

ABSTRACT
Growing mammalian oocytes accumulate substantial amounts of RNA, most of which are degraded during the subsequent maturation stage. The growth-to-maturation transition begins with nuclear envelope breakdown (GVBD) and is critical for oocyte quality. However, the concomitant changes in the transcriptome during GVBD as well as the underlying machinery remained unclear. Here, we report that an RNA exosome-associated RNase, EXOSC10, degrades poly(A) RNA to facilitate the oocyte growth-to-maturation transition. We establish an oocyte-specific knockout of Exosc10 in mice using CRISPR/Cas9 and find female subfertility due to failed GVBD. By performing single oocyte RNA-seq at GV, GVBD and MII stages, we document dysregulated transcriptomes in mutant oocytes, and many up-regulated RNAs that encode proteins important for endomembrane trafficking, meiotic cell cycle and RNA metabolism. EXOSC10-depleted oocytes have impaired endomembrane components including endosome, lysosome, ER and Golgi. In addition, CDK1 fails to be activated possibly due to persistent WEE1 activity, which blocked lamina phosphorylation and disassembly in mutant oocytes. Collectively, we propose that EXOSC10 promotes the growth-to-maturation transition in mouse oocytes by degrading growth-phase factors and sculpting the transcriptome to support the maturation phase of oogenesis.
INTRODUCTION
Improper oocyte maturation directly causes ovulatory disorders and leads to female infertility (1,2). Maturing oocytes dramatically alter their transcriptome due to active RNA degradation in the absence of transcription. A growing body of work documents that coordinated sequestration, deadenylation, translation and degradation collectively regulate RNA metabolism during oocyte maturation. A dysregulated transcriptome can impair oogenesis and post-fertilization embryogenesis (3–8).
For historic reasons, the oocyte nucleus is referred to as the germinal vesicle (GV). After their growth phase, GV-intact oocytes remain quiescent in pre-ovulatory follicles until induced to mature by luteinizing hormone. Critical events in the transition from growth to maturation include chromatin condensation, termination of transcription and nuclear envelope breakdown (GVBD). The first two are directly coupled and are dominated by an increase of repressive histone modifications including H3K9 methylation that modifies the oocyte chromatin (9,10). The third event, GVBD, requires precise coordination of the meiotic cell cycle and membrane trafficking. Meiotic cell cycle control is determined by the cyclin B/CDK1 activity through complex signaling pathways including cAMP-PKA and PKB/Akt (11). High levels of cAMP cause CDK1 inhibitory phosphorylation that favors GV arrest. Conversely, decreases of cAMP activate CDK1 to form the functional cyclin B/CDK1 complex that induces GVBD. The membrane trafficking machinery involves endomembrane components. For example, an exchange apparatus of ER-NE (endoplasmic reticulum - nuclear envelope) has been reported to facilitate NE formation post mitosis by reorganizing membrane structure around chromatin (12). The COPI-coated vesicles that normally traffic from the Golgi to the ER also promote GVBD upon recruitment by nucleoporin Nup153 (13). However, the molecular basis of the observed coordination of transcriptome sculpting with GVBD remains undetermined.
The RNA exosome is a highly conserved complex that degrades or processes cellular RNAs from the 3’ end. RNA exosome-related genetic mutations have been identified in a wide range of diseases, including diarrhea of infancy, neurodegenerative disorders and multiple myeloma (14–16). The association of the core complex with DIS3, DIS3L or EXOSC10 RNases provide the required enzymatic activity. EXOSC10 is a nuclear RNase, the absence of which causes RNA processing defects in yeast (17) and increased sensitivity to DNA damage in fly and human cells (18,19). EXOSC10 has been documented to promote mRNA turnover (20), 3’ pre-rRNA processing (21) and long noncoding/enhancer RNA degradation (22). It also has been reported to control the onset of spermatogenesis in male germ cells (23). However, whether EXOSC10 is essential for oogenesis has remained unclear. Published datasets (6,24) document that Exosc10 transcript are highly abundant in mouse oocytes and early embryos, raising the possibility of its participation in maternal RNA metabolism.
In the current study to evaluate EXOSC10 function in the oogenesis, we established oocyte-specific conditional Exosc10 knockout (cKO) mice. cKO females had decreased fecundity associated with defects in GVBD and impaired oocyte maturation. Using single oocyte RNA-seq and ERCC spike-in normalization, we identified dysregulated transcripts involved in endomembrane disfunction and abnormal CDK1 phosphorylation.
MATERIAL AND METHODS
Mice
Mice were maintained in compliance with the guidelines of the Animal Care and Use Committee of the National Institutes of Health under a Division of Intramural Research, NIDDK-approved animal study protocol.
Generation of Exosc10 floxed allele by CRISPR/Cas9
Two guide RNAs (gRNA, 50 ng/μl), two homology-directed repair (HDR, 100 ng/μl) templates and Cas9 cRNA (100 ng/μl) were microinjected into 1-cell mouse embryos. The two gRNA sequences are: 5’tcagtggagacctgcgatct3’ (left loxP) and 5’gaaattctgatgtctagcgg3’ (right loxP). Each gRNA was in vitro transcribed from a double stranded (ds) DNA template by MEGAshortscript™ T7 Transcription Kit (Thermo Fisher Scientific, AM1354) and purified by MEGAclear Transcription Clean-Up Kit (Thermo Fisher Scientific, AM1908). The dsDNA templates for sgRNAs were initially synthesized as single stranded (ss) DNA from Integrated DNA Technologies (IDT) and amplified by primers 5’GATCCCTAATACGACTCACTATAG3’ and 5’ AAAAAAAGCACCGACTCGGTGCCAC3’ into ds DNA: 5’gatccctaatacgactcactataggtcagtggagacctgcgatctgttttagagctagaaatagcaagttaaaataaggctagtccgt tatcaacttgaaaaagtggcaccgagtcggtgcttttttt3’ and 5’gatccctaatacgactcactatagggaaattctgatgtctagcgggttttagagctagaaatagcaagttaaaataaggctagtccgtt atcaacttgaaaaagtggcaccgagtcggtgcttttttt3’.
The two HDR templates for each edited locus were synthesized as ssDNA from IDT: 5’gagagagcacgtatggctcttgcagaggactggtactctaccccagcacccatgttaggtgggtcacaactgcttgtaactccagct ccaagaGCGGCCGCATAACTTCGTATAATGTATGCTATACGAAGTTATtcgcaggtctccactgacactg gcactcaggagcacgt3’ (left loxP) and 5’actcacactgtagaccagtctggcctcaaactcacaaagatccacctgcctctgcctcctaagtgctggggttaaatgggtactctac caccgGAATTCATAACTTCGTATAGCATACATTATACGAAGTTATctagacatcagaatttctaaatataaaa aggagaatg3’ (right loxP).
After linearization by Pmel, plasmid #42251 (Addgene) was used as a template to transcribe Cas9 cRNA in vitro by mMESSAGE mMACHINE™ T7 ULTRA Transcription Kit (Thermo Fisher Scientific, AM1345). The synthesized cRNA was purified by MEGAclear Transcription Clean-Up Kit (Thermo Fisher Scientific, AM1908).
For mouse embryo microinjections, hormonally stimulated B6D2F1 female mice were mated to B6D2F1 males. 1-cell zygotes were flushed from oviducts into M2 medium (CytoSpring, #M2114) and microinjected with the mixed components for gene-editing with CRISPR/Cas9. The injected embryos were cultured in KSOM (CytoSpring, #K0113) at 37 °C with 5% CO2 for 24 hr. 2-cell embryos were transferred into the oviducts of 0.5-day post coitus pseudopregnant ICR females.
To obtain Exosc10 oocyte-specific conditional KO mice (cKO), Exosc10 floxed mice were crossed to Zp3-cre mice (25). The genotyping primers for the Exosc10 floxed and deletion alleles are as follows:
left loxP: 5’atgagtcgggtaatgcagtac and 5’tgtgtgaggatgttgtgagc3’;
right loxP: 5’ccgactctgacattgagtgg3’ and 5’gcctctttcccacagttccag3’;
Deletion allele: 5’atgagtcgggtaatgcagtac and 5’gcctctttcccacagttccag3;
Cre: 5’gcggtctggcagtaaaaactatc3’ and 5’gtgaaacagcattgctgtcactt3’.
Oocyte collection and culture
Ovaries were dissected from female mice (6-10 w/o) into M2 medium plus milrinone (2.5 μM). The ovaries were pierced mechanically by 30-gauge-needles to release oocytes into M2 (milrinone) medium. Only fully-grown oocytes (GV oocytes) detaching easily from the granulosa cells were collected for further experiments. For oocyte ex vivo maturation, GV oocytes were washed (20 times) with M2 medium without milrinone and cultured in M2 at 37 °C with 5% CO2. The GVBD/GV ratio was determined at GV+3hr and meiosis II progression was evaluated at 14 hr.
cRNA in vitro transcription and microinjection
The Exosc10 coding sequence was inserted into plasmid #44118 (Addgene) to form an in-frame fusion with mVenus. Simultaneously a T7 promoter (TAATACGACTCACTATAGGG) was inserted into the 5’ end of the Exosc10 coding sequence. The plasmid was linearized by XbaI, purified and in vitro transcribed by mMESSAGE mMACHINE™ T7 ULTRA Transcription Kit (Thermo Fisher Scientific, AM1345). The cRNAs were purified by MEGAclear Transcription Clean-Up Kit (Thermo Fisher Scientific, AM1908) and diluted to a proper concentration (500 ng/μl, unless otherwise stated) for microinjection.
Immunofluorescence and confocal microscopy
Ex vivo cultured oocytes or embryos were fixed by 2% paraformaldehyde (Thermo Fisher Scientific, #50-980-492), diluted in PBS containing 0.1% Triton X-100) at 37 °C for 30 min. After fixation, oocytes were washed with PBVT (PBS, 3mg/ml polyvinylpyrrolidone-40 and 0.1% Tween-20), and permeabilized with 0.5% Triton X-100 in PBS for 30 min at room temperature. Oocytes were blocked by 5% normal goat serum in PBVT for 1 hr at room temperature followed by primary antibody incubation overnight at 4 °C. On the second day, the oocytes were washed (4X, 15 min) with PBVT and incubated with fluorescence conjugated secondary antibody overnight at 4 °C. On the third day, after washing by PBVT (4X, 15 min), oocytes were stained with DAPI and mounted with PBS for confocal microscopic imaging (LSM 780, Carl Zeiss).
Source and dilution of antibodies, staining reagents and live staining dyes
Anti-GM130 (1:200, BD Transduction Laboratories 610823); LysoTracker™ Green DND-26 (1:1000, ThermoFisher L7526); LysoTracker™ Blue DND-22 (1:1000, ThermoFisher L7525); ER tracker red (1:1000, Invitrogen E34250); anti-lamin B1 (B-10) (1:200, Santa Cruz sc-374015); anti-αtubulin (1:200, Sigma T5168); anti-lamin A/C (4C11) (1:200, CST 4777T); anti-phospholamin A/C (Ser22) (1:200, CST 2026); anti-RAB5 [EPR21801] (1:500, Abcam ab218624); anti-phosphoCDK1 (Thr14, Tyr15) (17H29L7) (1:200, ThermoFisher 701808); anti-cAMP (1:200, RD # MAB2146); anti-pericentrin (1:2000, Abcam #ab4448); anti-γtubulin (1:500, Abcam ab11316); goat anti-rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 546 (1:500, Invitrogen A-11010); goat anti-mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 (1:500, Invitrogen A-21235). DAPI (Sigma D9542-1MG); goat serum (Sigma G9023-10ML); Tween-20 (Sigma P1379-25ML).
Ovary histology
Ovaries were dissected from female mice of desired ages into PBS buffer. After removing the surrounding lipid and tissue, the ovaries were fixed in newly prepared 2.5% glutaraldehyde and 2.5% paraformaldehyde in 0.083 M sodium cacodylate buffer (pH 7.2) for 3-5 hr at 4 °C. Ovaries were washed with 0.1 M sodium cacodylate buffer (pH 7.2) and kept at 4 °C overnight. Finally, the ovaries were transferred into 70% ethanol and stored for more than 1 day at 4 °C. The sectioning (2 μm, every 20th section), periodic acid–Schiff (PAS) staining and mounting were performed by American HistoLab. The imaging of ovary histology was performed on a Nikon ECLIPSE Ti microscope using a 20x objective.
RNA FISH of oligo(dT) probe
FISH assays were performed in U-bottom 96-well plates. Oocytes at desired stages were fixed with 2% paraformaldehyde (Thermo Fisher Scientific, #50-980-492), diluted in PBS containing 0.1% Triton X-100) at 37 °C for 30 min. Oocytes were dehydrated stepwise at room temperature using 25%, 50%, 75%, 100% (methanol: PBS volume) and washed (3X) with 100% methanol. After −20 °C treatment >1 hr, the oocytes were rehydrated into PBT (PBS-0.1% Tween-20) through 75%, 50%, 25%, 0% (methanol: PBT volume) steps. Oocytes then were washed (3X) with PBT, treated with 0.5% SDS for 30 min at room temperature and re-washed (3X) with PBT. Oocytes were washed (2X) with newly prepared FISH wash buffer (10% formamide, 2x SSC, 0.1% Tween-20 in nuclease-free water), transferred into hybridization buffer (10% formamide, 2x SSC, 10% dextran sulfate in nuclease-free water) and incubated at 37 °C for 2 hr. The oocytes then were incubated with probe-containing hybridization buffer for 16 hr at 37 °C. On the second day, oocytes were incubated sequentially with hybridization buffer (30 min, 37 °C), FISH wash buffer (1X, 5 min; 4X, 20 min at 37 °C), and FISH wash buffer (1X, 20 min) at room temperature. Oocytes were further washed (2X) with PBT at room temperature, mounted onto ProLong™ Diamond Antifade Mountant (Thermo Fisher Scientific, P36970) and imaged by confocal microscopy (LSM 780, Carl Zeiss).
Single oocyte RNA-seq library preparation
Single oocyte RNA-seq libraries were prepared according to a published single-cell RNA seq pipeline with minor modifications (26). Briefly, oocytes at desired stages were collected individually into 2.5 μl RLT Plus (Qiagen) and stored at −80 °C until all samples were acquired. In the beginning of RNA purification, ERCC RNA spike-in mix (Thermo Fisher Scientific, 4456740) was diluted 105 fold, and 1 μl of the diluted ERCC mix was added to each sample. Poly(A) RNA was isolated by oligo (dT) beads, reverse transcribed, amplified and purified. Preliminary sequencing was performed with different amplification cycles (10, 12, 14, 16, 18) and different portions of oocytes as initial material (1/8, 1/4, 1/2, 1) to test the linear range of amplification. 14-cycles was chosen as the best condition based on the cDNA acquired and the regression analysis of ERCC. The purified cDNAs were analyzed by Bioanalyzer 2100 (Agilent) to confirm successful amplification and quality. Qualified cDNAs were used to construct sequencing libraries by Nextera DNA Sample Preparation Kits (Ilumina). The generated 72 sequencing libraries were evaluated by Bioanalyzer 2100 and pooled into 6 groups for purification. The sequencing was performed by NIDDK Genomic Core Facility.
RNA-seq analysis based on ERCC RNA spike-in mix
Raw sequence reads were trimmed with cutadapt 1.18 to remove adapters while performing light quality trimming with parameters “-a CTGTCTCTTATA -q 20 --minimum-length=25.” Sequencing library quality was assessed with fastqc v0.11.8 with default parameters. The presence of common sequencing contaminants was evaluated with fastq screen v0.13.0 with parameters “--subset 100000 --aligner bowtie2.” Trimmed reads were mapped to the Mus musculus mm10 reference genome plus ERCC.fasta using hisat2 2.1.0 with default parameters. Multi-mapping reads were filtered using samtools 1.9. Uniquely aligned reads were then mapped to gene features using subread featureCounts v1.6.2 as an unstranded library with default parameters. A gene/ERCC count was considered valid when present in at least 5 reads in at least 2 libraries. The oocyte libraries were normalized by ERCC counts by defining the ERCC genes as the controlGenes when estimating the sizeFactors. Differential expression between groups of samples was tested using R version 3.5.1 (2018-07-02) with DESeq2 1.20.0. Transcript quantification was performed with salmon 0.11.3 with parameters “--gcBias --libType A --seqBias --threads 1.”
Quantitative RT-PCR
Single oocytes were collected individually for poly(A) RNA enrichment, purification, reverse-transcription and amplification following a similar pipeline as single oocyte RNA-seq protocol. The purified cDNA was directly used as templates and the qRT-PCR was performed by iTaq Universal SYBR Green Supermix (Bio-Rad, #1725121) and QuantStudio 6 Flex Real-Time PCR System (Thermo Fisher Scientific). The primers for Exosc10 and Gapdh are:
Exosc10 primer 1: 5’ccgactctgacattgagtgg3’ and 5’gcctctttcccacagttccag3’;
Exosc10 primer 2: 5’ATCCCCCAGGGAAAGACTTC3’ and 5’GTCCGACTTTCCAACAGCAA3’;
Gapdh: 5’TGCACCACCAACTGCTTAGC3’ and 5’GGCATGGACTGTGGTCATGAG3’.
Electron Microscopy
Mouse GV oocytes were collected from unstimulated ovaries, washed (2X) with PBS, and fixed by newly prepared 2.0% glutaraldehyde, 2.5% PFA in 0.1 M cacodylate buffer (pH 7.4) for 10 min at room temperature followed by 2 hr on ice. Fixed oocytes were washed (3X) with 0.1 M cacodylate buffer. Oocytes were embedded in 1% agarose (Sigma, A2576) and stored in 0.1 M cacodylate buffer overnight at 4 °C. Following sample preparation for transmission electron microscopy, sectioning and imaging were performed by NIBIB Electron Microscopy Unit. Briefly, the embedded oocytes were post-fixed in 2% osmium tetroxide plus 0.8% potassium ferricyanide in 0.1 M cacodylate buffer. After several rinses with 0.1 M cacodylate buffer, the samples were dehydrated in methanol as described above. Samples were then infiltrated with Epon-Aradite (Electron Microscopy Sciences): 30% Epon-Aradite in ethanol for 2 hr, 50% for 4 hr, 75% overnight, and 100% for 1 day with 2X changes. Samples were polymerized at 60 °C for two days. Ultrathin sections (about 80 nm) were cut with a Reichert Ultracut E Microtome and collected on copper slot grids. Sections were counter-stained with uranyl acetate and lead citrate and examined under a FEI Tecnai12 transmission electron microscope operating with a beam energy of 120 keV. Images were acquired using a Gatan 2k × 2k cooled CCD camera.
Statistical analysis
The replicates of the oocytes, ovaries or mice were combined and analyzed by paired-sample t-test, which was performed by GraphPad Prism 8 software. The fluorescence quantification was performed in Image J (Fiji) software.
RESULTS
Oocyte-specific knockout of Exosc10 leads to female subfertility by disrupting oocyte maturation
Homozygous constitutive knockout of Exosc10 in mice is embryonic lethal (Figure S1A, C). To study the function of EXOSC10 in oocyte development, we generated an Exosc10 floxed allele by CRISPR/Cas9 and used Zp3-cre mice to specifically knock out Exosc10 in growing oocytes. Two loxP sites surrounding exons 4-10 resulted in a deletion and a frame shift of the remaining coding sequence (Figure 1A; Figure S1B). To increase loxP recombination efficiency, we used a mating strategy to present only one floxed allele for cre to obtain the desired oocyte-conditional knockout (Flox/-; Zp3-cre, cKO thereafter) (Figure 1B). qPCR of single oocytes confirmed the loss of Exosc10 transcripts in cKO oocytes from the GV stage onward (Figure 1C; Figure S1B, D).

(A) Schematic of strategy to generate an Exosc10 floxed allele using CRISPR/Cas9. Two loxP sites were inserted to bracket exons 4-10. (B) Mating strategy to obtain oocyte-specific conditional knockouts of Exosc10 (cKO). Siblings with other genotypes were used as controls. (C) qRT-PCR of Exosc10 in single oocytes obtained from controls and cKO mice. (D) Dot plot of individual litter sizes over 6-months of harem breeding of controls and cKO females with wildtype males. The sizes of the dots are normalized by the average litter number per female. The number in parenthesis is the number of females having the indicated genotypes. The number of pups born is indicated below each group. The horizontal lines represent the mean and standard deviation. (E) Bright-field images cKO and controls oocytes cultured ex vivo for 0 (GV) or 3 hr (GV+3hr). (F) Percentage of GVBD oocytes in E. Number of oocytes indicated below each group. (G-H) Confocal fluorescence and brightfield/DAPI images of oocytes after lamin B immunostaining at GV (G) and GV+3hr (H) stages. Lamin B and DAPI are maximum intensity projections and bright-field images are single optical sections containing the nucleus. Quantification of lamin B fluorescence is on the right. The horizontal lines inside the violins represent the median and the quartiles. The number of oocytes from at least 3 females are indicated below each group. **** P <0.0001 in D, G, H, two-tailed Student’s t-test. Scale bars: 100 μm in E; 20 μm in G, H.
To assess female fertility affected by oocyte-specific EXOSC10 depletion, we mated pairs of cKO and control females with wildtype males for 6 months. Combining the records of 7 harems including 8 cKO females and 9 controls, the cKO females exhibited substantial subfertility with reduced pups (8.8 vs. 30.1 total number), litter number (2.6 vs. 4.1) and litter size (3.3 vs. 7.3) per female (Figure 1D). Intrigued by the subfertility, we investigated possible defects in oocyte growth, oocyte maturation and early embryogenesis. At 12 weeks, cKO and control females had indistinguishable ovaries in terms of weight, histology and antral follicle number (Figure S1E-H) which suggests normal oocyte growth. Although the diameter of the cKO oocytes was modestly decreased (Figure S1I), we concluded that the subfertility of cKO females is likely due to defects after the growth phase.
Next, we examined oocyte maturation and determined that GV oocytes collected from cKO females had deficient meiotic progression after 20 hr ex vivo culture (Figure S1J). Correspondingly, the number of ovulated eggs recovered following gonadotropin stimulation also was significantly decreased in cKO females (Figure S1K). We narrowed the defect to GVBD (Figure 1E-F). Although the mitotic organization center protein of the spindle appeared normal (Figure S1L), lamin B persisted at GV+3hr in cKO oocytes and its intensity of immunostaining was substantially increased at both GV and GV+3hr stages compared to controls (Figure 1G-H). We in vitro synthesized Exosc10-mVenus cRNA and found that EXOSC10-mVenus had nuclear localization in both oocytes and embryos (Figure S1M). We also evaluated pre-implantation development of embryos derived from homozygous cKO females. In ex vivo culture of 1-cell embryos, many had developmental delay from embryonic day 1.5 (E1.5) to E3.5, and some of them arrested at the 2-cell stage. There was decreased blastocyst formation at E3.5 (Figure S1N-O). In sum, oocyte-specific EXOSC10 depletion causes substantial subfertility due to defective GVBD and embryos derived from cKO females progress abnormally during pre-implantation development.
EXOSC10 depletion dysregulates poly(A) RNA profile
To investigate the molecular basis of this phenotype, we sought to identify the types of RNA regulated by EXOSC10 in oocytes. EXOSC10 exhibits ribonuclease activity toward a range of poly(A) RNA molecules, including mRNA and rRNA (27,28). We therefore employed RNA FISH to quantify the poly(A) RNA with an oligo(dT) probe. In wildtype oocytes, there was a modest decrease during maturation from GV to GV+3hr maturation and then a sharp decrease during progression to the MII stage (Figure S2A-B) which is consistent with RNA degradation during oocyte maturation (6). Oocytes, over-expressing Exosc10-mVenus, had significantly decreased poly(A) compared to mVenus alone over-expression at both GV and GV+3hr stages, suggesting a role for EXOSC10 in degrading poly(A) RNA. However, when the ribonuclease catalytic sites were mutated (D313N and E315Q in EXOSC10), over-expression of the dExosc10-mVenus lost the ability to accelerate poly(A) RNA degradation (Figure S2C-D). On the other hand, the overall poly(A) RNA intensity in cKO oocytes showed no obvious change compared to controls, probably due to little transcription from the GV stage onward (Figure S2E-F). These results indicate that EXOSC10 participates in poly(A) RNA elimination during mouse oocyte maturation.
To characterize the dysregulated poly(A) RNA that account for the phenotype, we performed single oocyte RNA-seq at GV, GV+3hr and MII stages. The poly(A) RNA were isolated by oligo-dT beads (Figure 2A) and an equal amount of ERCC Spike-In Mix was added to each oocyte lysate to ensure library quality and to compare initial RNA quantities (Figure S3). In total, 71 oocytes (from 7 controls and 8 cKO mice) were sequenced, and 64 passed quality control. In normalizing oocyte total RNA with ERCC, we observed dependence on library size (Figure 2B) and used ERCC as the normalization reference for later differential analysis of gene expression. In addition to an overall mild delay in RNA degradation (Figure 2C), cKO oocytes exhibited distinct gene expression patterns as determined by pan-transcriptome expression heatmap (every 100th transcript after being ranked by expression level) and principal component analysis (Figure 2D-E). As expected, the nearly complete loss of Exosc10 transcript in cKO oocytes was validated by RNA-seq (Figure 2F).
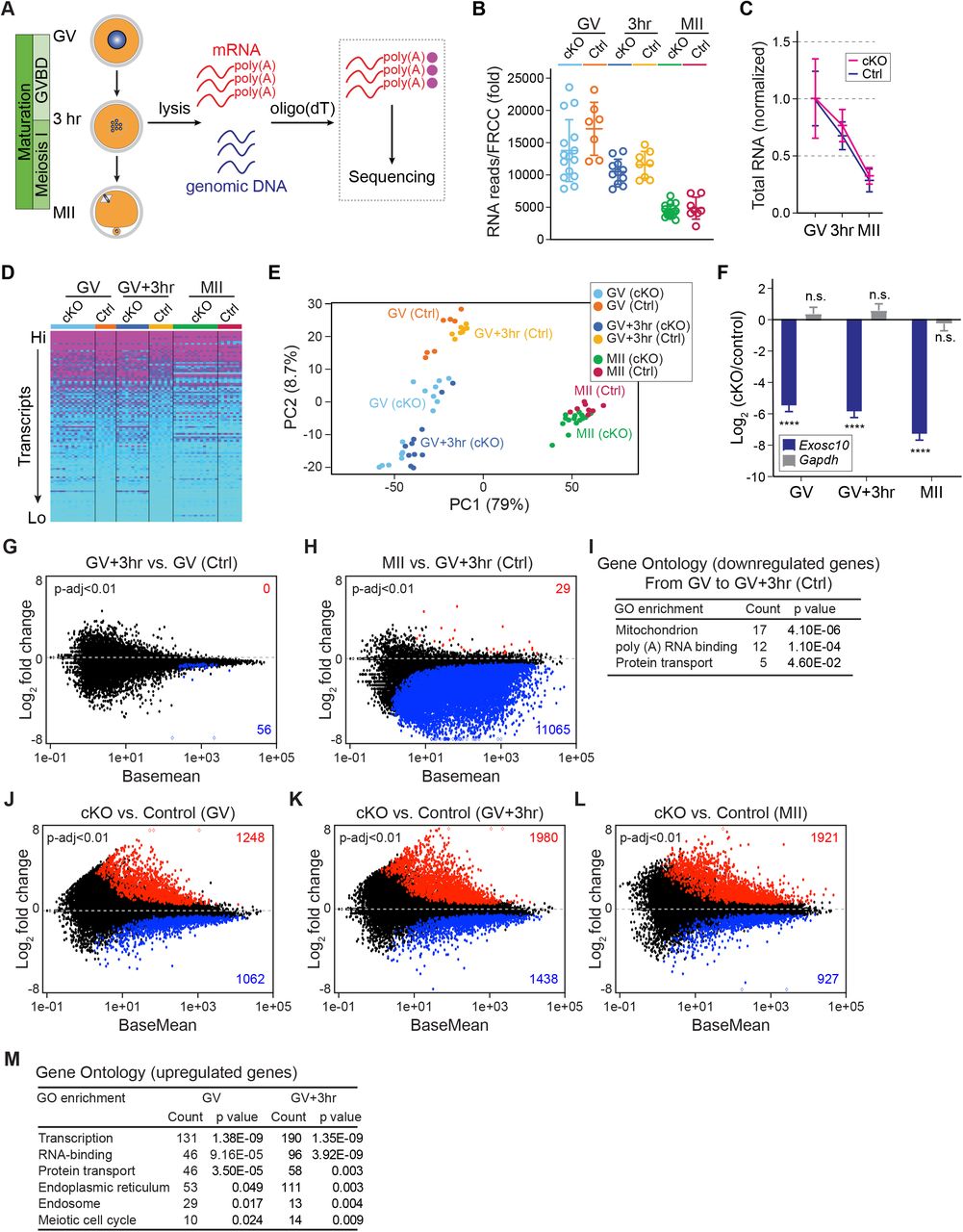
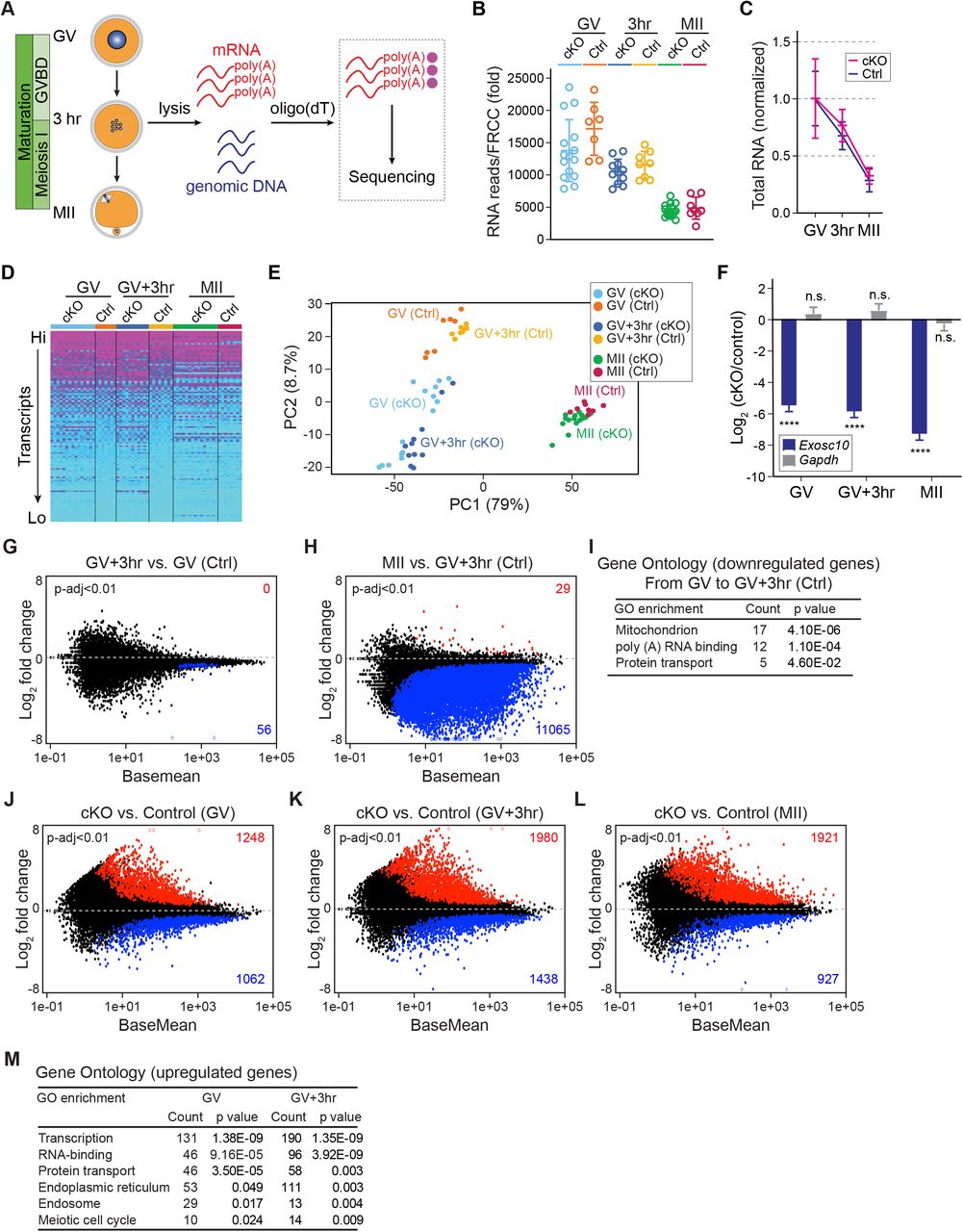
(A) Schematic illustrating the pipeline of single oocyte RNA-seq. After individual oocyte lysis, oligo-dT beads captured poly(A) RNAs for library construction and sequencing. Genomic DNA determines genotype. (B) Total RNA levels are normalized for each library by ERCC RNA spike-in mix. (C) Further normalization of total RNA level in B by the mean value of GV stage within each genotype. (D) Heatmap of all libraries (genes are ranked from highest to lowest expression level and every 100th transcript was selected to represent the top half of the entire transcriptome, color-coded according to the transcription level). (E) Principal component analysis (PCA) of the 64 libraries. Each dot represents one library, color coded by genotype and stage. (F) Log2 fold change of Exosc10 and Gapdh with cKO vs. control. The bars and lines are log2 fold change and standard error of the mean from DESeq2 analyses. **** P-adjust <0.0001, n.s. no significance, which are the P-adjust values in DESeq2 analysis. (G-H) MA-plots of transcript changes from GV to GV+3hr, and from GV+3hr to MII stage in control groups. The up-regulated and down-regulated gene numbers in each comparison are labeled by red and blue, respectively (both have P-adjust <0.01). (I) Gene ontology of up-regulated genes in G. (J-L) MA-plots of transcript changes in cKO vs. control oocytes at GV, GV+3hr and MII stages. The up-regulated and down-regulated gene numbers in each comparison are labeled by red and blue, respectively (P-adjust <0.01). (M) Gene ontology of the up-regulated genes with log2 fold-change more than 1 in J-K.
ERCC normalization allowed comparison of transcriptomes between stages of development. Within control oocytes, we identified 56 genes (P-adjust <0.01) down-regulated from GV to GV+3hr. Gene Ontology (GO) analysis revealed these transcripts were associated with mitochondria, poly(A) RNA binding and protein transport through the endomembrane system. From GV+3hr to MII stage, we identified 11,065 down-regulated genes. There were almost no genes up-regulated which is consistent with minimal transcription during oocyte maturation (Figure 2G-I). Differentially expressed genes in cKO and controls were further analyzed. At each stage, we identified more up-regulated genes than down-regulated genes (Figure 2J-L; Table S1). Considering that EXOSC10 directly eliminates poly(A) RNA (Figure S2), the up-regulated transcripts were more likely to be EXOSC10’s substrates responsible for the phenotype. We performed GO analysis of the up-regulated genes at GV and GV+3hr. The most prominent terms were associated with transcriptional control, RNA metabolism, endomembrane transport and meiosis (Figure 2M). All of these biologic processes occur during nuclear envelope breakdown (29) which links the dysregulated transcriptome in cKO oocytes with the failure of GVBD.
Heterogeneity of the cKO oocytes underlies subfertility
The RNA FISH and the original PCA of the RNA-seq both suggest high similarity between GV and GV+3hr stages of the control oocytes. Thus, we combined the GV and GV+3hr stages together as growth-to-maturation transition period to investigate the effect of the EXOSC10 depletion on GVBD. The original PCA documented substantial variance within cKO oocytes during this period (Figure 2E) suggesting oocyte heterogeneity. It seemed plausible that greater dysregulation of the transcriptome would correlate with more reduced fertility of individual oocytes. Therefore, we performed PCA again using the 41 samples of control oocytes and cKO oocytes at both GV and GV+3hr stages and employed k-means clustering to define three groups (k=3). As expected, there was one group that included all the control oocytes which is consistent with the high similarity between GV and GV+3hr of control oocytes analyzed in the original PCA (Figure 2E). The two other groups were designated cKO (major) and cKO (minor) according to their greater and lesser distances from the control group on the PCA plot (Figure 3A-B). By differential analysis of cKO (major) and cKO (minor) vs. control, we found that the cKO (major) group exhibited more up-regulated genes than the cKO (minor) group (Figure 3C-D). The differentially expressed genes between cKO (major) and cKO (minor) groups were enriched for GO terms including transcription regulation, mitochondrion, cell cycle and endomembrane vesicles, which recapitulated the observed differences in the original PCA result of cKO vs. control (Figure 3E-F; Figure 2M; Table S1). To confirm the stronger phenotype in cKO (major) compared to cKO (minor), we examined six genes that were markedly up regulated in the 6 GO terms enriched by cKO vs. control (Figure 2M), including Zfp668, Hormad1, Nox4, Xpot, Exoc8 and Dio3. The abundance of transcripts from all six genes was significantly higher in the cKO (major) compared to the cKO (minor) group (Figure 3G). Thus, greater dysregulation of the transcriptome in individual oocytes decreased their potential fecundity.

(A) Principal component analysis (PCA) of 41 libraries of control oocytes and cKO oocytes at both GV and GV+3hr stages. Subsequently the 41 libraries were sub-clustered by k-means algorithm into 3 groups, including control, cKO (major) and cKO (minor). Dots are color-coded to indicate library source. (B) Sample distance matrix of the 41 libraries in A. (C-D) MA-plots of cKO (major) vs. control and cKO (minor) vs. control libraries. (E) MA-plot of cKO (major) vs. cKO (minor). The up-regulated genes and down-regulated gene numbers in each comparison are labeled by red and blue, respectively (P-adjust <0.01). (F) Gene ontology of up-regulated genes with log2 fold change more than 1 in E. (G) PCA plots of A that is color-coded by the expression level of each transcript in individual oocytes. The control, cKO (major) and cKO (minor) groups in each plot are the same as labeled in the first plot.
cKO oocytes have disrupted endomembrane system
Nuclear envelope breakdown involves multiple components of the endomembrane system, including the ER, Golgi, endosomes and lysosomes (30–32). Consistent with these findings, the GO analysis of the cKO RNA-seq highlighted intracellular vesicle trafficking. Therefore, we examined a range of cytoplasmic vesicles including endosomes, ER, lysosomes and Golgi. RAB5-labeled early endosomes displayed abnormal aggregation and increased abundance in cKO oocytes (Figure 4A-B). In contrast, RAB7-labeled late endosomes were reduced (Figure 4C-D) suggesting insufficient endosome maturation (33). Live imaging of oocytes treated by ER-Tracker had a reduced signal, whereas the Lyso-Tracker treated oocytes had increased signal (Figure 4E). The Golgi apparatus normally exhibits a pan-cytoplasmic distribution with slight increases in perinuclear and pericytoplasmic regions, a distribution pattern that becomes accentuated after GVBD. However, the cKO oocytes showed a much stronger pericytoplasmic enrichment of Golgi and reduced signal in the perinuclear region which remained unchanged followed by 3 hr culture (Figure 4F-G). In sum, formation of the endomembrane system was disrupted after EXOSC10 depletion. This was confirmed by electron microscopy showing vesicle aggregation close to the cytoplasmic membrane which contain endosome vesicles and mitochondria (Figure 4H).

(A) Immunostaining of oocyte RAB5A/C (early endosome) at the GV stage. (B) Quantification of RAB5A/C vesicle fluorescence intensity in A. (C) Same as A, but for RAB7 (late endosome). (D) Quantification of RAB7 vesicle intensities in C. For the violin plots in B, D, the horizontal lines inside the violins represent the median and the dash lines represent the quartiles. **** P <0.0001, two-tailed Student’s t-test. Number of oocytes is indicated below each group. (E) Live imaging of oocytes derived from control and cKO incubated with ER Tracker and LysoTracker. The untreated group is in the right column. (F) Immunostaining of GM130 for Golgi apparatus at GV and 3 hr stages. (G) Radial profile quantification of GM130 in F. Each curve shows the mean and standard error of oocytes with the oocyte number in the parenthesis. For each oocyte, the radial profile is performed by measuring the integrated intensity at each radius from the center to the cytoplasmic membrane which has 20 bins. (H) Electron microscopy showing vesicle aggregation close to the plasma membrane. Red dashed line indicates the clustered vesicles. (I) A bar graph showing mean and standard error of log2 fold change of Rab5A, Rab5C and Rab7 transcripts from single oocyte RNA-seq. **** P <0.0001, n.s. no significance, which are the P-adjust values by DESeq2 analysis. (J) PCA plots of Rab5 (left) and Rab7 (right) that is color-coded by the expression level of each transcript in individual oocytes. The control, cKO (major) and cKO (minor) groups are labeled in the Rab5 plot. Scale bars: 20 μm in A, C, F; 100 μm in H; 1 μm in E. Arbitrary fluorescence units (au) in B, D.
In the RNA-seq analyses, Rab5a transcripts were significantly increased in the cKO oocytes whereas Rab7 transcripts were decreased (Figure 4I). The accumulative Rab5a up-regulation and Rab7 down-regulation from cKO (minor) to cKO (major) was confirmed by their expression levels on the PCA plot (Figure 4J). We tried to phenocopy the cKO oocytes by over-expressing Rab5a-mVenus/Rab5c-mVenus cRNA in GV oocytes. However, there was no obvious delay or arrest in Rab5a-mVenus/Rab5c-mVenus over-expressed oocytes compared with the mVenus over-expression controls (Figure S4A-B). Nor did we observe defects in endosome maturation (Figure S4C-D) indicating that the endosome failure was coupled to other impaired vesicle components. To conclude, the cKO oocytes exhibited extensive defects in the endomembrane system.
Inhibitory phosphorylated CDK1 persists in cKO oocytes to block lamina disassembly
The driving force of nuclear envelope breakdown is the phosphorylation of nuclear lamina protein and nuclear pore components by active mitotic/meiotic kinases (34,35). To obtain better insight into GVBD, we divided oocytes into three groups based on lamin B integrity: intact, GVBD (early) and GVBD (late). The intact phase had an even and continuous distribution of lamin B surrounding the nucleus and was widely present among oocytes after 0 to 3 hr ex vivo incubation. The GVBD (early) group emerged ~1 hr, during which lamin B was still continuous (XY optical sections) but exhibiting much smaller enclosed areas and an unevenly shrunken pattern. Finally, after the nuclear envelop was breached by the spindle, lamin B became discontinuous and quickly disappeared, which defined the GVBD (late) phase. Lamin A/C staining precisely recapitulated that of lamin B (Figure S5).
Given the well-studied control of CDK1 phosphorylation in meiosis, we hypothesized that the inhibitory phosphorylated CDK1 favors GV arrest while its elimination eventually results in GVBD. To demonstrate this, we explored the change of inhibitory phosphorylated CDK1 (pT14/Y15CDK1) during wildtype GVBD. In oocytes at the intact phase, the pT14/Y15CDK1 formed nuclear puncta. When entering GVBD (early) phase, the puncta pattern underwent a constant decrease and eventually disappeared during the GVBD (late) phase (Figure 5A-B). We then hypothesized that the pT14/Y15CDK1 remained constant in cKO oocytes to block GVBD. At the GV stage, pT14/Y15CDK1 in cKO oocytes was similar to the control group, At GV+3hr when most control oocytes finished GVBD and exhibited a low level of pT14/Y15CDK1, the cKO oocytes retained pT14/Y15CDK1 puncta of high intensity (Figure 5C-D). Thus, failure in transiting CDK1 phosphorylation from inhibitory to active status results in the GV arrest in cKO oocytes.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
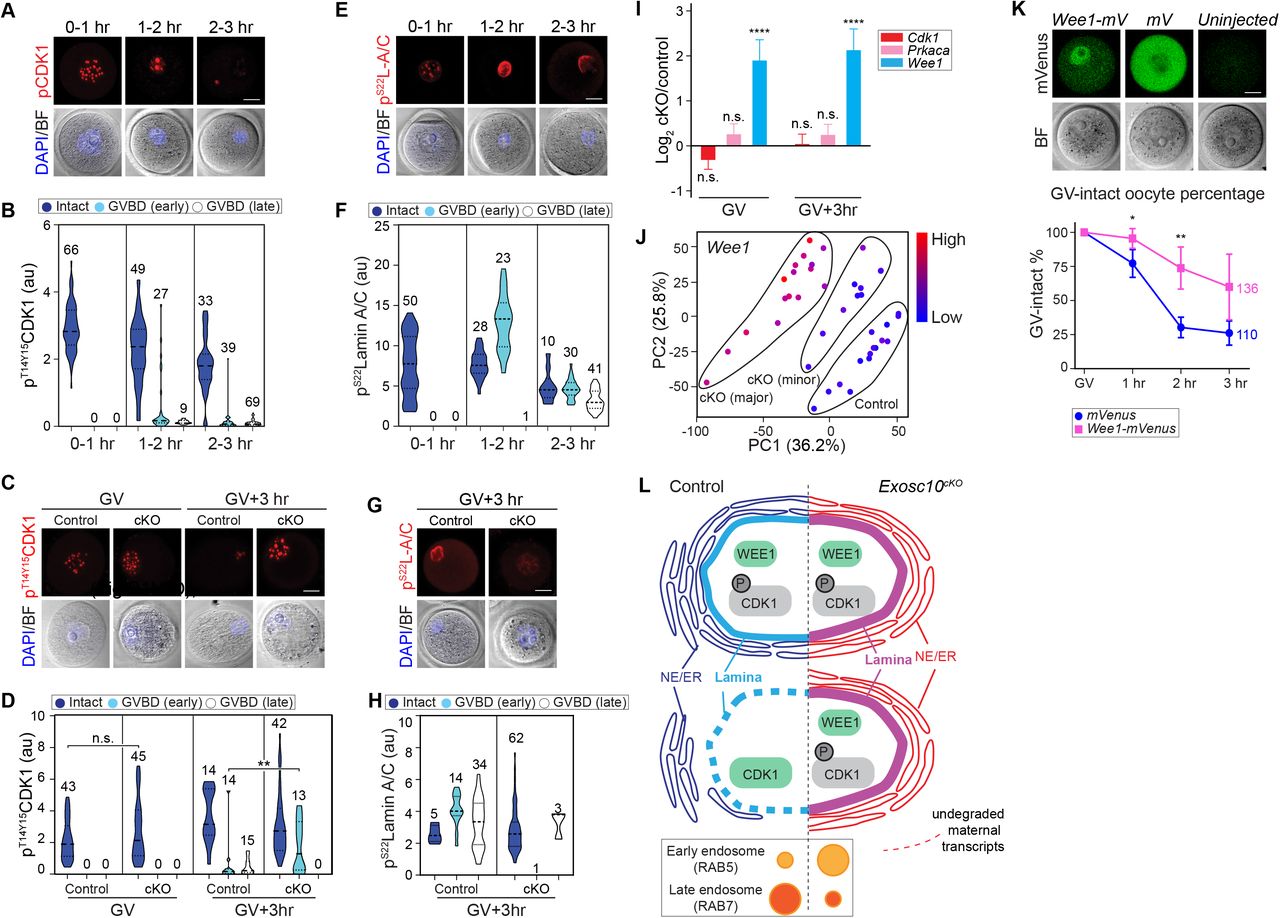
(A) Confocal fluorescence images of inhibitory phosphorylated CDK1 (pT14/Y15CDK1), bright-field and DAPI in wildtype oocytes at different time points during ex vivo culture and GVBD. Oocytes were collected every 30 min and combined into three stages: 0-1 hr includes 0 and 0.5 hr stages; 1-2 hr includes 1 and 1.5 hr stages; 2-3 hr includes 2, 2.5 and 3 hr stages. (B) Quantification of pT14/Y15CDK1 in A. Number of oocytes are indicated above each group. At each time points, oocytes are clustered as Intact, GVBD-early or GVBD-late based on lamin B integrity. (C) Representative images of pT14/Y15CDK1 in oocytes at GV and GV+3hr stages derived from control and cKO mice. (D) Quantification of pT14/Y15CDK1 fluorescence intensities in C. The numbers of oocytes from at least three experiments are indicated above each group. (E) Same as A, but for pS22lamin A/C (pS22L-A/C). (F) Same as B, but for pS22lamin A/C. (G) Same as C, but for pS22lamin A/C only at GV+3hr. (H) Same as D, but for pS22lamin A/C only at GV+3hr. (I) Bar graph of mean and standard error of log2 fold change of Cdk1, Prkaca, Wee1 transcripts from the single oocyte RNA-seq. **** P <0.0001, n.s. no significance, which are the P-adjust values by DESeq2 analysis. (J) PCA plot of Wee1 that is color-coded by the expression level in each oocyte. The control, cKO (major) and cKO (minor) groups are labeled in the plot. (K) Fluorescence of Wee1-mVenus and mVenus over-expression in GV-intact oocytes. The quantification of GV-intact oocyte percentage in 3 hr ex vivo culture is in the bottom. Each dot shows mean and standard deviation. ** P <0.01, * P <0.05 by two-tailed Student’s t-test. The horizontal lines in B, D, F, Hrepresent the median and quartiles. Scale bars: 20 μm in A, C, E, G, K. (L) Working model of the characterized defects in Exosc10cKO oocytes in which the absence of the RNase delays degradation of maternal transcripts which prevents dephosphorylation of CDK1. In the absence of this active cell cycle component, degradation of nuclear lamin B is delayed which preserves the nuclear envelope and delays GVBD leading to decrease female fecundity.
Next, we analyzed phosphorylation of lamin A/C (pS22lamin A/C) which is the direct substrate of active phosphorylated CDK1 (35). After 1 hr ex vivo culture, pS22lamin A/C displayed a nuclear puncta pattern in oocytes. Upon entering the GVBD (early) phase, pS22lamin A/C increased its abundance with concomitant loss of punctate loci and co-localized with lamin A/C. After progressing to the GVBD (late) phase, lamin A/C and pS22lamin A/C co-localized on the dissolving nuclear envelope albeit with decreased abundance (Figure 5E-F). These observations suggest that the activation of pT14/Y15CDK1 increases pS22lamin A/C, which results in nuclear envelope disassembly. In cKO oocytes, pS22lamin A/C failed to change from puncta to perinuclear localization at GV+3hr and failed to accumulate, both of which appear necessary for entering the GVBD (late) phase (Figure 5G-H).
The inhibitory CDK1 phosphorylation may be regulated by upstream kinase activity, including PKA and WEE1/2 (36,37). Our RNA-seq data documented that Wee1 was up-regulated significantly in cKO oocytes (log2FC 1.92 and 2.16 at GV and GV+3 hr, respectively, P-adjust<0.01), potentially contributing the higher pT14/Y15CDK1 level (Figure 5I). The significantly increased abundance of Wee1 during the transition from cKO (minor) to cKO (major) was confirmed by visualizing its expression level on the PCA plot (Figure 5J). To test whether the up-regulation of Wee1 may contribute to the GVBD failure in the cKO, we over-expressed Wee1-mVenus cRNA in wildtype GV oocytes. Nuclear expression of WEE1-mVenus was significantly increased in GV-intact oocytes within 3 hr ex vivo culture (Figure 5K) suggesting a delay and block of GVBD by WEE1 up-regulation. We also examined cAMP signaling which functions upstream of CDK1 phosphorylation to maintain the oocyte in a GV-intact state. However, cAMP levels and its known activators did not increase in cKO oocytes. These observations are consistent with signaling pathways downstream of cAMP facilitating persistent inhibitory CDK1 phosphorylation (Figure S6A-B). In conclusion, cKO oocytes had inappropriate CDK1 and lamin A/C phosphorylation regulation associated with the block to GVBD.
DISCUSSION
RNA degradation plays a critical role in shaping the maternal transcriptome for functional transitions, perturbation of which can result in disrupted meiosis or failed pre-implantation development. In our study, we generated oocyte-specific, conditional Exosc10 knockout mice in which the maternal transcriptome was dysregulated. We document an important role for EXOSC10 in orchestrating endomembrane components and meiotic cell cycle control through poly(A) RNA degradation. The absence of EXOSC10 directly leads to a decline in female fecundity.
Fully grown GV and GVBD oocytes are transcriptionally quiescent and genetic defects in normal degradation of RNA result in transcriptome dysregulation (6,10). In gene-edited mouse models that affect RNA metabolism, overall RNA profile is changed as a result of direct and secondary responses to the mutant genes. For example, Zfp36l2 is an activator of mRNA decay. The oocyte-specific knockout of Zfp36l2 disturbs a set of histone modifiers necessary to inhibit chromatin silencing at the end of oocyte growth (10). Knockout of Btg4, a scaffold protein bridging translation and deadenylation components, disrupts the translation of many RNAs and prevents morphologically normal egg from being fertilized (6). To obtain information on RNAs that undergo regulated degradation during oocyte maturation, we selected a gene with known RNase activity, EXOSC10, and confirmed poly(A) RNA as EXOSC10 substrates through both RNA FISH and RNA-seq. Combining phenotypic characterization and sequence analysis, we conclude that the EXOSC10-depleted GV oocytes have less potential to organize germinal vesicle breakdown because of defects in endomembrane trafficking and CDK1 phosphorylation activation which disrupts meiosis I resumption.
In our study, we introduced ERCC spike-in normalization for single oocyte RNA-seq analysis. This method not only provides a stringent criterion for library quality evaluation, but also allows comparison among libraries that vary considerably in size. In control oocytes, we observed a decrease in the abundance of 56 transcripts as oocytes progressed from the GV to the GV+3hr stage. Among them, 12 transcripts encoded ribosomal proteins, potentially preparing the poly(A) RNA for degradation and/or translation to provide proteins necessary for the growth-to-maturation transition. The other transcripts encoded proteins that were related to mitochondria and protein transport which are involved extensively in oocyte quality control and aging (38). We also observed a dramatic elimination of RNAs during oocyte progression from GV+3hr to the MII stage, consistent with known biological shrinkage of total RNA in maturing oocytes. Similar to the mutants of the other RNA regulators, we observed an overall change in RNA abundance in Exosc10 cKO oocytes, including a dramatic up-regulation of many genes, and a relatively milder down-regulation which may reflect secondary effects. The immediate dysregulation of the GV oocyte transcriptome after EXOSC10 depletion indicates its importance in oogenesis.
Mechanistically, our study shows that EXOSC10 sculpts the transcriptome to affect endomembrane components necessary for nuclear envelope breakdown. The endomembrane components participate in the maturation of oocytes by regulating: endosomes that balance GPR-cAMP signaling for meiosis arrest control (39); Golgi that fragment for assisting spindle initiation (40); ER that reorganizes from cytoplasmic network into cortical clusters for meiosis division (41); and lysosomes along with autophagosomes that actively degrade proteins, lipids and sugars (42). In control oocytes, we identified the initial reframing of the endomembrane system in the growth-to-maturation transition (GV to GV+3hr), including the down regulation of Sec61g and Sec61b. It has been reported that over expression of Sec61 does not affect post-mitotic nuclear envelope reorganization (12). However, given its close association with NE-ER exchange, our result indicates that Sec61 decrease may be required for NE disassembly. In EXOSC10 depleted oocytes, endomembrane components exhibit substantial defects. We identified several Rab family members up regulated in cKO oocytes, including Rab5a, Rab5b, Rab34 and Rab37, which are known as molecular switches in regulating intracellular membrane trafficking. The enlarged early endosome (Rab5) suggests the defects of endosome maturation. Rab34 up-regulation is known to promote phagosome fusion with lysosome; concomitantly, up-regulated Rab37 and Atg16l2 can also directly activate the phagosome formation (43,44), collectively suggesting that the enlarged lysosome in cKO oocytes may originate from activated phagocytosis (45). An important future study should address coupling between different vesicles which will add to the understanding of the role of the endomembrane system in oocyte quality control.
Meanwhile, the CDK1/lamin phosphorylation pathway that directly determines nuclear envelope breakdown is also impaired by depleting EXOSC10 in the oocytes. Previous studies demonstrate that WEE1 and WEE2 (also named WEE1B) both controls CDK1 inhibitory phosphorylation, down-regulation of which could promote GVBD, suggesting their roles in maintaining the meiotic arrest (37,46,47). In this study, Wee2 remains unchanged in the Exosc10 cKO oocytes whereas Wee1 is up-regulated about four folds, and the overexpression of Wee1 cRNA could reduce GVBD ratio, which collectively support that EXOSC10 could degrade Wee1 RNA to allow CDK1 activation for meiosis I resumption.
From the subfertility phenotype of the Exosc10 cKO mutant, we suspected heterogeneity among the cKO oocytes which is confirmed by the single oocyte RNA-seq results. The cKO (minor) to cKO (major) difference successfully demonstrates an exacerbation of the phenotype, including a growing number of differentially expressed genes that affect important pathways for maturation. The mechanism underlying this heterogeneity remains unclear. One possibility is that other RNA exosome-associated RNases, such as DIS3, could compensate for the loss of EXOSC10. In control groups, Dis3 transcript increases from GV to GV+3hr, potentiating its functions for oocyte maturation. However, Exosc10 cKO oocytes lack an increase in Dis3 abundance which makes it unlikely that DIS3 compensates for the loss of EXOSC10. Thus, there may be another RNase functioning redundantly for EXOSC10. Another possibility could come from stochastic variations of oocyte development. For example, variance in chromosome condensation at the end of oocyte growth may differentially affect gene silencing and affect nuclear envelope breakdown. Once key transcripts are below a functional threshold, oocytes may be able to proceed to meiosis with minimal amounts of EXOSC10.
In conclusion, our data document transcriptome remodeling in the transition from oocyte growth to maturation. We demonstrate that EXOSC10-mediated degradation of active poly(A) RNA underlies the transition by coordinating the transcriptome for endomembrane trafficking and resumption of meiosis. These results are consistent with a growing body of experimental data that RNA degradation governs the later stages of oogenesis while putting the oocyte gain of developmental potential into a more refined time window.
DATA AVAILABILITY
The sequencing data reported in this study has been deposited in the Gene Expression Omnibus website with accession code GSE133748.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
This research was supported by the Intramural Research Program of NIH, National Institute of Diabetes and Digestive and Kidney Diseases (1ZIADK015603).
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGEMENTS
We appreciate the critical reading of the manuscript by Shaohe Wang, Ph.D. and Yangu Zhao, Ph.D. as well as useful discussions with all the members of the Jurrien Dean lab. D.W. and J.D conceived the project.
D.W. performed the experiments and C.P. provided bioinformatic expertise. D.W. and J.D. wrote manuscript with input from C.P.
Footnotes
Figures updated. Supplemental figures updated. One more supplemental table added.
REFERENCE




