

Abstract
The domestic pig (Sus scrofa) is important both as a food source and as a biomedical model with high anatomical and immunological similarity to humans. The draft reference genome (Sscrofa10.2) represents a purebred female pig from a commercial pork production breed (Duroc), and was established using older clone-based sequencing methods. The Sscrofa10.2 assembly was incomplete and unresolved redundancies, short range order and orientation errors and associated misassembled genes limited its utility. We present two genome assemblies created with more recent long read technologies and a whole genome shotgun strategy, one for the same Duroc female (Sscrofa11.1) and one for an outbred, composite breed male animal commonly used for commercial pork production (USMARCv1.0). Both assemblies are of substantially higher (>90-fold) continuity and accuracy compared to the earlier reference, and the availability of two independent assemblies provided an opportunity to identify large-scale variants and to error-check the accuracy of representation of the genome. We propose that the improved Duroc breed assembly (Sscrofa11.1) become the reference genome for genomic research in pigs.
Introduction
High quality, richly annotated reference genome sequences are key resources and provide important frameworks for the discovery and analysis of genetic variation and for linking genotypes to function. In farmed animal species such as the domestic pig (Sus scrofa) genome sequences have been integral to the discovery of molecular genetic variants and the development of single nucleotide polymorphism (SNP) chips1 and enabled efforts to dissect the genetic control of complex traits, including responses to infectious diseases2.
Genome sequences are not only an essential resource for enabling research but also for applications in the life sciences. Genomic selection, in which associations between thousands of SNPs and trait variation as established in a phenotyped training population are used to choose amongst selection candidates for which there are SNP data but no phenotypes, has delivered genomics-enabled genetic improvement in farmed animals3 and plants. From its initial successful application in dairy cattle breeding, genomic selection is now being used in many sectors within animal and plant breeding, including by leading pig breeding companies4,5.
The domestic pig (Sus scrofa) has importance not only as a source of animal protein but also as a biomedical model. The choice of the optimal animal model species for pharmacological or toxicology studies can be informed by knowledge of the genome and gene content of the candidate species including pigs6. A high quality, richly annotated genome sequence is also essential when using gene editing technologies to engineer improved animal models for research or as sources of cells and tissue for xenotransplantation and potentially for improved productivity7,8.
The highly continuous pig genome sequences reported here are built upon a quarter of a century of effort by the global pig genetics and genomics research community including the development of recombination and radiation hybrid maps9,10, cytogenetic and Bacterial Artificial Chromosome (BAC) physical maps11,12 and a draft reference genome sequence13. The previously published draft pig reference genome sequence (Sscrofa10.2), developed under the auspices of the Swine Genome Sequencing Consortium (SGSC), has a number of significant deficiencies14–17. The BAC-by-BAC hierarchical shotgun sequence approach18 using Sanger sequencing technology can yield a high quality genome sequence as demonstrated by the public Human Genome Project. However, with a fraction of the financial resources of the Human Genome Project, the resulting draft pig genome sequence comprised an assembly, in which long-range order and orientation is good, but the order and orientation of sequence contigs within many BAC clones was poorly supported and the sequence redundancy between overlapping sequenced BAC clones was often not resolved. Moreover, about 10% of the pig genome, including some important genes, were not represented (e.g. CD163), or incompletely represented (e.g. IGF2) in the assembly19. Whilst the BAC clones represent an invaluable resource for targeted sequence improvement and gap closure as demonstrated for chromosome X (SSCX)20, a clone-by-clone approach to sequence improvement is expensive notwithstanding the reduced cost of sequencing with next-generation technologies.
The dramatically reduced cost of whole genome shotgun sequencing using Illumina short read technology has facilitated the sequencing of several hundred pig genomes17,21,22. Whilst a few of these additional pig genomes have been assembled to contig level, most of these genome sequences have simply been aligned to the reference and used as a resource for variant discovery.
The increased capability and reduced cost of third generation long read sequencing technology as delivered by Pacific Biosciences and Oxford Nanopore platforms, have created the opportunity to generate the data from which to build highly contiguous genome sequences as illustrated recently for cattle23,24. Here we describe the use of Pacific Biosciences (PacBio) long read technology to establish highly continuous pig genome sequences that provide substantially improved resources for pig genetics and genomics research and applications.
Results
Two individual pigs were sequenced independently: a) TJ Tabasco (Duroc 2-14) i.e. the sow that was the primary source of DNA for the published draft genome sequence (Sscrofa10.2)13 and b) MARC1423004 which was a Duroc/Landrace/Yorkshire crossbred barrow (i.e. castrated male pig) from the USDA Meat Animal Research Center. The former allowed us to build upon the earlier draft genome sequence, exploit the associated CHORI-242 BAC library resource (https://bacpacresources.org/ http://bacpacresources.org/porcine242.htm) and evaluate the improvements achieved by comparison with Sscrofa10.2. The latter allowed us to assess the relative efficacy of a simpler whole genome shotgun sequencing and Chicago Hi-Rise scaffolding strategy25. This second assembly also provided data for the Y chromosome, and supported comparison of haplotypes between individuals. In addition, full-length transcript sequences were collected for multiple tissues from the MARC1423004 animal, and used in annotating both genomes.
Sscrofa11.1 assembly
Approximately sixty-five fold coverage (176 Gb) of the genome of TJ Tabasco (Duroc 2-14) was generated using Pacific Biosciences (PacBio) single-molecule real-time (SMRT) sequencing technology. A total of 213 SMRT cells produced 12,328,735 subreads of average length 14,270 bp and with a read N50 of 19,786 bp (Supplementary Table ST1). Reads were corrected and assembled using Falcon (v.0.4.0)26, achieving a minimum corrected read cutoff of 13 kb that provided 19-fold genome coverage for input resulting in an initial assembly comprising 3,206 contigs with a contig N50 of 14.5 Mb.
The contigs were mapped to the previous draft assembly (Sscrofa10.2) using Nucmer27. The long range order of the Sscrofa10.2 assembly was based on fingerprint contig (FPC)12 and radiation hybrid physical maps with assignments to chromosomes based on fluorescent in situ hybridisation data. This alignment of Sscrofa10.2 and the contigs from the initial Falcon assembly of the PacBio data provided draft scaffolds that were tested for consistency with paired BAC and fosmid end sequences and the radiation hybrid map13. The draft scaffolds also provided a framework for gap closure using PBJelly28, or finished quality Sanger sequence data generated from CHORI-242 BAC clones from earlier work13,20.
Remaining gaps between contigs within scaffolds, and between scaffolds predicted to be adjacent on the basis of other available data, were targeted for gap filling with a combination of unplaced contigs and previously sequenced BACs, or by identification and sequencing of BAC clones predicted from their end sequences to span the gaps. The combination of methods filled 2,501 gaps and reduced the number of contigs in the assembly from 3,206 to 705. The assembly, Sscrofa11 (GCA_000003025.5), had a final contig N50 of 48.2 Mb, only 103 gaps in the sequences assigned to chromosomes, and only 583 remaining unplaced contigs (Table 1). Two acrocentric chromosomes (SSC16, SSC18) were each represented by single, unbroken contigs. The SSC18 assembly also includes centromeric and telomeric repeats (Supplementary Tables ST5, ST6; Supplementary Figures SF9, SF10), albeit the former probably represent a collapsed version of the true centromere. The reference genome assembly was completed by adding Y chromosome sequences from other sources (GCA_900119615.2)20 because TJ Tabasco (Duroc 2-14) was female. The resulting reference genome sequence was termed Sscrofa11.1 and deposited in the public sequence databases (GCA_000003025.6) (Table 1).
Summary statistics for assembled pig genome sequences and comparison with current human reference genome§
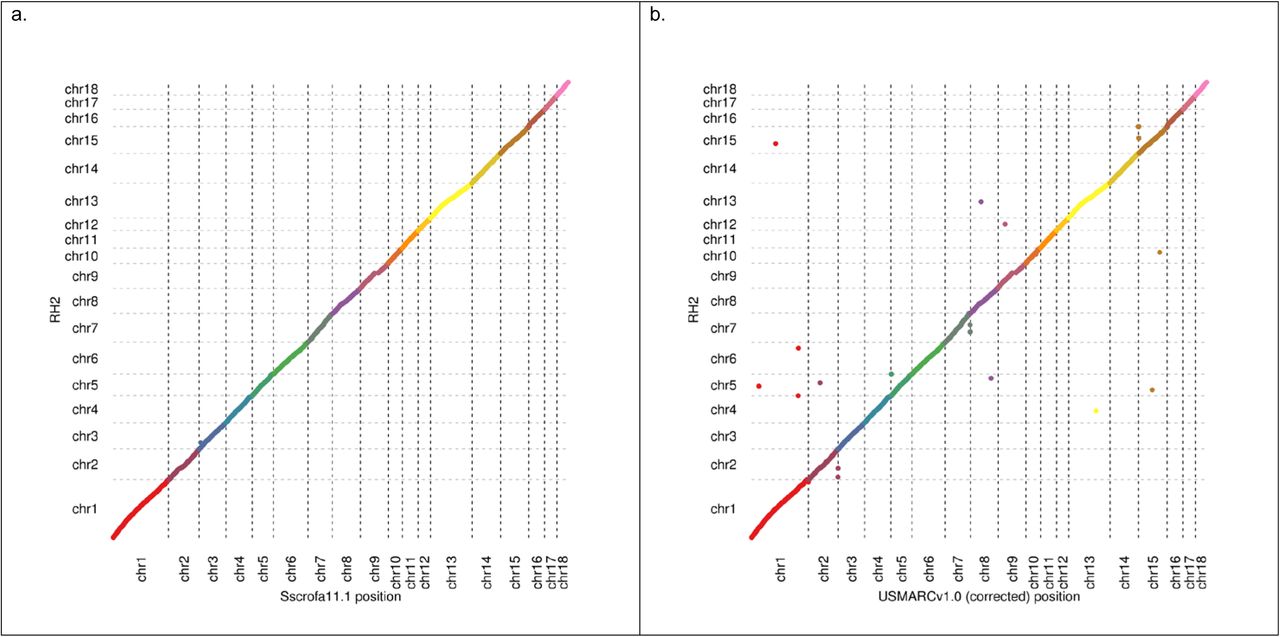
The medium to long range order and orientation of Sscrofa11.1 assembly was assessed by comparison to an existing radiation hybrid (RH) map9. The comparison strongly supported the overall accuracy of the assembly (Figure 1a), despite the fact that the RH map was prepared from a cell line of a different individual. There is one major disagreement between the RH map and the assembly on chromosome 3, which will need further investigating. The only other substantial disagreement on chromosome 9, is explained by a gap in the RH map9. The assignment and orientation of the Sscrofa11.1 scaffolds to chromosomes was confirmed with fluorescent in situ hybridisation (FISH) of BAC clones (Supplementary Table ST2, Supplementary Figure SF1). The BAC end sequences and in some cases complete BAC clone sequences from the BAC clones used as probes for FISH analyses were aligned with the Sscrofa11.1 assembly in order to establish the link between the FISH results and the assembly.

Plot illustrating co-linearity between radiation hybrid map and a) Sscrofa11.1 and b) USMARCv1.0 assemblies (autosomes only)
The quality of the Sscrofa11 assembly, which corresponds to Sscrofa11.1 after the exclusion of SSCY, was assessed as described previously for the existing Sanger sequence based draft assembly (Sscrofa10.2)14. Alignments of Illumina sequence reads from the same female pig were used to identify regions of low quality (LQ) or low coverage (LC) (Table 2). The analysis confirms that Sscrofa11 represents a significant improvement over the Sscrofa10.2 draft assembly. For example, the Low Quality Low Coverage (LQLC) proportion of the genome sequence has dropped from 33.07% to 16.3% when repetitive sequence is not masked, and falls to 1.6% when repeats are masked prior to read alignment. The remaining LQLC segments of Sscrofa11 may represent regions where short read coverage is low due to known systematic errors of the short read platform related to GC content, rather than deficiencies of the assembly.
Summary of quality statistics for SSC1-18, SSCX
The Sscrofa11.1 assembly was also assessed visually using gEVAL29. The improvement in short range order and orientation as revealed by alignments with isogenic BAC and fosmid end sequences is illustrated for a particularly poor region of Sscrofa10.2 on chromosome 12 (Supplementary Figure SF12). The problems in this area of Sscrofa10.2 arise from failures to order and orient the sequence contigs and resolve the redundancies between these sequence contigs within BAC clone CH242-147O24 (FP102566.2). The improved contiguity in Sscrofa11.1 not only resolves these local order and orientation errors, but also facilitates the annotation of a complete gene model for the ABR locus. Further examples of comparisons of Sscrofa10.2 and Sscrofa11.1 reveal improvements in contiguity, local order and orientation and gene models (Supplementary Figure SF13-15).
USMARCv1.0 assembly
Approximately sixty-five fold coverage of the genome of the MARC1423004 barrow was generated on a PacBio RSII instrument. The sequence was collected during the transition from P5/C3 to P6/C4 chemistry, with approximately equal numbers of subreads from each chemistry. A total of 199 cells of P5/C3 chemistry produced 95.3 Gb of sequence with mean subread length of 5.1 kb and subread N50 of 8.2 kb. A total of 127 cells of P6/C4 chemistry produced 91.6 Gb of sequence with mean subread length 6.5 kb and subread N50 of 10.3 kb, resulting in an overall average subread length, including data from both chemistries, of 6.4 kb. The reads were assembled using Celera Assembler 8.3rc230 and Falcon (https://pb-falcon.readthedocs.io/en/latest/about.html). The resulting assemblies were compared and the Celera Assembler result was selected based on better agreement with a Dovetail Chicago® library25, and was used to create a scaffolded assembly with the HiRise™ scaffolder consisting of 14,818 contigs with a contig N50 of 6.372 Mb (GenBank accession GCA_002844635.1; Table 1). The USMARCv1.0 scaffolds were therefore completely independent of the existing Sscrofa10.2 or new Sscrofa11.1 assemblies, and they can act as supporting evidence where they agree with those assemblies. However, chromosome assignment of the scaffolds was performed by alignment to Sscrofa10.2, and does not constitute independent confirmation of this ordering. The assignment of these scaffolds to individual chromosomes was confirmed post-hoc by FISH analysis as described for Sscrofa11.1 above. The FISH analysis revealed that several scaffold assemblies (SSC1, 5, 6-11, 13-16) are inverted with respect to the chromosome (Supplementary Table ST2, Supplementary Figures SF1, 3-5). After correcting the orientation of these inverted scaffolds, there is good agreement between the USMARCv1.0 assembly and the RH map9 (Figure 1b).
Sscrofa11.1 and USMARCv1.0 are co-linear
The alignment of the two PacBio assemblies reveals a high degree of agreement and co-linearity, after correcting the inversions of several USMARCv1.0 chromosome assemblies (Supplementary Figure SF2). The agreement between the Sscrofa11.1 and USMARCv1.0 assemblies is also evident in comparisons of specific loci (Supplementary Figures SF13-15) although with some differences (e.g. Supplementary Figure SF14). The whole genome alignment of Sscrofa11.1 and USMARCv1.0 (Supplementary Figure SF2) masks some inconsistencies that are evident when the alignments are viewed on a single chromosome-by-chromosome basis (Supplementary Figures SF3-5). It remains to be determined whether the small differences between the assemblies represent errors in the assemblies, or true structural variation between the two individuals (see discussion of the ERLIN1 locus below).
Repetitive sequences, centromeres and telomeres
The repetitive sequence content of the Sscrofa11.1 and USMARCv1.0 was identified and characterised as described in the Supplementary Materials. These analyses allowed the identification of centromeres and telomeres for several chromosomes. The previous reference genome (Sscrofa10.2) that was established from Sanger sequence data and a minipig genome (minipig_v1.0, GCA_000325925.2) that was established from Illumina short read sequence data were also included for comparison.
Completeness of the assemblies
The Sscrofa11.1 and USMARCv1.0 assemblies were assessed for completeness using two tools, BUSCO (Benchmarking Universal Single-Copy Orthologs)31 and Cogent (https://github.com/Magdoll/Cogent). BUSCO uses a database of expected gene content based on near-universal single-copy orthologs from species with genomic data, while Cogent uses transcriptome data from the organism being sequenced, and therefore provides an organism-specific view of genome completeness. BUSCO analysis suggests both new assemblies are highly complete, with 93.8% and 93.1% of BUSCOs complete for Sscrofa11.1 and USMARCv1.0 respectively, a marked improvement on the 80.9% complete in Sscrofa10.2 (Supplementary Table ST3).
Cogent is a tool that identifies gene families and reconstructs the coding genome using high-quality transcriptome data without a reference genome, and can be used to check assemblies for the presence of these known coding sequences. The PacBio transcriptome (Iso-Seq data, from nine adult tissues)32 used for the Cogent analyses originated from the MARC1423004 animal. Thus, it is possible that genes flagged as absent or fragmented genes by the Cogent analysis of Sscrofa11.1 are missing due to true deletion events in the Duroc 2-14 genome rather than errors in the assembly. There were five genes that were present in the Iso-Seq data, but missing in the Sscrofa11.1 assembly. In each of these five cases, a Cogent partition (which consists of 2 or more transcript isoforms of the same gene, often from multiple tissues) exists in which the predicted transcript does not align back to Sscrofa11.1. NCBI-BLASTN of the isoforms from the partitions revealed them to have near perfect hits with existing annotations for CHAMP1, ERLIN1, IL1RN, MB, and PSD4.
ERLIN1 is missing in Sscrofa11.1, in its expected location there is a tandem duplication of the neighbouring gene CYP2C33 (Supplementary Figure SF16), which the Illumina and BAC data in this region support, suggesting this area may represent a true haplotype. Indeed, a copy number variant (CNV) nsv1302227 has been mapped to this location on SSC1433 and the ERLIN1 gene sequences present in BAC clone CH242-513L2 (ENA: CT868715.3) were incorporated into the earlier Sscrofa10.2 assembly. However, an alternative haplotype containing ERLIN1 was not found in any of the assembled contigs from Falcon and this will require further investigation. The ERLIN1 locus is present on SSC14 in the USMARCv1.0 assembly (30,107,823 – 30,143,074; note the USMARCv1.0 assembly of SSC14 is inverted relative to Sscrofa11.1) as determined with a BLAST search with the sequence of pig ERLIN1 mRNA (NM_001142896.1).
The other 4 genes are annotated in neither Sscrofa10.2 nor Sscrofa11.1. Two of these genes, IL1RN and PSD4, are present in the original Falcon contigs, however they were trimmed off during the contig QC stage because of apparent abnormal Illumina, BAC and fosmid mapping in the region which was likely caused by the repetitive nature of their expected location on chromosome 3 where a gap is present. CHAMP1 is expected to be in the telomeric region of chromosome 11, and is present in an unplaced scaffold of USMARCv1.0, so it is likely the gene is erroneously missing from the end of chromosome 11. Genes expected to neighbour MB, such as RSD2 and HMOX1, are annotated in Sscrofa11.1, but are on unplaced scaffolds AEMK02000361.1 and AEMK02000361.1, respectively. A gene annotated in MB’s expected position (ENSSSCG00000032277) appears to be a fragment of MB, but as there is no gap in the assembly it is likely that the incomplete MB is a result of a misassembly in this region. This interpretation is supported by a break in the pairs of BAC and fosmid end sequences that map to this region of the Sscrofa11.1 assembly. The MB gene is present in the USMARCv1.0 assembly flanked as expected by HMOX1 and RBFOX2. Cogent analysis also identified 2 cases of potential fragmentation in the Sscrofa11.1 genome assembly that resulted in the isoforms being mapped to two separate loci, though these will require further investigation. In summary, the BUSCO and Cogent analyses indicate that the Sscrofa11.1 assembly captures a very high proportion of the expressed elements of the genome.
Improved annotation
Annotation of Sscrofa11.1 was carried out with the Ensembl annotation pipeline and released via the Ensembl Genome Browser34 (http://www.ensembl.org/Sus_scrofa/Info/Index) (Ensembl release 90, August 2017). Statistics for the annotation are listed in Table 3. This annotation is more complete than that of Sscrofa10.2 and includes fewer fragmented genes and pseudogenes.
Ensembl annotation of pig (Sscrofa10.2, Sscrofa11.1, USMARCv1.0), human (GRCh38.p12) and mouse (GRCm38.p6) assemblies
The annotation pipeline utilised extensive short read RNA-Seq data from 27 tissues and long read PacBio Iso-Seq data from 9 adult tissues. This provided an unprecedented window into the pig transcriptome and allowed for not only an improvement to the main gene set, but also the generation of tissue-specific gene tracks from each tissue sample. The use of Iso-Seq data also improved the annotation of UTRs, as they represent transcripts sequenced across their full length from the polyA tract.
In addition to improved gene models, annotation of the Sscrofa11.1 assembly provides a more complete view of the porcine transcriptome than annotation of the previous assembly (Sscrofa10.2; Ensembl releases 67-89, May 2012 – May 2017) with increases in the numbers of transcripts annotated (Table 3). However, the number of annotated transcripts remains lower than in the human and mouse genomes. The annotation of the human and mouse genomes and in particular the gene content and encoded transcripts has been more thorough as a result of extensive manual annotation.
Efforts were made to annotate important classes of genes, in particular immunoglobulins and olfactory receptors. For these genes, sequences were downloaded from specialist databases and the literature in order to capture as much detail as possible (see supplementary information for more details).
These improvements in terms of the resulting annotation were evident in the results of the comparative genomics analyses run on the gene set. The previous annotation had 12,919 one-to-one orthologs with human, while the new annotation of the Sscrofa11.1 assembly has 15,543. Similarly, in terms of conservation of synteny, the previous annotation had 11,661 genes with high confidence gene order conservation scores, while the new annotation has 15,958. There was also a large reduction in terms of genes that were either abnormally short or split when compared to their orthologs in the new annotation.
The Sscrofa11.1 assembly has also been annotated using the NCBI pipeline (https://www.ncbi.nlm.nih.gov/genome/annotation_euk/Sus_scrofa/106/). We have compared these two annotations. The Ensembl and NCBI annotations of Sscrofa11.1 are broadly similar (Supplementary Table ST14). There are 18,722 protein coding genes and 811 non-coding genes in common. However, 1,625 of the genes annotated as protein-coding by Ensembl are annotated as pseudogenes by NCBI and 1,378 genes annotated as non-coding by NCBI are annotated as protein-coding by Ensembl. The NCBI RefSeq annotation can be visualised in the Ensembl Genome Browser by loading the RefSeq GFF3 track and the annotations compared at the individual locus level. Similarly, the Ensembl annotated genes can be visualised in the NCBI Genome Browser. More recently, we have annotated the USMARCv1.0 assembly using the Ensembl pipeline and this annotation is due for release with Ensembl release 97 (expected July 2019; see Table 3 for summary statistics).
Discussion
We have assembled a superior, extremely continuous reference assembly (Sscrofa11.1) by leveraging the excellent contig lengths provided by long reads, and a wealth of available data including Illumina paired-end, BAC end sequence, finished BAC sequence, fosmid end sequences, and the earlier curated draft assembly (Sscrofa10.2). The pig genome assemblies USMARCv1.0 and Sscrofa11.1 reported here are 92-fold to 694-fold respectively, more continuous than the published draft reference genome sequence (Sscrofa10.2)13. The new pig reference genome assembly (Sscrofa11.1) with its contig N50 of 48,231,277 bp and 506 gaps compares favourably with the current human reference genome sequence (GRCh38.p12) that has a contig N50 of 57,879,411 bp and 875 gaps (Table 3). Indeed, considering only the chromosome assemblies built on PacBio long read data (i.e. Sscrofa11 - the autosomes SSC1-SSC18 plus SSCX), there are fewer gaps in the pig assembly than in human reference autosomes and HSAX assemblies. Most of the gaps in the Sscrofa11.1 reference assembly are attributed to the fragmented assembly of SSCY. The capturing of centromeres and telomeres for several chromosomes (Supplementary Tables ST5, ST6; Supplementary Figures SF9, SF10) provides further evidence that the Sscrofa11.1 assembly is more complete. The increased contiguity of Sscrofa11.1 is evident in the graphical comparison to Sscrofa10.2 illustrated in Figure 2.

{kind=link}
{kind=link}
Graphical visualisation of contigs for Sscrofa11 (top) and Sscrofa10.2 (bottom) as alternating dark and light grey bars
The improvements in the reference genome sequence (Sscrofa11.1) relative to the draft assembly (Sscrofa10.2)13 are not restricted to greater continuity and fewer gaps. The major flaws in the BAC clone-based draft assembly were i) failures to resolve the sequence redundancy amongst sequence contigs within BAC clones and between adjacent overlapping BAC clones and ii) failures to accurately order and orient the sequence contigs within BAC clones. Although the Sanger sequencing technology used has a much lower raw error rate than the PacBio technology, the sequence coverage was only 4-6 fold across the genome. The improvements in continuity and quality (Table 2; Supplementary Figures SF13-15) have yielded a better template for annotation resulting in better gene models. The Sscrofa11.1 and USMARCv1.0 assemblies are classed as 4|4|1 and 3|5|1 [10X: N50 contig (kb); 10Y: N50 scaffold (kb); Z = 1|0: assembled to chromosome level] respectively compared to Sscrofa10.2 as 1|2|1 and the human GRCh38p5 assembly as 4|4|1 (see https://geval.sanger.ac.uk).
The improvement in the complete BUSCO (Benchmarking Universal Single-Copy Orthologs) genes indicates that both Sscrofa11.1 and USMARCv1.0 represent superior templates for annotation of gene models than the draft Sscrofa10.2 assembly (Supplementary Table ST3). Further, a companion bioinformatics analysis of available Iso-seq and companion Ilumina RNA-seq data across the nine tissues surveyed has identified a large number (>54,000) of novel transcripts32. A majority of these transcripts are predicted to be spliced and validated by RNA-seq data. Beiki and colleagues identified 10,465 genes expressing Iso-seq transcripts that are present on the Sscrofa11.1 assembly, but which are unannotated in current NCBI or Ensembl annotations.
We demonstrate moderate improvements in the placement and ordering of commercial SNP genotyping markers on the Sscrofa11.1 reference genome which will impact future genomic selection programs. The reference-derived order of SNP markers plays a significant role in imputation accuracy, as demonstrated by a whole-genome survey of misassembled regions in cattle that found a correlation between imputation errors and misassemblies35. We identified 1,709, 56, and 224 markers on the PorcineSNP60, GGP LD and 80K commercial chips that were previously unmapped and now have coordinates on the Sscrofa11.1 reference (Supplementary Table ST8). These newly mapped markers can now be imputed into a cross-platform, common set of SNP markers for use in genomic selection. Additionally, we have identified areas of the genome that are poorly tracked by the current set of commercial SNP markers. The previous Sscrofa10.2 reference had an average marker spacing of 3.57 kbp (Stdev: 26.5 kb) with markers from four commercial genotyping arrays. We found this to be an underestimate of the actual distance between markers, as the Sscrofa11.1 reference coordinates consisted of an average of 3.91 kbp (Stdev: 14.9 kbp) between the same set of markers. We also found a region of 2.56 Mbp that is currently devoid of suitable markers on the new reference. These gaps in marker coverage will inform future marker selection surveys, which are likely to prioritize regions of the genome that are not currently being tracked by marker variants in close proximity to potential causal variant sites.
The cost of high coverage whole-genome sequencing (WGS) precludes it from routine use in breeding programs. However, it has been suggested that low coverage WGS followed by imputation of haplotypes may be a cost-effective replacement for SNP arrays in genomic selection36. Imputation from low coverage sequence data to whole genome information has been shown to be highly accurate37,38. At the 2018 World Congress on Genetics Applied to Livestock Production Aniek Bouwman reported that in a comparison of Sscrofa10.2 with Sscrofa11.1 (for SSC7 only) for imputation from 600K SNP genotypes to whole genome sequence overall imputation accuracy on SSC7 improved considerably from 0.81 (1,019,754 variants) to 0.90 (1,129,045 variants) (Aniek Bouwman, pers. comm). Thus, the improved assembly may not only serve as a better template for discovering genetic variation but also have advantages for genomic selection, including improved imputation accuracy.
Advances in the performance of long read sequencing and scaffolding technologies, improvements in methods for assembling the sequence reads and reductions in costs are enabling the acquisition of ever more complete genome sequences for multiple species and multiple individuals within a species. For example, in terms of adding species, the Vertebrate Genomes Project (https://vertebrategenomesproject.org/) aims to generate error-free, near gapless, chromosomal level, haplotyped phase assemblies of all of the approximately 66,000 vertebrate species and is currently in its first phase that will see such assemblies created for an exemplar species from all 260 vertebrate orders. At the level of individuals within a species, smarter assembly algorithms and sequencing strategies are enabling the production of high quality truly haploid genome sequences for outbred individuals24. The establishment of assembled genome sequences for key individuals in the nucleus populations of the leading pig breeding companies is achievable and potentially affordable. However, 10-30x genome coverage short read data generated on the Illumina platform and aligned to a single reference genome is likely to remain the primary approach to sequencing multiple individuals within farmed animal species such as cattle and pigs21,39.
There are significant challenges in making multiple assembled genome resources useful and accessible. The current paradigm of presenting a reference genome as a linear representation of a haploid genome of a single individual is an inadequate reference for a species. As an interim solution the Ensembl team are annotating multiple assemblies for some species such as mouse (https://www.ensembl.org/Mus_musculus/Info/Strains)40. We are currently implementing this solution for pig genomes, including an annotated USMARCv1.0 that will facilitated the detailed comparison of the two assemblies described here.
The current human genome reference already contains several hundred alternative haplotypes and it is expected that the single linear reference genome of a species will be replaced with a new model – the graph genome41,42,43. These paradigm shifts in the representation of genomes present challenges for current sequence alignment tools and the ‘best-in-genome’ annotations generated thus far. The generation of high quality annotation remains a labour-intensive and time-consuming enterprise. Comparisons with the human and mouse reference genome sequences which have benefited from extensive manual annotation indicate that there is further complexity in the porcine genome as yet unannotated (Table 3). It is very likely that there are many more transcripts, pseudogenes and non-coding genes (especially long non-coding genes), to be discovered and annotated on the pig genome sequence32. The more highly continuous pig genome sequences reported here provide an improved framework against which to discover functional sequences, both coding and regulatory, and sequence variation. After correction for some contig/scaffold inversions in the USMARCv1.0 assembly, the overall agreement between the assemblies is quite high and illustrates that the majority of genomic variation is at smaller scales of structural variation. However, both assemblies still represent a composite of the two parental genomes present in the animals, with unknown effects of haplotype switching on the local accuracy across the assembly.
Future developments in high quality genome sequences for the domestic pig are likely to include: (i) gap closure of Sscrofa11.1 to yield an assembly with one contig per (autosomal) chromosome arm exploiting the isogenic BAC and fosmid clone resource as illustrated here for chromosome 16 and 18; and (ii) haplotype resolved assemblies of a Meishan and White Composite F1 crossbred pig currently being sequenced. Beyond this haplotype resolved assemblies for key genotypes in the leading pig breeding company nucleus populations and of miniature pig lines used in biomedical research can be anticipated in the next 5 years. Unfortunately, some of these genomes may not be released into the public domain. The first wave of results from the Functional Annotation of ANimal Genomes (FAANG) initiative (Andersson et al., 2015; Foissac et al., 2018), are emerging and will add to the richness of pig genome annotation.
In conclusion, the new pig reference genome (Sscrofa11.1) described here represents a significantly enhanced resource for genetics and genomics research and applications for a species of importance to agriculture and biomedical research.
Author contributions
A.L.A. and T.P.L.S. conceived, coordinated and managed the project; A.L.A., P.F., D.A.H., T.P.L.S. M.W. supervised staff and students performing the analyses; D.J.N., L.R., L.B.S., T.P.L.S. provided biological resources; R.H., K.S.K. and T.P.L.S. generated PacBio sequence data; H.A.F., T.P.L.S. and R.T. generated Illumina WGS and RNA-Seq data; N.A.A., C.A.S., B.M.S. provided SSCY assemblies; D.J.N, and T.P.L.S. generated Iso-Seq data; G.H., R.H., S.K., A.M.P., A.S.S, A.W. generated sequence assemblies; A.W. polished and quality checked Sscrofa11.1; W.C., G.H., K.H., S.K., B.D.R., A.S.S., S.G.S., E.T. performed quality checks on the sequence assemblies; R.E.O’C. and D.K.G. performed cytogenetics analyses; L.E. analysed repeat sequences; H.B., H.L., N.M., C.K.T. analysed Iso-Seq data; D.M.B. and G.A.R. analysed sequence variants; B.A., K.B., C.G.G., T.H., O.I., F.J.M. annotated the assembled genome sequences; A.W. and A.L.A drafted the manuscript; all authors read and approved the final manuscript.
Acknowledgements
We are grateful for funding support from the i) Biotechnology and Biological Sciences Research Council (Institute Strategic Programme grants: BBS/E/D/20211550, BBS/E/D/10002070; and response mode grants: BB/F021372/1, BB/M011461/1, BB/M011615/1, BB/M01844X/1); ii) European Union through the Seventh Framework Programme Quantomics (KBBE222664); iii) University of Cambridge, Department of Pathology; iv) Wellcome Trust: WT108749/Z/15/Z; v) European Molecular Biology Laboratory; and vi) the Roslin Foundation. In addition HL and HB were supported by USDA NRSP-8 Swine Genome Coordination funding; SK and AMP were supported by the Intramural Research Program of the National Human Genome Research Institute, US National Institutes of Health; D.M.B was supported by USDA CRIS projects 8042-31000-001-00-D and 5090-31000-026-00-D. B.D.R was supported by USDA CRIS project 8042-31000-001-00-D. T.P.L.S. was supported by USD CRIS project 3040-31000-100-00-D. This work used the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov); and the Iowa State University Lightning3 and ResearchIT clusters. The Ceres cluster (part of the USDA SCInet Initiative) was used to analyse part of this dataset.
We are grateful to Chris Tyler-Smith (Wellcome Trust Sanger Institute) for sharing the SSCY sequence data for Sscrofa11.1.
References




