

Abstract
The insomniac (inc) gene is required for normal sleep in Drosophila and encodes a conserved BTB protein that is a putative adaptor for the Cullin-3 (Cul3) ubiquitin ligase. Here we test whether Inc serves as a Cul3 adaptor by generating mutant forms of Inc and assessing their biochemical properties and physiological activity in vivo. We show that the N-terminal BTB domain of Inc is necessary and sufficient for Inc self-association and interactions with Cul3. Inc point mutations that weaken interactions with Cul3 impair the ability of Inc to rescue the sleep deficits of inc mutants, indicating that Cul3-Inc binding is critical for Inc function in vivo. Deletions of the conserved Inc C-terminus preserve Inc-Inc and Inc-Cul3 interactions but abolish Inc activity in vivo, implicating the Inc C-terminus as an effector domain that recruits Inc substrates. Mutation of a conserved C-terminal arginine similarly abolishes Inc function, suggesting that this residue is vital for the recruitment or ubiquitination of Inc targets. Mutation of the same residue in the human Inc ortholog KCTD17 is associated with myoclonic dystonia, indicating its functional importance in Inc family members. Finally, we show that Inc assembles into multimeric Cul3-Inc complexes in vivo and that depleting Cul3 causes accumulation of Inc, suggesting that Inc is negatively regulated by Cul3-dependent autocatalytic ubiquitination, a hallmark of Cullin adaptors. Our findings implicate Inc as a Cul3 adaptor and provide tools to identify the targets of Inc family proteins that impact sleep and neurological disorders.
Introduction
Sleep is a conserved animal behavior regulated by genetic and molecular mechanisms that remain elusive. Elucidating these mechanisms is a longstanding goal in biology, given that the purpose of sleep is still not well understood and because alterations in these mechanisms may cause sleep disturbances, including those associated with neurodegenerative and neurodevelopmental disorders. Various genes that strongly influence the duration and characteristics of sleep have been identified by unbiased genetic screens in flies (Afonso et al., 2015; Cirelli et al., 2005; Koh et al., 2008; Pfeiffenberger and Allada, 2012; Rogulja and Young, 2012; Stavropoulos and Young, 2011; Toda et al., 2019), zebrafish (Chiu et al., 2016; Singh et al., 2017), worms (Iannacone et al., 2017), and mice (Funato et al., 2016). While many of these genes function in pathways governing neuronal excitability or neurotransmission (Chiu et al., 2016; Cirelli et al., 2005; Koh et al., 2008; Singh et al., 2017; M. Wu et al., 2014; M. N. Wu et al., 2010), others influence sleep by mechanisms that remain poorly understood.
insomniac (inc) mutations strongly curtail the duration and consolidation of sleep but do not alter its circadian regulation (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). inc activity is required in neurons for normal sleep, and conversely, restoring inc solely to neurons is largely sufficient to rescue the sleep deficits of inc mutants (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). While these findings indicate that inc influences sleep primarily through neurons, the molecular mechanisms by which inc functions remain poorly defined. inc encodes a conserved protein of the Bric-a-brac, Tramtrack, Broad Complex (BTB) domain superfamily (Stavropoulos and Young, 2011), which comprises more than 80 proteins in Drosophila (Stogios et al., 2005). The BTB domain mediates protein-protein interactions including homomeric self-associations and binding to heterologous proteins (Stogios et al., 2005). BTB-domain containing proteins cluster into distinct subfamilies based on sequence variation within the BTB domain and the presence of additional domains that underlie diverse cellular functions (Stogios et al., 2005). BTB proteins include transcriptional regulators (DiBello et al., 1991; Godt et al., 1993; Harrison and Travers, 1990), ion channels (Butler et al., 1989; Papazian et al., 1987; Pongs et al., 1988; Wei et al., 1990), auxiliary subunits of GABAB receptors (Schwenk et al., 2010), and adaptors for the Cullin-3 (Cul3) ubiquitin ligase (Furukawa et al., 2003; Pintard et al., 2003; Xu et al., 2003).
Indirect evidence suggests that Inc may serve as a substrate adaptor for the Cul3 ubiquitin ligase complex (Li et al., 2017; Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). BTB proteins that function as Cul3 adaptors form homomultimers that bind Cul3 and recruit substrates to Cul3 complexes for ubiquitination (Furukawa et al., 2003; Geyer et al., 2003; Pintard et al., 2003; Xu et al., 2003). In cultured cells, Inc is able to bind Cul3 (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011) and to self-associate (Li et al., 2017; Pfeiffenberger and Allada, 2012), but whether these molecular interactions occur in vivo or are necessary for the physiological activity of Inc is unknown. Neuronal depletion of either inc or Cul3 causes short sleep (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011), consistent with the notion that Cul3 and inc influence sleep through a common pathway, yet the modular nature of Cul3 complexes leaves unclear whether Cul3 impacts sleep through Inc. Cul3 assembles with tens of different BTB adaptors to ubiquitinate hundreds of substrates (Emanuele et al., 2011), and may impinge upon sleep through multiple adaptor and substrate pathways. In the absence of functional evidence linking Inc and Cul3 in a concerted mechanism that influences sleep, the role of Inc as Cul3 adaptor remains speculative.
Here, we have generated mutant forms of Inc to test the hypothesis that Inc impacts sleep by functioning as a Cul3 adaptor. Analysis of these Inc mutants in cultured cells and in vivo reveals that Inc has the biochemical properties and molecular interactions expected of a Cul3 adaptor. The N-terminal BTB domain of Inc mediates interactions with Cul3 and homomeric self-associations. Weakening Cul3-Inc associations impairs Inc activity in vivo, indicating that Inc function requires the assembly of Cul3-Inc complexes. Deletion of the conserved C-terminal domain of Inc abolishes Inc function in vivo, as does a missense mutation of a conserved, disease-associated arginine residue in the C-terminal domain. These findings identify the Inc C-terminus as a putative substrate binding domain and define a region within the C-terminus likely to bind substrates. Mutation of the same residue in KCTD17, a human ortholog of Inc, is associated with myoclonic dystonia (Mencacci et al., 2015), indicating its functional importance across Inc family proteins. Our findings reveal that Inc influences sleep by functioning as an adaptor for the Cul3 ubiquitin ligase complex. More broadly, our studies provide structural and functional insights into Inc family proteins and tools to identify their substrates that impact neuronal function and behavior.
Results
The Inc BTB domain mediates Cul3 binding and Inc homomultimerization
Inc and its orthologs have two conserved domains, an N-terminal BTB domain and a C-terminal domain unique to Inc family members (Figures 1A and S1A) (Dementieva et al., 2009; Stavropoulos and Young, 2011). The BTB domains of Cul3 adaptors mediate Cul3 binding and adaptor multimerization, while distal domains recruit substrates (Furukawa et al., 2003; Geyer et al., 2003; Pintard et al., 2003; Xu et al., 2003). To test whether Inc domains have the biochemical properties expected of a Cul3 adaptor, we generated a deletion series in which Inc is truncated progressively from its N- or C-terminus (Figure 1A). We first assessed the stability of these Inc derivatives and their associations with Cul3 by expressing epitope-tagged forms of these proteins in S2 cells and performing co-immunoprecipitations. Deleting residues preceding the BTB domain (Inc22-211) did not significantly alter interactions with Cul3 or the stability of Inc (Figure 1B). In contrast, removing part of the BTB domain (Inc31-211) or deleting the BTB domain entirely (Inc124-211) abolished Cul3 binding and resulted in a destabilized Inc protein (Figure 1B). In contrast, C-terminal truncations of Inc did not alter Cul3 binding or Inc stability. Inc proteins lacking 25 C-terminal residues (Inc1-186), most of the C-terminal domain (Inc1-156), or all residues following the BTB domain (Inc1-123) associated with Cul3 similarly to full-length Inc (Figure 1B), indicating that the Inc C-terminus is dispensable for Inc-Cul3 interactions. The isolated Inc BTB domain (Inc22-123) was sufficient for Inc-Cul3 interactions, although these associations were weaker than those of Inc1-123 (Figure 1B), suggesting that Inc N-terminal residues contribute to Cul3 binding. Alternatively, weaker binding of Inc22-123 to Cul3 might reflect an inhibitory effect of fusing the 3×FLAG tag to the isolated BTB domain. Together, these results indicate that the Inc BTB domain is necessary and sufficient for Cul3 binding, a key property expected of a Cul3 adaptor.

A) Schematic of N- and C-terminally truncated Inc proteins. CTD, C-terminal domain. B and C) Co-immunoprecipitation of 3×Myc-tagged Inc or Inc truncation proteins with 3×FLAG-Cul3 (B) or Inc-HA (C) from transiently transfected S2 cells. LC, immunoglobulin light chain.
Next, we assessed the ability of truncated Inc proteins to associate with full length Inc. Partial deletion of the Inc BTB domain (Inc31-211) or its complete removal (Inc124-211) abolished Inc-Inc interactions (Figure 1C). The reduction in Inc stability caused by truncating the BTB domain (Figures 1B and 1C) suggests that Inc is an obligate homomultimer and that Inc monomers are intrinsically unstable. In contrast, C-terminal truncations of Inc did not detectably alter Inc-Inc associations (Figure 1C), indicating that the Inc C-terminus is dispensable for Inc multimerization. The Inc BTB domain (Inc22-123) was sufficient to bind Inc (Figure 1C), albeit more weakly than Inc1-123, suggesting that N-terminal Inc residues contribute to Inc-Inc interactions or that interactions of the isolated BTB domain are occluded by the 3×FLAG tag. We conclude that the Inc BTB domain mediates Inc self-association and Cul3 binding and that the Inc C-terminus is dispensable for these interactions.
Identification of Inc point mutants that selectively weaken Inc-Cul3 and Inc-Inc interactions
We next sought to identify Inc point mutants that selectively perturb Inc-Cul3 and Inc-Inc interactions, in order to test the necessity of these interactions for Inc activity in vivo. Our efforts to mutagenize Inc were informed by the crystal structure of human KCTD5, an Inc ortholog that forms homopentamers (Dementieva et al., 2009), and by the structures of Cul3 complexed with BTB-MATH and BTB-BACK-Kelch adaptors (Canning et al., 2013; Errington et al., 2012; Ji and Privé, 2013), in which adaptors form homodimers and assemble with Cul3 in a 2:2 stoichiometry (Canning et al., 2013; Errington et al., 2012; Ji and Privé, 2013). While Inc and its orthologs are divergent from BTB-MATH and BTB-BACK-Kelch adaptors and likely bind Cul3 with a different mechanism and stoichiometry (Balasco et al., 2014; Ji et al., 2015), comparison of these structures suggested a region of Inc that may bind Cul3. We selected eight residues in the Inc BTB domain that are conserved between Inc and its three human orthologs and that reside on the solvent-accessible surface of KCTD5 (Figures S1A and S1B), reasoning that these residues may contribute to Cul3 binding. We mutated these residues individually to alanine (F47A, D57A, D61A, F105A, Y106A, N107A) or to oppositely charged residues (R50E, E104K), and assessed the consequences for Inc-Inc and Inc-Cul3 interactions in cultured S2 cells. Most mutants did not detectably alter Inc self-association or Cul3 binding (Table 1). Two mutants, F47A and F105A, weakened interactions with Cul3 but did not significantly affect Inc self-association or stability (Figures 2A and 2B). A double mutant combining F47A and F105A behaved similarly and selectively impaired Cul3 binding (Figures 2A and 2B). These phenylalanine residues cluster in a hydrophobic patch on the surface of KCTD5 that may be buried upon Cul3 binding (Figure S1B). This patch is distinct from the interface of adjacent KCTD5 subunits, consistent with the lack of a measurable impact of mutating F47 and F105 on Inc-Inc interactions (Figure 2B).

(A) Alignment of Inc and its human orthologs. Identical and similar residues are shaded in black and gray, respectively. Locations of Inc truncations are indicated by arrowheads. Inc point mutants are indicated by closed circles. (B) Crystal structure of the BTB domains of adjacent subunits of human KCTD5. Residues that do not impair Inc-Cul3 association when mutated are highlighted in cyan. Mutations that weaken Inc-Cul3 associations (F47A and F105A) are highlighted in blue. (C) Crystal structure of the BTB domains of adjacent subunits of human KCTD5. Residues mutated to impair Inc-Inc association are indicated. The T36A/D71A/R85E triple mutation weakens Inc homomultimerization and decreases Inc stability. (D) Crystal structure of human KCTD5. Inc C-terminal point mutants are indicated.

A-D) Co-immunoprecipitation analysis of 3×HA-tagged Inc or Inc point mutants and 3×FLAG-Cul3 (A and C) or 3×FLAG-Inc (B and D) from transiently transfected S2 cells.
Summary of co-immunoprecipitation analysis of Inc point mutants designed to weaken Inc-Cul3 interaction from transfected S2 cells
To identify Inc mutants that impair Inc-Inc homomultimerization, we mutated seven conserved residues that form a network of polar and charged interactions between adjacent KCTD5 subunits (Dementieva et al., 2009) (Figures S1A and S1C). We mutated these residues to alanine (T36A, D71A, D73A, N82A) or to oppositely charged residues (R85E, K88D, E101K) in single and double mutant combinations. None of these single or double mutants significantly altered Inc self-associations in S2 cells (Table 2). We reasoned that Inc may form homopentamers, like KCTD5, and that cooperative interactions between Inc subunits might limit the effects of these mutations. We therefore generated a triple point mutant predicted to disrupt interactions of Inc subunits with both flanking neighbors in a putative Inc pentamer (T36A, D71A, R85E) (Figure S1C). This triple mutant exhibited significantly weakened interactions with both Inc and Cul3 and a markedly reduced stability (Figures 2C and 2D), resembling the consequences of deleting the Inc BTB domain (Figures 1B and 1C). These findings support the conclusion that multimerization-deficient Inc proteins are unstable and suggest that Inc multimerization is required for efficient Cul3 binding. These results furthermore suggest that Cul3 binds cooperatively to adjacent Inc subunits and that Cul3-Inc complexes have an architecture distinct from Cul3-adaptor complexes formed by BTB-MATH and BTB-BACK-Kelch proteins (Canning et al., 2013; Errington et al., 2012; Ji and Privé, 2013).
Summary of co-immunoprecipitation analysis of Inc point mutants designed to weaken Inc-Inc interaction from transfected S2 cells
The assembly of Inc-Cul3 complexes in vivo is required for the function of Inc
Inc mutations that selectively impair Cul3 binding represent key tools to assess whether Inc impacts sleep by functioning as a Cul3 adaptor. To assess the physiological activity of Inc mutants in vivo, we generated UAS transgenes expressing 3×FLAG-Inc or -Inc point mutants that weaken Cul3 interactions: IncF47A, IncF105A, and IncF47A/F105A. We integrated these transgenes at the same genomic site (attP2) and backcrossed them to generate an isogenic allelic series, enabling careful comparisons of their activity. We first assessed the expression of these proteins using inc-Gal4, a driver that fully rescues the sleep deficits of inc mutants when used to restore inc expression (Li et al., 2017; Stavropoulos and Young, 2011) and which therefore recapitulates inc expression in cells relevant for sleep. Inc and Inc point mutants were expressed at similar levels under inc-Gal4 control in inc mutants (Figure 3A) or inc+ animals (Figure S2A), indicating that these point mutants do not alter Inc stability in vivo, recapitulating findings from cultured cells (Figures 2A and 2B). Next, we assessed the impact of expressing Inc point mutants on sleep-wake behavior. Because adaptor proteins deficient for Cul3 interactions might sequester substrates and thus function as dominant negatives, we first tested whether the sleep of inc+ animals was altered by the expression of Inc point mutants. Animals expressing Inc point mutants under the pan-neuronal elavc155-Gal4 control slept indistinguishably from those expressing wild-type Inc and from control animals lacking UAS transgenes (Figure S2B). Thus, expression of Inc point mutants in neurons does not elicit dominant negative effects or antagonize endogenous Inc function.

(A) Biochemical analysis of inc-Gal4 animals expressing 3×FLAG-tagged Inc or Inc point mutants. (B) Behavioral analysis of elavc155-Gal4 animals expressing 3×FLAG-tagged Inc or Inc point mutants. Mean ± SEM is shown. n = 5-24; ns, not significant (p>0.05) compared to elavc155-Gal4 control. For all panels, animals are heterozygous for UAS transgenes.
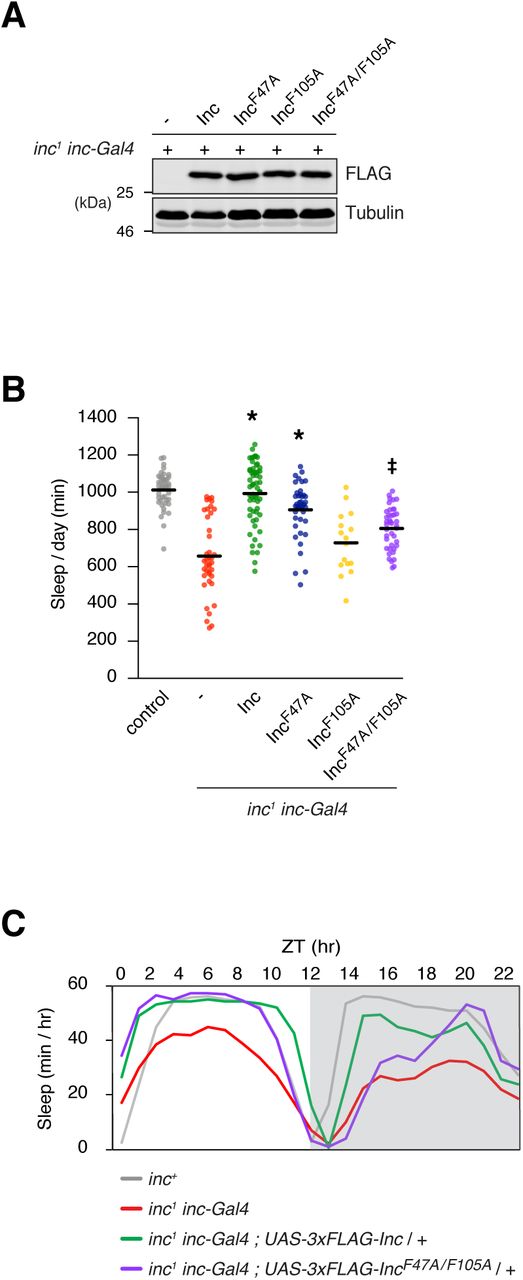
Biochemical and behavioral analysis of inc1 inc-Gal4 animals expressing 3×FLAG-tagged Inc or Inc point mutants that weaken Cul3 associations. A) Immunoblot analysis of whole animal lysates. B) Total sleep duration. Mean ± SEM is shown. n = 16-58; *p < 0.01 for comparison to inc1 inc-Gal4 animals and not significantly different from wild-type controls; ‡p < 0.01 for comparisons to inc1 inc-Gal4 animals and wild-type controls. C) Population average sleep traces summed hourly for indicated genotypes. n = 41-58. For all panels, animals are heterozygous for UAS transgenes.
We next tested whether Inc-Cul3 interactions are required for Inc function in vivo. If so, attenuating these interactions should impair the ability of Inc to rescue the sleep phenotypes of inc mutants. While Inc expressed with inc-Gal4 fully rescued the short sleep of inc1 null mutants, Inc mutants that weaken Inc-Cul3 interactions provided only a partial rescue (Figures 3B and 3C), supporting the interpretation that Inc activity in vivo requires binding to the Cul3 ubiquitin ligase complex. Rescue was more strongly impaired for IncF105A and IncF47A/F105A than IncF47A (Figures 3B and S3A-D). Because these Inc mutants are expressed at similar levels in vivo (Figure 3A), we speculate that the F47A mutation might impair Cul3 binding in vivo to a lesser degree than in S2 cells. Collectively, the impaired rescuing activity of this trio of Inc mutants suggests that Inc activity requires the assembly of Cul3-Inc complexes in vivo and that disruption of these complexes inhibits sleep.

(A-D) Sleep parameters for inc1 inc-Gal4 animals expressing 3×FLAG-tagged Inc or Inc point mutants. Mean ± SEM is shown. n = 16-58 as in Fig 3B; *p < 0.01 for comparison to inc1 inc-Gal4 animals and not significantly different from wild-type controls; ‡p < 0.01 for comparisons to inc1 inc-Gal4 animals and wild-type controls. A) Nighttime sleep. B) Daytime sleep. C) Sleep bout duration. D) Sleep bout number. For all panels, animals are heterozygous for UAS transgenes.
The conserved C-terminal domain of Inc is essential for Inc activity in vivo
While the Inc C-terminus is dispensable for Inc-Inc and Inc-Cul3 associations (Figures 1B and 1C), its evolutionary conservation suggests an essential function. If Inc serves as a Cul3 adaptor, its C-terminus is predicted to bind substrates and recruit them to the Cul3 complex for ubiquitination. To test whether the C-terminus is required for physiological function of Inc in vivo, we generated UAS transgenes expressing 3×FLAG-tagged Inc C-terminal truncations: Inc1-186, Inc1-156, and Inc1-123. These proteins were expressed at similar levels under inc-Gal4 control in inc mutants (Figure 4A), indicating that these proteins are stable in vivo, as in cultured cells (Figures 1B and 1C). Neuronal expression of Inc C-terminal truncations using elavc155-Gal4 did not significantly alter sleep, indicating that these proteins do not elicit dominant negative effects or antagonize endogenous inc function (Figure S4).

Behavioral analysis of elavc155-Gal4 animals expressing 3×FLAG-tagged Inc or C-terminally truncated Inc proteins. Mean ± SEM is shown. n = 23-54; ns, not significant (p>0.05) compared to elavc155-Gal4 control. For all panels, animals are heterozygous for UAS transgenes.

Biochemical and behavioral analysis of inc1 inc-Gal4 animals expressing 3×FLAG-tagged Inc or C-terminally truncated Inc proteins. A) Immunoblot analysis of whole animal lysates. B) Total sleep duration. Mean ± SEM is shown. n = 27-30; *p < 0.01 for comparison to inc1 inc-Gal4 animals and not significantly different from wild-type controls. C) Population average sleep traces summed hourly for indicated genotypes. n = 27-30. For all panels, animals are heterozygous for UAS transgenes.
To determine whether the Inc C-terminus is essential for Inc function, we tested whether C-terminally truncated Inc proteins rescue the sleep deficits of inc mutants. Wild-type Inc expressed under inc-Gal4 control fully rescued the sleep deficits of inc1 mutants (Figures 4B and 4C). In contrast, animals expressing Inc1-156 or Inc1-123 slept indistinguishably from inc mutants, indicating that these proteins failed to rescue the inc phenotype (Figures 4B and 4C). Expression of Inc1-186 partially rescued the sleep phenotypes of inc mutants as assessed by several sleep parameters, including total sleep duration (Figure 4B), nighttime and daytime sleep (Figures S5A and S5B), and sleep bout duration and number (Figures S5C and S5D). These findings indicate that the Inc C-terminus is essential for Inc activity in vivo and that removing the terminal 25 residues of Inc curtails Inc function. The stability of C-terminally truncated Inc proteins and their ability to engage normally in Inc-Cul3 and Inc-Inc interactions (Figures 1B and 1C), supports the hypothesis that the Inc C-terminus recruits Inc binding partners including substrates whose ubiquitination influences sleep.

(A-D) Sleep parameters for inc1 inc-Gal4 animals expressing 3×FLAG-tagged Inc or C-terminally truncated Inc proteins. Mean ± SEM is shown. n = 27-30 as in Fig 4B; *p < 0.01 for comparison to inc1 inc-Gal4 animals and not significantly different from wild-type controls; ‡p < 0.01 for comparisons to inc1 inc-Gal4 animals and wild-type controls. A) Nighttime sleep. B) Daytime sleep. C) Sleep bout duration. D) Sleep bout number. For all panels, animals are heterozygous for UAS transgenes.
Mutation of a conserved disease-associated arginine in the Inc C-terminus abolishes Inc function and defines a region of Inc likely to recruit substrates
To further dissect the function of the Inc C-terminus, we mutated several conserved residues whose attributes suggested they might contribute to Inc function. A missense mutation of a conserved C-terminal arginine in the human Inc ortholog KCTD17 (R145H) is associated with a dominant form of myoclonic dystonia, a neurological movement disorder (Mencacci et al., 2015). We reasoned that this arginine residue might be critical for the function of Inc and its orthologs. In particular, the analogous residue in KCTD5 (R159) resides on a lateral solvent-accessible surface of the C-terminal domain that might bind and orient substrates for Cul3-dependent ubiquitination (Figure S1D). We therefore generated the analogous mutation in Inc (IncR135H) to assess whether it impacts Inc function. We also mutated three conserved Inc residues (N167, Y168, G169) to alanine (IncAAA), reasoning that their location within a surface-exposed loop on the bottom surface of the KCTD5 C-terminus and their conservation amid flanking non-conserved residues (Figures S1A and S1D) might reflect an important function. In cultured S2 cells, both IncR135H and IncAAA were stably expressed and associated with Cul3 and Inc indistinguishably from wild-type Inc, indicating that these mutations do not alter Inc multimerization or Cul3 binding (Figure 5).
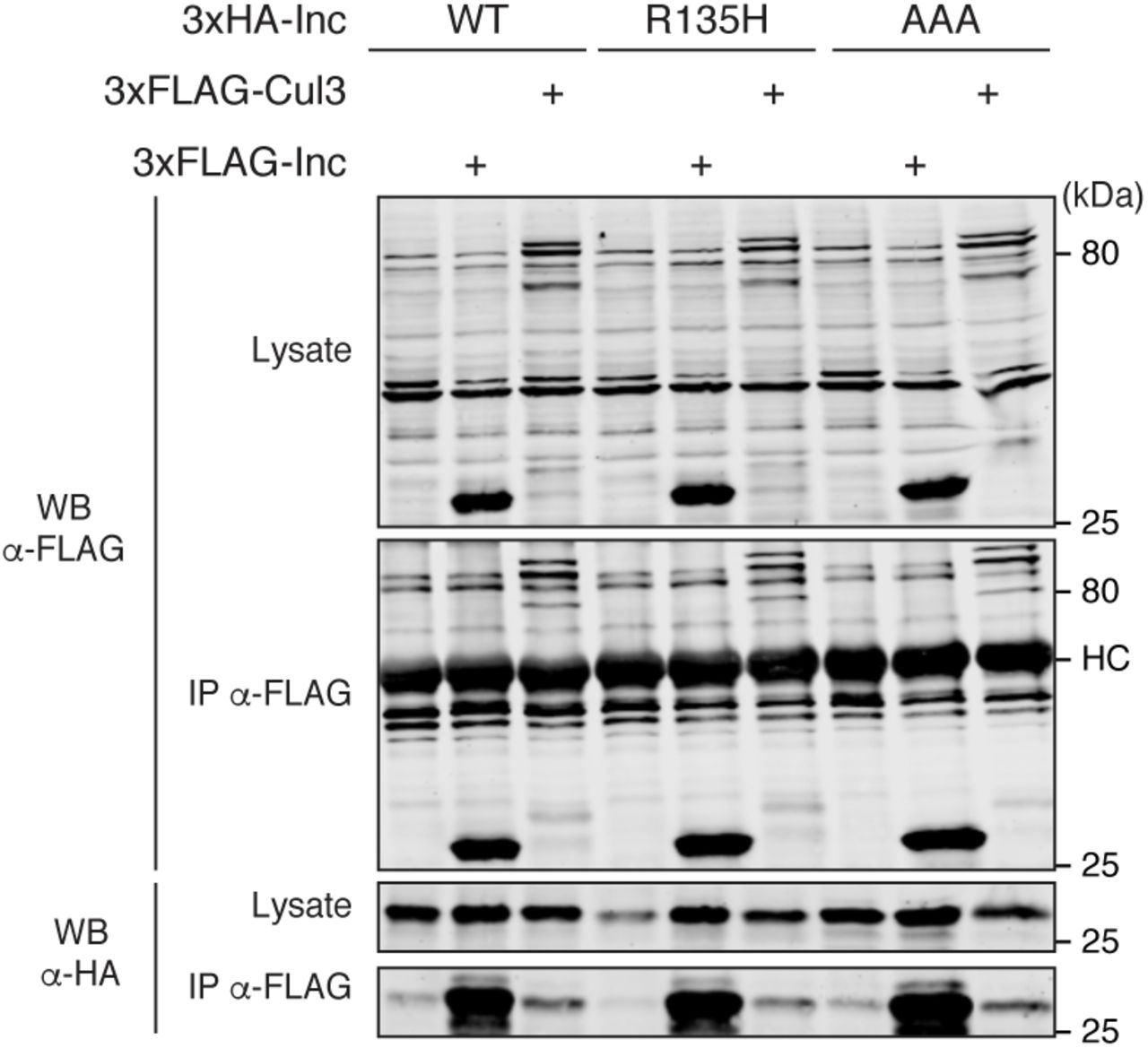
Co-immunoprecipitation analysis of 3×HA-tagged Inc or Inc point mutants and 3×FLAG-Cul3 or 3×FLAG-Inc from transiently transfected S2 cells.
To determine whether IncR135H and IncAAA alter the physiological activity of Inc, we generated UAS transgenes to express these mutants in vivo. As in cultured cells, IncR135H and IncAAA were stably expressed in vivo (Figure 6A). Expression of IncR135H and IncAAA with elavc155-Gal4 did not alter sleep in inc+ animals, indicating that these proteins do not have dominant negative activity or inhibit endogenous Inc function (Figure S6). Next, we tested whether IncR135H and IncAAA were able to rescue the sleep deficits of inc mutants. IncR135H was unable to restore sleep to inc1 mutants, indicating that the R135H mutation abolishes Inc function (Figures 6B-C and S7A-D). In contrast, IncAAA completely rescued inc1 mutants and behaved indistinguishably from wild-type Inc (Figures 6B-C and S7A-D). These data indicate that R135 is critical for Inc function and suggest that mutation of this residue impairs the recruitment of Inc binding partners including substrates. The analogous mutation in KCTD17 may similarly alter its ability to engage targets, suggesting that deficient substrate ubiquitination is a cause of KCTD17-associated myoclonic dystonia.

Behavioral analysis of elavc155-Gal4 animals expressing 3×Myc-tagged Inc or Inc point mutants. Mean ± SEM is shown. n = 18-65; ns, not significant (p>0.05) compared to elavc155-Gal4 control. For all panels, animals are heterozygous for UAS transgenes.

(A-D) Sleep parameters for inc1 inc-Gal4 animals expressing 3×Myc-tagged Inc or Inc point mutants. Mean ± SEM is shown. n = 24-46 as in Fig 4B; *p < 0.01 for comparison to inc1 inc-Gal4 animals and not significantly different from wild-type controls; ‡p < 0.01 for comparisons to inc1 inc-Gal4 animals and wild-type controls. A) Nighttime sleep. B) Daytime sleep. C) Sleep bout duration. D) Sleep bout number. For all panels, animals are heterozygous for UAS transgenes.
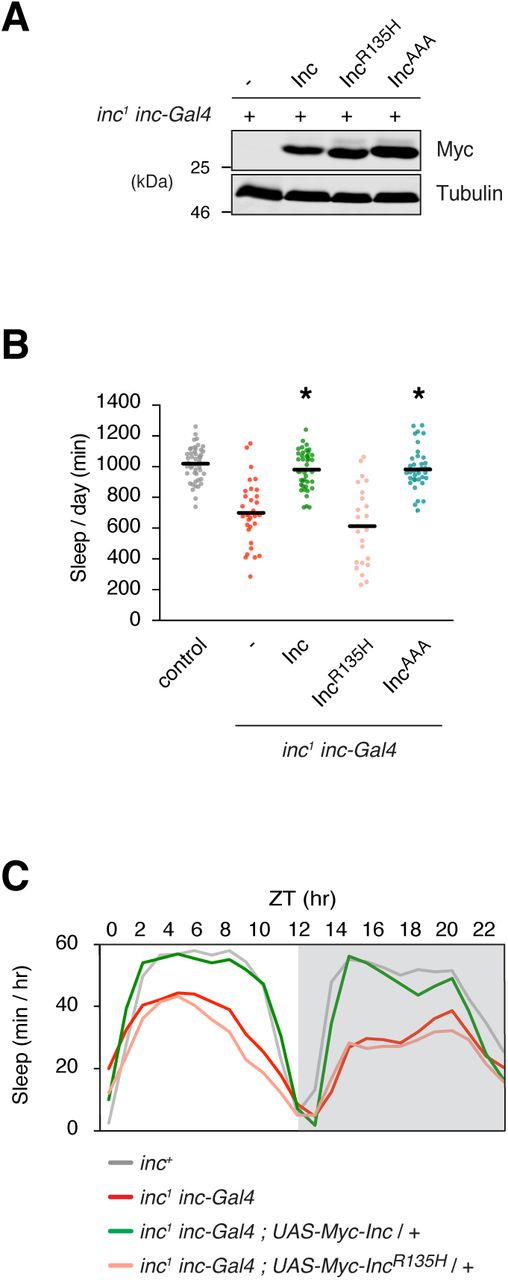
Biochemical and behavioral analysis of inc1 inc-Gal4 animals expressing 3×Myc-tagged Inc or Inc point mutants. A) Immunoblot analysis of whole animal lysates. B) Total sleep duration. Mean ± SEM is shown. n = 24-46; *p < 0.01 for comparison to inc1 inc-Gal4 animals and not significantly different from wild-type controls. C) Population average sleep traces summed hourly for indicated genotypes. n = 24-46. For all panels, animals are heterozygous for UAS transgenes.
Inc exhibits properties of a Cul3 adaptor in neurons in vivo
Neuronal depletion of inc or Cul3 shortens sleep, indicating that the activity of both genes is required in neurons for normal sleep (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). We performed a series of experiments to determine whether Inc exhibits the properties of a Cul3 adaptor in vivo. First, we tested whether Cul3-Inc complexes assemble in vivo, by co-expressing epitope-tagged Inc and Cul3 in neurons using the elavc155-Gal4 driver and performing co-immunoprecipitations. We observed that Cul3 and Inc associate in neurons in vivo (Figure 7A). Second, we tested whether Inc homomultimerizes in vivo, by co-expressing HA-Inc and Myc-Inc in neurons and assessing their physical interactions. We observed strong self-association of Inc in neurons (Figure 7B), indicating that Inc forms homomultimeric complexes in vivo, as in cultured cells (Figure 1C) (Li et al., 2017; Pfeiffenberger and Allada, 2012). Third, we tested whether Inc abundance is regulated by Cul3 in vivo. Cul3 adaptors are often regulated by autocatalytic ubiquitination and degradation in Cul3 complexes (Djagaeva and Doronkin, 2009; Geyer et al., 2003; Hudson and Cooley, 2010; Pintard et al., 2003; Wee et al., 2005; Zhang et al., 2005). To test whether Inc abundance is regulated by Cul3 activity, we expressed neuronal RNAi against Cul3 and assessed Inc levels in head lysates. We observed an increase in Inc protein levels upon depletion of Cul3 (Figure 7C), indicating that Cul3 negatively regulates Inc in adult neurons. qRT-PCR revealed that neuronal Cul3 RNAi did not increase inc transcript levels, indicating that elevated Inc levels arise by a post-transcriptional mechanism (Figure 7D). We conclude that Inc is regulated endogenously in neurons by Cul3-dependent autocatalytic ubiquitination and subsequent degradation. Together, these results indicate that Inc binds Cul3 in vivo and has the characteristics expected of a Cul3 adaptor in neurons, a cell type through which inc influences sleep (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Co-immunoprecipitation analysis of 3×Myc-tagged Inc and 3×FLAG-3×HA-Cul3 (A) or 3×HA-Inc (B) from head lysates prepared from indicated genotypes. C) Western blots of head lysates prepared from indicated genotypes. D) qRT-PCR analysis of inc mRNA levels of indicated genotypes. Mean ± SEM is shown. *p < 0.05; ns, not significant (p>0.05).
Discussion
The neuronal activity of Insomniac is vital for sleep, yet the molecular mechanism underlying its function has remained speculative. Our analysis implicates Inc as a Cul3 adaptor in vivo and provides insight into the mechanism by which Inc binds Cul3 and recruits substrates to ultimately impact behavior. Inc has a modular organization and conserved domains that fulfill the properties expected of a Cul3 adaptor (Furukawa et al., 2003; Geyer et al., 2003; Pintard et al., 2003; Xu et al., 2003). The Inc BTB domain mediates the homomeric assembly of Inc and interactions with Cul3, while the Inc C-terminus is dispensable for these interactions but is vital for Inc function in vivo, consistent with its putative function in recruiting substrates to Cul3-Inc complexes for ubiquitination. Inc assembles into Cul3-Inc complexes in neurons and importantly, reducing the affinity of Inc-Cul3 associations impairs the activity of Inc in vivo. These findings link the biochemical associations of Cul3 and Inc to prior findings that normal sleep-wake cycles require the activity of both proteins in neurons (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). The negative regulation of Inc by Cul3 in neurons is characteristic of autocatalytic degradation exhibited by Cul3 adaptors (Djagaeva and Doronkin, 2009; Geyer et al., 2003; Hudson and Cooley, 2010; Pintard et al., 2003; Wee et al., 2005; Zhang et al., 2005), providing further support for the function of Inc as a Cul3 adaptor in cells through which Inc impacts sleep.
Our data and prior studies (Balasco et al., 2014; Dementieva et al., 2009; Ji et al., 2015) suggest that Inc forms an obligate homopentamer in which neighboring Inc subunits interact cooperatively. Consistent with a pentameric structure for Inc, a triple mutation altering opposite sides of Inc and thus its interactions with both neighboring subunits severely compromised Inc multimerization. Inc multimerization is furthermore required for efficient Cul3 binding, suggesting that Cul3 binds cooperatively to the BTB domains of neighboring Inc subunits. Such a mechanism is consistent with the consequences of mutating conserved phenylalanine residues (F47 and F105) near this interface, which impair Cul3 binding and Inc function in vivo. It is perhaps surprising that Inc mutants that impair Cul3 interactions do not elicit dominant negative phenotypes when overexpressed, although we note that these mutants retain some ability to bind Cul3. Similarly, Inc overexpression might be expected to sequester substrates but does not alter sleep (Li et al., 2017; Stavropoulos and Young, 2011). We speculate that robust Inc homomultimerization may buffer Cul3-Inc complexes against perturbations in Inc abundance and contribute, along with additional mechanisms that regulate Inc activity, to the permissive influence that Inc exerts on sleep.
Our functional analysis of the Inc C-terminus provides insight into the mechanism by which Inc is likely to bind substrates and orient them for ubiquitination in Cul3 complexes. The Inc C-terminus is dispensable for Cul3 binding and Inc multimerization, yet is absolutely essential for Inc function in vivo, implicating it as a substrate recruitment domain. We furthermore identified a conserved, surface-exposed arginine residue (R135) within the Inc C-terminus required for Inc activity in vivo. Mutation of this residue is behaviorally indistinguishable from deleting the Inc C-terminus and does not alter Inc stability, suggesting that it does not disrupt the structure of the C-terminal domain. While our results do not exclude the possibility that this residue is essential for Inc trafficking or other mechanisms that might regulate Inc activity, the simplest possibility suggested by its position on the lateral surface of the Inc C-terminus is that it contributes directly to binding targets that impact sleep. A missense mutation of the same residue in the human Inc ortholog KCTD17 (R145H) is associated with myoclonic dystonia (Mencacci et al., 2015), suggesting a similar functional importance for substrate recruitment in Inc orthologs. The partial functional impairment of Inc caused by removing C-terminal residues (Inc1-186) is consistent with the possibility that they contribute to substrate binding. These residues are not resolved in the KCTD5 crystal structure (Dementieva et al., 2009), suggesting that they are flexible in the absence of substrates and may become structured upon substrate binding. While a parsimonious model is that Inc directly binds substrates, our findings do not rule out more complex models of substrate recruitment that require co-adaptor proteins, analogous to the mechanism recently described for KLHL12, a BTB-Kelch family adaptor (McGourty et al., 2016). Further studies are required to elucidate Inc substrates and distinguish among these models.
Our recent findings revealed that orthologs of Inc can substitute for Inc in flies and restore sleep to inc mutants, suggesting that the functions and targets of Inc and its orthologs are evolutionarily conserved (Li et al., 2017). Our present studies provide evidence for the function of Inc as a Cul3 adaptor in vivo and the foundation for identifying Inc substrates. While substrates for Inc orthologs have been recently described in cultured cells (Brockmann et al., 2017; Kasahara et al., 2014; Kim et al., 2017), whether these targets or other proteins mediate the impact of Cul3 and Inc on sleep remains unknown. Elucidating Inc substrates and the downstream pathways will advance our understanding of how protein ubiquitination pathways contribute to the regulation of nervous system function and mechanisms underlying behavior.
Methods
Plasmids and molecular cloning
Vectors for expression in S2 cells were as follows:
pAc5.1–Inc-HA (pNS277) encodes Inc fused to a C-terminal 1×HA epitope (GSYPYDVPDYA) and was generated by ligating EcoRI-XhoI digested pAc5.1-v5-HisA backbone and an EcoRI-XhoI Inc-HA fragment from pNS273 (as described below).
pAc5.1–3×Myc-Inc (pNS351) encodes an N-terminal 3×Myc epitope (MEQKLISEEDLGSEQKLISEEDLGSEQKLISEEDLAS) fused to Inc as previously described (Stavropoulos and Young, 2011).
pAc5.1–3×Myc-Inc22-211 (pNS370) was generated by ligating NheI-XhoI digested pNS309 (Stavropoulos and Young, 2011) and the NheI-XhoI fragment liberated from the PCR amplification product of pNS351 template and primers oNS285 and oNS315.
pAc5.1–3×Myc-Inc31-211 (pNS371) was generated similarly to pNS370, substituting primers oNS285 and oNS316.
pAc5.1–3×Myc-Inc124-211 (pNS372) was generated similarly to pNS370, substituting primers oNS285 and oNS317.
pAc5.1–3×Myc-Inc1-123 (pNS374) was generated similarly to pNS370, substituting primers oNS277 and oNS319.
pAc5.1–3×Myc-Inc1-156 (pNS375) was generated similarly to pNS370, substituting primers oNS277 and oNS320.
pAc5.1–3×Myc-Inc1-186 (pNS376) was generated similarly to pNS370, substituting primers oNS277 and oNS321.
pAc5.1–3×HA-Inc (pNS402) encodes an N-terminal 3×HA epitope (MYPYDVPDYAGSYPYDVPDYAGSYPYDVPDYAAS) fused to Inc and was generated by ligating NheI-XhoI digested pNS310 (Stavropoulos and Young, 2011) and a NheI-XhoI inc fragment liberated from pNS351.
pAc5.1–3×FLAG-Cul3 (pNS403) encodes an N-terminal 3×FLAG epitope (MDYKDDDDKGSDYKDDDDKGSDYKDDDDKAS) fused to Drosophila Cul3 and was generated by ligating NheI-NotI digested pNS311 and a NheI-NotI Cul3 fragment liberated from pNS314 (Stavropoulos and Young, 2011). pNS311 contains an N-terminal 3×FLAG tag and was generated from pNS298, a derivative of pAc5.1/V5-HisA that contains a C-terminal 3×FLAG tag. To construct pNS298, oligonucleotides oNS234 and oNS235 were phosphorylated, annealed, and cloned into XhoI-XbaI digested pAc5.1/V5-HisA. To construct pNS311, EcoRI-NotI digested pAc5.1/V5-HisA was ligated to the EcoRI-NotI fragment liberated from the PCR amplification product of pNS298 template and primers oNS240 and oNS241.
pAc5.1–3×FLAG-Inc (pNS408) encodes an N-terminal 3×FLAG epitope (MDYKDDDDKGSDYKDDDDKGSDYKDDDDKAS) fused to Inc and was generated by ligating NheI-NotI digested pNS311 and a NheI-XhoI inc fragment liberated from pNS351. pAc5.1–3×HA-IncF47A (pNS409) encodes an N-terminal 3×HA epitope (MYPYDVPDYAGSYPYDVPDYAGSYPYDVPDYAAS) fused to IncF47A and was generated by ligating NheI-XhoI digested pNS310 and an NheI-XhoI IncF47A fragment. The NheI-XhoI IncF47A fragment was liberated from the fusion PCR amplification product of primers oNS683 and oNS684 and an equimolar mix of overlapping 5’ and 3’ IncF47A fragments as template. The 5’ and 3’ fragments were generated by PCR amplification of pNS408 template with primers oNS683/oNS695 and oNS684/oNS694, respectively.
pAc5.1–3×HA-IncR50E (pNS410) was generated similarly to pNS409, substituting primers oNS683/oNS697 and oNS684/oNS696 for generating the 5’ and 3’ IncR50E fragments, respectively.
pAc5.1–3×HA-IncD57A (pNS411) was generated similarly to pNS409, substituting primers oNS683/oNS699 and oNS684/oNS698 for generating the 5’ and 3’ IncD57A fragments, respectively.
pAc5.1–3×HA-IncD61A (pNS412) was generated similarly to pNS409, substituting primers oNS683/oNS701 and oNS684/oNS700 for generating the 5’ and 3’ IncD61A fragments., respectively.
pAc5.1–3×HA-IncE104K (pNS413) was generated similarly to pNS409, substituting primers oNS683/oNS703 and oNS684/oNS702 for generating the 5’ and 3’ IncE104K fragments, respectively.
pAc5.1–3×HA-IncD73A (pNS430) was generated similarly to pNS409, substituting primers oNS683/oNS686 and oNS684/oNS685 for generating the 5’ and 3’ IncD73A fragments, respectively.
pAc5.1–3×HA-IncN82A (pNS431) was generated similarly to pNS409, substituting primers oNS683/oNS686 and oNS684/oNS685 for generating the 5’ and 3’ IncN82A fragments, respectively.
pAc5.1–3×HA-IncK88D (pNS432) was generated similarly to pNS409, substituting primers oNS683/oNS688 and oNS684/oNS687 for generating the 5’ and 3’ IncK88D fragments, respectively.
pAc5.1–3×HA-IncE101K (pNS433) was generated similarly to pNS409, substituting primers oNS683/oNS688 and oNS684/oNS687 for generating the 5’ and 3’ IncE101K fragments, respectively.
pAc5.1–3×HA-IncT36A (pNS434) was generated similarly to pNS409, substituting primers oNS683/oNS693 and oNS684/oNS689 for generating the 5’ and 3’ IncT36A fragments, respectively.
pAc5.1–3×HA-IncD71A (pNS435) was generated similarly to pNS409, substituting primers oNS683/oNS693 and oNS684/oNS689 for generating the 5’ and 3’ IncD71A fragments, respectively.
pAc5.1–3×HA-IncR85E (pNS436) was generated similarly to pNS409, substituting primers oNS683/oNS692 and oNS684/oNS691 for generating the 5’ and 3’ IncR85E fragments, respectively.
pAc5.1–3×HA-IncD73A/N82A (pNS414) was generated similarly to pNS409, substituting pNS430 template and primers oNS683/oNS686 for generating the 5’ IncD73A/N82A fragment; and pNS431 template and primers oNS684/oNS685 for generating the 3’ IncD73A/N82A fragment.
pAc5.1–3×HA-IncK88D/E101K (pNS415) was generated similarly to pNS409, substituting pNS432 template and primers oNS683/oNS688 for generating the 5’ IncK88D/E101K fragment; and pNS433 template and primers oNS684/oNS687 for generating the 3’ IncK88D/E101K fragment.
pAc5.1–3×HA-IncT36A/D71A (pNS416) was generated similarly to pNS409, substituting pNS434 template and primers oNS683/oNS693 for generating the 5’ IncT36A/D71A fragment; and pNS435 template and primers oNS684/oNS689 for generating the 3’ IncT36A/D71A fragment.
pAc5.1–3×HA-IncD71A/R85E (pNS417) was generated similarly to pNS409, substituting pNS435 template and primers oNS683/oNS692 for generating the 5’ IncD71A/R85E fragment; and pNS436 template and primers oNS684/oNS691 for generating the 3’ Inc D71A/R85E fragment.
pAc5.1–3×HA-IncT36A/D71A/R85E (pNS418) was generated by ligating EcoRV-XhoI digested pNS434 backbone and an EcoRV-XhoI 3×HA-IncD71A/R85E fragment from pNS417.
pAc5.1–3×HA-IncF105A (pNS419) was generated similarly to pNS409, substituting primers oNS683/oNS1123 and oNS684/oNS1122 for generating the 5’ and 3’ IncF105A fragments, respectively.
pAc5.1–3×HA-IncY106F (pNS420) was generated similarly to pNS409, substituting primers oNS683/oNS1146 and oNS684/oNS1145 for generating the 5’ and 3’ IncY106F fragments, respectively.
pAc5.1–3×HA-IncN107A (pNS421) was generated similarly to pNS409, substituting primers oNS683/oNS1125 and oNS684/oNS1124 for generating the 5’ and 3’ IncN107A fragments, respectively.
pAc5.1–3×HA-IncF47A/F105A (pNS422) was generated similarly to pNS409, substituting pNS409 template, primers oNS683/oNS1123, and oNS684/oNS1122 for generating the 5’ and 3’ IncF47A/F105A fragments, respectively.
pAc5.1–3×HA-IncR135H (pNS426) was generated by ligating HindIII-XhoI digested pNS402 backbone and a HindIII-XhoI IncR135H fragment from pNS428 (as described below).
pAc5.1–3×HA-IncAAA (pNS427) was generated similarly to pNS426, substituting a HindIII-XhoI IncAAA fragment from pNS429 (as described below).
Vectors for Drosophila transgenesis were as follows:
pUASTattB–Myc-Inc (pNS346) encodes an N-terminal Myc epitope (MEQKLISEEDLAS) fused to Inc, as previously described (Li et al., 2017).
pUASTattB–Myc-IncR135H (pNS428) was generated by ligating a HindIII-XhoI digested pNS346 backbone and a HindIII-XhoI IncR135H fragment. The IncR135H fragment was liberated from the fusion PCR amplification product of primers oNS1126 and oNS1127 and an equimolar mix of overlapping 5’ and 3’ IncR135H fragments as template. The 5’ and 3’ fragments were generated by PCR amplification of pNS346 template with primers oNS1126/oNS1555 and oNS1127/oNS1554, respectively.
pUASTattB–Myc-IncAAA (pNS429) was generated similarly as pNS428, substituting a HindIII-XhoI IncAAA fragment. The IncAAA fragment was generated similarly as IncR135H, substituting primers oNS1126/oNS1557 and oNS1127/oNS1556 for generating the 5’ and 3’ IncAAA fragments, respectively.
pUAST–Inc-HA (pNS273) encodes Inc fused to a C-terminal HA epitope (GSYPYDVPDYA) and was generated by three piece ligation of pUAST BglII-XhoI, a BglII-EcoRI inc fragment liberated from pNS272 (Stavropoulos and Young, 2011), and an EcoRI-XhoI HA fragment generated by phosphorylating and annealing oligonucleotides oNS191 and oNS192.
pUASTattB–3×FLAG-Inc (pNS404) encodes an N-terminal 3×FLAG epitope (MDYKDDDDKGSDYKDDDDKGSDYKDDDDKAS) fused to Inc and was generated by three piece ligation of EcoRI-XhoI digested pUASTattB, an EcoRI-NheI 3×FLAG fragment liberated from the PCR amplification product of pNS311 template and primers ACF and oNS241, and a NheI-XhoI inc fragment liberated from pNS351.
pUASTattB–3×FLAG-Inc1-186 (pNS405) was generated similar to pNS404, substituting a NheI-XhoI Inc fragment prepared as for pNS376.
pUASTattB–3×FLAG-Inc1-156 (pNS406) was generated similar to pNS404, substituting a NheI-XhoI Inc fragment prepared as for pNS375.
pUASTattB–3×FLAG-Inc1-123 (pNS407) was generated similar to pNS404, substituting a NheI-XhoI Inc fragment prepared as for pNS374.
pUASTattB–3×FLAG-IncF47A (pNS423) was generated similar to pNS404, substituting a NheI-XhoI Inc fragment prepared as for pNS409.
pUASTattB–3×FLAG-IncF105A (pNS424) was generated similar to pNS404, substituting a NheI-XhoI Inc fragment prepared as for pNS419.
pUASTattB–3×FLAG-IncF47A/F105A (pNS425) was generated similar to pNS404, substituting a NheI-XhoI Inc fragment prepared as for pNS422.
Oligonucleotides
Oligonucleotides used in this work, listed 5’ to 3’, are as follows:
Cell culture and biochemistry
S2 cells were cultured in S2 media containing 10% FBS, penicillin, and streptomycin, and were transfected with Effectene (Qiagen) as described previously (Stavropoulos and Young, 2011). Transfections were performed in 6 well plates for ∼24 hr, after which liposome-containing media was replaced with fresh culture media. 400 ng of total DNA was used for each transfection. For transfections involving two plasmids, an equal amount of each was used. Empty vector lacking insert was used to equalize DNA amounts as indicated in Figures. Cells were harvested 36-48 hr after transfections were initiated, washed twice in PBS, and lysed in ice-cold NP40 lysis buffer (50 mM Tris pH 7.6, 150mM NaCl, 0.5% NP40) containing protease inhibitors. Protein extracts were quantitated in duplicate (BioRad, 5000111).
For co-immunoprecipitations of truncated Inc proteins from S2 cells, 700-1000 µg total protein was incubated overnight at 4°C with 1:100 anti-FLAG (Sigma, F1804) or anti-Myc (Sigma, C3956) antibody. Complexes were precipitated by incubation with Gammabind G sepharose beads (Life Technologies, 10-1243) for 1 hr at 4°C on a nutator, washed 4×5 min at 4°C with lysis buffer, and denatured in SDS sample buffer, separated on Tris SDS-PAGE gels, and transferred to nitrocellulose. For co-immunoprecipitations of Inc point mutant proteins from S2 cells, 450-1000 µg of total protein was immunoprecipitated overnight with 20 µl (50% slurry) of anti-FLAG (Sigma, F2426) affinity gel at 4°C on a nutator. Samples were then washed 4×5 min at 4°C with lysis buffer, denatured in SDS sample buffer, separated on Tris SDS-PAGE gels, and transferred to nitrocellulose. Membranes were blocked for 1-1.5 hr at room temperature in LI-COR Odyssey buffer (LI-COR, 927-40000). Membranes were subsequently incubated in blocking buffer containing 0.1% Tween 20 and the appropriate primary antibodies for 1-2 hr at room temperature or 4°C overnight: rabbit anti-Myc (1:2,000, Sigma, C3956), mouse anti-FLAG (1:2,000, Sigma, F1804), rat anti-HA (1:2,000, Roche, 11867431001), and rabbit anti-HA (1:2,000, Bethyl Laboratories, A190-208A). After washing 4×5 min in a solution containing 150 mM NaCl, 10mM Tris pH 7.6, and 0.1% Tween 20 (TBST), membranes were incubated in the dark for 30-60 min at room temperature with appropriate secondary antibodies, all diluted 1:15,000 or 1:30,000 in blocking buffer containing 0.1% Tween 20 and 0.01% SDS: Alexa 680 donkey anti-rabbit (Life Technologies, A10043), Alexa 680 donkey anti-mouse (Life Technologies, A10038), Alexa 790 anti-mouse (Life Technologies, A11371), Alexa 790 anti-rat (Jackson ImmunoResearch, 712-655-153). Membranes were then washed 4×5 min in TBST, 1×5 min in TBS, and imaged on a Li-Cor Odyssey CLx instrument.
Fly protein extracts were prepared from whole animals or from sieved heads by manual pestle homogenization in ice-cold NP40 lysis buffer supplemented with protease inhibitors. For co-immunoprecipitation from fly head protein extracts, complexes were immunoprecipitated with 30 µl (50% slurry) of anti-Myc (Sigma, E6654) affinity gel for 1.5 hr at 4°C on a nutator. Samples were then washed 4×5 min at 4°C with lysis buffer, denatured in SDS sample buffer, separated on Tris SDS-PAGE gels and blotted as described above. Primary antibodies were rabbit anti-Myc (1:2,000, Sigma, C3956), mouse anti-FLAG (1:2,000, Sigma, F1804), and rat anti-HA (1:2,000, Roche, 11867431001). Secondary antibodies were Alexa 680 donkey anti-rat (Jackson ImmunoResearch, 712-625-1533), Alexa 680 donkey anti-mouse (Life Technologies, A10038), Alexa 790 anti-rabbit (Life Technologies, A11374). For assessing in vivo expression of Inc proteins, 30 µg was separated on Tris-SDS-PAGE gels and blotted as described above. Primary antibodies were rabbit anti-Myc (1:2,000, Sigma, C3956), mouse anti-FLAG (1:2,000, Sigma, F1804), mouse anti-tubulin (1:10,000, DSHB, 12G10), and rabbit anti-tubulin (1:60,000, VWR, 89364-004). Secondary antibodies were Alexa 680 anti-mouse (Life Technologies, A10038) and Alexa 790 anti-rabbit (Life Technologies, A11374).
qRT-PCR
Total RNA was isolated using TRIZOL (Life Technologies, 15596-026). 5 µg of RNA was reverse transcribed with a poly-T primer (oNS337) and SuperScript II reverse transcriptase. qPCR was performed using SYBR Green Supermix (Bio-Rad, 1725272) and the following primers: oNS98 and oNS184 (inc); oNS810 and oNS811 (Cul3); RPS3A and RPS3B (rps3). All real-time PCR reactions were performed using a BioRad DNA Engine Opticon 2 System.
Fly stocks and transgenes
elavc155-Gal4 (Lin and Goodman, 1994), inc1 (Stavropoulos and Young, 2011), inc-Gal4 (Stavropoulos and Young, 2011), inc1 inc-Gal4 (Li et al., 2017), attP2: UAS-Myc-Inc (Li et al., 2017), attP2: UAS-3×FLAG-Inc (Li et al., 2017), and UAS-3×FLAG-3×HA-Cul3 (Hudson and Cooley, 2010) are previously described. UAS-Cul3-RNAi is 11861R-2 obtained from the NIG-Fly stock center. Transgenic flies generated in this study using pUASTattB-based vectors were integrated at attP2 (Groth et al., 2004) with phiC31 recombinase (BestGene); integration was verified by PCR using primer attP2-5’ paired with oNS277. All transgenes were backcrossed six to eight generations to Bloomington stock 5905, an isogenic w1118 stock described elsewhere as iso31 (Ryder et al., 2004).
Sleep analysis
Crosses were set with five virgin females and three males on cornmeal, agar, and molasses food. One to four day old male flies eclosing from LD-entrained cultures raised at 25°C were loaded in glass tubes containing cornmeal, agar, and molasses food. Animals were monitored for 5-7 days at 25°C in LD cycles using DAM2 monitors (Trikinetics). The first 36-48 hours of data were discarded and an integral number of days of data (3-5) were analyzed using custom Matlab code. Locomotor data were collected in 1 min bins. Sleep was defined by locomotor inactivity for 5 min or more; all minutes within inactive periods exceeding 5 min were assigned as sleep. This definition classifies more sleep than the definition used in our prior studies (Li et al., 2017; Li and Stavropoulos, 2016; Stavropoulos and Young, 2011), in which sleep is scored as beginning on the fifth minute of locomotor inactivity and the preceding four minutes are classified as quiet wakefulness. Dead animals were excluded from analysis by a combination of automated filtering and visual inspection of locomotor traces.
Statistics
One-way ANOVA and Tukey post-hoc tests were used for comparisons of total sleep, daytime sleep, nighttime sleep, and sleep bout number. Nonparametric Kruskal-Wallis tests and Dunn’s post hoc tests were used for comparisons of sleep bout length. Unpaired two-sided Student’s t-tests were used for comparisons of inc mRNA levels in vivo.
Sequence alignments
Alignments were performed with Clustal Omega 2.1 and BOXSHADE. GenBank accession numbers for proteins in Figure S1A are: Inc, NP_001284787; KCTD2, NP_056168; KCTD5, NP_061865; KCTD17.3, NP_001269614; KCTD17.4, NP_001269615.
Acknowledgements
We thank Zachary Zuchowski for assistance in constructing plasmids. This work was supported by an International Student Research Fellowship from the Howard Hughes Medical Institute (HHMI) to Q.L., and by grants from the National Institutes of Health (NS111304), the Mathers Foundation, Whitehall Foundation grant 2013-05-78, fellowships from the Alfred P. Sloan and Leon Levy Foundations, a NARSAD Young Investigator Award from the Brain and Behavior Foundation, the J. Christian Gillin, M.D. Research Award from the Sleep Research Society Foundation, and a Career Scientist Award from the Irma T. Hirschl/Weill-Caulier Trust to N.S.
References




