

Abstract
Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and CRISPR associated (Cas) effector proteins enable the direction of DNA double-strand breaks at defined loci based on a variable length RNA guide specific to each effector. The guides are generally similar in size and form, consisting of a ∼20 base sequence homologous to the DNA target and a secondary structure used by the effector for guide/nuclease recognition. However, the effector proteins vary in size, DNA binding kinetics, nucleic acid hydrolyzing activities, and Protospacer Adjacent Motif (PAM) requirements. Recently, a Cas12a family member protein named Mad7 was identified that is most similar to the Cas12a from Acidaminococcus sp. Here, we report for the first time Mad7 activity in zebrafish and human cells. We utilize a fluorescent reporter system to demonstrate CRISPR/Mad7 elicits strand annealing mediated DNA repair more efficiently than CRISPR/Cas9. Finally, we use CRISPR/Mad7 with our previously reported gene targeting method GeneWeld in order to integrate reporter alleles in both zebrafish and human cells. Together, this work provides methods for deploying an additional CRISPR/Cas system, increasing the flexibility researchers have in applying genome engineering technologies.
Introduction
CRISPR systems have been widely adopted in zebrafish research due to their efficacy and the ease of reprogramming DNA binding activity, which is mediated by a chimeric single guide RNA (sgRNA or gRNA) molecule (Jinek et al. 2012) (Cong et al. 2013) (Jao, Wente, and Chen 2013). The CRISPR toolbox continues to expand with the identification of systems that display varying PAM requirements, differing nuclease size, and DSB architecture (Adli 2018). For example, while Cas9 often hydrolyzes DNA leaving a blunt-ended cut three bp 5’ of the PAM sequence, Cas12a proteins from Acidaminococcus sp and Lachnospiraceae hydrolyze DNA in a staggered fashion 3’ of their 5’ PAM and leave four nucleotide 5’ overhangs (Zetsche et al. 2015). As DSB architecture and subsequent end resection is a critical determinant in DNA repair pathway activation, there is demand for CRISPR variants that elicit more predictable repair outcomes for precision genome engineering (Truong et al. 2013). For example, inducing DNA overhangs with staggered CRISPR/Cas9 nickases targeted to opposite strands can stimulate precision genome engineering using oligonucleotides (Ran et al. 2013). This supports the need for identifying CRISPR systems that show differing DSB architectures.
CRISPR/Cas12a activity has been reported in zebrafish (Moreno-Mateos et al. 2017) (Fernandez et al. 2018). In these studies, Cas12a activity is enhanced by the injection of ribonucleoprotein (RNP) complexes. Further, Cas12a-mediated DNA cleavage was enhanced by either a 34°C heat shock or the co-targeting of nuclease dead Cas9 (dCas9) to the region where Cas12a cleavage was desired, indicating that DNA melting is a potential rate limiting step for Cas12a. Cas12a mediates enhanced oligonucleotide incorporation as compared to Cas9, perhaps indicating mechanistically distinct enzymatic activities and resulting genomic DNA end availability (Moreno-Mateos et al. 2017). Recently, a distantly related CRISPR/Cas12a family member was described called Mad7 (Ryan T. Gill 2017). Mad7 is a 1262 amino acid protein that recognizes a 5’-YTTN PAM and uses a 42 or 56 base RNA guide to recognize and catalyze site-specific DNA cleavage (Ryan T. Gill 2017). While CRISPR/Mad7 is active in human cells (Ryan T. Gill 2017), the cut site has not been reported and its application in gene targeting has yet to be described.
In the early zebrafish embryo, microhomology mediated end joining (MMEJ) is an active DNA repair pathway that generates predictable alleles after nuclease targeting (Thyme and Schier 2016) (He et al. 2015) (Ata et al. 2018). MMEJ-based gene editing has been described in zebrafish and mammalian cells, though the homology arms used for these events may be more similar in length to those employed in Single Strand Annealing (SSA) (Aida et al. 2016) (Hisano et al. 2015) (Nakade et al. 2014) (Bhargava, Onyango, and Stark 2016). In this method, nuclease targeting of a donor plasmid in vivo liberates a donor cassette, exposing homology arms that are used to direct integration. A more general term for using this method for gene targeting is Homology-Mediated End Joining (HMEJ) (Yao et al. 2017).
We recently reported an advanced methodology with high efficiency gene targeting using HMEJ methodology with short homologies (Wierson et al. 2018). This method, dubbed GeneWeld, involves the simultaneous in vivo targeting of a genomic target and a donor plasmid with designer nucleases to reveal 24 or 48 bp homology arms to be used with MMEJ or SSA machinery. GeneWeld works with both TALENs and CRISPR/Cas9-mediated genomic double-strand DNA break events, but other nucleases have not yet been tested. We report here the use of Mad7 in a vertebrate model organism and its applications for gene targeting in human cells. Consistent with its similarity to Cas12a, high Mad7 activity requires a 34°C heat shock treatment in zebrafish. However, injection of mRNA encoding for Mad7 is sufficient to induce DSB activity, and pre-crRNA is an effective RNA guide for Mad7 activity, in contrast to reports using previously described Cas12a and related systems (Moreno-Mateos et al. 2017; Liu et al. 2019). We developed a Universal pre-crRNA (U-pre-crRNA) for Mad7 that can be used in zebrafish and human cells, and we show that CRISPR/Mad7 potently induces Strand Annealing Mediated Repair (SAMR) in a genomic reporter at efficiencies greater than CRISPR/Cas9. Additionally, CRISPR/Mad7 promotes GeneWeld activity at rates similar to CRISPR/Cas9. Finally, we demonstrate Mad7 activity at multiple human loci, including the safe harbor locus AAVS1 and apply GeneWeld with Mad7 in human cells.
Results
Mad7 induces indels in zebrafish embryos
We synthesized a zebrafish codon optimized Mad7 and added dual SV40 nuclear localization signals at the N- and C-termini, as dual NLS was shown to increase efficacy of Cas9 in zebrafish (Supplementary Table 1) (Ryan T. Gill 2017) (Jao, Wente, and Chen 2013). Following the workflow outlined in Figure 1a, we targeted the notochord-specific transcription factor noto to gain a rapid phenotypic read-out of Mad7 mutagenic activity in zebrafish. Three pre-crRNAs and one crRNA were designed to target exon 1 of noto (Figure 1b). Injection of mRNA encoding zebrafish codon optimized, dual NLS-Mad7 (nMad7n) with noto-pre-crRNA1 and incubation at 28° C after injection did not elicit any activity, similar to Cas12a (Moreno-Mateos et al., 2017 and data not shown). However, injection of nMad7n mRNA with noto-pre-crRNA1 or noto-pre-crRNA3 and a 4-hour 34° C heat shock treatment following injection elicits phenotypes characteristic of biallelic loss of noto including loss of notochord and a shortened tail at efficiencies ranging from 7-61% (Supplementary Figure 1a) (Talbot et al. 1995). PCR across these individual target sites and subsequent heteroduplex mobility shift assays show the presence of indels compared to analyses from uninjected wild type DNA characteristic of NHEJ activity (Supplementary Figure 1b, 1c). noto-pre-crRNA2 and noto-crRNA1 do not elicit a phenotype, and heteroduplex mobility shift assays across those targets indicate they are inactive gRNAs (data not shown). However, efficiencies of biallelic noto inactivation at the same target varied considerably from injection to injection (Supplementary Figure 1), consistent with previous reports in human cells and zebrafish using CRISPR/Cas12a (Kim et al. 2016) (Kim et al. 2017) (Moreno-Mateos et al. 2017) (Liu et al. 2019). To confirm our results are not specific to a single locus, we injected nMad7n mRNA and pre-crRNA targeting two independent loci at cx43.4 (Figure 1c). PCR across the individual target sites and subsequent RFLP analysis demonstrates that cx43.4-pre-crRNA2 is active (Supplementary Figure 1d), while pre-crRNA1 is inactive (data not shown).

(a) Data plots showing ratio of injected animals displaying somatic noto phenotype. Whiskers represent mean +/− s.d. p values calculated with one-tailed Student’s t-test. (b, c) Gel showing heteroduplex mobility shift of injected animals vs wild type (WIK) for pre-crRNA1 and pre-crRNA3 at noto. (d) RFLP analysis showing pre-crRNA2 is active at cx43.4.

(a) Workflow showing dual NLS-Mad7 mRNA and pre-crRNA injection into single cell animals. Animals are heat shocked for 4 hours and then allowed to develop normally until DNA is isolated and analyzed. (b) Schematic and sequences of pre-crRNA used to target noto. (c) Schematic and sequences of pre-crRNA used to target cx43.4. (d) Results displaying the wildtype and top 5 mutated alleles after AmpliconEZ analysis of two noto pre-crRNAs and one cx43.4 pre-crRNA.
To gain a better understanding of Mad7 insertion/deletion (indel) events, we used next generation sequencing (NGS) to analyze the efficiencies of mutation at the active noto and cx43.4 target sites. DNA amplicons from five noto mutant embryos injected with Mad7 mRNA and noto-pre-crRNA1 or noto-pre-crRNA3 and five cx43.4 pre-crRNA1 injected embryos were selected randomly for next generation sequencing. As expected from biallelic inactivation at noto, ∼61% of alleles at noto-pre-crRNA1 show indels, characteristic of NHEJ and/or MMEJ after nuclease targeting (Figure 1d). The phenotypically more efficient noto-pre-crRNA3 showed ∼90% of alleles containing indels (Figure 1d). However, in agreement with the RFLP analysis, the majority (∼74%) of alleles sequenced at cx43.4 are wild type sequence, indicating not all targeting events with Mad7 are efficient enough for biallelic mutation of the target locus (Figure 1d). Taken together, these data indicate CRISPR/Mad7 encoded as an RNA system is active in zebrafish albeit dependent on a 34°C heat shock, and pre-crRNA is an active RNA guide for directing Mad7 activity to genomic target sites. CRISPR/Mad7 is thus a highly accessible Cas12a system for zebrafish researchers, and we explored its use with a variety of gene editing applications.
Mad7 elicits strand annealing mediated DNA repair more efficiently than Cas9 in a genomic reporter system in zebrafish
We next employed a stably integrated red fluorescent protein (RFP) reporter system to visually assay the efficiencies with which CRISPR/Cas9 and CRISPR/Mad7 differentially elicit DNA repair by strand-annealing mediated repair (SAMR). Upon targeting the universal sites in the reporter, end resection reveals 48 nucleotide direct repeats that can restore the reading frame of RFP, leading to a semi-quantitative read-out of repair efficiencies (Figure 2a). We used GeneWeld to create a line of single copy noto:RFP-DR48 reporter zebrafish (Supplemental Figure 1a-d). Injection into single-cell noto:RFP-DR48 transgenic embryos with Cas9 mRNA and UgRNA results in RFP expression in the notochord, indicative of SAMR after CRISPR/Cas9 targeting (Figure 2b).

(a) Schematic of noto:RFP-DR48 showing the location of the 48 bp direct repeats flanking both the Cas9 and Mad7 universal RNA cursor sites (underline) and PAM sites (red text). (b) Data plot showing the ratio of injected animals displaying RFP in the notochord out of total carrying the transgene. Whiskers represent mean +/− s.d. p values calculated with one-tailed Student’s t-test. (c) Qualitatively scored ratios of notochord converted to RFP+ after Cas9 induced SAMR. (d) Qualitatively scored ratios of notochord converted to RFP+ after Mad7 induced SAMR. (e-g) Representative embryos for broad, intermediate, and narrow conversion of RFP in the notochord.
We then assayed whether CRISPR/Mad7 promotes SAMR in the noto:RFP-DR48 assay. We designed a universal pre-crRNA (U-pre-crRNA), with no predicted off target sites in zebrafish or human cells, to direct Mad7 activity to noto:RFP-DR48. Injection of nMad7n mRNA and U-pre-crRNA results in RFP expression in the notochord (Figure 2b). As a percentage of injected animals with RFP+ notochords, Mad7 elicits SAMR statistically greater than Cas9 (Figure 2b). Repair events are mosaic and were qualitatively sorted into three classes of notochord expression pattern: broad, intermediate, and narrow (Figure 2e-i). While approximately ∼70% of Mad7-injected animals showed RFP+ cells in their notochords as opposed to ∼40% in their Cas9-injected counterparts, Mad7 repair events are equally mosaic (Figure 2f). These results indicate that noto:RFP-DR48 is a viable assay for screening the propensity of designer nucleases to elicit SAMR and that the enzymatic activity of Mad7 enhances activation of SAMR in vivo.
Using Mad7 for precise integrations in zebrafish
We leveraged the activity of the U-pre-crRNA to determine whether CRISPR/Mad7 can catalyze targeted integration of fluorescent reporters using GeneWeld (Wierson et al. 2018). First, we injected nMad7n, noto-pre-crRNA1, U-pre-crRNA, and a GFP reporter plasmid programmed with the U-pre-crRNA site and 24 bp of homology 5’ to noto-crRNA1 genomic site to promote gene targeting at noto (Supplementary Figure 3a, Supplementary Table 4). Fluorescent positive notochord cells were observed at the 18-somite stage, indicating in frame integration of the GFP cassette at noto (Supplementary Figure 3b, b’). On average, 24% of embryos injected display targeted noto integrations (Supplementary Figure 3c). However, notochords were highly mosaic, indicating a low efficiency of somatic integration activity in tissue types that form the notochord (Supplementary Figure 3b, b’). GFP was often observed outside of the notochord, yet in the mesoderm, as expected with biallelic disruption of noto and transfating of notochord cells (Talbot et al. 1995). Junction fragment PCR was conducted on GFP+ embryos to confirm integration using the programmed homology. As expected, a PCR band was recovered in GFP+ embryos, while no PCR band is detected in the same experiment performed without the genomic pre-crRNA (Supplementary Figure 3d, 3e). DNA sequencing confirmed precise junctions at the 5’ end of the integration (Supplementary Figure 3f).

(a) Schematic showing RFP-DR48 integrated at noto. The NBM (yellow) contains both Cas9 and Mad7 PAMs for universal targeting using the underlined RNA cursor target site. Red is the engineered stop codon. (b) Southern blot of three noto:RFP-DR48 lines. Band intensity is roughly single copy based on the copy number controls in the noto probed blot (top). Gel shifts are present indicating precise (line 1) and linear vector integration (lines 2 and 3). (c) Junction fragment analysis of integrations showing precision at the 5’ junction. Line 1 contains a precise 3’ integration, while line 2 and line 3 contain differing NHEJ events. (d) Schematic of integrations events as determined by Southern blot and DNA sequencing analysis.

(a) Schematic showing designed homology for precise 5’ integration using Mad7. Green is designed homology. The PAM for Mad7 targeting in the genome and donor is underlined. (b, b’) Representative confocal Z-stack image showing mosaic GFP expression in the notochord of an injected animal. Scale bar is 100 um. (c) Data plot showing the ratio of embryos with GFP expression in the notochord out of total injected embryos. Whiskers represent mean +/− s.d. (d) Gel showing junction fragment expected after precise integration using the homology domain. (e) Gel showing no junction, indicating there is no integration without the genomic pre-crRNA. (f) DNA sequencing 5’ junctions showing precise integration using the programmed homology.
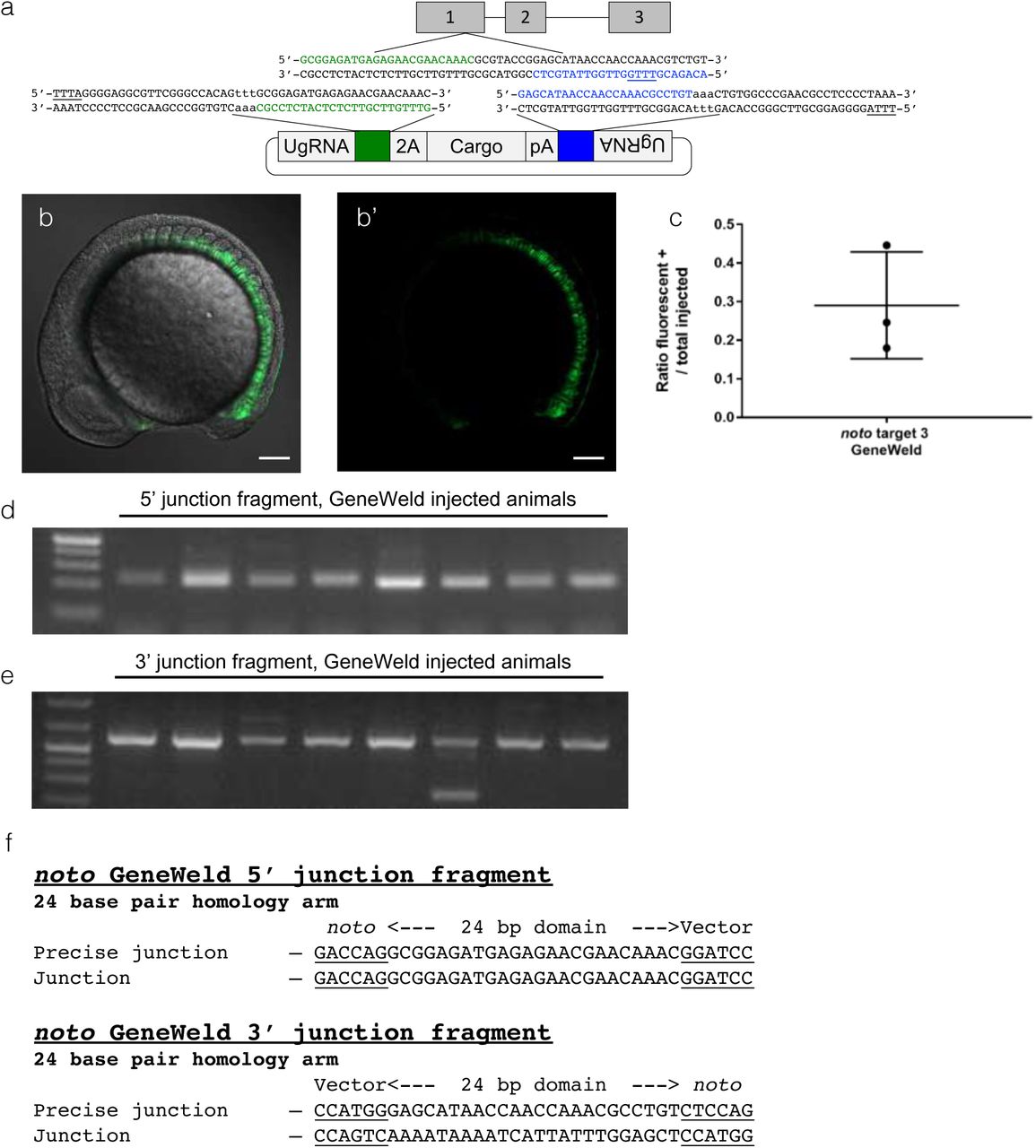
We next designed a GeneWeld donor with both 5’ and 3’ homology domains to integrate a DNA cassette without the vector backbone, as previously demonstrated (Figure 3a) (Wierson et al. 2018). Targeting noto using pre-crRNA3 with GeneWeld resulted in an average of 31% of embryos with GFP+ notochords (Figure 3b, 3b’, Figure 3c). Most notochords were highly mosaic and displayed GFP expression indicative of lost cell fate, as with noto target 1 targeting (Supplementary Figure 3b), though some events are recovered where greater than 90% of the notochord is expressing GFP (Figure 3b). To probe for integration of the cargo at the genomic target site, PCR was used to analyze the junctions between the genome and cargo. Using PCR, predicted 5’ and 3’ junction fragments were recovered in GFP+ embryos (Figure 3d, 3e). Junction fragment sequencing from a single embryo demonstrated precise integration at both ends of the cassette (Figure 3f). These data indicate that Mad7 is an effective nuclease for catalyzing GeneWeld integrations, but we also show that optimization is needed to enhance somatic integration efficiency, likely of the gRNAs themselves, to be comparable to Cas9-mediated integrations frequencies with GeneWeld in zebrafish.

(a) Schematic of noto showing designed homology for precise integration using Mad7 and the U-pre-crRNA. Green is designed 5’ homology. Blue is designed 3’ homology. The PAM for Mad7 targeting in the genome and donor is underlined. (b, b’) Confocal Z-stack image showing broad GFP expression in the embryo. Scale bar is 100 um. (c) Data plot showing the ratio of embryos with GFP expression in the notochord out of total injected embryos. Whiskers represent mean +/− s.d. (d) Gel showing 5’ junction fragment expected after precise integration using GeneWeld. (e) Gel showing 3’ junction fragment expected after precise integration using GeneWeld. (f) DNA sequencing of lane 5 in (d) and (e) showing a precise integration using the programmed homology.
Mad7 induces double-strand breaks in human cells
We developed an all-in-one expression vector for cis expression of dual nuclear localization signal Mad7 (nMad7n) and a pre-crRNA in vitro (Figure 4a). Using this expression vector and the workflow in Figure 4a, we targeted two therapeutically relevant “safe harbor” loci, AAVS1 and CCR5 (Papapetrou and Schambach 2016) (Figure 4b, d). We additionally targeted the TRAC locus due to its reported value in generating Chimeric Antigen Receptor T cells (Figure 4e) (Eyquem et al. 2017). To first assess the in vitro cleavage activity of Mad7 with our in vitro expression system, we utilized the T7 endonuclease (T7EI) assay to determine if a DSB and subsequent indels were induced at AAVS1. The characteristic banding pattern was apparent in pMad7-AAVS1-pre-crRNA1 transfected cell DNA, but not control cell DNA, indicating that Mad7 is active at the chosen locus (Figure 4c). No indel activity was detected at the AAVS1 pre-crRNA2 site by T7E1 assay (data not shown). These data suggest that this all-in-one expression system is active in vitro.
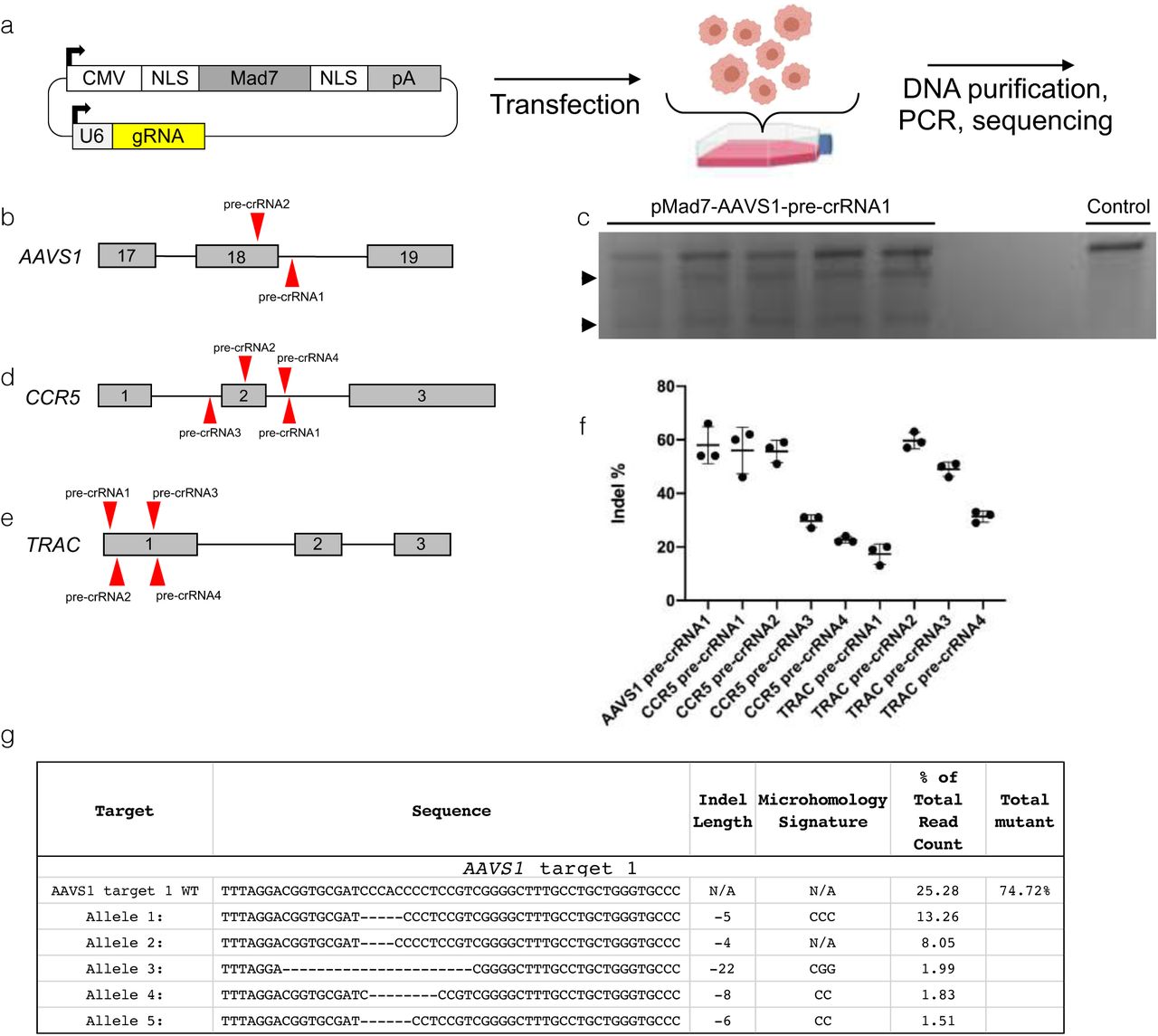
(a) Schematic showing the workflow for Mad7 and pre-crRNA expression in human cells from a cis expression plasmid. After transfection, cells are recovered and then analyzed. (b) Schematic of two pre-crRNAs designed to target AAVS1. (c) T7E1 assay showing AAVS1-pre-crRNA1 is active. Arrowheads are cleaved bands, indicating nuclease activity. (d) Schematic of four pre-crRNAs designed to target CCR5. (e) Schematic of 4 pre-crRNAs designed to target TRAC. (f) Graph showing percentage of indels after using pre-crRNAs to target AAVS1, CCR5, and TRAC in HEK293 cells as determined by ICE analysis. Whiskers represent mean +/− s.d. (f) Results displaying the wildtype and top 5 mutated alleles after AmpliconEZ analysis of AAVS1 targeted DNA.
To gain a more quantitative understanding of the cleavage activity of Mad7 at the chosen target loci, PCR amplicons of cells transfected with Mad7 targeting the respective loci (AAVS1, CCR5, TRAC) were submitted for Sanger sequencing and subjected to the bioinformatic ICE analysis program that infers CRISPR activity from sequencing trace reads (Hsiau et al. 2019). ICE analysis shows that targeting AAVS1 with Mad7 and pre-crRNA1 creates indels in 58% of sequenced amplicons (Figure 4f, Supplementary Table 6). Likewise, ICE analysis shows that targeting CCR5 with Mad7 and pre-crRNA1 creates indels in up to 60% of sequenced amplicons, with some target sites displaying markedly lower activity (Figure 4f, Supplementary Table 6). ICE analysis shows that the most active pre-crRNA targeting TRAC causes indels in almost 60% of sequenced amplicons (Figure 4f, Supplementary Table 6). Because ICE is based on Sanger sequencing data, which considers fewer reads than NGS, it is likely to underestimate the true percentage of indels. We therefore measured indel activity at the AAVS1-pre-crRNA1 target site using next generation sequencings (NGS), which shows that ∼74% of recovered alleles are edited (Figure 4g). Taken together, these data demonstrate that Mad7 is active in human cells at clinically relevant genomic loci.
Mad7 elicits strand annealing mediated DNA repair more efficiently than Cas9 in an episomal reporter system in human cells
Episomal reporters have been routinely used in cell culture systems to assay DNA repair pathways and decipher the proteins involved (Pierce et al. 1999) (Certo et al. 2011) (Gunn and Stark 2012) (Bennardo and Stark 2010). Here, we modified the donor molecule for noto:RFP-DR48 for use in mammalian cell culture and created pMiniCAAGs:RFP-DR48 (Figure 5a). Similar to the zebrafish reporter noto:RFP-DR48, expression of a nuclease and requisite gRNA should promote a double-strand DNA break at the nuclease binding site, resulting in a fluorescent readout of nuclease activity if SAMR is used to repair the break. Transfection of both pMiniCAAGs:RFP-DR48 and pMad7-U-pre-crRNA or pCas9-UgRNA resulted in RFP+ HEK293 cells. We performed flow cytometry for RFP+ cells transfected with either the Mad7 or Cas9 systems and demonstrated that Mad7 induces SAMR significantly greater than Cas9 in human cells (Figure 5b). These data indicate that the cleavage activity of Mad7 in zebrafish and human cells is similar, and that the U-pre-crRNA is active in human cells. Further, these data suggest that Mad7 displays a cross-model advantage over Cas9 in mediating SAMR.

(a) Schematic showing the workflow for determining strand annealing mediated repair in human cells. An all in one expression plasmid for Mad7 or Cas9 is transfected along with pMini-CAAGs::RFP-DR48. Cells are allowed to recover and then FAC sorted for RFP+ cells. (b) Quantification of RFP+ cells per total cells analyzed by FACs. Whiskers represent mean +/− s.d. p values calculated with one-tailed Student’s t-test.
Mad7 promotes precise integration in human cells
We next wanted to determine whether Mad7 could mediate precise transgene insertion at the AAVS1 pre-crRNA1 site due to Mad7’s high activity at this locus and AAVS1’s use in the therapeutic expression of transgenes (Smith et al. 2008), (Yang et al. 2008), (Zou et al. 2011). To this end, we used the U-pre-crRNA in human cells to mediate GeneWeld integrations (Figure 6a). We transfected the pGFP:Zeo-48 GeneWeld (Figure 6b) donor and pMad7-AAVS1-U into HEK293 cells with an all-in-one expression plasmid making nMad7n, AAVS1 pre-crRNA1, and U-pre-crRNA (pMad7-AAVS1-U) or the pGFP:Zeo-48 GeneWeld donor and pMad7-AAVS1 pre-crRNA1 only (Figure 6a). GFP+ cells were isolated by flow assisted cell sorting (FACS) one day post electroporation as a control for plasmid delivery and screened for stable integration by flow cytometry 2 weeks post transfection to measure dropout of GFP+ cells (Figure 6c). These data show a roughly 2.5 fold increase in number of cells that remained GFP+ when transfected with donor molecule, Mad7 and the pre-crRNA, as compared to donor alone (Figure 6d). Positive cell populations were screened for insertion by junction fragment PCR, which showed that the precise fragment appeared in the population transfected with pMad7-AAVS1-U and the donor (Figure 6e,f), but was absent in the donor alone population. These data suggest that Mad7 is a tractable nuclease for GeneWeld-mediated precise insertions at AAVS1 in human cells.
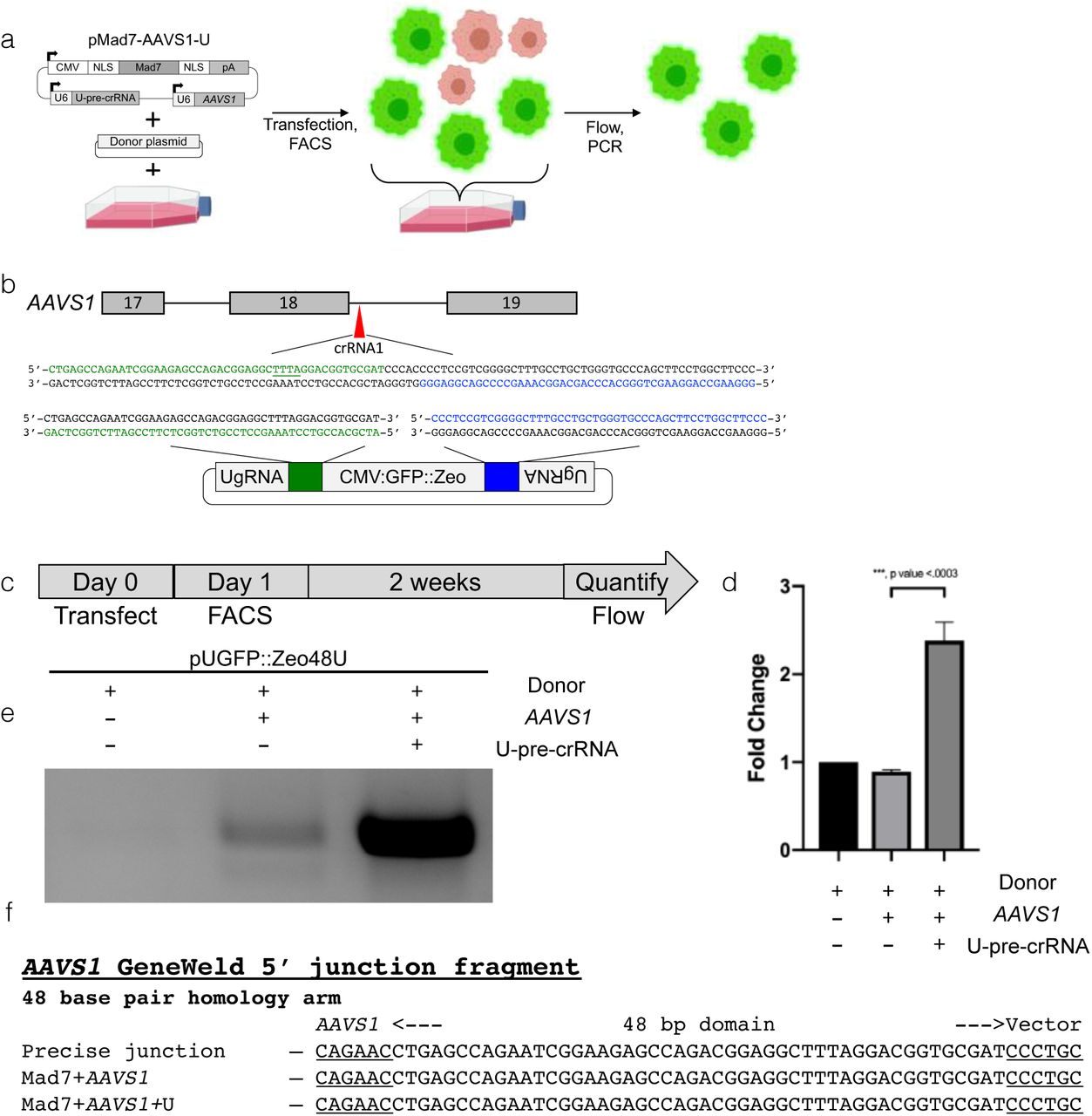
(a) Schematic showing the workflow for making targeted integrations in HEK293 cells. (b) Schematic of AAVS1 showing designed homology for precise integration using Mad7 and the U-pre-crRNA to liberate the cassette. Green is designed 5’ homology. Blue is designed 3’ homology. The PAM for Mad7 targeting in the genome and donor is underlined. (c) Schematic for flow analysis after transfection of GeneWeld reagents for targeting AAVS1. (c’) Data plot showing the ratio of cells with GFP expression as determined by flow cytometry. Whiskers represent mean +/− s.d. p values calculated with one-tailed Student’s t-test. (d) Junction fragment gel showing the expected 5’ integration amplicon. No amplicon is seen when transfecting donor alone. (e) DNA sequencing of lanes in (d) showing a precise integration using the programmed homology.
Discussion
In this report, we demonstrate CRISPR/Mad7’s activity in zebrafish and its application as an efficient alternative nuclease for generating HMEJ-based gene targeting events in vivo and in vitro. We employed nMad7n mRNA injection using pre-crRNAs to create frame shift knock-out alleles in noto and cx43.4 at ∼54% and ∼35%, respectively, based on next generation sequencing analysis. Mad7 activity is temperature-dependent and requires a 34°C heat shock for activity in zebrafish. We demonstrate that Mad7 elicits SAMR in a reporter assay at levels nearly 2-fold greater than Cas9 in both zebrafish and human cells. We also demonstrate that Mad7 is a viable nuclease for mediating gene targeting using the GeneWeld strategy in zebrafish. In addition, we show robust double-strand break induction at multiple therapeutically relevant human loci and demonstrate Mad7 is effective for GeneWeld integrations in human cells at AAVS1. These data suggest the capacity for Mad7 to be employed as a tractable nuclease for gene editing applications across multiple species for both research and clinical purposes.
In prior reports of Cas12a CRISPR system activity in zebrafish, appreciable nuclease activity was only observed after RNP delivery and heat shock, “proxy-CRISPR” to relax chromatin structure, or targeting a gene with multiple crRNAs at once (Moreno-Mateos et al. 2017) (Liu et al. 2019). Nuclease activity was shown to be dependent on the stability of the pre-crRNA or crRNA complexing with Cas12a, protecting it from degradation. While pre-crRNA was ineffective for nuclease activity under normal conditions with Cas12a, longer heat shocks rescued activity, indicating a likely kinetics issue with the use of pre-crRNA. It is interesting to note that in our experiments, pre-crRNA injection with nMad7n mRNA permitted activity, displaying a stark contrast between these Cas12a and related proteins and Mad7 in either their ability to complex with the RNA guide and access DNA or the stability of the differing pre-crRNA structures. Further, the use of multiple crRNAs per target gene in tandem dramatically increases the possibility of off target effects, while our study displays sufficient knock-out activity with only a single pre-crRNA per target gene.
Levels of mosaicism in gene targeting using HMEJ in zebrafish vary greatly in the reported literature (Hisano et al. 2015) (Wierson et al. 2018). Consistent with the observation that SAMR in noto:RFP-DR48 is mosaic, it was recently reported that translation of Cas9 mRNA and subsequent gene editing after one-cell stage injection is not complete until the 16 or 32 cell stage while RNP injections result in appreciable nuclease activity by the 2-4 cell stage (Zhang, Zhang, and Ge 2018). Thus, noto:RFP-DR48 is an assay system where injection conditions can be optimized to enhance somatic gene targeting and decrease mosaicism in cell types that arise from the mesoderm.
While Mad7 showed enhanced activation of SAMR in our noto:RFP-DR48 assay vs Cas9, our gene targeting experiments with Mad7 in zebrafish displayed no substantive difference compared to our previous reports (Wierson et al. 2018). It could be predicted that repair of DNA in cis is more efficient than in trans, and thus the outcome of strand annealing in a genomic reporter will not translate to targeted integrations. However, noto:RFP-DR48 represents a novel reporter in zebrafish for experimenting with small molecules, dominant-negative proteins, and other strategies that may alter double-strand break repair pathway choice. Additionally, while Mad7 is an effective catalyst for GeneWeld integrations, mosaicism of expression is qualitatively higher than when using Cas9 as the GeneWeld nuclease (Wierson et al. 2018).
To date, Cas12a orthologs have displayed relatively poor editing efficiency in mammalian systems (Tu et al. 2017),(Kim et al. 2016), (Kleinstiver et al. 2016), (Kim et al. 2017). AsCpf1 and LbCpf1 are among the best characterized Cas12 systems and have very modest activity in human cells (Kim et al. 2016; Kleinstiver et al. 2016). To address this issue, additional Cas12a systems with enhanced activity and differential targeting ability such as FnCpf1 were engineered and characterized. Though FnCpf1 uses a more common TTN PAM sequence, its cleavage activity is still modest (Tu et al. 2017). Editing efficiency of the Cas12a systems has since been enhanced by artificially engineering the crRNA to increase activity without a loss of specificity (Bin Moon et al. 2018). Therefore, it is of interest to test various modifications of crRNAs and note their effect on mutagenesis and gene targeting going forward.
Though Cpf1 has been shown to be amendable to HDR-mediated integration, these transgene integration events have been shown to be highly dependent on using RNP complexes in concert with modified crRNAs to show appreciable activity (Kim et al. 2017). Here we demonstrate Mad7’s utility in generating robust editing efficiency without chemically modified crRNAs, showing its utility in streamlined and accessible gene editing for most research applications. Further, we show Mad7 mediates precise integration without engineering of the nuclease or the corresponding guide RNA with mRNA and plasmid DNA.
While we show that Mad7 is compatible with the GeneWeld system in vitro as demonstrated by the prominent 5’ junction PCR band and retention of GFP expression indicating stable integration, we also showed that in the absence of the UgRNA liberating the donor there is still cryptic integration of the transgene at AAVS1. When GeneWeld is used with Cas9, the donor homology arms can include a portion of the genomic target, but exclude the 3’ PAM sequence and prevent nuclease targeting at the homology arms. However, because Mad7 utilizes a 5’ PAM and induces a distal DSB, it is likely that the homology arm is being targeted by Mad7 and linearizing the donor at the 5’ end. In our design the homology arms contain up to 17bp of target region that may be sufficient for a Cas12a-like seed region for the crRNA and facilitate a DSB (Chen et al. 2018) (Swarts, van der Oost, and Jinek 2017). With further optimization of both the crRNA and donor constructs, Mad7 can readily be adapted for precise integration in mammalian systems.
Conclusion
Here, we demonstrate effective genome editing in vertebrate models in vivo and in vitro using the newly described CRISPR/Mad7 system. CRISPR/Mad7 is active in zebrafish and promotes efficient somatic mutation and HMEJ-mediated integration. Integration of donor cassettes is achieved at levels up to 44% in zebrafish, demonstrating the utility of this system for generating precise genome modifications. In human cells, Mad7 represents an additional tool to target therapeutically relevant loci, including the safe harbor loci AAVS1 and CCR5. Alternative nuclease systems are of interest to the field of precision therapeutics to expand the available toolbox for creating a desired genome editing outcome. Mad7 increases the flexibility that researchers have in generating gene targeting events beyond the canonical CRISPR/Cas9 by increasing the number of accessible regions in the genome due to its AT-rich PAM and could better facilitate transgene integration by leveraging the variable staggered DNA break without the need of engineered nuclease variants or chemically modified crRNAs. Upon further optimization, our data suggest that Mad7 may be an invaluable tool for gene editing in both research and future clinical gene therapy applications.
Author Contributions
WAW, BWS, SCE, JJE conceived the study. WAW designed nlsMad7nls and conducted the zebrafish work. BWS conducted the human cell work. WAW and BWS wrote the manuscript with input from ZWJ, DD, KJC, MM, SCE, and JJE. JMW created the vector backbone for noto:RFP-DR48. CM analyzed the NGS data. WAG and MAB designed the in vitro knock in backbone and 5’ junction primer. All authors approved of the manuscript and signed off on the study.
Declaration of interests
WAW, ZWJ, BWS, SCE, JJE have a financial interest with LEAH Laboratories, a licensee from Mayo Clinic/Iowa State University for the filed GeneWeld patents. WAW, ZWJ, SCE, JJE have a financial interest with LifEngine Biotechnologies, a licensee from Mayo Clinic/Iowa State University for the filed GeneWeld patents. JJE has financial interests in Recombinetics, Inc.
Acknowledgements
The authors would like to acknowledge Inscripta for providing Mad7 as an open source nuclease for the gene editing community. pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA;U6::BsaI-sgRNA is a gift from Feng Zhang. H.A for experimental design discussions, K.M for providing insight into in vitro knock in experimental results, and G.M.G in assisting in analyzing NGS data. We would further like to thank the Mayo Clinic Microscopy and Cell Analysis Core for conducting flow cytometry and fluorescence activated cell sorting. This work was supported by NIH grants OD020166 and GM63904.
Materials and methods
Zebrafish husbandry
Zebrafish were maintained in Aquatic Habitats (Pentair) housing on a 14 hour light/10 hour dark cycle. Wild-type WIK were obtained from the Zebrafish International Resource Center. All experiments were carried out under approved protocols from Iowa State University IACUC.
nMad7n cDNA cloning
gBlocks were ordered from IDT with zebrafish codon optimized Mad7 cDNA sequences based on Inscripta public disclosure and the addition of dual NLS sequences at the 5’ and 3’ end of the cDNA (Supplementary Table 1). Three gBlock dsDNA templates were amplified with KOD HotStart DNA polymerase (EMD Millipore) using primers mad7f1/mad7r1, mad7f2/mad7r2, mad7f3/mad7r3, cut with respective restriction enzymes, and four-part restriction cloning into NcoI/SacII cut pT3TS vector backbone was performed (Plasmid #46757 Addgene). Sequence of Mad7 using primer walking and a full annotation was confirmed.
Injection protocol
Linear, purified pT3TS-nCas9n or pT3TS-nMad7n was used as template for in vitro transcription of capped, polyadenylated mRNA with the Ambion T3TS mMessage mMachine Kit. mRNA was purified using Qiagen miRNeasy Kit. The Cas9 universal sgRNAs were generated using cloning free sgRNA synthesis as described in Varshney et al., 2015 and purified using Qiagen miRNeasy Kit. All Mad7 pre-crRNA and crRNA was ordered as custom RNA oligos from Synthego with sequences described in Figure 1a.
Heat shock protocol
Immediately after injection, embryos were placed in a 34°C incubator for 4 hours. At 4 hours, embryos were sorted for fertilization and fertilized embryos were moved to 28°C incubator as normal.
DNA isolation and PCR genotyping
Genomic DNA for PCR was extracted by digestion of single embryos in 50mM NaOH at 95°C for 30 minutes and neutralized by addition of 1/10th volume 1M Tris-HCl pH 8.0. GoTaq Green was used as DNA polymerase master mix with the primers listed in Supplemental Table 2. AmpliconEZ from GeneWiz was used for NGS sequencing (see below) using primers mad7noto1fEZ and mad7noto1rEZ for noto target 1, mad7noto3fEZ and mad7noto3rEZ for noto target 3, and cxm7gRNA2fEZ, cxm7gRNA2rEZ for cx43.4 listed in Supplemental Table 2. GFP+ embryo 5’ junction fragments for noto target 1 and target 3 were PCR-amplified with primer notojxnf and gfp5’r listed in Supplemental Table 2. GFP+ embryo 3’ junction fragments for noto target 3 were PCR-amplified with primer GFP3’F and notojxnr listed in Supplemental Table 2. All junction fragment products were cloned into pCR4-TOPO vector and sequenced (Invitrogen).
Donor preparation
Donors were prepared and purified as described previously (Wierson et al., 2018). Homology arms are built as follows: One arm begins 13 bp 3’ of the PAM while the other arm begins immediately outside of the 3’ end of the crRNA target site. See Figure 3 and Figure 4 for homology arm design, and Supplemental Table 3 for GeneWeld homology arm oligos used for Golden Gate cloning. Gene targeting oligos and donor vector sequences are listed in Supplemental Table
Generating noto:RFP-DR48
Zebrafish RFP assay generation, injection, and line isolation pPRISM-V3 was PCR amplified with v3f and v3r to remove the ocean-POUT terminator and add SgrAI and SpeI cloning sites (Supplemental Table 2). Bactin 3’ UTR was PCR amplified using KOD polymerase with primers bactinf and bactinr to add SgrAI and SpeI enzyme sites for sticky end cloning. pPRISM-V3(pout negative) amplicon and bactin 3’ UTR were cut with SgrAI and SpeI. After ligation with Fisher Optizyme T4 Ligase and sequence verification to create pPRISM-V3-bactin, pPRISM-V3-bactin-SSA-DR48 was created by simultaneously adding the NBM and creating a direct repeat with phosphorylated primers DRf and DRr using pPRISM-V3-bactin with KOD polymerase followed by Fisher Optizyme T4 Ligation and sequence verification. To target these constructs to noto, homology domains up and downstream of a genomic CRISPR/Cas9 target site were chosen as described in Wierson et al., 2018. Oligos flhv35aflh, flhv35bflh, flhv33aflh, flhv33bflh, containing the gene targeting information, were added to pPRISM-V3-bactin RFP-DR48 using Golden Gate cloning as described in Wierson et al., 2018. The RFP cassette was liberated from the donor using the same noto gRNA used to cut the genome. Gamma-crystalin:eGFP positive embryos were sorted and raised to adulthood, outcrossed to generate the F1 generation, and outcrossed again to generate lines of F2s.
Southern blot analysis
Genomic Southern blot and copy number analysis was performed as described previously (McGrail et al. 2010). PCR primers used for genomic and donor specific probes are listed in Supplementary Table 2.
Cell culture
HEK293 cells were obtained from ATCC (CRL-3216). Cells were maintained in Dulbecco’s Modified Eagle Medium (Gibco #11995-040) supplemented with 10% fetal bovine serum (Gibco #26140079) and 1% Penicillin Streptomycin (Gibco #15140-122). Media was changed every 2-3 days and replated at final dilution of 1:10 maintained at about 750,000 cells/ml.
DNA isolation and PCR analysis
DNA from whole cell populations was purified using Qiagen DNeasy Blood & Tissue Kit (Qiagen 69504). PCR amplification was performed with MyTaq DNA Polymerase(Bioline BIO-21108) and purified with Qiagen QIAquick PCR Purification kit (Qiagen 28104). Samples used for ICE analysis were submitted to GeneWiz Sanger Sequencing service.
Cloning in vitro Mad7 construct targeting AAVS1 (pMad7-AAVS1)
Due to redundant restriction sites in the guide scaffold and Mad7 protein, the pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA;U6::BsaI-sgRNA plasmid (Addgene #61591) was digested with BsaI and NotI to first insert the Mad7 secondary structure and sgRNA targeting AAVS1 with Mad7 sgRNA AAVS1 top and Mad7 sgRNA AAVS1 bottom (termed AAV: Mad7 AAVS1 sgRNA) (Table 3). Following this Mad7 as well as the Xenopus globin 5’ UTR and both N and C termini SV40 NLS signals were amplified from “T3TS nMadn” using PCR primers Mad7 AgeI forward and Mad7 BamHI reverse (Supplementary Table 1). The resulting 3.9kbp PCR fragment was cloned into an Agilent pSC Strataclone PCR cloning vector (termed Mad7 Strataclone) to amplify the fragment with the desired restriction sites. Mad7 Strataclone was digested with AgeI and BamHI to isolate Mad7 with ends compatible with the AAV:Mad7 AAVS1 sgRNA construct. The px601 plasmid was likewise digested with AgeI and BamHI to remove SaCas9 and replace it with Mad7. Plasmids were screened for insertion of both Mad7 as well as the AAVS1 targeting sgRNA (pMad7-AAVS1) and amplified with Qiagen Endotoxin Free Maxiprep kit (Qiagen 12362).
pMad7-AAVS1-U
pMad7-AAVS1-U was generated by inserting Mad7 pre-U-crRNA into AAV 601 by digesting AAV601 with BsaI+ NotI and inserting annealed oligos Mad7 UgRNA+Scaffold Top and Mad7 UgRNA+Scaffold Bottom. The U6 promoter, the cr-RNA scaffold and the U-pre-crRNA were PCR amplified from AAV 601 using Mad7 U6+ugRNA Primer PfoI Fw and Mad7 U6+ugRNA Primer PfoI Rev. This PCR amplicon as well as pMad7-AAVS1 were digested with PfoI and ligated with T4 DNA ligase to generate pMad7-AAVS1-pre-crRNA1-U-pre-crRNA.
Generating knock-in cassette for AAVS1
The 24 and 48bp homology arm CMV/GFP/Zeocin resistance knock-in cassettes were generated by designing PCR primers complementary to the psiRNA-SV40 Early PolyA GFPzeo plasmid (Invivogen) flanked by the 48 base pairs of homology and the UgRNA sequence separated by a 3bp spacer. The left 48HA forward primer AAVSI T1 L48HA and the right 48HA reverse primer AAVS1 T1 R48HA (IDT) were used to amplify the CMV/GFP/Zeocin resistance cassette containing the homology arms and the UgRNA target sequence (Supplementary Table 2, Supplementary Table 3). This 2.6kbp PCR fragment was subsequently cloned into a Strataclone PCR cloning vector and screened for the insert. Sequence confirmed knock in constructs were amplified with Qiagen Maxiprep Kit and termed “pGFP::Zeo-48”.
Generating Mad7 EZ Clone
nMad7n was PCR amplified from pT3TSnMad7n with Platinium Taq DNA polymerase HiFi (Thermo Fisher #11304011) with Mad7 AgeI Fw and Mad7 BamHI Rev and PCR cloned into the Agilent Strataclone vector (Agilent #240205) to generate Mad7 Strataclone. Mad7 Strataclone was subjected to site directed mutagenesis with Mad7 SDM remove BsaI Top and Mad7 SDM remove BsaI bottom with the Q5 SDM kit (New England Biolabs #E0554S) to generate Mad7 Strataclone no BsaI. The nMad7n fragment lacking the BsaI site was removed from the Mad7 Strataclone no BsaI backbone by digesting with AgeI and BamHI. Likewise the pX601-AAV-CMV::NLS-SaCas9-NLS-3xHA-bGHpA;U6::BsaI-sgRNA plasmid (Addgene #61591) was digested with AgeI and BamHI and had nMad7n inserted into it to generate p601nMad7n no BsaI. The cr-RNA scaffold was inserted into the p601nMad7n no BsaI by digesting with BsaI and NotI and inserted the annealed oligos Mad7 EZ clone scaffold top and Mad7 EZ clone scaffold bottom to generate Mad7 EZ Clone.
Cloning in vitro Mad7 construct targeting CCR5 and TRAC
Cr-RNA oligos corresponding to the genomic target site (e.g. CCR5 sgRNA 1 Top+ CCR5 sgRNA 1 Bottom) were annealed in a thermocycler according to Zhang lab protocol. Briefly, oligos are annealed by incubating at 37°C for 30 minutes followed by 95°C for 5 minutes and then ramped down to 25°C at 5°C/min. The annealed oligo duplex was cloned into Mad7 EZ clone Digested with BsaI and ligated by T4 DNA ligase.
Generating pMiniCAAGs:RFP-DR48
RFP assay generation, transfection, and analysis pPRISM-V3-bactin SSA-DR48 was PCR amplified with Platinum Taq DNA polymerase HiFi (Thermo Fisher #11304011) with Broken RFP Transfer to Tol2 XhoI Fw and Broken RFP Transfer to Tol2 BglII Rev to add XhoI and BglII restriction sites to the RFP cassette. pkTol2C-EGFP as well as the RFP amplicon were digested with XhoI and BglII to create compatible cohesive ends and ligated with T4 DNA ligase to generate pkTol2CBrokenRFP. In order to for the construct to express episomally in cell culture systems a kozak sequence was added by digesting pkTol2CBrokenRFP with EcoRI and XhoI and restriction cloning the annealed oligos Kozak Seq Top and Kozak Seq Bottom to generate pMiniCAAGs:RFP-DR48.
Mad7 constructs targeting the UgRNA were generated by digesting Mad7 EZ clone with BsaI and cloning in the annealed oligos Mad7 UgRNA BsaI Top and Mad7 UgRNA BsaI bottom to generate pMad7-U-pre-crRNA. Cas9 constructs targeting the UgRNA were generated by digesting lentiCRISPR v2 (Addgene #52961) with BsmBI and cloning in the annealed oligos Cas9 UgRNA BsmBI Top and Cas9 UgRNA BsmBI Bottom to generate pCas9-UgRNA. To control for promoter expression between Mad7 and Cas9, the CMV promoter was added to pCas9-UgRNA by digesting Mad7 EZ clone with XbaI and AgeI to remove the CMV promoter and pCas9-UgRNA with NheI and AgeI to replace the native EF1 alpha promoter.
HEK 293T cells were transfected with 5ug pMiniCAAGs:RFP-DR48 and 5ug of either pMad7-U-pre-crRNA or pCas9-UgRNA with the Etta H1 electroporator as described above. Cells were assessed for RFP expression 48 hours later by flow cytometry with 584nm emission and 607nm detection.
Transfection
Cells used for indel acquisition assays were transfected with Liopofectamine 3000 (Thermo Fisher #L3000008) according to manufacturer’s protocol with 5ug of Mad7 plasmid targeting each site. Cells used for targeted integration assays were transfected with Etta H1 electroporator with the following parameters: 200V, 784ms interval, 6 pulses, 1000us pulse duration, at a concentration of 20E6 cells/ml at the volume of 100ul in Etta EB electroporation buffer. Cells are recovered post electroporation by incubating at 37°C for 5-10 minutes before being plated in a 6-well tissue culture plate at a density of about 1.5E6cells/ml.
GeneWiz AmpliconEZ
DNA Library Preparation and Illumina Sequencing
DNA library preparations, sequencing reactions, and initial bioinformatics analysis were conducted at GENEWIZ, Inc. (South Plainfield, NJ, USA). DNA Library Preparation, clustering, and sequencing reagents were used throughout the process using NEBNext Ultra DNA Library Prep kit following the manufacturer’s recommendations (Illumina, San Diego, CA, USA). End repaired adapters were ligated after adenylation of the 3’ends followed by enrichment by limited cycle PCR. DNA libraries were validated on the Agilent TapeStation (Agilent Technologies, Palo Alto, CA, USA), and were quantified using Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA) and multiplexed in equal molar mass. The pooled DNA libraries were loaded on the Illumina instrument according to manufacturer’s instructions. The samples were sequenced using a 2x 250 paired-end (PE) configuration. Image analysis and base calling were conducted by the Illumina Control Software on the Illumina instrument.
Data analysis
The raw Illumina reads were checked for adapters and quality via FastQC. The raw Illumina sequence reads were trimmed of their adapters and nucleotides with poor quality using Trimmomatic v. 0.36. Paired sequence reads were then merged to form a single sequence if the forward and reverse reads were able to overlap. The merged reads were aligned to the reference sequence and variant detection was performed using GENEWIZ proprietary Amplicon-EZ program.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}