccination.11, 95, 96 In fact, in our 2015 IIV cohort, the magnitude of the day 7 plasmablast and cTfh response did not correlate with the fold change in HA-IgG titer at day 120 after vaccination (Supplemental Fig 1J-K) In the absence of germinal centers, the long-lived ASC response is impaired.80 Consistent with this finding, network analysis of PBMC transcriptome datasets after IIV correlate early proliferation gene signatures with the development of an antibody response.97 Our data demonstrate a correlation between the circulating early HApos T-bethi response and long-lived HA IgG titer that is antigen specific. These data suggest that the HApos T-bethi Bmem subset reflects the induction of a new germinal center response by the IIV. Although we were unable to demonstrate, via incremental increases in mutation rates or increase in HA binding affinity, evidence that either HApos T-bethi or T-betlo Bmem are direct outputs of a new GC response to vaccine, we did find exclusive persistence of HApos T-bethi Bmem clonotypes in circulation and an enriched reactivity of these clonotypes to H1 viral variants (Figure 4G). These findings may reflect this subset’s origin from an ongoing GC response (Figure 5L), which is less stringent to the selection of B cells by affinity/epitope reactivity.98–100
It is as yet unclear if circulating HApos T-bethi Bmem simply serve as a biomarker of the long-lived ASC response or actually represent the direct precursors of bone marrow resident long-lived ASCs. In this study we used serial annual influenza vaccination of three subjects with the 2016 and 2017 IIV to follow the fate of circulating HA-specific Bmem clonotypes by T-bet expression after antigen re-challenge. The 2016 and 2017 IIV formulations were highly conserved as they shared the same H3 and influenza B antigens and only differed according to the H1 antigen (Ca-H1 vs Mi-H1). In fact, these two H1 antigens were also highly similar between formulations, distinguished only by a single K166Q mutation and an adjacent N-linked glycosylation site.39 Sequential influenza vaccination with conserved antigens has been shown to attenuate the vaccine-specific plasmablast101 and Bmem response17 possibly because pre-existing Ab mediate epitope masking or epitope clearance.102 We did not observe a reduction in the total or T-betlo HApos Bmem fraction after re-vaccination (Figure 5F). This discrepancy from prior reports may be due to our directly enumerating the antigen-specific Bmem compartment versus assaying the compartment indirectly via ELISPOT as was done previously. However, and in keeping with prior reports,17, 101 we observed relatively fewer accumulations of connections between PB clones across successive vaccine seasons than between T-bethi Bmem and PB clones in two of three donors tested [Figure 5I, J, Supplemental 3D].
It is interesting to note that large lineages from the circulating 2016 D7 HApos T-bet hi subset were not consistently recalled into the 2017 PB compartment after repeat IIV [Figure 5K]. The fate of these large clonotypes is not clear. They may have differentiated into short or long-lived ASCs or may have died in the intercurrent time period between sequential vaccines. Recent data demonstrate HA-specific Bmem in non-lymphoid tissues like the human lung as enriched in atypical memory marker expression.3 In fact, CXCR3+ flu-specific effector Bmem in the murine lung are broadly reactive against HA viral variants103 and directly differentiate into ASCs after antigen challenge for local immune protection.22 These data suggest raise the alternative possibility that circulating expanded HApos T-bethi Bmem clones migrate and are maintained in non-lymphoid tissues as resident effector memory against influenza viral variants [Figure 5L]. Elucidating clonotypic relationships among effector and central HA-specific Bmem at various human tissue sites (blood, lymphoid, mucosal) and with the circulating flu-specific Ig repertoire will be an important goal of future studies.
Author Contributions
F.E.L. conceived the idea for the project and secured the initial funding. F.E.L. and A.N. designed the experiments that were performed by A.N., C.D.S., R.G.K, C.M.T., E.Z., B.M. and K.M. B cell tetramers were developed and produced by J.E.B. Human samples used in this study were obtained via the Alabama Vaccine Research Clinic, directed by P.A.G. Bioinformatic analyses were performed by A.F.R, C.D.S., C.F. and T.M. All other data was analyzed by A.N and F.E.L. A.N., A.F.R. and F.E.L wrote the manuscript and prepared final figures. Critical feedback on the project and manuscript were provided by A.F.R, T.D.R, J.F.K., S., and J.M.B.
Materials and Methods
Human Subjects and Samples
The UAB Institutional Review Board approved all study protocols for influenza vaccinated subjects. All subjects gave written informed consent as part of participation prior to providing peripheral blood for analysis. Influenza vaccinated patients self-identified as healthy and were recruited through the Alabama Vaccine Research Clinic (AVRC). Subjects received either the 2015-2016 Fluzone (Sanofi-Pasteur), 2015-2016 FluMist (AztraZeneca) the 2016-2017 Fluvirin (Sequiris), the 2017-2018 Fluzone (Sanofi-Pasteur) and the 2018-2019 Fluzone (Sanofi-Pasteur). Blood was drawn on days 0, 7, 14, 21, 28 and 120 days +/− 1 week.
Lymphocyte and plasma isolation
Peripheral blood from human subjects was drawn into K2-EDTA tubes (BD Bioscience). Peripheral blood mononuclear cells (PBMCs) and plasma were isolated by density gradient centrifugation over Lymphocyte Separation Medium (CellGro). Red blood cells were lysed with ammonium chloride solution (StemCell). Plasma and PBMCs were either used immediately or aliquoted and stored in −80C freezers.
Human B cell purification
Total B cells were negatively selected from PBMCs by using EsaySep TM B cell enrichment kits (StemCell). Antigen-specific B cells were further sort-purified for sequencing experiments as outlined below.
Influenza Hemagglutinin Tetramer production and staining
The coding sequencing (encoding amino acids 18-524) of the hemagglutinin ectodomain were synthesized from the following influenza virus strains, A/California/VRDL7/2009, A/Switzerland/9715293/2013, A/Hong Kong/4801/204, and A/Michigan/45/2015, were synthesized (GeneArt, Regensburg, Germany) in frame with the human CD5 signal sequence located 5’ to the HA coding region. Two mammalian expression constructs were made with a 6XHIS tag or an AviTag located 3’ to the trimerization domain. HA-6X HIS and HA-AviTag were co-transfected in a 2:1 ration into FreeStyle TM 293-F Cells (ThermoFisher Scientific). Recombinant HA trimers with an average of 2 HIS6X monomers and 1AviTag monomer per trimer were purified from media by FPLC using a HisTrap HP column (GE Healthcare) for biotinylation in vitro using BirA biotin-protein ligase (Avidity). Tetramers of HA trimers were made by titrating in fluorochorome-conjugated streptavidin to biotinylated HA trimers until the volumetric ratio for saturation was reached. To detect HA-binding B cells, cells were treated at 37C with 0.5U/ml neuraminidase (C. perfringens, Sigma) to remove sialic acid, and then were washed, blocked and stained with HA tetramers.
Hemagglutinin ELISAs
Recombinantly generated hemagglutinin proteins (see above) were coated onto EIA/RIA ELISA plates (Costar) at 1:500 to 1:1000 dilution. Plasma from vaccinated samples were serially diluted onto these coated plates. HA-specific IgG antibodies from vaccine samples were detected using peroxidase-conjugated anti-human IgG secondary antibodies (Jackson ImmunoResearch) and were developed using ABTS development with acid stop. Absorbance was measured at 415nm using a SpectraMaxM2 (Molecular Devices).
Flow Cytometry
Single cell suspensions were blocked with 2% human serum before cell surface staining. Antibodies used to stain lymphocytes are listed in Supplemental File 2. 7AAD or LIVE/DEAD Fixable Dead Cell Stain Kits (Molecular Probes/ThermoFisher) were used to discriminate live cells. Intracellular staining was performed after staining with antibodies specific for cell surface markers. Cells were then fixed with formalin solution (neutral buffered, 10%; Sigma) and permeabilized with 0.1% IGEPAL (Sigma) in the presence of antibodies or fluorochrome labeled HA tetramers. Stained cells were analyzed using a FACSCanto II (BD Bioscience) or the Attune NxT flow cytometer (Invitrogen, ThermoFisher). Cells were sort-purified with a FACSAria (BD Biosciences) or Melody (BD Biosciences) in the UAB Comprehensive Flow Cytometry Core. FlowJo v9.9.3 or FlowJo v10.2 were used to perform analysis.
RNA-seq library preparation and analysis
RNA was isolated from HA-specific Bmem as well as ASCs and Naïve B cell by flow sorting these populations directly into RLT buffer (Qiagen) and snap freezing in liquid nitrogen. RNA was extracted using the QuickRNA Micro Prep Kit (Zymo). All resulting RNA from six biological replicates per B cell subset at the day 7 timepoint and four biological replicates per B cell subset at the day 14 timepoint was used as input for the SMART-seq v4 cDNA synthesis kit (Takara). Final libraries were constructed using 200 pg cDNA as input for the NexteraXT kit (Illumina) and quality assessed on a bioanalyzer. Libraries were pooled and sequenced using 50 bp paired-end chemistry on a HiSeq2500. Sequencing reads were mapped to the hg38 version of the human genome using STAR with the default settings and the UCSC KnownGene table as a reference transcriptome. Reads overlapping exons were tabulated using the GenomicRanges package in R/Bioconductor. Genes expressed at 3 reads per million or more in all samples from one group were considered detected and used as input for edgeR to identify differentially expressed genes. P-values were false-discovery rate (FDR) corrected using the Benjamin-Hochberg method with genes of an FDR <0.05 considered significant. Expression data was normalized to reads per kilobase per million mapped reads (RPKM).
GSEA analyses
Gene set enrichment analysis were submitted to the GSEA program (http://software.broadinstitute.org/gsea/index.jsp). Detected genes were ranked by multiplying the –log10 of the P-value from edgeR by the sign of the fold change for use as input in the GSEA PreRanked ananlysis.
Ingenuity Pathway Analysis (IPA)
IPA upstream regulator analysis (Qiagen Redwood City CA) was performed using the log2 fold change in gene expression between genes that were significantly differentially expressed as defined by corrected FDR of P <0.05, between T-bethi/FcRL5hi HA-specific Bmem over T-betlo/FcRL5lo HA-specific Bmem. Upstream regulators with an activation z-score of >/2 or </-2 were considered to activated or inhibited. Fischer’s exact test of p <1×10^-6 was used to determine significant overlap between a regulator’s downstream targets and our gene list.
ATAC-seq preparation and analysis
ATAC-seq was performed on HA-specific Bmem as follows. Cells were re-suspended in 25 ul of tagmentation reaction buffer (2.5 ul Tn5, 1xTagment DNA Buffer, 0.02% Digitonin, 0.01% Tween-20) and incubated for 1hr at 37C. Cells were then lysed with 25 ul 2x Lysis Buffer (composed of 300 mM NaCl, 100 mL EDTA, 0.6% SDS, 1.6 ug Proteinase-K) for 30 min at 40C, low molecular weight DNA purified by size-selection with SPRI-beads (Agencourt), and PCR amplified using Nextera primers with 2x HiFi Polymerase Master Mix (KAPA Biosystems). Amplified, low molecular weight DNA was isolated using another SPRI-bead size selection. Quality control was performed on a bioanalyzer. Libraries were sequenced using a 50bp paired end run at the UAB Heflin Genomics Center. Raw sequencing reads were mapped to the hg38 version of the human genome using Bowtie (Langmead et al 2009) with the default settings. Duplicate reads were annotated using the Picard Tools MarkDuplicates function (http://broadinstitute.github.io/picard/) and eliminated from downstream analysis. Enriched peaks were identified using MACS2 with the default settings. Genomic and motif annotations were computer for ATAC-seq peaks using the HOMER104 annotatePeaks.pl script. Read counts for all peaks were annotated for each sample from the bam file using the GenomicRanges105 R/Bioconductor package and normalized to reads per peak per million (rppm).106
Illumina MiSeq
HA-specific Bmem and plasmablasts were sort-purified into RLT buffer and snap frozen in liquid nitrogen. RNA was extracted using the quick start protocol from QIAGEN RNeasy Mini Kit. First strain cDNA synthesis was performed using iScript cDNA synthesis kit (BioRad) and 8ul of RNA following manufacturer protocol. First round amplification of IgG, IgA, and IgM was performed in a 25 ul reaction volume using 4-8 ul cDNA, Platinum PCR SuperMix High Fidelity (Invitrogen), and 1ul gene specific primers (120 nM) of Vn1-Vh7 FR1 (forward) and Ca, Cu, Cg (reverse). First round PCR conditions were: 95C for 3 min, 42 cycles of 30s 95C, 30s 58C, 30s 72C, and 72C for 3 minutes. Amplification was verified using 1.2% agarose gels (Lonza). Samples were ligated in a second round PCR with Nextera Index kit (Illumina). PCR2 conditions for indexing were: 72C for 3 minutes, 98C for 30s and 5 cycles of 98C for 10s, 63C for 30s, and 72 C for 3 minutes. Products were purified with Agencourt AMPure XP beads (Beckman) and nanodropped for final concentration before pooling into a final library. Library was denatured using 0.2N NaOH and quenched with cold HT1 per manufacturer (Illumina) instruction. Denatured libraries were diluted with 20% PhiX (Illumina) as an internal quality control and loaded onto a 600-cycle V3 MiSEQ cartridge (Illumina) for amplification.
Clonotype Assembly and Analysis
Raw sequence reads were processed using a combination of in-house and public analytic tools. Full methodology for this data processing has been described elsewhere.107 Pair-end reads were joined and filtered based on sequence length and quality thresholds. Alignment was performed using IMGT/Hi-Vquest, sequences and analyzed for clonality and for mutations in the V region by a custom program written by the authors (AFR, CF) in perl and Matlab that is available on request. Frequency and distribution of somatic hypermutation was determined on the basis of non-gap mismatches of expressed sequences with closest germline Vh sequence. For visualization, alluvial plots and lineage accumulation curves were constructed in Matlab. Detailed clonotype analysis is presented in Supplemental File 2.
BCR Cloning/Recombinant Antibody Screening
Antigen binding B cells were index sorted by Fcrl5 expression status as single cells into hypotonic lysis buffer in 384 well plates i and stored at −80deg. Lysates were used to generate cDNA using the High-capacity cDNA generation kit (Roche) following manufactures instructions. PCR was performed using primers specific for nucleotides encoding the amino terminus of the mature IGHV, IGKV, and IGLV proteins. The resulting amplicons were inserted into a mammalian expression plasmid containing the IGG1, IGKC, or IGLC gene sequence. To generate recombinant antibody, plasmids encoding Ig heavy light chain pairs were co-transfected into 293FreeStyle cells (Invitrogen) using standard Polyethylenimine transfection methods. Supernatant was assayed for recombinant antibody expression and for antigen specificity using the flow cytometric bead array (Spherotech). conjugated to IgG or to recombinant hemagglutinin antigen as has been described elsewhere (PLOS ONE, Kelsoe).76
Statistical Analysis
Detailed statistical analytic details are presented in Supplemental File 2. Analysis was performed using GraphPad Prism version 7.0a.
Supplemental File 1. Supplemental Figures 1-3 as outlined in Figure Legends.
Supplemental File 2. Detailed statistical and clonotype type data.
Supplemental File 3. Full RNA-seq, ATAC-seq, and PAGERANK network analysis for the data presented in Figures 2, 3, and Supplemental Figure 2.
Acknowledgements
We thank the Alabama Vaccine Research Clinic and particularly Pamela Cunningham, Heather Logan, Aeryn Peck, Catrena Johnson and Megan Oelschig for recruiting and consenting subjects for this study. We also thank Vidyasagar Hanumanthu, director of the UAB Flow Core Facility, for his assistance with flow sorts and ImageStream experiments. We are grateful to Amy S. Weinmann for her thoughtful commentary regarding this manuscript. Funding for the work was provided by the US National Institutes of Health (NIH): P01 AI125180 (I.S., F.E.L, J.M.B, C.D.S), U19 AI 109962 (F.E.L, T.D.R). A.N. received pilot grant support from the UAB AMC21 Immunology Autoimmunity and Transplantation Strategic Initiative as well as funding from the UAB CCTS (UL1 TR001417) to support this work. NIH P30 AR048311 and P30 AI027767 provided support for the UAB Consolidated Flow Cytometry Core. The authors have no known conflicts of interest to disclose.
References:
- 1.↵
- 2.↵
- 3.↵
- 4.
- 5.
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.
- 25.↵
- 26.
- 27.
- 28.
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.
- 84.
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.↵
- 90.
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.
- 100.↵
- 101.↵
- 102.↵
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵




