

Abstract
The Ribosome-associated Quality Control (RQC) pathway targets incomplete polypeptides from stalled translation for degradation. The primary route of degradation involves ubiquitylation of ribosome-tethered incomplete polypeptides by the E3 ligase Ltn1. To safeguard cells from Ltn1 failure, the protein Rqc2 directs the ribosome to append incomplete polypeptides with carboxy-terminal alanine and threonine residues (CAT tails) that act as degrons, marking RQC-evading polypeptides for degradation. Ltn1 disruption leads to neurodegeneration in animal models yet how this becomes toxic to cells is unclear. We show here that increased Rqc2 levels alter CAT tail composition and exacerbate the toxicity of Ltn1 failure by driving CAT tail aggregation, which inhibits CAT tail degron activity and disrupts proteostasis. Guanidinium hydrochloride exposure or RNA Polymerase III perturbation reverses these Rqc2-induced effects. Our work demonstrates that increased Rqc2 levels convert CAT tails from degrons into toxic aggregates and that pharmacologic or genetic interventions can reverse this process.
Introduction
Cells must continuously surveil their proteomes to protect themselves from defective proteins. Disruption of this process causes a buildup of defective proteins, a hallmark of many neurodegenerative diseases (Hartl, 2017; Ross and Poirier, 2004). Understanding how compromised protein surveillance can become toxic to cells has been a fundamental goal of the protein quality control field.
The earliest stage of surveillance occurs when cells monitor protein synthesis to detect failed rounds of translation and degrade potentially toxic incomplete polypeptides (Brandman and Hegde, 2016; Defenouillère and Fromont-Racine, 2017; Ikeuchi et al., 2018; Joazeiro, 2019). Protein synthesis can fail to complete due to inefficient messenger RNA (mRNA) decoding (Dimitrova et al., 2009; Doma and Parker, 2006; Gamble et al., 2016; Letzring et al., 2010; Meaux and Van Hoof, 2006), causing ribosomes translating the same mRNA to stall and subsequently collide with each other (Ikeuchi et al., 2019; Juszkiewicz et al., 2018; Simms et al., 2017). These collisions trigger a process called Ribosome-associated Quality Control (RQC), which recycles these ribosomes and targets their attached incomplete polypeptides (RQC substrates) for degradation (Bengtson and Joazeiro, 2010; Brandman et al., 2012; Defenouillère et al., 2013; Shao et al., 2013; Verma et al., 2013). RQC begins when a set of proteins detect the collided ribosomes and separate them into small (40S) and large (60S) subunits, leaving the incomplete polypeptide tethered to the 60S subunit (Juszkiewicz and Hegde, 2017; Lyumkis et al., 2014; Matsuo et al., 2017; Pisareva et al., 2011; Shao et al., 2013; Shoemaker et al., 2010; Sitron et al., 2017; Sundaramoorthy et al., 2017; Tsuboi et al., 2012).
The newly separated 60S subunit recruits a complex of proteins that collaborate to ubiquitylate the incomplete polypeptide and facilitate its degradation by the proteasome (Bengtson and Joazeiro, 2010; Brandman et al., 2012; Defenouillère et al., 2013; Lyumkis et al., 2014; Shao et al., 2013; Verma et al., 2013). A member of this complex, the RING E3 ubiquitin ligase Ltn1, ubiquitylates the incomplete polypeptide while it is still attached to the ribosome (Bengtson and Joazeiro, 2010; Brandman et al., 2012; Defenouillère et al., 2013; Lyumkis et al., 2014; Shao et al., 2013; Shao and Hegde, 2014; Verma et al., 2013). Rqc2 stabilizes Ltn1’s binding to the ribosome (Defenouillère et al., 2013; Shao et al., 2015) and directs the 60S subunit to extend the C-terminus of the incomplete polypeptide with alanine and threonine residues (“CAT tails”) (Osuna et al., 2017; Shen et al., 2015). This process (“CATylation”) enables Ltn1 to ubiquitylate lysine residues previously buried in the ribosome exit tunnel (Kostova et al., 2017; Sitron and Brandman, 2019) or confined to structured regions on incomplete polypeptides (Sitron and Brandman, 2019). Additionally, CAT tails act as degrons off the ribosome to mark polypeptides that escape Ltn1 for proteasomal degradation (Sitron and Brandman, 2019), a function conserved from prokaryotes (Lytvynenko et al., 2019). By targeting incomplete polypeptides for degradation, RQC protects cells from the proteotoxic consequences of failed translation.
In animal models, crippling RQC with hypomorphic LTN1 alleles (Chu et al., 2009) or mutations that compromise the ability to respond to increased ribosome stalling (Ishimura et al., 2014) leads to neurodegeneration. Cellular models of RQC failure display phenotypes that suggest how disruption of RQC might result in this pathology. Failure of RQC after CATylation occurs, for example by mutation of LTN1, leads to a buildup of CATylated proteins (Shen et al., 2015). CAT tails have amyloid-like properties, which can cause CATylated proteins to aggregate and form inclusions that sequester chaperones (Choe et al., 2016; Defenouillère et al., 2016; Yonashiro et al., 2016). Indeed, inclusions of stalled proteins with C-terminal extensions that depended on an Rqc2 ortholog were observed in a Drosophila model of Parkinson’s Disease (Wu et al., 2019). Despite these aberrant phenotypes, RQC failure is well-tolerated in cellular models and does not impair viability in the absence of strong additional stressors (Choe et al., 2016; Defenouillère et al., 2013; Kostova et al., 2017).
Toward the eventual goal of understanding the relationship between RQC failure and neurodegeneration, we sought to determine how RQC failure can become toxic in Saccharomyces cerevisiae. We found that the combination of Ltn1 disruption and elevated Rqc2 levels changed CAT tail composition, potentiated aggregation of CATylated proteins (aggregation previously observed by Yonashiro and colleagues [Yonashiro et al., 2016]), and antagonized the ability of CAT tails to act as degrons. These Rqc2-perturbed cells displayed phenotypes characteristic of proteostasis disruption such as sequestration of the chaperone Sis1 into CAT tail inclusions, induction of the heat shock response, and sensitivity to otherwise mild stressors. Thus, our work indicates that Rqc2, which ordinarily protects cells from RQC failure, increases the toxicity of RQC failure when expressed at high levels.
We additionally uncovered two interventions that reverse the toxic effects of RQC failure. First, improving the solubility of CATylated proteins by growing cells in the presence of the protein denaturant guanidinium hydrochloride reduced the proteostasis strain caused by increased Rqc2 levels. Second, we identified RNA Polymerase III (Pol III) in a screen for modulators of the heat shock response to RQC failure. Genetic disruption of Pol III depleted an insoluble species that converted CAT tails into their aggregated form; Pol III perturbation thereby restored CAT tail degron function and proteostasis. Our work demonstrates that interventions targeting CAT tail aggregation can reduce the toxicity of RQC failure.
Results
Elevated Rqc2 levels increase the toxicity of RQC failure
To study RQC failure in Saccharomyces cerevisiae, we first needed a model substrate that would enable us to visualize defects in the RQC pathway. We used a model substrate (“RQCsub”) that consists of green fluorescent protein (GFP) attached via an inert linker including a tobacco etch virus (TEV) protease site to twelve stall-inducing arginine CGN codons (Fig. 1A) (Letzring et al., 2010). Translation of RQCsub produces a stalled GFP-linker-arginine “arrest product” (Brandman et al., 2012). This RQCsub arrest product accumulated to low levels in wt strains by SDS-PAGE (Fig. 1A). This arrest product increased in abundance after we compromised RQC function by deleting two RQC genes: the CAT tail-elongating factor RQC2 or the E3 ubiquitin ligase LTN1 (Fig. 1A). This result indicates that Rqc2 and Ltn1 contribute to RQCsub degradation. While deletion of RQC2 or LTN1 stabilized RQCsub, the resultant protein products displayed different mobilities; RQCsub migrated as a crisp band in rqc2Δ and a higher molecular weight smear in ltn1Δ (Fig. 1A). This smear collapsed into a single band after additional deletion of RQC2 in the ltn1Δ strain (Fig. 1A) or cleavage of RQCsub’s C-terminus with TEV protease (Fig. 1B), consistent with the smear containing CATylated RQCsub. Taken together, these results confirm that the model substrate RQCsub enables visualization of RQC-mediated degradation and CATylation.

RQC2 overexpression enhances aggregation of CATylated proteins and Sis1 in ltn1Δ cells. (A) Left, whole cell immunoblots (IBs) of lysates containing RQCsub (a model RQC substrate; schematic above). Right, immunoblot of RQCsub immunoprecipitated (IPed) from RQC2 overexpression and LTN1 deletion (ROLD) lysate. Arrow indicates molecular weight of RQCsub without CAT tails. Asterisk denotes the full-length RQCsub protein product, produced when ribosomes translate through the stall sequence (region past the stall not pictured in schematic). GFP, green fluorescent protein. OE, overexpression. (B) Immunoblot of RQCsub immunoprecipitated from ltn1Δ and ROLD lysates with and without tobacco etch virus (TEV) protease treatment. Arrows and asterisks as in A. (C) Fluorescence microscopy of cells expressing RQCsub with percentages of cells containing observable GFP inclusions reported below. (D) Fluorescence microscopy of SIS1-RFP cells expressing RQCsub. (E) Whole-cell immunoblot of Sis1-RFP. Asterisk denotes a non-specific band detected by the anti-RFP antibody.
We hypothesized that overexpression of RQC2, which potentiates CAT tail aggregation (Yonashiro et al., 2016), would increase the cellular toxicity of RQC failure. As expected, when we overexpressed RQC2 in the RQC-compromised ltn1Δ strain, a large proportion of RQCsub remained in the well rather than migrating into an SDS-PAGE gel (Fig. 1A,B) despite prior boiling in dodecylsulfate detergent. This detergent-resistant species was more apparent in the RQC2 overexpression and LTN1 deletion (hereafter referred to as “ROLD”) strain than ltn1Δ (Fig. 1B). TEV treatment, which cleaves RQCsub’s C-terminus and removes its CAT tails, enabled RQCsub to migrate as a single band into the gel (Fig. 1B). The formation of this detergent-resistant species is consistent with RQC2 overexpression reducing the solubility of CATylated RQCsub in the ltn1Δ background. Microscopy of RQCsub under these conditions revealed that RQCsub had a diffuse localization in the majority of ltn1Δ cells (<1% of cells with inclusions) but became more punctate in ROLD (56.9% of cells with inclusions) (Fig. 1C). To test whether these inclusions depended on CATylation, we disrupted CATylation by deleting RQC2 or overexpressing a CATylation-incompetent rqc2aaa allele (Shen et al., 2015) instead of RQC2-WT. Neither of these perturbations (rqc2Δ ltn1Δ and rqc2aaa-OE ltn1Δ) induced the punctate RQCsub localization we observed in ROLD (Fig. 1C), confirming that formation of RQCsub inclusions requires CATylation. These results indicate that RQC2 overexpression produces aggregation-prone CAT tails that sequester RQC substrates into detergent-insoluble inclusions in cells with compromised Ltn1.
Cytosolic CAT tail aggregates recruit the Hsp40 chaperone Sis1 (Choe et al., 2016; Defenouillère et al., 2016; Yonashiro et al., 2016), thereby depleting Sis1 from the nucleus. In strains where RQCsub had diffuse localization (wt, ltn1Δ, and ltn1Δ rqc2Δ), there was little colocalization of Sis1 and the substrate (Fig. 1D). In contrast, RQCsub colocalized with Sis1 in inclusions at the periphery of most cells in the inclusion-containing ROLD strain (Fig. 1D). This peripheral localization of Sis1 did not overlap with a nuclear marker, consistent with the ROLD perturbation depleting Sis1 from the nucleus (Supplementary Fig. 1A). Furthermore, a larger portion of Sis1 was retained as a detergent-resistant species in the well of an SDS-PAGE gel in the ROLD condition compared to wt, ltn1Δ, and ltn1Δ rqc2Δ (Fig. 1E). Although perturbation of LTN1 compromises RQC, these data demonstrate that LTN1 deletion alone does not modify Sis1 localization. However, the combination of LTN1 deletion and increased RQC2 expression leads to depletion of Sis1 from the nucleus into detergent-resistant inclusions.

ROLD depletes Sis1 from the nucleus and impairs CAT tail degron activity. (A) Fluorescence microscopy of cells containing an RFP-tagged SIS1 allele in their genomes and expressing a nuclear marker, bipartite (bp) SV40 NLS-tagged GFP. (B) Additional data to support Fig. 2C. Stability measurements of RQCsubLONG expressed in ltn1Δ and ROLD cells after bortezomib (btz) treatment to inhibit the proteasome and HUL5 deletion to block CAT tail degron activity. Error bars represent s.e.m. from three independent cultures. P-values from statistical tests appear above bars. Thicker lines come from paired t-tests, measuring the significance of bortezomib-induced stabilization. Thinner lines come from t-tests for particular contrast, measuring how significantly different HUL5 deletion-induced stabilization is between ltn1Δ and ROLD backgrounds.
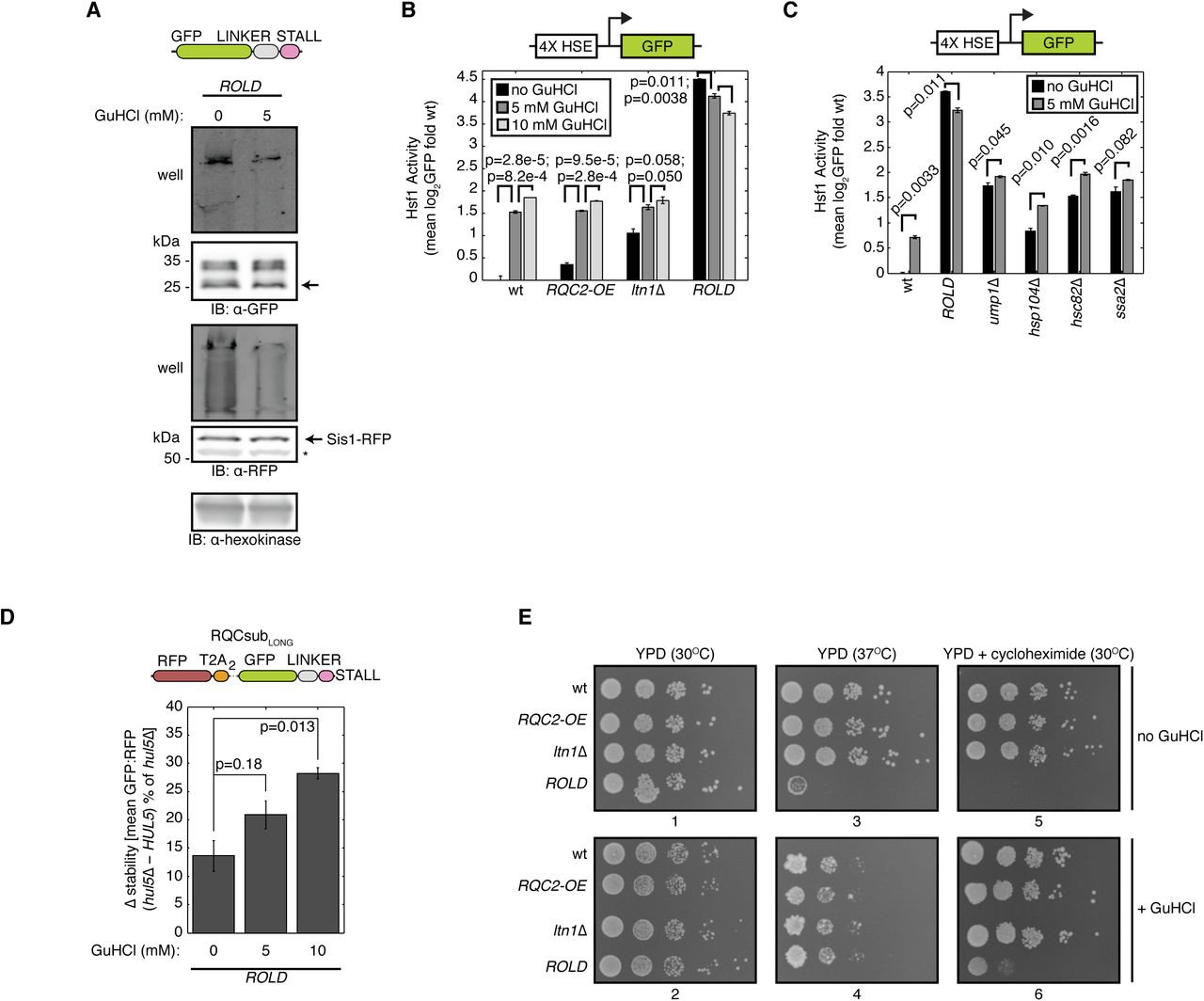
Sequestration of Sis1 and other chaperones into aggregates has been shown to impair proteostasis (Choe et al., 2016; Defenouillère et al., 2016; Park et al., 2013; Yonashiro et al., 2016). Proteostasis impairment activates Heat Shock Factor 1 (Hsf1), a transcription factor that orchestrates transcription of chaperones and other proteostasis-restoring factors (Åkerfelt et al., 2007; Morimoto, 2011). To assess the magnitude of proteostasis disruption caused by ROLD, we measured Hsf1 activity using a previously published reporter (Brandman et al., 2012), consisting of GFP under the control of an artificial promoter containing four heat shock elements (Sorger and Pelham, 1987), the consensus binding site for Hsf1 (Fig. 2A). Deletion of LTN1 alone activated the Hsf1 reporter 2.07-fold relative to wt, while additional RQC2 overexpression (ROLD) led to 22.6-fold activation (Fig. 2A). The potent activation observed in ROLD required LTN1 deletion, as RQC2 overexpression in the wt background weakly affected the reporter (Fig. 2A). These data establish that RQC2 overexpression and LTN1 deletion have a synthetic interaction that results in elevated Hsf1 transcriptional activity. This synergistic enhancement in Hsf1 activity implies that increased Rqc2 levels may exert additional proteotoxic stress on RQC-compromised cells.

ROLD induces Hsf1 activation, disrupts CAT tail composition and degron function, and sensitizes cells to stress. (A) Flow cytometry of cells containing an integrated reporter for Hsf1 activation (schematic above). Error bars represent the standard error of the mean (s.e.m.) derived from three independent cultures. P-values from indicated paired t-tests are indicated by lines between bars. HSE, heat shock element. (B) Amino acid analysis of RQCsub immunoprecipitated from ltn1Δ, ltn1Δ rqc2Δ, or ROLD lysates. (C) Left, schematic of expression-controlled model RQC substrate RQCsubLONG. Center, definitions of “stability,” “Δ stability,” and “CAT tail degron activity.” Right, Δ stability measurements of RQCsubLONG expressed in ltn1Δ and ROLD cells after HUL5 deletion to assess CAT tail degron activity. Error bars as in A. P-values above bars are derived from t-tests for particular contrast, measuring how significantly different HUL5 deletion-induced stabilization is between ltn1Δ and ROLD backgrounds. (D) Spot assay for strains grown under indicated conditions.
To investigate how RQC2 overexpression might increase the proteotoxicity of RQC failure, we analyzed the composition of CAT tails under this condition. As expected, RQCsub purified from the CATylation-competent ltn1Δ strain was enriched in alanine and threonine relative to the CATylation-incompetent ltn1Δ rqc2Δ strain (Fig. 2B). This result confirmed that this assay could detect changes in CAT tail composition. Unexpectedly, alanine content in RQCsub was enriched by 31% in ROLD relative to ltn1Δ (Fig. 2B). Thus, RQC2 overexpression, which induces a large disruption of proteostasis in ltn1Δ cells, increases alanine content in CAT tails.
One reason why cells tolerate LTN1 disruption is that CAT tails function as Ltn1-independent degrons, thereby targeting RQC substrates that escape Ltn1 for proteasomal degradation (Sitron and Brandman, 2019). To assess whether RQC2 overexpression impairs this degradation pathway, we quantified the stability of the model RQC substrate RQCsubLONG (Sitron and Brandman, 2019). RQCsubLONG enables quantification of its stability by including an internal expression control, red fluorescent protein (RFP) followed by viral T2A peptides, at the N-terminus of GFP-linker-arginine (Fig. 2C). Ribosomes translating RQCsubLONG produce RFP then skip formation of a peptide bond within the T2A sequence (Donnelly et al., 2001; Szymczak and Vignali, 2005), severing RFP from the GFP-linker-arginine RQC substrate (Fig. 2C). As a result, each round of translation produces RFP and GFP stoichiometrically, but only GFP becomes an RQC substrate (Fig. 2C). Thus, the GFP:RFP ratio serves as an internally-controlled readout for RQCsubLONG stability (Fig. 2C). CAT tail degron activity on RQCsubLONG requires the E3 ubiquitin ligase Hul5 and the proteasome (Sitron and Brandman, 2019). Therefore, comparing stability before and after HUL5 deletion quantifies CAT tail degron activity (Fig. 2C). RQCsubLONG stability did not change after treatment with the proteasome inhibitor bortezomib in ltn1Δ hul5Δ or ROLD hul5Δ (Supplementary Fig. 1B), supporting the requirement for Hul5 in CAT tail-mediated degradation of RQCsubLONG by the proteasome. HUL5 deletion stabilized RQCsubLONG by 30.2% in the ltn1Δ background and 13.6% in the ROLD background (Fig. 2C), consistent with RQC2 overexpression reducing RQCsubLONG degradation. These results demonstrate that RQC2 overexpression impairs CAT tail degron activity.
Given that RQC2 overexpression in ltn1Δ impaired degradation of CATylated proteins and potentiated their aggregation, we wondered whether this results in defective cellular fitness. At 30°C, all of the strains we measured grew equally well in a spot assay (Fig. 2D). This result demonstrates that ROLD does not impair growth in the absence of stress. We then introduced two mild stressors to perturb proteostasis: the thermal stress of 37°C incubation or introduction of 50 ng/ml cycloheximide to increase translational stalling. Neither of these stresses induced a growth effect in the RQC2 overexpression (RQC2-OE) or ltn1Δ strains relative to wt (Fig. 2D). Contrastingly, 37°C incubation and 50 ng/ml cycloheximide severely inhibited ROLD growth (Fig. 2D). Thus, RQC2 overexpression sensitizes RQC-compromised cells to otherwise mild stressors. We posit that this stress-sensitivity, in conjunction with elevated Hsf1 activation, reflects a fragile state of proteostasis in ROLD.
The denaturant guanidinium hydrochloride ameliorates the toxic effects of elevated Rqc2 levels
We have observed that ROLD induces several phenotypes that correlate with aggregation of CATylated proteins: co-aggregation of Sis1 (Fig. 1D,E), increased Hsf1 activity (Fig. 2A), impaired Ltn1-independent degradation of CATylated proteins (Fig. 2C), and sensitivity to mild stressors (Fig. 2D). These correlations prompted us to hypothesize that aggregation of CATylated proteins causes this constellation of phenotypes. To test this hypothesis, we treated live ROLD cells with guanidinium hydrochloride (GuHCl), a protein denaturant that suppresses aggregation of CAT tails and other aggregation-prone proteins at low concentrations (Defenouillère et al., 2016; Ness et al., 2002). Sis1 and RQCsub still localized to inclusions by microscopy after 5 mM GuHCl treatment in ROLD (Supplementary Fig. 2A). However, monitoring RQCsub mobility by SDS-PAGE assessed how GuHCl affected RQCsub and Sis1 more sensitively than microscopy, revealing that GuHCl treatment enabled a larger proportion of both RQCsub and Sis1 extracted from ROLD cells to migrate into an SDS-PAGE gel rather than remaining in the well (Fig. 3A). GuHCl thus reduces the detergent-insolubility of CATylated proteins and Sis1 caused by ROLD.

Effects of guanidinium hydrochloride on RQCsub inclusion formation and CAT tail degron activity. (A) Fluorescence microscopy of ROLD SIS1-RFP cells expressing RQCsub in the absence or presence of 5 mM guanidinium hydrochloride (GuHCl). (B) Additional data to support Fig. 3D. Stability measurements of RQCsubLONG expressed in ROLD cells grown in indicated GuHCl concentrations with additional bortezomib treatment to inhibit the proteasome and HUL5 deletion to measure CAT tail degron activity. Error bars represent s.e.m. from three independent cultures. P-values are indicated above bars. Thick lines indicate paired t-tests, probing the significance of bortezomib-induced stabilization. Thin lines denote t-tests for particular contrast, measuring how significantly different HUL5 deletion-induced stabilization is at different GuHCl concentrations.

Guanidinium hydrochloride solubilizes CAT tails and mitigates ROLD-induced phenotypes. (A) Whole cell immunoblot (IB) of ROLD cells expressing genomically RFP-tagged Sis1 and RQCsub with and without guanidinium hydrochloride (GuHCl) treatment. Arrow next to the anti-GFP IB indicates molecular weight of RQCsub without CAT tails. Asterisk next to the anti-RFP IB points to a non-specific band detected by the anti-RFP antibody. (B and C) Flow cytometry of cells containing an integrated reporter for Hsf1 activation with and without GuHCl treatment. Error bars represent s.e.m. of three independent cultures. Lines between bars indicate p-values from paired t-tests. (D) Δ stability measurements of RQCsubLONG to assess how growth in GuHCl affects CAT tail degron activity (defined in Fig. 2C) for ROLD cells. Error bars as in B and C. P-values indicated above bars are the result of a t-test for particular contrast, measuring how significantly different CAT tail degron activity is at different GuHCl concentrations. (E) Spot assay of strains grown at 30°C, 37°C, and in 50 ng/ml cycloheximide with and without the presence of 5 mM GuHCl. Plate numbers indicated below images.
We then tested whether improving CATylated protein solubility with GuHCl would relieve the Hsf1 activation observed in ROLD. GuHCl treatment activated the Hsf1 reporter in a dose-dependent manner in wt, RQC2-OE, and ltn1Δ strains (Fig. 3B). This result might indicate that GuHCl, a protein denaturant, exerts some proteotoxic stress in cells. Despite this, GuHCl decreased Hsf1 reporter activation in ROLD (Fig. 3B), suggesting that solubilization of CAT tails alleviates the Hsf1 activation caused by ROLD. We tested the specificity of this response by measuring the effect of GuHCl on the Hsf1 reporter in other genotypes with high Hsf1 activity (Brandman et al., 2012) (Fig. 3C). Hsf1 reporter deactivation was unique to the ROLD condition, as GuHCl increased Hsf1 reporter activation in ump1Δ, hsp104Δ, hsc82Δ, and ssa2Δ strains (Fig. 3C). These results are consistent with aggregation of CAT tails exerting proteotoxic stress on cells.
Given that short, soluble CAT tails act as Ltn1-independent degrons, we questioned whether solubilizing aggregated CAT tails in ROLD might rescue deficits in CAT tail degron activity (Fig. 2C). We again compared the stability (GFP:RFP) of RQCsubLONG in ROLD relative to ROLD hul5Δ, the condition where RQCsubLONG degradation is blocked (Sitron and Brandman, 2019), to measure CAT tail degron activity. GuHCl had two effects on degradation of RQCsubLONG. First, increasing concentrations of GuHCl increased the magnitude that HUL5 deletion stabilized RQCsubLONG (from 13.6% to 20.9% to 28.2% at 0, 5, and 10 mM GuHCl; Fig. 3D), suggesting that treating ROLD cells with GuHCl enhances degradation of RQCsubLONG. Second, the magnitude of bortezomib-induced RQCsubLONG stabilization increased with increasing GuHCl when Hul5 was intact (Supplementary Fig. 2B). This result indicates that the degradation that GuHCl enhanced involved both Hul5 and the proteasome. Taken together, these data have two implications: 1) the insolubility of CAT tails in ROLD impairs CAT tail degron activity, and 2) solubilization of CAT tails rescues this impairment.
We next investigated whether GuHCl could dampen the sensitivity of ROLD cells to mild stressors. GuHCl did not generally affect growth (Fig. 3E, plate 1 compared to 2), but additional GuHCl partially rescued ROLD growth in the presence of cycloheximide (Fig. 3E, plate 5 compared to plate 6). While the combination of GuHCl and 37°C incubation led to poorer growth of all strains relative to 37°C alone, ROLD grew comparably to wt, RQC2-OE, and ltn1Δ when exposed to both GuHCl and 37°C (Fig. 3E, plate 3 compared to plate 4). Thus, CAT tail-solubilizing GuHCl treatment ameliorates the stress sensitivity caused by RQC2 overexpression in cells with impaired RQC.
RNA Polymerase III modulates the toxicity of RQC failure
To identify pathways responsible for the proteotoxicity of RQC failure, we screened for mutants that had weak activation of the Hsf1 reporter in an RQC-compromised strain. After crossing a genome-wide collection of hypomorphs into the Hsf1-inducing rqc1Δ rps0aΔ strain (Brandman et al., 2012), we found that all hits other than RQC2 (whose deletion blocks CATylation) were related to RNA Polymerase III (Pol III). These hits included a non-essential polymerase biogenesis factor, BUD27 (Mirón-García et al., 2013), as well as essential Pol III subunits, RPC17 and RPC160. Disruption of each of these by deletion or a “decreased abundance by mRNA perturbation” allele (DAmP) (Yan et al., 2008) nearly eliminated activation of the Hsf1 reporter in ROLD compared to wt (Fig. 4A). To assess the specificity of this reduction in Hsf1 reporter induction, we measured the effect of BUD27 and RPC17 perturbations in a panel of strains with elevated Hsf1 signaling (Fig. 4B). Perturbations to BUD27 and RPC17 reduced Hsf1 reporter activity in wt, ump1Δ, hsp104Δ, hsc82Δ, and ssa2Δ strains (Fig. 4B, comparison between black and grey bars). In each strain, the magnitude of this reduction was substantially less than the 20.7-fold and 13.7-fold reduction we observed in ROLD after disruption of BUD27 and RPC17, respectively (Fig. 4B). Furthermore, Hsf1 reporter activation increased in the Pol III-perturbed backgrounds after the ROLD perturbation or upon deletion of UMP1, HSP104, HSC82, and SSA2 (Fig. 4B; Supplementary Fig. 3A). Taken together, these data indicate that Pol III impairment lessens proteotoxicity caused by ROLD more than it does for other genetic stressors and does not generally prevent Hsf1 activation.

Effects of Pol III perturbation on Hsf1 activation, ribosome stalling, CAT tail composition, and Sis1 localization. (A) Flow cytometry of Pol III-perturbed cells containing an integrated reporter for Hsf1 activation. These data are also contained in Fig. 4B, but are reordered here to simplify comparisons within two Pol III-perturbed genetic backgrounds. Error bars indicate s.e.m. from three independent cultures. (B) Left, schematic of a stalling reporter similar to that used in Juszkiewicz and Hegde 2017 and Sundaramoorthy et al. 2017. Right, schematic of stalling reporter with the same (CGN)12 stalling sequence contained in RQCsub or a non-stalling (Ser-Thr)6 sequence. Error bars as in A. (C) Quantification of the stalling reporter from B containing various lengths of CGA codon stretches rather than (CGN)12. Error bars as in A. (D) Amino acid analysis of RQCsub immuno-purified from ROLD bud27Δ cells compared to ROLD. (E) Fluorescence microscopy of SIS1-RFP cells expressing GFP with a nuclear localization sequence, bipartite SV40.

Pol III perturbation reverses ROLD-induced effects on heat shock, CAT tail aggregation, and CAT tail degron activity. (A and B) Flow cytometry of cells containing an integrated reporter for Hsf1 activation with and without perturbations to RNA Polymerase III. Error bars indicate s.e.m. from three independent cultures. P-values from paired t-tests are indicated with lines connecting bars. (C) Whole-cell immunoblot of RQCsub. Arrow indicates molecular weight of RQCsub without CAT tails. (D) Fluorescence microscopy of cells expressing RQCsub. The percentages of cells containing inclusions are indicated below. (E) Fluorescence microscopy of SIS1-RFP cells expressing RQCsub. (F) Whole-cell immunoblot of Sis1-RFP. (G) Δ stability measurements of RQCsubLONG expressed in indicated strains after HUL5 deletion to quantify CAT tail degron activity. Error bars as in A and B. P-value indicates the result of a t-test for particular contrast; this assesses how significantly different HUL5 deletion-induced stabilization is in ROLD compared to ROLD bud27Δ.
One mechanism by which Pol III disruption might alleviate proteotoxicity in ROLD is by preventing ribosome stalling. Translation initiation, which involves Pol III target genes such as 5S rRNA and tRNAs (Turowski and Tollervey, 2016), modulates stalling by controlling the amount of ribosomes on an mRNA (Juszkiewicz et al., 2018; Simms et al., 2017). When fewer ribosomes translate an mRNA (low initiation), ribosomes stuck at stalling sequences collide less frequently, thereby preventing recognition of stalls and subsequent RQC (Juszkiewicz et al., 2018; Simms et al., 2017). We therefore speculated that lower levels of translation components in Pol III-perturbed strains might reduce the generation of RQC substrates, thereby preventing their toxic build-up. To evaluate the effect of Pol III perturbation on translation of stalling sequences, we used a quantitative stalling reporter similar to one previously developed (Juszkiewicz and Hegde, 2017; Sundaramoorthy et al., 2017) (Supplementary Fig. 3B). BUD27 deletion did not alleviate stalling induced by (CGN)12 (the stalling sequence in RQCsub) in wt or ROLD relative to control (Supplementary Fig. 3B). Thus, the effects of Pol III perturbation on translation should not confound our analysis of RQCsub by preventing ribosomes translating RQCsub from stalling. We then titrated the strength of the stalling sequence by varying the number of difficult-to-decode CGA codons (Letzring et al., 2010) it contained. In the ROLD background, BUD27 deletion had no effect on stalling induced by short stretches of CGA codons (one or two CGA codons) or a more severe stretch (eight CGA codons); BUD27 deletion weakly reduced stalling on sequences that contained four or six CGA codons (Supplementary Fig. 3C). Thus, ribosomes still stall when translating difficult-to-decode codons after Pol III perturbation. This result suggests that prevention of stalling is not the mechanism by which Pol III perturbation alleviates proteotoxicity in ROLD.
Given that stalling still occurred after disruption of Pol III, we wondered whether CATylation remains intact under this condition. To analyze the effect of Pol III perturbation on CATylation, we monitored RQCsub mobility by SDS-PAGE (Fig. 4C). As in ltn1Δ, RQCsub expressed in ROLD bud27Δ migrated as a smear which was higher in molecular weight than RQCsub expressed in the CATylation-deficient ltn1Δ rqc2Δ strain (Fig. 4C). Furthermore, amino acid analysis revealed that RQCsub CAT tails had similar composition in ROLD and ROLD bud27Δ cells (Supplementary Fig. 3D). Pol III perturbation thereby has no effect on CATylation in ROLD.
We next investigated whether a reduction in co-aggregation of CATylated proteins and Sis1 could explain the dampened Hsf1 activation observed after Pol III disruption in ROLD. ROLD cells carrying hypomorphic BUD27 or RPC17 alleles formed RQCsub inclusions less frequently (3.57% and 4.37% relative to 56.9% in ROLD), consistent with a reduction in CAT tail aggregation (Fig. 4D). Similarly, BUD27 deletion in ROLD prevented colocalization of RQCsub with Sis1 (Fig. 4E) and led to stronger Sis1 nuclear localization than in ROLD (Supplementary Fig. 3E). Compared to ROLD, a smaller proportion of Sis1 in ROLD bud27Δ migrated as a detergent-resistant species during SDS-PAGE (Fig. 4F). Taken together, these results indicate that Pol III disruption limits aggregation of both CATylated proteins and Sis1 caused by RQC2 overexpression in cells with compromised RQC.
Because we previously observed that solubilization of CAT tails with GuHCl improved their degron strength, we wondered whether Pol III perturbation would have the same effect. To quantify CAT tail degron strength, we measured degradation of RQCsubLONG in the ROLD and ROLD bud27Δ backgrounds. The difference in RQCsubLONG stability (GFP:RFP) in each of these backgrounds with (degradation intact) and without Hul5 (degradation blocked) defined the degree of degradation (Sitron and Brandman, 2019). We validated this approach by demonstrating that the proteasome inhibitor bortezomib did not significantly stabilize RQCsubLONG in hul5Δ strains (Supplementary Fig. 4A), demonstrating that HUL5 deletion still blocked degradation. HUL5 deletion stabilized RQCsubLONG in the ROLD bud27Δ background more than in ROLD (33.7% compared to 13.6%; Fig. 4G), consistent with improved degradation. Thus, perturbing Pol III in the ROLD condition improves CAT tail solubility and degradation of CATylated proteins.

Effects of mutations on an RQC substrate and hard-coded CAT tail constructs. (A) Additional data in support of Fig. 4G. Stability measurements of RQCsubLONG expressed in indicated strains with bortezomib (btz) treatment to inhibit the proteasome and HUL5 deletion to block CAT tail degron activity. Error bars indicate s.e.m. from three independent cultures. P-values are given above bars. Results of paired t-tests measuring the significance of bortezomib-induced stabilization are indicated with thick lines. The result of a t-test for particular contrast is indicated with thin lines; this assesses how significantly different HUL5 deletion-induced stabilization is in ROLD compared to ROLD bud27Δ. (B) Flow cytometry of RFP with appended hard-coded CAT tails 20 (AT20) or 60 (AT60) amino acids in length. Error bars as in A.
Pol III perturbation renders cells refractory to RQC stress
Because disruption of Pol III allowed for CATylation in ROLD but the resultant CAT tails did not form inclusions in cells, we hypothesized that Pol III impairment depletes a factor that nucleates aggregation of CAT tails. To test this hypothesis, we added lysate from the ROLD strain to lysate from an RQCsub-expressing ROLD bud27Δ strain and monitored formation of the detergent-resistant RQCsub species in vitro (Fig. 5A). In agreement with microscopy from these strains (Figs. 1D and 5D), deletion of BUD27 in ROLD reduced the abundance of a detergent-resistant RQCsub species that failed to migrate into an SDS-PAGE gel (Fig. 5A). Addition of ROLD lysate that lacked RQCsub to ROLD bud27Δ lysate containing RQCsub converted a population of the substrate into its detergent-resistant form (Fig. 5A), suggesting that aggregate nucleation had taken place. Lysate from wt cells lacked this apparent nucleation activity (Fig. 5A). We then tested whether the nucleating factor from ROLD was an RNA species (e.g. a Pol III-dependent transcript) by first Rnase A-treating the ROLD lysate. This RNAse A-treated lysate still stimulated the appearance of detergent-resistant RQCsub (Fig. 5A). However, the 95,000 g supernatant of the ROLD lysate displayed less ability to convert RQCsub into its detergent-resistant form compared to total ROLD lysate (Fig. 5A). Taken together, these data indicate that the ROLD condition leads to accumulation of an insoluble species that converts CAT tails into a detergent-insoluble form. We posit that perturbation of Pol III reduces the abundance of this species and thereby limits CAT tail aggregation.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Pol III perturbation renders cells resistant to nucleation of CAT tail aggregates and insensitive to increased ribosome stalling. (A) Upper, schematic of lysate mixing experiment. RQCsub-expressing lysate from ROLD bud27Δ was mixed with various RQCsub-free lysates. Lower, immunoblot of immunoprecipitated mixed lysates. Arrow and asterisk indicate molecular weight of non-CATylated RQCsub and RQCsub product derived from complete translation through the RQCsub stall sequence, respectively. (B) Fluorescence microscopy of cells expressing RFP with a hard-coded CAT tail appended to its C-terminus (RFP-AT20 and RFP-AT60 indicate 20 and 60 amino acid-long appendages, respectively). Quantification of cells containing inclusions indicated below images. (C) Fluorescence microscopy of cells co-expressing RQCsub and RFP-AT60. (D) Fluorescence microscopy of ROLD bud27Δ SIS1-RFP cells co-expressing RQCsub and GST with hard-coded CAT tails. (E) Flow cytometry of cells containing an integrated reporter for Hsf1 activation expressing indicated constructs. EV, empty vector. Error bars indicate s.e.m. from three independent cultures. P-values from paired t-tests indicated above bars. (F) Spot assay of cells grown under indicated conditions.
We then sought to determine whether forcing nucleation of CAT tails into inclusions would revert Pol III-perturbed ROLD cells into a stressed state. To accomplish this, we first tested whether expression of “hard-coded” CAT tails, proteins containing genetically-encoded stretches of alanine and threonine, would nucleate aggregation of endogenous CAT tails. These proteins were not substrates for RQC, as depletion of RQC components failed to increase levels of RFP constructs containing hard-coded CAT tails (Supplementary Fig. 4B). RFP with a C-terminal sequence containing a 20 amino acid hard-coded CAT tail (RFP-AT20) displayed diffuse localization in wt and ROLD bud27Δ (Fig. 5B), two strains where few cells contain inclusions of CATylated RQC substrates (Figs. 1C, 4D). In the ROLD strain, wherein many cells carry inclusions of CATylated RQC substrates (Fig. 1C), RFP-AT20 formed peripheral inclusions (Fig. 5B). These results suggest that a 20 amino acid-long CAT tail alone cannot nucleate aggregation, but can form inclusions in strains with existing CAT tail aggregates. RFP attached to a 60 amino acid-long hard-coded CAT tail (RFP-AT60) localized to inclusions in all strains we tested (Fig. 5B). Expressing this protein in ltn1Δ and ROLD bud27Δ, strains with few endogenous CAT tail inclusions (Figs. 1C, 4D), led to colocalization of RFP-AT60 and endogenously CATylated RQCsub in peripheral inclusions (Fig. 5C). Similarly, expression of the non-fluorescent hard-coded CAT tail construct GST-AT60 in ROLD bud27Δ led to colocalization of Sis1 and RQCsub in puncta, while the shorter GST-AT20 hard-coded CAT tail did not (Fig. 5D). Taken together, these results demonstrate that hard-coded CAT tails can force nucleation of endogenously CATylated proteins and Sis1 into inclusions.
Using hard-coded CAT tails, we monitored whether restoring nucleation of CAT tail inclusions disrupts proteostasis in Pol III-perturbed ROLD cells. We measured the activity of an Hsf1 reporter as a proxy for proteostasis, reasoning that high Hsf1 activity indicated a disruption in proteostasis. ROLD bud27Δ cells displayed Hsf1 reporter activity lower than ROLD (Fig. 5E). Expression of the non-fluorescent GST-AT20 hard-coded CAT tail, which did not nucleate inclusions of RQCsub and Sis1 (Fig. 5D), did not activate the Hsf1 reporter in ROLD bud27Δ cells (Fig. 5E). Surprisingly, despite its ability to induce formation of inclusions (Fig. 5D), GST-AT60 expression failed to activate the Hsf1 reporter (Fig. 5E). These data imply that: 1) re-localization of Sis1 from the nucleus into CAT tail inclusions does not suffice for Hsf1 activation, and 2) formation of CAT tail aggregates does not disrupt proteostasis in the Pol III-perturbed ROLD condition.
We reasoned that this lack of Hsf1 activation after Pol III perturbation implies that Pol III perturbation decreases the fragility of proteostasis caused by ROLD. To test this hypothesis, we analyzed cells’ sensitivity to mild stress under these conditions. As previously published, BUD27 deletion slightly lowered growth rate at 30°C and strongly impaired growth at 37°C in the wt background (Deplazes et al., 2009) as well as in ROLD (Fig. 5F). Thus, the sensitivity to mild thermal stress caused by Pol III impairment renders this perturbation unable to rescue heat sensitivity in ROLD. Despite causing a growth defect in wt cells, BUD27 deletion enabled faster growth of ROLD cells in the presence of cycloheximide (Fig. 5F). Unexpectedly, the growth rate of ROLD bud27Δ cells upon cycloheximide exposure was faster than the bud27Δ strain (Fig. 5F). We thus posit that Pol III perturbation desensitizes ROLD cells to stress caused by increased ribosome stalling.
Discussion
Here we discovered that the combination of high RQC2 expression and compromised RQC function altered CAT tail composition and converted them into an aggregated form that compromised CAT tail degron activity and disrupted proteostasis (Figs. 1-2). This proteostasis-disrupted state featured re-localization of Sis1 from the nucleus into CAT tail inclusions (Fig. 1D), activation of heat shock signaling (Fig. 2A), and hypersensitivity to mild stressors (Fig. 2D). Guanidinium hydrochloride (GuHCl) treatment and Pol III perturbation ameliorated the effects of RQC2 overexpression, restoring CAT tail degron function and decreasing proteotoxic stress (Figs. 3-5), thereby mitigating the toxicity of RQC failure.
Although CAT tails have amyloid-like character (Choe et al., 2016; Defenouillère et al., 2016; Yonashiro et al., 2016), we observed little CAT tail aggregation at endogenous Rqc2 levels (Fig. 1B). However, increased RQC2 expression led to formation of CAT tail inclusions (Fig. 1C). These results imply that high Rqc2 levels alter the properties of CAT tails to drive their aggregation. We posit that this alteration may occur in cis or in trans. In cis, the increased alanine content in CAT tails caused by RQC2 overexpression (Fig. 2B) could increase their amyloid-like character. Polyalanine tracts have been shown to form amyloid and render proteins detergent-insoluble (Albrecht and Mundlos, 2005; Oma et al., 2007; Rankin et al., 2000; Shinchuk et al., 2005), similarly to what we observed for CAT tails after RQC2 overexpression (Fig. 1A,B). In trans, increased Rqc2 levels could enable CATylation of more endogenous RQC substrates that nucleate aggregation of other CATylated proteins. This explanation assumes that endogenous Rqc2 is limiting for CAT tail aggregation, as has been hypothesized previously (Yonashiro et al., 2016). In support of this trans-acting explanation, we demonstrated that transacting factors can nucleate CAT tail aggregation: addition of exogenous lysate converted non-aggregated CAT tails into a detergent-resistant state (Fig. 5A) and short CAT tails that did not typically aggregate formed inclusions in cells with abundant CAT tail aggregates (Fig. 5B). These cis and trans mechanisms are not mutually exclusive, and could both contribute to how elevated Rqc2 levels promote CAT tail aggregation.
In addition to potentiating CAT tail aggregation, RQC2 overexpression in RQC-compromised (ltn1Δ) cells disrupted proteostasis more than LTN1 deletion alone (Figs. 1-2). Yet, in this same ltn1Δ background, we showed previously that the absence of Rqc2 prevents degradation of escaped RQC substrates and sensitizes cells to stress (Sitron and Brandman, 2019). This dichotomy implies that cells must strike a balance in setting Rqc2 levels to avoid toxicity when RQC fails, maintaining Rqc2 expression at a level that supports CAT tail degron function without leading to proteostasis disruption. Rqc2 levels may thereby modify the pathology of RQC failure. For instance, our results predict that tissues with high Rqc2 levels relative to Ltn1 or low expression of both would be most susceptible to impaired RQC. Future studies will evaluate the role of RQC2 expression in the neurodegenerative pathology of RQC-compromised animals.
CAT tail degron activity safeguards cells from RQC failure. We show here that high levels of aggregation caused by RQC2 overexpression and LTN1 deletion (ROLD) co-occur with disruption of this back-up degradation pathway. This co-occurrence highlights a potential pathologic positive feedback loop wherein impaired degradation of CATylated proteins causes them to accumulate, increasing their aggregation, and further sequestering them from degradation. Two possibilities exist for how RQC2 overexpression triggers such a feedback loop: 1) either the altered composition of CAT tails caused by increased Rqc2 (Fig. 2B) creates weaker degrons that start to build up and aggregate, or 2) a decrease in solubility (Fig. 1A,B) due to altered amino acid composition (Fig. 2B) or nucleation by trans-acting factors (Fig. 5A) (e.g. other CATylated proteins) causes CAT tails to become inaccessible to degradation. The observation that GuHCl both improved the solubility of CAT tails (Fig. 3A) and restored their degron functionality (Fig. 3D) argues for the latter possibility, although these models are not mutually exclusive. This inverse relationship between the function of CAT tails as degrons and CAT tail aggregation implies that CAT tail aggregation is toxic, rather than being adaptive as previously hypothesized (Yonashiro et al., 2016). Thus, improving the solubility of CAT tails may prove to be an effective intervention for combating the toxicity of RQC failure.
We describe here two interventions that increase the solubility of CAT tails, restore CAT tail degron function, and reduce the toxicity of RQC failure (Figs. 3-5). GuHCl is a chaotrope which may disfavor interactions between CAT tails and thereby disrupt aggregate nucleation, as has been shown for high concentrations of chaotropic agents on amyloid-like proteins (Rubin et al., 2013). Alternatively, GuHCl could improve CAT tail solubility though an indirect mechanism, such as Hsp104 inhibition mediated by low concentrations of GuHCl (Ness et al., 2002). Pol III impairment reversed RQC failure toxicity more potently than GuHCl. Pol III plays many roles in cells (Turowski and Tollervey, 2016), so perturbation of Pol III is likely pleiotropic. Our results suggest that Pol III regulates CAT tail aggregation at the level of nucleation, as Pol III-perturbed lysates lacked the ability to nucleate CAT tail aggregates (Fig. 5A). Investigating the mechanism by which Pol III inhibition relieves toxicity from RQC failure and leveraging this mechanism to develop therapies are promising areas for future research. Indeed, genetic and small-molecule disruption of Pol III has been shown to be well tolerated in Drosophila and C. elegans, leading to lifespan extension (Filer et al., 2017). Our work demonstrates that pharmacologic and genetic interventions can mitigate the toxic effects of RQC failure.
Materials and Methods
Yeast Strains and Culturing
The parental wild-type yeast strain used in this study was BY4741. All gene deletions, genomic integrations, and plasmid transformations were done in this background using standard methods. For a complete list of strains used, see Supplementary Table 1. For a complete list of plasmids used, see Supplementary Table 2.
Yeast strains used in this study.
Plasmids used in this study.
Yeast cultures were grown at 30°C in YPD or synthetic defined (SD) media with appropriate nutrient dropouts. SD media used for growth of yeast cultures contained: 2% w/v dextrose (Thermo Fisher Scientific, Waltham, MA), 13.4 g/L Yeast Nitrogen Base without Amino Acids (BD Biosciences, San Jose, CA), 0.03 g/L L-isoleucine (Sigma-Aldrich, St. Louis, MO), 0.15 g/L L-valine (Sigma-Aldrich), 0.04 g/L adenine hemisulfate (Sigma-Aldrich), 0.02 g/L L-arginine (Sigma-Aldrich), 0.03 g/L L-lysine (Sigma-Aldrich), 0.05 g/L L-phenylalanine (Sigma-Aldrich), 0.2 g/L L-Threonine (Sigma-Aldrich), 0.03 g/L L-tyrosine (Sigma-Aldrich), 0.018 g/L L-histidine (Sigma-Aldrich), 0.09 g/L L-leucine (Sigma-Aldrich), 0.018 g/L L-methionine (Sigma-Aldrich), 0.036 g/L L-tryptophan (Sigma-Aldrich), and 0.018 g/L uracil (Sigma-Aldrich). Media additionally contained 5 mM or 10 mM guanidinium hydrochloride (Sigma-Aldrich) where indicated and bortezomib (LC Laboratories, Woburn, MA) treatment lasted 4 hours.
Plasmids
Plasmids were constructed using NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs, Ipswich, MA). All plasmids used in this study are described in Supplementary Table 2.
Two variants of RQCsub were used that had identical construction except for the region following the stall: one had a fluorescent protein (RFP) and the other had a series of affinity tags. The version without the post-stall RFP was used for microscopy and the version with the post-stall RFP was used in immunoblots; the one exception to this is that the version without the post-stall RFP was used in an immunoblot in Fig. 3A.
Hard-coded CAT tail constructs contained stretches of alanine and threonine that were encoded by the DNA sequences that follow. AT20: 5’-GCAACCGCAACAGCTACCGCTACAGCCACGGCGACCGCCACTGCAACAGCGACAGCGAC TTAA-3’. AT60: 5’-GCCGCCGCAACAACAGCCACTACCACAGCTGCCACTGCTGCGACGACCACAGCCGCAGC GACGGCTACAGCAGCGACAGCGACTACCGCAACGGCTGCAGCGACAGCCACGGCGACTG CAGCTACTGCCACGACGACGGCAACCGCCACGGCAACAACAACTACAACCGCGACTGCC GCATAA-3’.
Flow cytometry
Fluorescence was measured using a BD Accuri C6 flow cytometer (BD Biosciences). Three independent yeast cultures were grown to log phase overnight in appropriate SD dropout media. Data analysis was performed with MATLAB (MathWorks, Natick, MA).
For measurements of the Hsf1 activity reporter, raw fluorescence values were first normalized to side scatter, log2-transformed, and then the value obtained from the wt strain was subtracted from each condition. These calculations are reported as log2 fold wt in the figures.
For measurements of RQCsubLONG, a detailed description of the analysis workflow can be found in Sitron and Brandman, 2019. Briefly, yeast expressing the plasmid were gated according to above-background RFP fluorescence. Then, bleedthrough from the RFP channel was calculated using an RFP-only control strain and subtracted from signal in the GFP channel. The resultant bleedthrough-corrected GFP:RFP values were normalized to the corresponding untreated hul5Δ strain for each background. To quantify the change in GFP:RFP upon HUL5 deletion, the normalized value from the HUL5 condition was subtracted from that of the hul5Δ condition.
For the stalling reporter, the analysis workflow matched that of RQCsubLONG with the order of the fluorophores being reversed, i.e. plasmid-expressing yeast selected based on GFP, bleedthrough from GFP subtracted from RFP. Where indicated, corrected RFP:GFP values were normalized to a non-stalling reporter that contained a non-stalling sequence encoding serine and threonine (Sitron et al., 2017) in place of the stalling CGN codons.
Statistical Analysis
A paired t-test (using the “ttest” function in MATLAB) was used to assess whether mean measurements differed between two different conditions, using the null hypothesis: µcondition 1 = µcondition 2. To calculate the s.e.m. for Δ stability measurements (Figs. 2C, 3D, 4G), a propagation of error formula was used: SEMhul5Δ - HUL5 = sqrt(SEMHUL52 + SEMhul5Δ2). To analyze the significance of Δ stability measurements, a t-test for particular contrast was performed using the “lm” and “linearHypothesis” functions in R (R Foundation for Statistical Computing). The null hypothesis for this test was: µhul5Δ+ condition 1 – µHUL5 + condition 1 = µhul5Δ+ condition 2 – µHUL5 + condition 2.
Immunoblots
For whole-cell immunoblots, 0.375/OD600 x mL of yeast culture (OD600 = 0.4-0.8) grown overnight were pelleted and resuspended then boiled for 5 min at 95°C in 15 µL 4x NuPage LDS Sample Buffer (Thermo Fisher Scientific) with 5% β-mercaptoethanol.
Samples were loaded into gels and transferred to nitrocellulose using one of two systems. First, samples were loaded into a NuPAGE Novex 4-12% Bis-Tris 1.5 mm protein gel (Thermo Fisher Scientific) and run in MOPS buffer. Gels were semi-dry transferred to 0.45 µm nitrocellulose membranes (Thermo Fisher Scientific) using the Trans-Blot Turbo system (Bio-Rad, Hercules, CA). Second, samples were loaded into hand-poured 10% Tris-Glycine protein gels with a 4% stacking gel and run in Tris-Glycine buffer. Gels were wet transferred to .45 µm nitrocellulose membranes (Thermo Fisher Scientific) in the Trans-Blot Cell (Bio-Rad) in Towbin Buffer.
Membranes were blocked in TBST with 5% fat-free milk (Safeway, Pleasanton, CA) for 1 hour, incubated overnight at 4°C or at room temperature for 4 hrs in one of the following primary antibodies: 1:3000 mouse α-GFP (MA5-15256, Thermo Fisher Scientific), 1:3000 rabbit α-hexokinase (H2035-01, US Biological, Salem, MA), or 1:1000 rabbit α-RFP (AB233, Evrogen, Moscow, Russia). The following secondary antibodies were then used at 1:5000 dilution: IRDye 800CW donkey anti-mouse, IRDye 800CW goat anti-rabbit, IRDye 680RD goat anti-rabbit, or IRDye 680RD goat anti-mouse (LiCor Biosciences, Lincoln, NE). Blots were scanned on a Licor Odyssey (LiCor Biosciences).
Lysate Preparation and Immunoprecipitation (IP)
Yeast were grown in SD media and harvested at OD600 0.8 to 1.0. Cells were harvested using either 1) centrifugation followed by resuspension in IP buffer (100 mM KOAc, 10 mM MgCl2, 25 mM HEPES-KOH pH 7.4) and freezing of cell droplets in liquid nitrogen; or 2) vacuum filtration and flash freezing in liquid nitrogen. Frozen yeast were cryogenically pulverized into powder using a Freezer/Mill (SPEX SamplePrep, Metuchen, NJ) and stored at −80°C.
Frozen yeast powder was thawed and solubilized at 4°C in IP buffer supplemented with Pierce Protease Inhibitor Tablets, EDTA-free (Thermo Fisher Scientific). Crude lysate was clarified by a single spin at 5000 x g, incubated with GFP-Trap_A (ChromoTek, Planegg-Martinsried, Germany) resin for 1 hour at 4°C with rotation, then washed 10 times in IP buffer. For immunoblotting, washed GFP-Trap resin was boiled in 4X NuPAGE LDS Sample Buffer (Thermo Fisher Scientific) with 5% β-mercaptoethanol and analyzed as described above in “Immunoblots.”
Amino Acid Analysis
To isolate RQCsub for amino acid analysis, a GFP IP was performed as described above, except elution was performed with IP buffer adjusted to pH 2.0 to acid-elute proteins. Amino acid analysis was performed by UC Davis Proteomics Core using the following protocol, adapted from (Nakazawa and Manabe, 1992): samples were dried in a glass hydrolysis tube. The samples were then hydrolyzed in 6N HCl, 1% phenol at 110°C for 24 hrs under vacuum. After hydrolysis, the samples were cooled, unsealed, and dried again. The dried samples were dissolved in 1mL sodium diluent (Pickering, Mountain View, CA) with 40 nmol/mL NorLeucine added as an internal standard. For each run, 50 µL of sample was injected onto the ion-exchange column. An amino acid standards solution for protein hydrolysate on the Na-based Hitachi 8800 (Sigma,A-9906) is used to determine response factors, and thus calibrate the Hitachi 8800 analyzer for all of the amino acids. In addition this standard has been verified against the National Institute of Standards and Technology (NIST) standard reference material 2389a.
TEV Protease Digestion
GFP IP was performed as described above, but instead of immediate elution, the rein was equilibrated in IP buffer with 1 mM DTT and incubated overnight at 4°C with 5 µL ProTEV Plus (Promega, Sunnyvale, CA) in 100 µL total reaction volume. The resin was then washed with fresh IP buffer with 1 mM DTT and boiled in 4X NuPAGE LDS Sample Buffer (Thermo Fisher Scientific) with 5% β-mercaptoethanol and analyzed as described above.
Lysate mixing
Yeast lysate was prepared as described above. Crude lysate from ROLD bud27Δ cells expressing RQCsub was mixed with crude lysate from ROLD or wt cells (lacking RQCsub) and then incubated for 2 hours at 30°C with agitation. For RNAse treatment, ROLD lysate was treated with 100 µg/mL RNAseA (Thermo Fisher Scientific) for 20 minutes at room temperature prior to lysate mixing. To remove insoluble elements, ROLD lysate was centrifuged at 95,000 x g for 20 minutes and only the upper 50% of the supernatant was used for lysate mixing. After lysate mixing, GFP IP and immunoblot analysis was performed as described above.
Fluorescence Microscopy
Live cell fluorescence imaging was performed using two set-ups: 1) a Nikon Eclipse Ti-E inverted fluorescence microscope (Nikon, Melville, NY) using a 100X/1.4NA oil objective lens and µManager (Edelstein et al., 2010), and 2) a Nikon Eclipse 80i microscope using a 100X/1.4NA oil objective lens and Metamorph (Molecular Devices, San Jose, CA). Yeast were grown in SD media to log phase, centrifuged briefly, resuspended in 5 µL fresh SD media and mounted on a clean coverslip and slide. Images were prepared and analyzed with ImageJ (NIH). For quantification, images were randomly acquired until N > 300 cells. Independent cultures were imaged on different days and error represents standard deviation of 3 independent cultures.
Yeast spot assay
Yeast were grown to log-phase in YPD, then diluted to OD600 = 0.1. 200 µL of this diluted culture was diluted 1:10 into YPD four times in a sterile 96-well plate (Greiner Bio-one, Kremsmünster, Austria). 5 µL of the serial dilutions were transferred from the 96-well plate onto YPD agar plates with or without additonal 5 mM guanidinium hydrochloride (Sigma-Aldrich) and/or 50 ng/mL cycloheximide (Sigma-Aldrich). Plates were then incubated at 30°C or 37°C. Plates grown at 30°C with or without 5 mM guanidinium hydrochloride as well as 37°C plates were imaged after two days. Plates containing 50 ng/mL cycloheximide with or without 5 mM guanidinium hydrochloride grown at 30°C were imaged after three days. Plates with 5 mM guanidinium hydrochloride grown at 37°C were imaged after six days.
Author Contributions
O.B., C.S.S., and J.H.P. conceived of the study and designed the experiments. C.S.S., J.H.P., and J.M.G. performed the experiments. O.B., C.S.S., and J.H.P. analyzed and interpreted the data. O.B., C.S.S., and J.H.P. wrote the manuscript. O.B. supervised the study.
Competing Interests
The authors declare no competing interests.
Acknowledgments
We thank D. Pincus, D. Jarosz, R. Kopito, B. Lu, Z. Wu, and the members of the Brandman lab for helpful suggestions. We thank the Theriot and Straight labs (Stanford University) for the use of their microscopes. J. Schulze (University of California at Davis, Davis, CA, USA) performed the amino acid analysis at the UC Davis Genome Center Molecular Structure Facility. We thank J. Weissman and D. Wong for facilitating the screen for suppressors of RQC-mediated Hsf1 activation. This work was supported by Stanford University (O.B.), the US National Institutes of Health (grant No. R01GM115968 to O.B.), and the National Institute of General Medical Sciences of the US National Institutes of Health (grant No. T32GM007276 to C.S.S.).
References




