

Abstract
Excitatory neurons of the mammalian cerebral cortex are organized into six functional layers characterized by unique patterns of connectivity, as well as distinctive physiological and morphological properties. Developmentally, cortical layers emerge after a highly regulated neuronal migration process from the ventricular zone, where proliferating cells reside, toward the superficial layers, where the early cortex starts to develop. Importantly, defects in this radial migration process have been implicated in neurodevelopmental and psychiatric diseases. Here we report that during the final stages of migration, transcription factor Neurogenic Differentiation 2 (Neurod2) contributes to terminal cellular positioning within the cortical plate and alignment of nascent dendrites toward the marginal zone. In mice, in utero knockdown of Neurod2 results in misaligned primary dendrites of migrating neurons within the nascent cortical plate. Our ChIP-Seq and RNA-Seq analyses of NEUROD2-regulated genes in the developing cortex identify Reelin (Reln), a critical regulator of neuronal migration, as a key downstream target. NEUROD2 binds to conserved E-box elements in multiple introns of the Reln gene, and binding is associated with suppression of gene expression. These data reveal a new role for NEUROD2 during neuronal migration and suggest that fine-tuning of Reln gene expression in migrating neurons is important for terminal cellular positioning of neurons upon arrival to the cortical plate.
Introduction
The cerebral cortex of higher animals is composed of myriad excitatory and inhibitory neuron types organized into six anatomical and functional layers. Developmentally, excitatory neurons originate from neural stem cells that divide in the ventricular zone of the anterior neural tube, then migrate radially to the overlying cortical plate. While neural stem cells initially divide symmetrically during an increase of the proliferative population, the division mode transitions to asymmetric division as embryogenesis progresses. One daughter cell from each cell division remains at the ventricular zone, while the other daughter exits cell cycle and initiates a complex mode of radial migration to the border between the cortical plate and the marginal zone. As development progresses, each wave of migrating neurons reaches the surface by passing the nascent cortex in an inside-out manner, eventually giving rise to the layered structure of the cerebral cortex (Kwan et al., 2012; Tan and Shi, 2013). Consequently, early-born neurons populate the deep layers, and late-born neurons settle in the superficial layers. The precise control of the radial migration process is essential for the proper formation of the six cortical layers. Importantly, in mice and humans, mutations, infections or environmental agents that perturb cortical migration cause cortical structural malformations, intellectual disability, neuropsychiatric diseases and seizures (Buchsbaum and Cappello, 2019; Hu et al., 2014; Romero et al., 2018).
A closer examination of the radial migration process reveals a series of different modes of cellular motility, as opposed to a uniform directed movement from the ventricular zone toward the superficial marginal zone. Initially neurons are multipolar in morphology as they emerge from the ventricular zone, which is followed by transition to a bipolar morphology and locomotion on radial glia fibers through the intermediate zone and much of the cortical plate (Hippenmeyer, 2014; Ohtaka-Maruyama and Okado, 2015). Upon approaching the border between the cortical plate and the marginal zone (MZ), they terminally translocate from the radial glia fibers. Terminal translocation is accompanied by positioning of neuronal cell bodies in the uppermost layer of the cortex, also termed as the primitive cortical zone (Sekine et al., 2011). During this developmental period, the marginal zone is populated by Cajal-Retzius cells which secrete the extracellular glycoprotein REELIN, considered a stop signal for migrating neurons (Hirota and Nakajima, 2017). Migrating neurons express the REELIN receptors VLDLR and APOER2 (LRP8), as well as the intracellular adaptor molecule DAB1, which conveys receptor activation to downstream signaling pathways. Ligand binding induces the endocytic internalization of REELIN and phosphorylation of DAB1 (Hirota and Nakajima, 2017). Activation of REELIN signaling in neurons induces anchorage of nascent primary dendrites to the extracellular matrix of the marginal zone, stabilization of the neuronal skeleton, and termination of migration (Chai et al., 2015; Franco et al., 2011; Matsuki et al., 2010; Olson et al., 2006; Sanada et al., 2004; Sekine et al., 2012). Loss of REELIN, VLDLR, APOER2 or DAB1 function causes profound defects in cortical lamination; the cortical layers develop in an inverted pattern, with early- and late-born neurons being located in superficial and deep layers, respectively (Caviness and Sidman, 1973; D’Arcangelo et al., 1995; Gonzalez et al., 1997; Howell et al., 1997; Ogawa et al., 1995; Sanada et al., 2004; Trommsdorff et al., 1999).
Individual steps of radial migration are controlled by the combinatorial expression of specific transcription factors (TFs) (Kwan et al., 2012). Prominent among these transcription factors are the bHLH family members, including the Neurogenins and the NeuroDs. In addition to their roles in neuronal cell fate specification and neuronal differentiation, these factors have also been shown to control specific steps of cortical radial migration described above (Hand et al., 2005; Heng et al., 2008; Pacary et al., 2011). A key member of the NeuroD family of neurogenic bHLH TFs is NEUROD2. The Neurod2 gene is highly expressed in the developing cortex and its expression persists, albeit at low levels, into adulthood in cortical excitatory neurons (Guner et al., 2017; Olson et al., 2001). Interestingly, several recent studies by our group and others have implicated NEUROD2 in the radial migration process of cortical neurons and have provided a general overview of its downstream genetic targets (Bayam et al., 2015; Telley et al., 2016). However, how NEUROD2 regulates the expression of its downstream targets and how this regulation impacts cortical migration remain largely unknown.
We previously characterized the genetic targets of NEUROD2 in the cerebral cortex at two developmental timepoints: embryonic day 14.5 (E14.5), representing the peak of neurogenesis and migration in the mouse cortex; and postnatal day 0 (P0), representing the onset of neuronal differentiation, dendritic growth and synaptogenesis (Bayam et al., 2015; Guner et al., 2017). Here, we carry out a comparative analysis of these two datasets and overlay it with transcriptomics analysis of primary neurons where Neurod2 expression is knocked down. We find that NEUROD2 exhibits quantitative differences in target-selectivity at these two developmental timepoints. Specifically, our genome-wide comparative analysis lead to the identification of significantly higher levels of NEUROD2 association with several introns located in the Reln gene at P0. Interestingly, although NEUROD2 has long been assumed to act as a transcriptional activator, we find that NEUROD2 binding to Reln introns is associated with a suppression of gene expression. Furthermore, consistent with previously described roles for REELIN protein as a stop signal for migration, suppressing Neurod2 expression in cortical neurons causes a defect in their cellular positioning to the primitive cortical zone and is accompanied by aberrant primary dendrite orientation. Our results point to NEUROD2 as a regulator of radial migration that acts, at least in part, by fine-tuning levels of Reln expression.
Results
NEUROD2 binds to overlapping and unique gene-sets in embryonic and postnatal cerebral cortex tissue
To evaluate whether NEUROD2 interactions with specific genomic sites are regulated developmentally, we compared NEUROD2 binding profiles between embryonic day 14.5 (E14.5) and postnatal day 0 (P0). We retrieved binding scores from our previously published NEUROD2 ChIP-Seq datasets that represent the amount of relative TF binding to target sequences and carried out a comparative analysis using the E14.5 and P0 results (Bayam et al., 2015; Guner et al., 2017). Based upon this comparison, we identified a large number of binding sites that were exclusively targeted at E14.5, and fewer sites that were targeted at both developmental stages or were specific to P0 (Supplemental Material 1) (Fig. 1A). For both datasets, we had previously reported that a majority of NEUROD2 binding sites were not located at promoters, but rather at distant regulatory elements. Therefore, we decided to employ a target gene prediction approach, called the Closest Gene Method, which generates a score based on the number of TF binding sites and their proximity to the transcription start site of each gene (Sikora-Wohlfeld et al., 2013). We calculated Closest Gene scores for the P0 dataset and compared it to the E14.5 dataset which we had previously published (Supplemental Material 2) (Bayam et al., 2015). Genes with a Closest Gene score above a threshold value were predicted as targets. Comparison of these predicted targets between two developmental stages revealed a large number of E14.5-specific genes, as well as a smaller number of target genes either only in the P0 developmental stage or at both stages (Fig 1A). In order to determine whether NEUROD2 gene targets at embryonic and postnatal timepoints represented functionally distinct gene groups, we carried out gene ontology analysis. Not surprisingly, at both tested developmental stages, genes related to nervous system development were significantly enriched. However, genes associated with migration of different types of neurons were specifically enriched at E14.5 (Fig. 1B). Collectively these data suggested that NEUROD2, a TF that is expressed throughout the lifetime of cortical excitatory neurons, may regulate distinct functions during different developmental events.
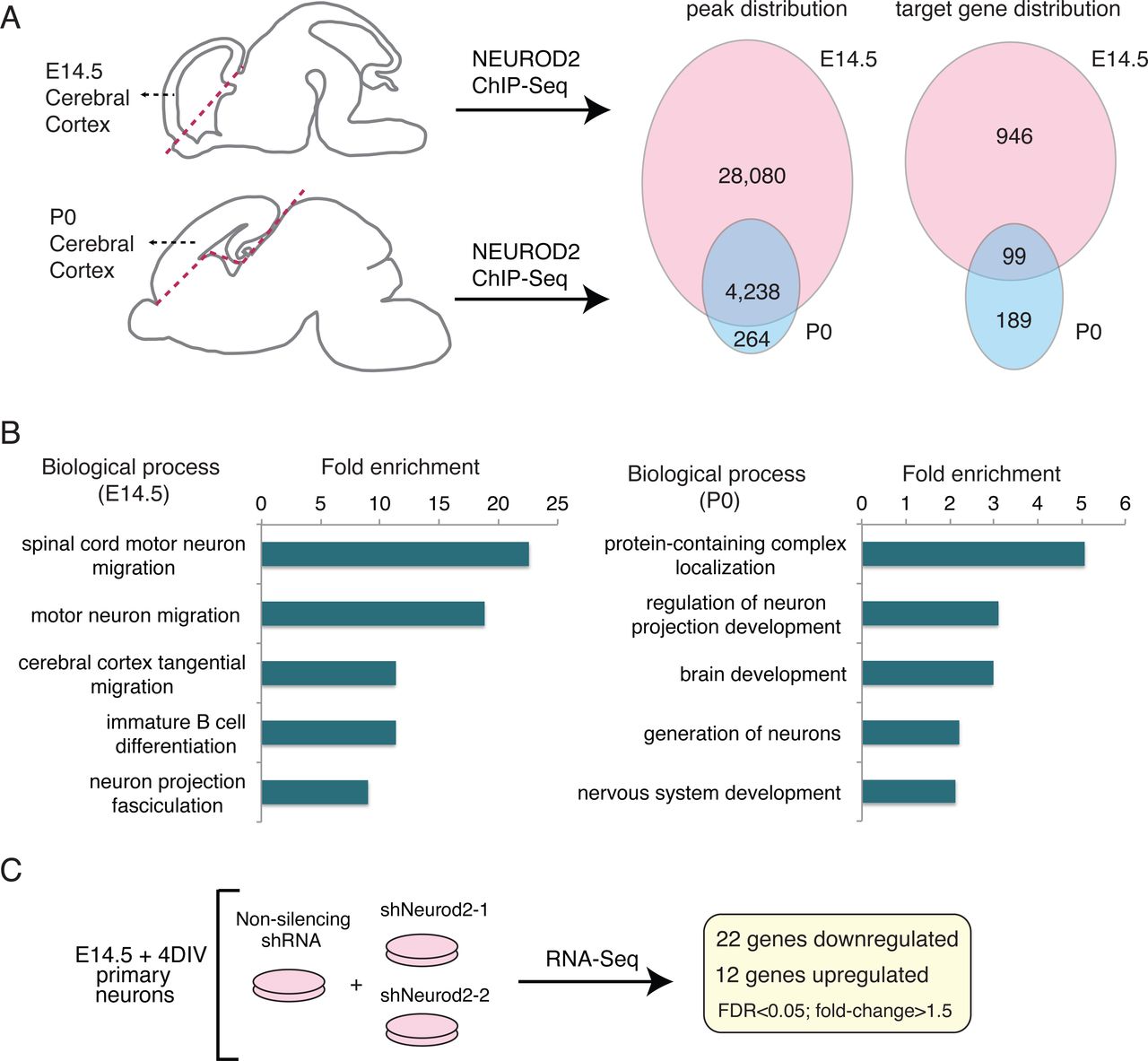
NEUROD2 binds to overlapping and unique target sites in embryonic and postnatal cerebral cortex. (A) NEUROD2 binding sites and target genes were compared between embryonic day 14.5 (E14.5) and postnatal day 0 (P0) cerebral cortical tissue. Numbers of genome-wide binding sites and target genes are based upon previously acquired ChIP-Seq data (Bayam et al., 2015; Guner et al., 2017). Target genes were identified based on the total number of binding sites proximal to the transcription start sites of individual genes (Sikora-Wohlfeld et al., 2013). (B) Gene ontology analysis of NEUROD2 target genes from P0 and E14.5 datasets (geneontology.org). (C) RNA-Seq analysis was carried out on primary cortical neurons electroporated with a non-silencing shRNA or one of two independent shRNAs against Neurod2. 22 genes were down-regulated and 12 were up-regulated genes upon knockdown of Neurod2.
To further focus our attention upon genes whose expression is regulated by NEUROD2, we silenced Neurod2 expression in primary cortical cultures using two validated shRNAs (Guner et al., 2017) and analyzed gene expression changes upon Neurod2 knockdown. When gene expression was compared to cells treated with a control shRNA, we found that 22 genes were upregulated and 12 genes were downregulated upon silencing of Neurod2 (Supplemental Material 3) (Fig. 1C). As expected, we detected Neurod2 as one of the significantly downregulated genes. Genes perturbed by Neurod2 depletion included those expressing DNA binding proteins (Arid5a, Ddit4, Ubn2, Trim66, Zglp1, and Sp8); regulators of extracellular matrix, adhesion and synaptogenesis (Thbs1, Reln, Col3a1, Col1a1, and Ank1), metabolic genes (Shmet2, Tkt, Pck2, and Pde12); signaling molecules (Chac1 and Sesn2) and a translational regulator (Rps6ka4). Next, we asked whether any of the regulated targets were also directly bound by NEUROD2 as revealed by our ChIP-Seq data. We found that 14/34 genes harbored binding sites for NEUROD2, with the remaining (20/34) potentially represented secondary or off-target effects (Supplemental Material 3). Among the direct and regulated targets of NEUROD2, we were intrigued to identify Reln, a gene with essential roles in neuronal migration, dendrite development and synaptogenesis. Our previous findings (Bayam et al., 2015) had also implicated NEUROD2 in regulating the signaling pathway activated by this extracellular matrix protein. Therefore, we decided to further investigate how NEUROD2 regulates Reln expression.
NEUROD2 regulates the expression of Reln gene in cortical neurons
In mice, Reln gene consists of 65 exons, spanning approximately 460 kb of genomic sequence (O’Leary et al., 2016). Upon inspection of NEUROD2 binding sites along this gene we identified four prominent peaks at introns 21, 22, 55 and 63 (Fig. 2A). Initially, we confirmed binding to all four peak sequences by ChIP followed by quantitative PCR (ChIP-qPCR) using cortical tissue from E14.5 and P0 pups (Fig. 2C). We normalized our qPCR signal individually from all four peaks to the amount of input DNA and compared to negative control samples where immunoprecipitation was carried out with an antibody against GFP. Our results suggested quantitatively more binding to all four intronic sequences from E14.5 cortex when compared to samples from P0 cortex (Fig. 2C).

NEUROD2 binding to introns along the Reln gene is associated with suppression of Reln gene expression. (A, B) Four prominent intronic NEUROD2 binding sites are plotted on the Reln gene. Several different histone modifications corresponding to promoters (H3K4me3), enhancers (H3K4me1), actively transcribed (H3K27ac and H3K36me3) or repressed (H3K9me3 and H3K27me3) chromatin are not enriched in NEUROD2 binding sites. A slight enrichment of CTCF binding within intron 3 of Reln is detected. (C) NEUROD2 binding to all four intronic regions is confirmed by ChIP-qPCR. Immunoprecipitation with an unrelated GFP antibody is used as a negative control. n= 12 (three biological x four technical replicates). Bars represent S.E.M. *p<0.05, **p<0.005, ***p<0.0001 by two-tailed unpaired Student’s t-test. (D) RT-qPCR analysis of Reln gene expression reveals significant upregulation upon knockdown of Neurod2 with two separate shRNAs. n= 9 (three biological x three technical replicates). Bars represent S.E.M. *p<0.0001 by two-tailed unpaired Student’s t-test.
In order to gain insight into the transcriptional activity of these sites in the embryonic cortex, we overlaid our NEUROD2 binding profiles with those representing different histone marks associated with promoters (H3K4me3), enhancers (H3K4me1), actively transcribed (H3K27ac and H3K36me3) or silenced (H3K9me3 and H3K27me3) chromatin (Fig. 2B). We did not observe a significant enrichment of either of these histone marks at NEUROD2 binding sites. Our observation might be indicative of a true lack of enrichment of specific histone marks at these loci or might be due to the dilution of ChIP-Seq signals of histone marks acquired from bulk cortical tissue containing many cells that do not express Neurod2. Interestingly, we did observe a modest enrichment of CTCF (CCCTC-binding factor) (Fig. 2B). This TF has previously been shown to act as a chromatin insulator and as either a transcriptional repressor or activator in a context-dependent manner (Arzate-Mejia et al., 2018; Chen and Lei, 2019; Prickett et al., 2013). Hence, beyond functioning as a typical promoter-associated TF, NEUROD2 might have additional roles in formation of higher-order chromatin structures. Future experiments conducted with purified Neurod2-expressing neurons will resolve how histone codes and binding of other factors are regulated at NEUROD2 binding sites.
During radial migration, Reln is expressed and secreted by a transient group of neurons located in the marginal zone called Cajal-Retzius (CR) cells (D’Arcangelo et al, 1995; Ogawa et al, 1995), which do not express Neurod2 (Yamazaki et al, 2004). On the other hand, migrating neurons express Neurod2, genes encoding REELIN receptors Lrp8 (Apoer2) and Vldlr, as well as components of their downstream signaling pathway, such as the adaptor protein Dab1. Recent single cell RNA-Seq analysis also point to a mutually exclusive pattern of expression of Reln and Neurod2 within the CR cells and within excitatory neurons of the developing cortex (Fan et al., 2018). Interestingly, our RNA-Seq results also suggested NEUROD2 as a negative regulator of Reln expression (Fig. 1C, Supplemental Material 3). To further explore the possibility that NEUROD2 represses Reln, we knocked down Neurod2 by two different shRNAs in primary cortical neurons. Indeed, we observed a significant upregulation in Reln expression (Fig. 2D) with the more efficient shNeurod2-1 RNA (Guner et al, 2017) leading to a more robust upregulation in Reln transcript levels compared to those neurons transfected with shNeurod2-2 (Fig. 2D). Taken together, our results suggest that NEUROD2 suppresses expression of Reln by binding to intragenic sites.
NEUROD2 is required for the terminal stages of cortical radial migration
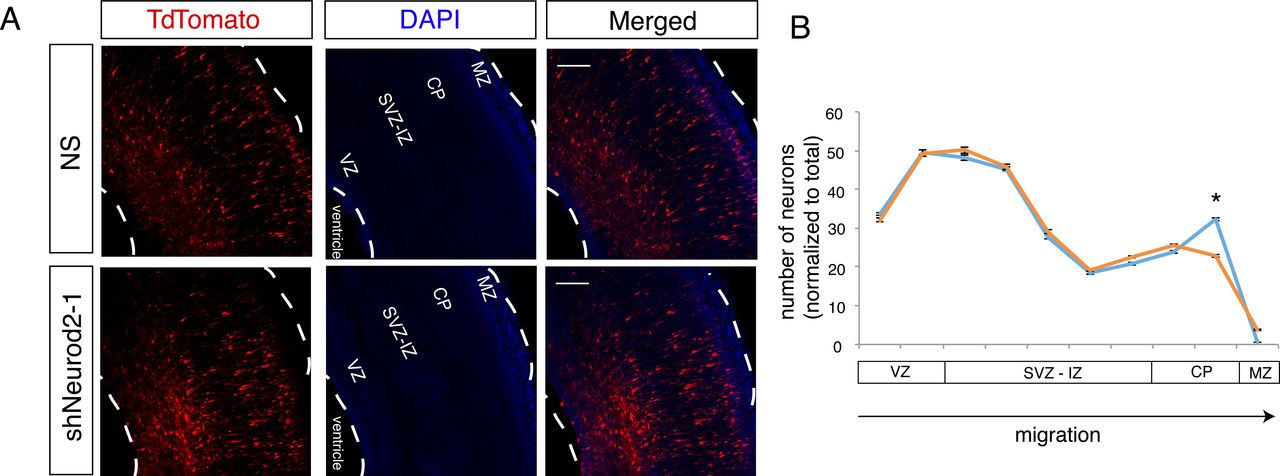
Here, we have demonstrated that Reln expression is negatively regulated by NEUROD2, and our previous results had implicated NEUROD2 in controlling the expression of the REELIN receptor (Lrp8) and its downstream signaling components (Bayam et al., 2015). Since the REELIN signaling pathway determines the proper migration of cortical neurons, we decided to test whether loss of NEUROD2 would affect this process. Toward this aim, we silenced Neurod2 in neural progenitor cells by in utero transfection of shNeurod2-1 and a tdTomato marker in E14.5 embryos, then quantified the number of tdTomato-positive neurons in different cortical layers at E17.5 by confocal microscopy (Fig. 3A and B). In control samples, we observed tdTomato-positive neurons at expected locations within the ventricular zone, along the subventricular and intermediate zones, and at the cortical plate. In samples in which we silenced Neurod2 expression, we did not observe gross defects in migration, and neurons generally appeared to migrate from the ventricular zone toward the cortical plate in a manner comparable to control samples. Specifically, multipolar migration and glial-guided migration appeared intact. However, we observed a striking reduction in the number of tdTomato-positive neurons in the primitive cortical zone (right below the marginal zone), suggesting that NEUROD2 depletion leads to a defect specific to the terminal stage of migration (Fig. 3A and B).

NEUROD2 is essential for neuronal soma positioning to the primitive cortical zone. (A) Embryos (at E14.5) were in utero electroporated with tdTomato fluorescent marker and with non-silencing shRNA or with shNeurod2-1. Embryos were retrieved at E17.5, coronally sectioned and tdTomato signal was subsequently imaged by confocal microscopy. VZ (ventricular zone), SVZ-IZ (subventricular-intermediate zone), CP (cortical plate), and MZ (marginal zone) are labeled. Scale bar: 100 µm. (B) Quantification of images from (A). Numbers of tdTomato-positive neurons as a function of distance from the ventricle are plotted. Significantly less neurons are localized to the upper cortical plate in neurons where Neurod2 is knocked down. Bars represent S.E.M. *p<0.05 by two-tailed unpaired Student’s t-test.
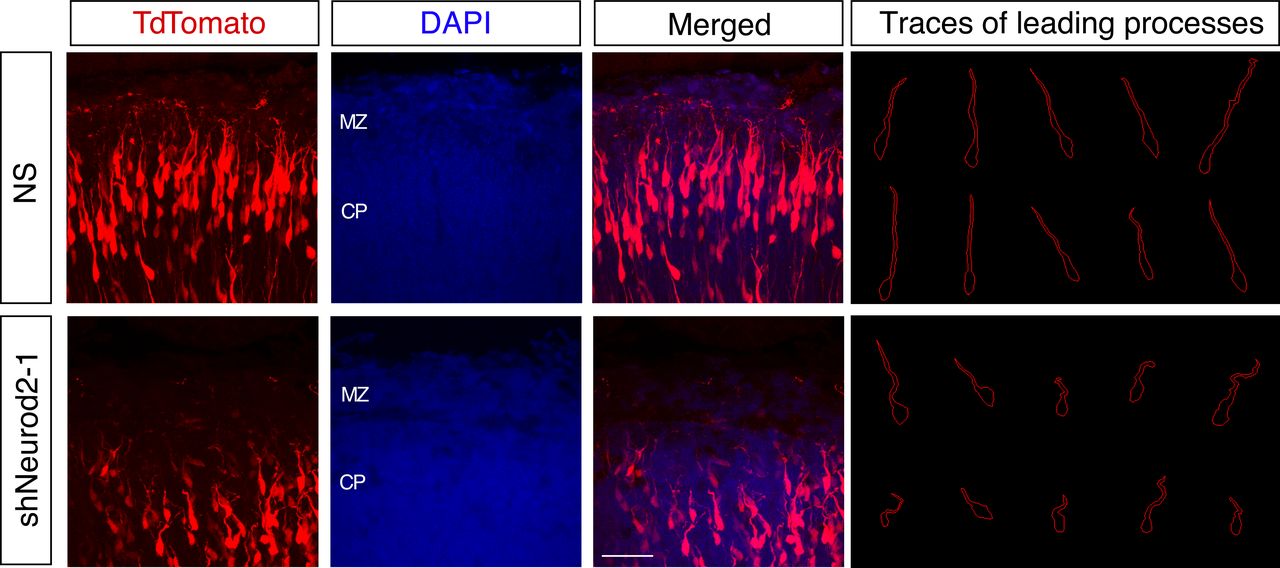
During the end stage of migration, neurons lose contact with radial glial processes and position their cell bodies in the upper cortical plate by a process called terminal translocation. Simultaneously they anchor their leading processes to the extracellular matrix of the marginal zone, which harbors REELIN-expressing CR cells (D’Arcangelo et al, 1995; Hirotsune et al, 1995; Ogawa et al, 1995). Past studies have demonstrated an essential role for REELIN signaling in all of these processes (O’Dell et al, 2015; Franco et al, 2011; Olson et al, 2006). Therefore, we decided to examine how cell bodies were positioned and how leading processes were oriented in neurons characterized by reduced levels of NEUROD2. Upon examination of higher magnification images of embryonic cortices from control samples, we observed leading processes that were properly oriented toward the pial surface and that arborized the target MZ. However, in neurons which were in utero electroporated with shNeurod2-1, we observed stunted and misaligned leading processes which did not reach the MZ, as well as cell bodies that were inappropriately positioned below the MZ border (Fig. 4). Taken together, our results point to a mechanism where NEUROD2 suppresses ectopic expression of Reln in migrating neurons and consequently inhibits premature somal positioning before neurons reach the primitive cortical zone.

NEUROD2 is important for leading process orientation of migrating neurons in the upper cortical plate. E14.5 embryos were in utero electroporated with non-silencing or shNeurod2-1, retrieved at E17.5, and cortical plates were imaged by confocal microscopy. Traces of leading edges of migrating neurons in the upper cortical plate reveal disorganized and stunted processes when Neurod2 expression is suppressed. Representative images from three embryos (from three independent pregnancies) were traced from controls and neurons where Neurod2 expression was knocked down. Scale bar: 50 µm.
NEUROD2 is required for dendritic differentiation of cortical neurons
Upon completion of neuronal migration, the leading processes transform into apical dendrites of the cortical pyramidal neurons (Hatanaka and Murakami, 2002; O’Dell et al., 2015). REELIN signaling has been shown to regulate the arborization and branching of these apical dendrites in the developing cortex, as well as dendrites of dissociated hippocampal primary neurons from postnatal cortices (Gladyshev et al., 1996; Niu et al., 2004; O’Dell et al., 2015). Therefore, we asked whether neurons silenced for Neurod2 also exhibited generalized dendritic arborization defects. Toward this aim, we cultured primary cortical neurons and transfected them with an EGFP marker and shNeurod2-1 at low efficiency to achieve Neurod2 knockdown in isolated neurons (Fig. 5A). We then quantified dendritic arborization by Sholl analysis, an approach that reports upon the number of dendrite branches found at various distances from the neuronal soma (Ferreira et al., 2014). Our results demonstrated that while the number of primary dendrites protruding from the soma were comparable between the two conditions, a significant reduction was observed in higher order dendritic branches in neurons where Neurod2 expression was silenced (Fig. 5A and 5B). Together, in vitro results from primary cortical cultures, as well as in vivo results obtained from embryonic cortex, indicate that NEUROD2 is essential for proper dendrite arborization.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
NEUROD2 is required for normal dendrite development in primary cortical neurons. (A) Primary cortical neurons from E14.5 embryos were transfected with non-silencing shRNA or shNeurod2-1 at 2 days DIV and fixed at 5 DIV. Images were captured by confocal microscopy, scale bar: 20 µm. (B) Dendrite development was quantified by Sholl analysis. n=35 neurons derived from two separate cultures. Bars indicate S.E.M. p=0.0026 by two-way ANOVA.
Discussion
Here we report that by fine-tuning expression of the Reln gene, NEUROD2 regulates cell body positioning and primary dendrite orientation in neurons during the terminal stage of radial migration. NEUROD2 is a neurogenic TF that is highly expressed by early neurons while they are radially migrating from the ventricular zone toward the marginal zone in the developing cortical plate. However how NEUROD2 regulates neuronal migration was unknown.
We had previously reported genomic targets of NEUROD2 both in embryonic and postnatal cerebral cortices (Bayam et al., 2015; Guner et al., 2017) and here we complemented these earlier studies by a comparative analyses of these two developmental timepoints. Furthermore, in order to focus upon targets whose expression is regulated by NEUROD2, we silenced its expression in primary cortical neurons and carried out an RNA-Seq analysis. Our comprehensive and comparative analyses of these two ChIP-Seq and RNA-Seq datasets converged upon the Reln gene. We show that in mice, NEUROD2 binds to several different introns of Reln both embryonically and postnatally to suppress its expression. Even though NEUROD2 has typically been considered as a transcriptional activator, our studies point to a dual function for NEUROD2; in addition to its previously reported transcription activating function (Aizawa et al., 2004; Ince-Dunn et al., 2006; Olson et al., 2001), it can also act as a repressor in a context-dependent manner. In support of our findings, Reln expression was shown to be strongly upregulated in the embryonic cortex of Neurod2/6 double knockout mice (Bormuth, 2015). NEUROD2’s capacity to function as a transcriptional repressor agrees well with our previous results demonstrating that NEUROD2 inhibits expression of the Stim1 gene to control calcium signaling in cortical neurons (Guner et al., 2017). Furthermore, several of the NEUROD2 binding peaks both in Reln and Stim1 genes have also been identified in ChIP-Seq experiments carried out for the chromatin insulating factor CTCF (encodeproject.org), suggesting that NEUROD2 might be involved in establishing or maintaining higher order architecture of chromatin loops. In support of this hypothesis, NEUROD2 was recently shown to function as a major organizer of cell-type specific chromatin folding in cortical neurons, suggesting that its transcription activating or repressing functions might in fact be directly related to its role in establishing or maintaining transcriptionally isolated domains (Bonev et al., 2017). The mechanistic details of how NEUROD2 regulates transcriptional activity in distinct genetic loci remain interesting problems for future investigation.
Past studies have suggested that NEUROD2 might be crucial for regulating neuronal migration (Bayam et al., 2015; Bormuth et al., 2013; Telley et al., 2016). In addition to our current finding demonstrating NEUROD2 regulation of Reln gene expression, our past work had shown that it also binds to promoters of several genes encoding downstream signaling proteins that transduce REELIN signals, such as Dab1 and Fyn, as well as the receptor gene Lrp8 (Bayam et al., 2015). These findings suggest that NEUROD2 might impact cortical lamination by regulating multiple components of the REELIN signaling pathway. Here we silenced Neurod2 expression in ventricular neural progenitors by in utero electroporation in mice and observed that NEUROD2 is important for proper positioning of the neuronal soma upon arrival at the primitive cortical zone and primary dendrite arborization within the MZ. Hence, we hypothesize that by suppressing Reln expression in migrating neurons, NEUROD2 contributes to a mechanism where primary dendrites of neurons that have reached the upper cortical plate are exposed to high levels of REELIN only in the MZ and not prematurely within deeper layers. Such a mechanism would ensure compartmentalization of REELIN signaling within the apical dendrite and contribute to precise spatial regulation of terminal translocation and apical dendrite anchorage in the MZ.
The control of Reln gene expression within a precise spatiotemporal window in the developing cortex appears to be a conserved mechanism regulated by several TFs. For example, double-knockout mice for the TFs Pbx1/2 also exhibit ectopic Reln expression, leading to a dramatic inversion of cortical layering reminiscent of mouse models with defective REELIN signaling (Golonzhka et al., 2015). Similarly, knockdown of histone methyl transferase Ezh2 and deletion of TF Foxg1 result in derepression of Reln gene expression, associated with radial migration and cortical development defects (Cargnin et al., 2018; Zhao et al., 2015). Finally, ectopic expression of Reln cDNA by in utero electroporation into neural progenitors causes formation of neuronal aggregates in deep layers that are incapable of migrating towards the cortical plate (Kubo et al., 2010). Taken together, these findings highlight the importance of maintaining a precise level of Reln gene expression for the proper development of cortical layers, and our results place NEUROD2 as a novel transcriptional regulator of the REELIN signaling pathway, primary dendrite orientation and somal positioning within the developing cortical plate.
Methods
Bioinformatic analyses of ChIP-Seq data
Previously published NEUROD2 ChIP-Seq data were used to identify differential NEUROD2 binding to genomic regions at timepoints E14.5 and P0 (Bayam et al., 2015; Guner et al., 2017). Histone ChIP-Seq data from E14.5 mouse embryonic cortex were produced by Bing Ren’s Laboratory, UCSD, USA (Consortium, 2012) (www.encodeproject.org). ChIP-Seq peaks were visualized using Trackster (Goecks et al., 2012) embedded within the Galaxy Project (Giardine et al., 2005). Details of Closest Gene score calculations were previously described (Bayam et al., 2015).
ChIP-qPCR
Chromatin immunoprecipitation with NEUROD2 antibody was carried out using cortical tissue collected from E14.5 and P0 pups of both sexes using an antibody raised against NEUROD2 (Abcam, ab109406) and against GFP as a negative control (Santa Cruz, sc-8334). Cross-linking, followed by chromatin DNA isolation from input and immunoprecipitated samples, was performed as described previously (Bayam et al., 2015). Cq values from immunoprecipitated DNA were normalized to those from input DNA as described (Guner et al., 2017). Primer sequences used for qPCR experiments were as follows: Reln-peak1-F (AATGGAAACTGGCTCGCATG); Reln-peak1-R (ATCCTGAGCAATGAGTGGCT); Reln-peak2-F (TGTTTCTGTGGTCTGCTTGC); Reln-peak2-R (AAAACAATCACAGGCGAGCC); Reln-peak3-F (GCCGGACTACCCTGATGATT); Reln-peak3-R (TGGAGGAAATGGATGGCTCT); Reln-peak4-F (GGCCTCCTGTCTTACTAGCC); Reln-peak4-R (CCTGACAGATGGAGCGTTTG).
RNA-Seq and RT-qPCR
Primary cultures were prepared from E14.5 embryos as described (Guner et al., 2017). Cortical neuron suspensions were electroporated with shNeurod2-1, shNeurod2-2, or a non-silencing plasmid immediately before plating (Amaxa™ P3 Primary Cell 4D-Nucleofector X Kit L; Lonza, CU133 program). The generation and knockdown efficiencies of shRNA plasmids were described previously (Guner et al., 2017). Total RNA was isolated with Absolutely RNA Microprep Kit (Agilent Technologies) at 5 days in vitro (DIV). The RNA samples were further used for either RNA-seq-directed library construction or for RT-qPCR. Library construction and sequencing were carried out at Genewiz Inc. (New Jersey, USA). RNA-Seq libraries were sequenced on the Illumina HiSeq2500 platform in 50 bp single-end format. After quality filtering of reads, sequences were mapped onto the genome with TopHat (Kim et al., 2013), assembled onto transcripts with Cufflinks (Trapnell et al., 2010) and differential expression was determined with Cuffdiff (Trapnell et al., 2010). Raw and processed RNA-Seq data are available at Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE131494. For RT-qPCR, cDNA was synthesized with Transcriptor high-fidelity cDNA synthesis kit (Roche) and RT-qPCR was performed using Luminaris HiGreen qPCR Master Mix (Thermo scientific) in a CFX Connect Real-Time PCR Detection System (Bio-Rad). The primer sequences were as follows: Reln-F (TTTACACTGAGGCTGGGGAG); Reln-R (TGCCACCATCTGAACTGGAT); Gapdh-F (AATGTGTCCGTCGTGGATCTGACGTGC); Gapdh-R (TTGCTGTTGAAGTCGCAGGAGACAACC).
Primary cortical cultures and Sholl analysis
Primary cultures were prepared from E14.5 embryos as described (Guner et al., 2017). For Sholl analysis, primary cortical neurons were transfected with shNeurod2-1 (Guner et al., 2017) at 3 DIV with Lipofectamine 2000 transfection reagent (Invitrogen). Neurons were fixed with 4% PFA at 5 DIV, immunostained with an antibody recognizing EGFP (Aves Lab, GFP 10-20) and imaged with a Nikon 90i Eclipse confocal microscope affixed with a 60x oil objective. Dendritic development was quantified by the Sholl analysis plug-in in ImageJ software (Ferreira et al., 2014; Schindelin et al., 2012).
Animals
All animal experiments were performed in accordance with the guidelines for the care and use of laboratory animals of Koç University (institutional ethics approval number 2014-07). For in utero electroporation and primary neuronal cultures timed-pregnant mice (Balb/c strain) were generated by The Koç University Experimental Animal Laboratory.
In utero electroporation and image analysis
Overall, our in utero electroporation method was based upon a previously published protocol (Saito, 2006). Briefly, 14.5 day pregnant mice were anesthetized by inhalation of isoflurane. 1 µl of DNA solution (composed of 500 ng of shRNA plasmid, 500 ng of pCAGGS-IRES-tdTomato and 0.1% Fast Green FCF) was injected unilaterally into the lateral ventricular of each embryo. 0.5 pulses at 30 V with 400 ms intervals were applied with a 5 mm tweezer electrode using an electroporator (BTX, Harvard Apparatus, ECM830). Following electroporation, embryos were placed back into the uterine cavity, and the pregnant female was sutured and placed into incubation chamber for recovery. Embryos were retrieved at developmental age E17.5, followed by brain removal and placement into 4 % PFA. Embryos were coronally sectioned to 50 µm with a cryostat (Leica CM1950). Confocal images of tdTomato-positive neurons were captured (Nikon Eclipse 90i) and maximum intensity projection images were generated using NIS Elements AR software. A grid of ten bins was overlaid onto the cortical cross-section images, and the number of tdTomato-positive neurons in each bin was counted using the cell counting tool integrated into ImageJ (Schindelin et al., 2012). For leading process tracing analysis, images were acquired with a LEICA DMI8 SP8 CS/DLS microscope fixed with a 63x objective and traces were created using ImageJ software.
Funding sources
The work was funded by following grants to G. Ince-Dunn: European Commission FP7 International Reintegration Grant (PIRG07-GA-2010-268433), Turkish Academy of Sciences Young Scientist Program (TUBA-GEBIP) and Koç University Seed Fund, Istanbul, Turkey, to C. Akkaya: TUBITAK-BIDEB 2211/E Scholarship Program and to C. Dunn: EMBO Installation Grant 2138.
Conflict of interest
Authors declare no conflict of interest.
Acknowledgements
We thank Ali C. Taşkin, Ahmet Kocabay and Mehmet Yücel (Koç University Animal Research Facility, Turkey) for their help in providing timed pregnant mice; Efil Bayam (Institute of Genetics and Cellular and Molecular Biology, Strasbourg, France) for assistance with the in utero electroporation method; Tamer Önder (Koç University School of Medicine, Turkey) for generously sharing the Lonza nucleofector equipment, and the Molecular Imaging Core Facility of Koç University Research Center for Translational Medicine (KUTTAM).
References




