

Abstract
Eukaryotic genomes are duplicated from thousands of replication origins that fire sequentially forming a defined spatiotemporal pattern of replication clusters. The importance of the organization of replisomes into functional clusters, called replication factories, is still poorly understood. Here we show that the isolation of Proteins On Nascent DNA (iPOND) method is strongly dependent on the organization level of replication factories. We find that RIF1 is a component of replication factories. The purification of replication-associated proteins from RIF1-depleted cells using iPOND reveals a major defect in the clustering of replication factors upon mild replicative stress. The loss of organization caused by RIF1 depletion leads to defects in replication forks dynamic and yields DNA lesions. We propose a model whereby RIF1 encases replication factories to prevent the formation of DNA lesions in response to replicative stress. The data highlight the importance of the organization of replication factories for the maintenance of genome integrity.
Introduction
The duplication of a complete genome is a formidable task that must be perfectly controlled to avoid the transmission of mutations or chromosomal rearrangements to daughter cells. Two meters of DNA are packed and replicated in a human cell of about 10 μm of diameter. Hence, the spatiotemporal program of DNA replication is largely defined by the global organization of the nucleus (Marchal et al., 2019). DNA replication is initiated from defined regions of the genome called origins of replication. More than 30000 replication origins are required for the duplication of the human genome (Mechali, 2010). Origins are organized in clusters of early and late origins. In response to replication impediments, backup origins (also known as dormant origins), are activated within each cluster to rescue stalled replication forks (Blow et al., 2011). The timing of replication is influenced by the organization of chromatin loops (Courbet et al., 2008). Cohesins generate loops by extrusion (Davidson et al., 2019; Kim et al., 2019). Consistent with this, cohesins influence origins firing locally (Guillou et al., 2010), yet without determining replication timing globally (Oldach and Nieduszynski, 2019). RIF1, a conserved protein involved in telomeres capping, DNA double-strand break repair and chromatin organization, has been shown to control the timing of DNA replication (Cornacchia et al., 2012; Foti et al., 2016; Hayano et al., 2012; Mattarocci et al., 2016; Yamazaki et al., 2012). RIF1 may determine replication timing by at least two means that are related. First, RIF1 interacts with PP1 phosphatase and could directly impact on origin licensing by counteracting DDK kinases (Dave et al., 2014; Hiraga et al., 2014; Mattarocci et al., 2014). Second, RIF1 interacts with G-quadruplexes and may thereby contribute to the formation of higher-order chromatin structures (Kanoh et al., 2015; Yamazaki et al., 2013).
DNA replication operates within superstructures called replication factories that have been described extensively using fluorescence and electron microscopy (Hozak et al., 1993; Jackson and Pombo, 1998). Different nuclear patterns of replication factories throughout S phase reflect the orderly and sequential replication of chromatin domains (Dimitrova and Berezney, 2002). To date, the benefit of clustering replication forks into a relative low number of DNA replication factories is not really understood. Replications forks encounter a variety of impediments from both endogenous and exogenous sources (Lambert and Carr, 2013; Zeman and Cimprich, 2014). The slowing or stalling of replication forks by these impediments induces the activation of the checkpoint kinase ATR, which ensures that DNA synthesis within actively replicating chromosomal domains is completed before the duplication of a new chromosomal domain has started. ATR signaling delays the activation of late replication clusters while promoting the firing of dormant origins within active replication clusters (Blow et al., 2011). This suggests that the organization of replisomes into clusters may be important for the cellular response to replicative stress. In support to this, some proteins involved in nuclear organization are required for the response to replicative stress. For example, Lamin A/C is required for the maintenance of chromosome integrity when the progression of replication forks is impeded by DNA lesions or upon nucleotide depletion (Singh et al., 2013). Furthermore, the association of Lamin A/C with PCNA is critical for replication forks stability (Cobb et al., 2016). Hutchinson-Gilford progeria syndrome is caused by a mutation of the LMNA gene that leads to an aberrant Lamin A protein named progerin. The association of progerin with PCNA alters the nuclear distribution of PCNA, induces ATR activation and the formation of γH2A.X (Wheaton et al., 2017). In budding yeast, cohesins accumulate in the vicinity of replication forks upon treatment with hydroxyurea and are required for replication fork restart (Tittel-Elmer et al., 2012). These few examples illustrate the links between replicative stress and nuclear structures, which are still incompletely described.
The isolation of Proteins on Nascent DNA coupled with Mass Spectrometry (iPOND-MS) allows the identification of proteins localized in the vicinity of active replication forks (Aranda et al., 2014; Dungrawala et al., 2015; Lopez-Contreras et al., 2013; Lossaint et al., 2013; Sirbu et al., 2011; Sirbu et al., 2013). iPOND experiments performed under various experimental conditions have revealed components of the replication machinery (e.g. PCNA and DNA polymerases), proteins that accumulate near forks under stressful conditions (e.g. ATR and FANCD2), and proteins that are required for the restoration of chromatin structures after passage of the replication fork (e.g. histones). Since iPOND involves formaldehyde protein crosslinking, we reasoned that proteins indirectly associated with replication factories (i.e. playing a structural role) may also be captured and identified by iPOND such as Lamin A (Wheaton et al., 2017).
Here we show that the efficacy of the iPOND method is strongly dependent on the organization of replication factories. We demonstrate that RIF1 is required to ensure the organization of replication factories in presence of replication stress. We propose that the clustering of replication forks into replication factories limits the formation of DNA lesions caused by DNA replication impediments.
Results
iPOND efficacy is dependent on replication organization
To test if proteins involved in the organization of replication can be identified by iPOND, we performed iPOND-MS using a highly sensitive last generation mass spectrometer (Sciex TripleTOF 5600+) and quantified the results using MaxQuant (Cox and Mann, 2008). We calculated the LogRatio of the average intensity of peptides isolated immediately after a 5 minutes pulse with EdU divided by the average intensity of peptides isolated after 120 minutes chase with thymidine. As expected, known replisome components such as PCNA, RFC5, Pol Alpha or FEN1 have a LogRatio largely superior to zero indicating that they are indeed associated with ongoing replisomes (Figure 1A). By contrast, histone H4 has a LogRatio close to zero because it does not accumulate near replication forks. We also isolated Lamins A/C and Lamins B1 with nascent DNA, both immediately and 120 minutes after EdU labelling of replicated DNA, consistent with a general role for lamins in the organization of nuclear structures. The iPOND-MS data also revealed an enrichment of several cohesins subunits (SMC3, SMC1A, STAG2 and PDS5A) near forks (Figure 1A). Since cohesins are thought to play an architectural role at replication foci (Guillou et al., 2010), it is likely that they are not associated directly with each replication fork. Therefore, their association with EdU is likely to be indirect and may reflect the structural organization of replication factories, suggesting that iPOND efficiency could be biased by the organization of individual replisomes. In accordance with this, methods that are using formaldehyde crosslinking such as ChIP or chromosome conformation capture are indeed dependent on nuclear organization. To test if iPOND efficiency is biased by replisome organization, we took advantage of the distinct and characteristic patterns formed by replication factories labelled with EdU (Dimitrova and Berezney, 2002). In early S-phase (replication of euchromatin), the EdU pattern is poorly clustered. Clusterization then increases in mid-S phase (replication of facultative heterochromatin) and is even stronger in late S-phase (replication of constitutive heterochromatin). We compared iPOND samples from cells synchronized using thymidine block with iPOND samples performed in asynchronous conditions (Figure 1B). The PCNA signal at T0 (just before releasing the cells into S-phase) was barely detectable, as expected, and comparable with the minus click control of the asynchronous conditions (Figure 1C). After 2 hours (T2, early) following release into S-phase, the PCNA signal became detectable, increased significantly after 4 hours (T4, mid) and was maximal after 8 hours (T8, late). Similar results were observed for MCM7 or histone H3 (Figure 1C). Importantly, the number of EdU-positive cells were similar in asynchronous conditions (8.5%), at T2 (8.9%), T4 (8.7%) and T8 (5.3%), indicating that the efficacy of protein isolation on nascent DNA did not correlate with the level of EdU incorporation. Since the clustering of replication forks is increasing during S-phase, we conclude that the recovery of replisomes components is strongly dependent of the organization of replication factories. We surmise that when replication clustering is low (T2, early) the isolation of EdU captures mainly the replisomes components that are directly associated with the EdU-labeled replication fork. When replication fork clustering is high (T4, mid and T8, late) the EdU molecules incorporated at individual forks are isolated along with replisomes components directly associated with this fork and proteins associated with neighboring replication forks (Figure 1D), hence explaining the apparent increased efficacy of the iPOND procedure. In conclusion, the efficacy of iPOND is strongly impacted by the organization of replication forks and therefore might be used as a new tool to explore how replication factories are organized.

A. iPOND coupled with mass spectrometry (label-free quantification using MaxQuant). HeLa S3 cells were pulse-labelled with EdU for 5 min and chased with thymidine for 120 min. The bar plot is showing the LogRatio (pulse/chase) of average peptides intensities corresponding to indicated proteins. Pulse experiments have been repeated 3 times and chase experiment 2 times. B. HeLa S3 cells were submitted to thymidine block during 20 hours and released into S-phase. Cells were collected at T0 (G1), T2 (Early-S), T4 (Mid-S) and T8 (Late-S) after 15 min EdU pulse. Replication patterns showing the different phases are represented. C. iPOND experiment performed on unsynchronized and synchronized cells. In no click samples, biotin-TEG azide was replaced by DMSO. D. Scheme explaining how replication organization is impacting iPOND efficiency.
RIF1 protects the integrity of replication forks upon prolonged replicative stress
We identified RIF1 as a protein associated with nascent DNA (Figure 1A), consistent with previous studies (Alabert et al., 2014; Munden et al., 2018). We detected RIF1 specifically in EdU pulldowns of newly synthesized DNA (Sup Figure 1A). The RIF1 signal was strongly reduced after 120 minutes chase with thymidine, indicating that RIF1 localizes specifically near active replication forks, like PCNA (Sup Figure 1A). iPOND performed in synchronized cells also confirmed the presence of RIF1 at replication forks (Figure 1C). We also detected RIF1 in immune-precipitates of endogenous PCNA (Sup Figure 1B). Although RIF1 is associated with active replisomes, suppression of RIF1 did not alter the progression of replication forks (Sup Figure 1C), consistent with previous studies (Cornacchia et al., 2012; Ray Chaudhuri et al., 2016). However it should also be noted that a higher frequency of stalled forks was observed in rif1-/- DT40 cells (Xu et al., 2010), suggesting that RIF1 could be important for fork progression in some contexts. Consistent with this, several studies have detected the activation of the checkpoint effector kinase Chk1 in RIF1-depleted cells (Chapman et al., 2013; Foti et al., 2016). We confirmed that Chk1 was phosphorylated on Serine 345 in the absence of RIF1 (Sup Figure 1D). We observed also that RPA32 was phosphorylated on Ser4/8, suggesting that RIF1 depleted cells accumulate DNA lesions (Sup Figure 1D). Interestingly, RIF1 recruitment at replication forks is slightly increased upon hydroxyurea (HU) treatment to limit DNA2-mediated DNA resection and DNA lesions (Garzón et al., 2019; Mukherjee et al., 2019; Ray Chaudhuri et al., 2016). Consistent with this, DNA lesions, genetic instability and HU sensitivity are increased upon RIF1 impairment (Buonomo et al., 2009; Mukherjee et al., 2019; Xu et al., 2010). This raises the possibility that RIF1 could be required for replication in stressful conditions. To test this we analyzed if RIF1 loss had any impact on replication fork dynamics in the presence of aphidicolin (APH). We labelled cells for 30 minutes with IdU and then for 30 minutes with CldU in presence of a low dose (0.05 μM) of APH. As expected, the ratio of the lengths of CldU versus IdU tracts was close to 1 in control conditions and reduced by half in presence of APH (Figure 2A). The status of RIF1 did not change the ratios of CldU/IdU tracts (Figure 2A) indicating that RIF1 depletion does not play any major role in early responses to APH. As RIF1 is protecting HU-stalled forks from nucleases degradation (Garzón et al., 2019; Mukherjee et al., 2019), we checked if this was also the case when replication forks were blocked with APH. To do so we treated cells 6 hours with a high dose (1 μM) of APH after 30 min sequential labelling of IdU and CldU and measured the ratio between the lengths of CldU and IdU tracts. The ratio was close to 1 in cells treated with a control siRNA, and bellow 1 in RIF1 depleted cells, confirming that RIF1 is indeed protecting APH-stalled forks (Figure 2B). Consistent with this, prolonged treatment (24 hours) with APH, increased the percentage of γ-H2A.X-positive cells to almost 2 fold (Figure 2C) and decreased by two fold the ability of replication forks to restart (Sup Figure 1E). Altogether, these data indicate that RIF1 is constitutively associated with replication forks and limits the formation of DNA lesions under stressful conditions.

A. DNA fibers labelling. HeLa S3 cells were labelled for 30 min with IdU and then for 30 min with CldU in the absence or presence of 0.05 μM aphidicolin (APH) in the cell culture medium. Graphic representation of the ratios of CldU versus IdU tract length. The horizontal bar represents the median with the value indicated in red. 50 replication tracts were measured for each experimental condition. The second graphic representation is showing the medians of three independent experiments. B. Analysis of DNA resection using DNA fibers labelling. HeLa S3 cells were labelled for 30 min with IdU and then for 30 min with CldU. 1 μM aphidicolin (APH) was added in the cell culture medium for 6 hours. Graphic representation of the ratios of CldU versus IdU tract length. The horizontal bar represents the median with the value indicated in red. 50 replication tracts were measured for each experimental condition. C. Immunofluorescence analysis of γH2A.X and RIF1 in HeLa S3 cells with siRNA against control or RIF1 in presence or absence of aphidicolin (APH) for 24 hours. Graphic representation of the percentage of γH2A.X positive cells based on 3 independent experiments.
RIF1-dependent loss of replication organization induces DNA lesions
Despite its role in protection of stalled replication forks (see above), RIF1 recruitment at forks is not tremendously increased in response to HU (Mukherjee et al., 2019) or APH (Figure 4A and data not shown) compared to proteins such as ATR, 9-1-1, TopBP1 or FANCD2/FANCI (Dungrawala et al., 2015; Lossaint et al., 2013). Therefore, we hypothesize that the impact of RIF1 on nascent DNA protection may reflect a structural role rather than a direct role at stalled replication forks. This is supported by several articles showing that RIF1 is crucial for the organization of higher-order chromatin domains and for the establishment of the replication timing program (Foti et al., 2016; Moriyama et al., 2018; Yamazaki et al., 2012). As already stated, replication factories form distinct patterns during S-phase reflecting the level of organization. Remarkably, in cells synchronized with thymidine block and released into S-phase, the mid-S pattern is selectively loss upon RIF1 impairment (Yamazaki et al., 2012). We confirmed this result in synchronized cells (Figure 3A, B) but not in asynchronous cells for which we found that RIF1 depletion did not alter the occurrence of the mid-S pattern (Figure 3A, B). This result indicates that the disappearance of the mid-S pattern in absence of RIF1 is a consequence of the synchronization and therefore cannot be solely explained by the difference in replication timing since it should be also observed in asynchronous cells. It is well established that synchronization with thymidine block perturbs the pool of nucleotides and induces DNA damage (Kurose et al., 2006). Thus, we hypothesized that the absence of mid-S pattern in RIF1-depleted cells synchronized using a thymidine block could reflect a defect in the organization of replication clusters that manifests during DNA replicative stress. To check this, we analyzed the level of the marker of DNA damage γ-H2A.X. In an asynchronous population of cells, the depletion of RIF1 had no impact on the percentage of γ-H2A.X positive cells (Figure 3A, C). As expected, the percentage of γ-H2A.X positive cells increased 2 hours after release from the thymidine block. Strikingly, suppression of RIF1 tripled the percentage of γ-H2A.X positive cells in the same conditions (6.9% in control versus 24.1% if shRIF1 (1) and 19.1% in shRIF1 (2)). We conclude that the disappearance of the mid-S pattern upon RIF1 depletion correlates with the formation of DNA lesions. The thymidine block procedure is affecting the pools of dNTP and therefore should have a direct impact on the progression of replication forks that might be exacerbated in the absence of RIF1. To test this, we monitored the phosphorylation of Chk1 on Serine 345. In control condition, we observed a mild phosphorylation of Chk1 on Serine 345, in line with the higher level of γ-H2A.X (Figure 3D). Interestingly, we observed a strong level of Chk1 phosphorylation in RIF1-depleted cells 2 hours after release from the thymidine block (Figure 3D). In asynchronous conditions, suppression of RIF1 did not significantly alter the progression of replication forks (Sup Figure 1C). Two hours after release from a thymidine block, however, replication tracts were longer in the absence of RIF1 (Figure 3E). This could be due to either unscheduled restart of stalled replication forks and/or reflects a defect in dormant origins activation as suggested recently (Hiraga et al., 2017). These phenotypes are consistent with higher level of Chk1 phosphorylation and the formation of DNA lesions. We propose that the occurrence of DNA lesions during prolonged replicative stress observed in RIF1 depleted cells is a consequence of a defect in the structural organization of replication factories.

A. Immunofluorescence analysis of γH2A.X and EdU in asynchronous cells and in cells synchronized with thymidine block followed by 2 hours release in HeLa S3 cells with shRNA against luciferase or RIF1. Efficiency of depletion were assessed by Western-blotting B. Graphic representation of the frequency of replication patterns (Late-S, Mid-S and Early-S) based on at least three independent experiments for each conditions. C. Quantification of γH2A.X intensity within nucleus stained with Hoechst using CellProfiler based on at least three independent experiments for each conditions. D. Analysis of Chk1 phosphorylation on Serine 345 upon RIF1 depletion in asynchronous conditions or upon thymidine block followed by 2 hours release. E. DNA fibers assay upon RIF1 depletion in asynchronous conditions or upon thymidine block followed by 2 hours release. HeLa S3 cells were labelled for 30 min with IdU and then for 30 min with CldU. Graphic representation of the ratios of CldU tract length. The horizontal bar represents the median with the value indicated in red. At least 50 replication tracts were measured for each experimental condition. The second graphic representation is showing the medians of three independent experiments.

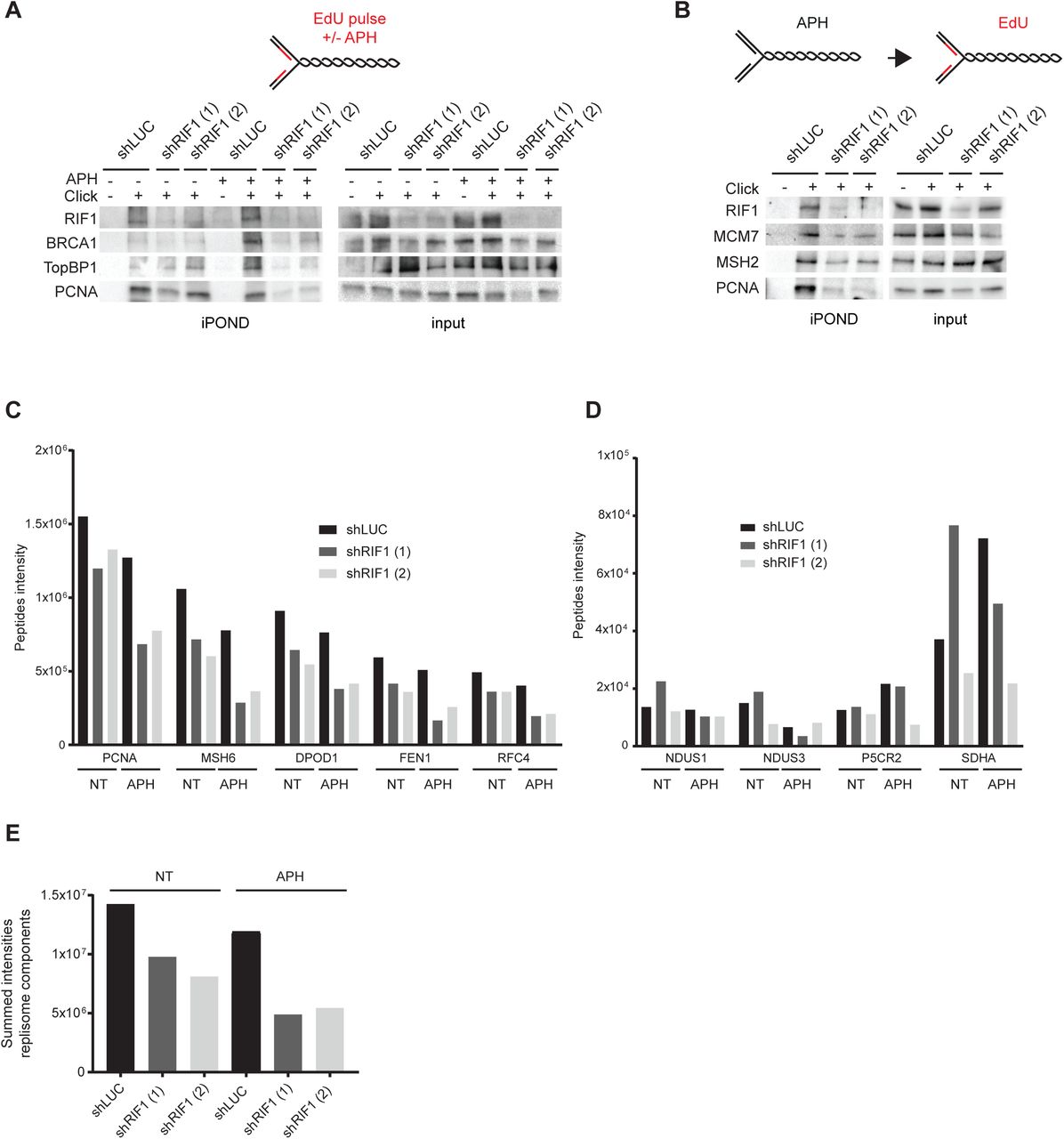
A. iPOND experiment. HeLa S3 cells (with shLUC or two different shRIF1) were labelled with EdU for 15 min or for 30 min with 0.1 μM aphidicolin (APH). Indicated proteins were analyzed by Western-blotting. In no click samples biotin-TEG azide was replaced by DMSO. B. iPOND experiment. HeLa S3 cells (with shLUC or two different shRIF1) were treated 30 min with 0.1 μM aphidicolin (APH) then washed and labelled with EdU for 30 min. Indicated proteins were analyzed by Western-blotting. In no click samples, biotin-TEG azide was replaced by DMSO. C, D. iPOND-MS analysis. HeLa S3 cells (with shLUC or two different shRIF1) were labelled with EdU for 15 min or for 30 min EdU with 0.1 μM aphidicolin (APH). Quantification of peptides intensity corresponding to the indicated proteins is represented. E. Summed intensities of peptides corresponding to replisomes components.
RIF1 is required to maintain the organization of replication factories in presence of replicative stress
Prolonged treatment with both APH and thymidine yield high level of γH2A.X in RIF1-depleted cells. We proposed that this is caused by a change in organization due to thymidine block. Since APH has also been widely used for cell synchronization, it is highly probable that prolonged APH treatment would have the same consequences on organization (Kurose et al., 2006). In any case this may be interpreted as simple correlation and more direct evidence is required. Since we show that iPOND can be used for assaying replication organization (Figure 1), we used it to test if RIF1 loss alters the structural organization of replication factories during treatment with APH. We first focused on the efficacy of PCNA recovery, which depends on the clustering of replication factories (Figure 1C). Under standard cell culture conditions, the efficacy of PCNA isolation with nascent DNA in RIF1-depleted cells was similar to that of control cells (Figure 4A). As expected, a 30 min treatment with a low dose of APH (0.1 μM) induced the recruitment of BRCA1 and TopBP1 on nascent DNA (Figure 4A). Strikingly, in RIF1-depleted cells treated with APH, the efficacy of PCNA, BRCA1 and TopBP1 purification with nascent DNA diminished dramatically. Importantly, in RIF1-depleted cells treated with APH, DNA fiber labelling did not reveal any major defect in replication forks progression or EdU incorporation (Sup Figure 2A & 2B). Thus, a defect in DNA synthesis does not account for the reduced isolation of EdU-bound proteins from RIF1-depleted cells. Furthermore, we detected similar levels of the replisome-associated proteins MSH2 and MCM7 in PCNA immune-precipitates from control and RIF1-depleted cells (Sup Figure 3C 2C), suggesting that RIF1 is not required for replisome stability and replication fork progression. The results of Figure 4A are consistent with a role for RIF1 in the maintenance of replisome organization under stressful conditions. To confirm this observation, we isolated EdU-bound proteins after recovery from an APH treatment (Figure 4B). We treated cells with 0.1 μM APH for 30 minutes and then washed the cells with drug-free medium and labelled nascent DNA for 30 minutes with EdU. In comparison with control cells (shLUC), the isolation of PCNA, MCM7 and MSH2 with nascent DNA from RIF1-depleted cells was dramatically reduced (Figure 4B). Finally, to have a broader view of replisome composition, we coupled iPOND to mass spectrometry and used MaxQuant for label-free quantification analyses. Quantification of mass spectrometry data confirmed western blot analyses (Figure 4C). In comparison with control cells, the treatment of RIF1-depleted cell with APH markedly reduced the abundance of the replication factors PCNA, MSH6, DPOD1, FEN1 and RFC4 captured by iPOND (Figure 4C). By contrast, changes in the efficacy of streptavidin pulldowns were not observed for mitochondrial proteins such as NDUS1, NDUS3, P5CR2 and SDHA, which are also isolated by iPOND (Figure 4D). To generalize this observation to the whole replisome, we summed the peptides intensities of all replisome proteins listed in a previous study (Lopez-Contreras et al., 2013). In control cells (shLUC), APH treatment moderately affected the recovery of replisome components (Figure 4E). By contrast, APH had a severe impact on the recovery of replisome components from RIF1-depleted cells (∼50% decrease for shRIF1 (1) and ∼33% decrease for shRIF1 (2)). We conclude that APH treatment reduces the probability to capture proteins associated with EdU-labelled DNA in RIF1-depleted cells. Thus, we propose that RIF1 is required to maintain the organization of replication factories in response to replicative stress to prevent the formation of DNA lesions.
Discussion
RIF1 was originally discovered more than 25 years ago in budding yeast as a negative regulator of telomere elongation (Hardy et al., 1992). It is now clearly established that RIF1 is a highly conserved protein (Sreesankar et al., 2012) involved in telomeres protection, DNA replication, DNA double-strand break repair, transcription and heterochromatin formation (Mattarocci et al., 2016). The links between the seemingly disparate functions of RIF1, however, remain elusive. Here we propose that RIF1 is a key organizing component of replication factories. This model is based on the following findings: (1) RIF1 is associated with replication factories in basal conditions (2) DNA replication stress in RIF1-depleted cells modifies S phase patterns and increases the level of the DNA damage marker γH2A.X (3) Suppression of RIF1 strongly affects the organization of replication in response to replicative stress. Our model is consistent with the finding that RIF1 bridges DNA molecules localized in proximity (Mattarocci et al., 2017) and that RIF1 (with 53BP1) creates a protective structure around DBSs (Ochs et al., 2019). Thus, we propose that the main function of RIF1 is to protect the integrity of higher-order structures during replication is particular in response to replicative stress (Figure 5). By analogy with its function at yeast telomeres, we would like to propose that RIF1 is encasing replication factories (Figure 5).

RIF1, thanks to its capacity to interact with DNA, is maintaining the replication factories encased thus preventing DNA resection by nucleases or excessive origins activation. In its absence, the replication factories are unprotected leading to DNA resection, DNA lesions and activation of DNA damage response.
The association of RIF1 with the replication forks has been previously observed by other groups (Alabert et al., 2014; Her et al., 2018; Munden et al., 2018). We confirmed that RIF1 impairment has no measurable effect on replication forks progression under standard conditions or in response to short treatment with replicative stress (Cornacchia et al., 2012; Ray Chaudhuri et al., 2016; Xu et al., 2010) despite the fact that RIF1 loss induces Chk1 phosphorylation on Ser345 (Chapman et al., 2013; Foti et al., 2016). Interestingly, we found that two hours after release from a thymidine block, replication tracts are longer in the absence of RIF1 and phosphorylation levels of Chk1 on Ser345 and H2A.X on Ser139 are increased. One possibility is that in the absence of RIF1, the disorganization of replication factories delays dormant origin activation which leads to longer replication tracts and increased resection at stalled replication forks. The increase of DNA lesions likely underpins the increased sensitivity of RIF1 defective cells to inhibitors of DNA replication (Buonomo et al., 2009; Feng et al., 2013; Xu et al., 2010; Xu et al., 2017).
RIF1 is recruited by 53BP1 at DSBs to prevent homologous recombination and favor NHEJ (Chapman et al., 2013; Di Virgilio et al., 2013; Escribano-Diaz et al., 2013; Zimmermann et al., 2013). Based on this, it has been proposed that RIF1 could be recruited by 53BP1 to protect stalled forks independently of BRCA1 (Xu et al., 2017). These data are raising the possibility of a role of 53BP1 in the recruitment of RIF1 at replication forks in basic conditions and in response to replicative stress. However, RIF1 recruitment is not impacted by 53BP1 depletion (Her et al., 2018) and RIF1, but not 53BP1, protects nascent DNA from degradation (Ray Chaudhuri et al., 2016) suggesting that the presence of RIF1 at replication forks is independent of 53BP1. Therefore, we speculate that RIF1 alone is sufficient to maintain the organization of replication factories, in accordance with it capacity to form higher order structures in budding yeast (Mattarocci et al., 2017).
Our model is compatible with the fact that RIF1 is protecting stalled replication forks from resection by nucleases, perhaps by creating a structure that prevent their recruitment (Garzón et al., 2019; Mukherjee et al., 2019; Ray Chaudhuri et al., 2016). A role for RIF1 in safeguarding the integrity of replication factories may also explain how RIF1 controls the activation of dormant origins in response to replicative stress (Hiraga et al., 2017) and prevents the formation of anaphase bridges (Hengeveld et al., 2015; Zaaijer et al., 2016). RIF1 depletion has a strong impact on replication timing (Cornacchia et al., 2012; Foti et al., 2016; Yamazaki et al., 2012), possibly by regulating DDK kinase activation through an interaction with the PP1 phosphatase (Dave et al., 2014; Hiraga et al., 2014; Mattarocci et al., 2014). Alternatively, it has been proposed that the function of RIF1 in the regulation of the replication-timing program could stem from its ability to bind G-quadruplexes and organize chromatin structures (Kanoh et al., 2015). Since the loss of RIF1 induces drastic changes in nuclear organization revealed by chromosome conformation capture methods (Foti et al., 2016), we favor the hypothesis that the impact of RIF1 on replication timing is a consequence of impaired nuclear organization rather than of a defect in the control of DDK kinases.
We would like to propose that RIF1 is a key organizer of the nucleus, which could depend on its association with Lamin B1 (Foti et al., 2016). This is also based on the fact that RIF1 and 53BP1 are forming a protective structure at DSBs repaired by NHEJ (Ochs et al., 2019) and that RIF1 is able to form higher order structures (Mattarocci et al., 2017). This model could explain why suppression of RIF1 perturbs transcription and heterochromatin formation (Dan et al., 2014; Li et al., 2017).
Finally this study illustrates an as yet unforeseen application of iPOND (or iPOND-related methods based on formaldehyde crosslinking). It is generally assumed that the iPOND method captures proteins associated with individual replisomes distributed along a linear DNA template. Here we show that the iPOND method is not only efficient to isolate replisome components but also to capture structural components of replication factories stabilized by formaldehyde crosslinking. Future studies using iPOND and other method should provide new insights into the role of the nuclear organization in DNA replication.
Author Contributions
Cyril Ribeyre: conceptualization, data curation, supervision, formal analysis, funding acquisition, investigation, visualization, methodology, project administration and writing original draft, review and editing.
Rana Lebdy: conceptualization, data curation, formal analysis, investigation, visualization, methodology and draft review and editing
Julie Patouillard: formal analysis, investigation, visualization and methodology.
Marion Larroque: data curation, formal analysis, investigation and methodology.
Christian Larroque: data curation, formal analysis and methodology.
Raghida Abou-Merhi: supervision, funding acquisition, project administration.
Angelos Constantinou: conceptualization, data curation, supervision, funding acquisition, visualization, methodology, project administration and writing original draft, review and editing.
Conflict of Interest Statement
The authors declare that they have no conflict of interest.
Methods
Cell lines
HeLa S3 (obtained from ATCC) cells were cultured in Dulbecco’s modified Eagle’s media (DMEM). Culture media was supplemented with 10% fetal bovine serum (Biowest) and penicillin/streptomycin (Sigma-Aldrich). Cells were incubated in a 5% CO2 at 37°C. For thymidine block experiments cells were treated 20 hours with 2 mM thymidine, washed then release into normal media.
Gene silencing
For RIF1 depletion siRNA oligonucleotides were purchased from Dharmacon (M-027983-01-0005) and transfected using INTERFERin (Polypus transfection). Anti-RIF1 shRNAs (1) and (2) and anti-luciferase shRNA were cloned in pSUPER-EBV and transfected using Lifofectamine 2000 (Thermo-Fisher). Stable cell lines were selected using puromycin.
Western-blot
The proteins were resolved by SDS-PAGE using home-made or precast gels (Bio-Rad) and transferred to a nitrocellulose membrane (GE Healthcare or Bio-Rad). Antibodies against the following proteins were used: Ser345 Phospho-Chk1 (Cell Signaling Technology 2348), Chk1 (Santa Cruz sc-8408), PCNA (Sigma-Aldrich P8825), Ser4/8 Phospho-RPA32 (A300-245A), RPA32 (Calbiochem NA18), TopBP1 (Bethyl A300-111A), histone H3 (Abcam ab62642) BRCA1 (Santacruz sc-642), RIF1 (Bethyl A300-568A-M), MSH2 (Calbiochem NA27), MCM7 (Abcam ab2360).
Co-Immunoprecipitation
Cells were incubated for 30 min in ice in high salt buffer (50 mM Tris Ph 7.5, 300 mM NaCl, 1% Triton, 1 mM DTT). After 10 min centrifugation at 14000g, supernatant were incubated with anti PCNA antibody (Sigma-Aldrich, P8825) or IgG Rabbit (Calbiochem NI01) overnight at 4°C. Magnetic beads coupled with protein G (Life 10004D) were added for 1 hour and washed 5 times with washing buffer (10 mM Hepes, 100 mM KOAc, 0.1 mM MgOAc). Beads were boiled in Laemmli buffer and supernatants were analyzed by Western-blot.
Isolation of proteins on Nascent DNA (iPOND)
iPOND was performed largely as described in (Lossaint et al., 2013; Ribeyre et al., 2016). Briefly, HeLa S3 cells were pulse labeled with 10 μM EdU for 5 min and a 120 min chase was performed with 10 μM thymidine. Cells were fixed with 1% formaldehyde for 5 min followed or not by quenching of formaldehyde by 5 min incubation with 0.125 M glycine. Fixed samples were collected by centrifugation at 2000 rpm for 3 min, washed three times with PBS and stored at −80°C. Cells were permeabilized with 0.5% triton and click chemistry was used to conjugate biotin-TEG-azide (Eurogentec) to EdU-labelled DNA. Cells were re-suspended in lysis buffer and sonication was performed using a Qsonica sonicator. Biotin conjugated DNA-protein complexes were captured using streptavidin beads (Ademtech). Captured complexes were washed with lysis buffer and high salt. Proteins associated with nascent DNA were eluted under reducing conditions by boiling into SDS sample buffer for 30 min at 95 °C.
DNA fibers labelling
DNA fibers labelling was performed as previously described (Lossaint et al., 2013; Ribeyre et al., 2016). Cells were labeled with 25μM IdU, washed with warm media and exposed to 50 μM CldU. Cells were lysed and DNA fibers were stretched onto glass slides. The DNA fibers were denatured with 2.5 M HCl for 1 hour, washed with PBS and blocked with 2% BSA in PBS-Tween for 60 minutes. IdU replication tracts were revealed with a mouse anti-BrdU/IdU antibody from BD Biosciences (347580) and CldU tracts with a rat anti-BrdU/CldU antibody from Eurobio (ABC117-7513). The following secondary antibodies were used: alexa fluor 488 anti-mouse antibody (Life A21241) and Cy3 anti-rat antibody (Jackson Immunoresearch 712-166-153). Replication tracts lengths were analyzed using ImageJ software. For statistical analysis we used a non-parametrical Mann-Whitney with Prism software.
Immunofluorescence
Cells were plated on glass coverslips and fixed with 4% paraformaldehyde in PBS for 20 min at room temperature. When indicated cells were incubated with EdU (5-ethynyl-2’-deoxyuridine) for the indicated times. PFA-fixed cells were permeabilized with 0.2% Triton X-100 in PBS for 5 min. Primary (Ser139 Phospho-H2A.X; Millipore 05-636 and RIF1; Bethyl A300-568A-M) and secondary antibodies (anti-mouse Alexa 488 and anti-rabbit alexa 546) were prepared in PBS with 0.1% Tween and incubations were carried out in a humidified chamber at room temperature (60 min and 30 min, respectively). EdU was coupled with Alexa fluor 555 using Click chemistry. DNA was stained with Hoechst. The cells were mounted on glass slides with Prolong (Life). Cells were analyzed by fluorescence microscopy and quantification of various signals was performed using CellProfiler software (Carpenter et al., 2006).
Mass Spectrometry Analysis
Mass spectrometry was performed as indicated in (Kumbhar et al., 2018). Analysis of raw files was performed using MaxQuant (Cox and Mann, 2008) version 1.5.6.5 using default settings with label-free quantification option enabled. Raw file spectra were searched against the human UniProt reference database. Protein, peptide, and site false discovery rate (FDR) were adjusted to < 0.01.

A. Indicated proteins were isolated by iPOND and detected by Western blotting. HeLa S3 cells were pulse-labelled with EdU for 15 min and chased with thymidine for 120 min. In no click biotin-TEG azide was replaced by DMSO. B. Western-blot analysis of indicated proteins after immunoprecipitation with an antibody directed against PCNA or against mouse IgG. C. DNA fibers labelling. HeLa S3 cells were labelled for 30 min with IdU and then for 30 min with CldU. Graphic representation of the ratios of CldU tract length. The horizontal bar represents the median with the value indicated in red. At least 50 replication tracts were measured for each experimental condition. The second graphic representation is showing the medians of three independent experiments. D. Western-blot analysis of the indicated proteins upon transfection with siRNA directed against RIF1 or a control target. E. Analysis of replication restart upon APH treatment using DNA fibers labelling. HeLa S3 cells were labelled for 30 min with IdU, then treated 16 hrs with 10μM APH and then for 30 min with CldU. Graphic representation of the percentage of restart based on 3 independent experiments.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A. DNA fibers labelling. HeLa S3 cells were labelled for 30 min with IdU and then for 30 min with CldU in the absence or presence of 0.05 μM aphidicolin (APH) in the cell culture medium. Graphic representation of the ratios of CldU versus IdU tract length. The horizontal bar represents the median with the value indicated in red. At least 50 replication tracts were measured for each experimental condition. B. Analysis of EdU incorporation using microscopy in HeLa S3 cells with shRNA against luciferase or RIF1. EdU was incorporated in cells during 15 min with or without 0.1 μM aphidicolin (APH). Quantification of EdU intensity within nucleus stained with Hoechst was performed using CellProfiler and is represented on the histogram. Error-bars corresponds to the average values of three independent experiments. C. Western-blot analysis of indicated proteins after immunoprecipitation with an antibody directed against PCNA or against mouse IgG. When indicated HeLa S3 cells (shLUc or shRIF1) were treated for 30 min with 0.1 μM aphidicolin (APH).
Acknowledgements
We thank all the lab members for comments and suggestions on the project and on the manuscript. We are grateful to Antoine Aze for critical reading of the manuscript. We thank Marie-Pierre Blanchard and the MRI microscopy platform for their support. We acknowledge the support of the Site de Recherche Intégrée sur le Cancer Montpellier Cancer grant INCa_INSERM_DGOS_12553. This work was supported by a Jeunes Chercheuses Jeunes Chercheurs (JCJC) grant (REPLIBLOCK ANR-17-CE12-0034-01) from the Agence National de la Recherche (ANR) to Cyril Ribeyre and by grants from la Fondation ARC pour la Recherche sur le Cancer (PGA1 RF20180206787) and Merck Sharp and Dohme Avenir (GnoSTic) to Angelos Constantinou. Rana Lebdy is funded by a fellowship from Azm & Saade Association.
References




