

Abstract
P-bodies (PBs) are cytoplasmic mRNA-protein (mRNP) granules conserved throughout eukaryotes which are implicated in the repression, storage and degradation of mRNAs. PB assembly is driven in part by proteins with self-interacting and low-complexity protein domains. Non-translating mRNA is also required for PB assembly, however no studies to date have explored whether particular mRNA transcripts are more critical than others in facilitating PB assembly. A previous genome-wide microscopy screen in yeast revealed that rps28bΔ (Ribosomal protein subunit-28B) mutants do not form PBs under normal growth conditions. Here, we demonstrate that the RPS28B 3’UTR is important for PB assembly, consistent with the fact that this is a known binding site for the PB assembly protein Edc3. However, expression of the RPS28B 3’UTR in isolation is insufficient to drive normal PB assembly. Intriguingly, chimeric mRNA studies revealed that Rps28 protein, translated in cis from an mRNA bearing the RPS28B 3’UTR, physically interacts more strongly with Edc3 than Rps28 protein synthesized in trans. This Edc3-Rps28 interaction in turn also facilitates PB assembly. In summary, our work indicates that PB assembly may be preferentially nucleated by specific RNA “scaffolds”, which may be a common theme in RNP granule assembly. Furthermore, this is the first description in yeast to our knowledge of a cis-translated protein interacting with another protein in the 3’UTR of the mRNA which encoded it, which in turn has functional consequences for assembly of cellular structures.
Introduction
Post-transcriptional processes are crucial regulators of gene expression. In the cytoplasm, mRNAs can have multiple fates. They can either be actively translated or be subject to translational repression followed by storage or decay. All of these non-translating mRNP states have been associated with P-bodies (PBs)[1,2].
PBs are membrane-less cytoplasmic RNA granules conserved from yeast to humans[3][4]. They are present in all cells growing under normal growth conditions but typically increase in size and number during stress[5]. They are composed of non-translating mRNAs and harbor numerous mRNA decay proteins including many involved in 5’-3’ mRNA decay, as well as nonsense-mediated decay (NMD) proteins[6]. miRNA and siRNA-associated proteins also localize in PBs[7]. While earlier understanding of PB composition largely derived from fluorescence microscopy approaches, recently, PBs were isolated from HEK293 cells and subject to mass spectrometry and RNA-Seq analysis, greatly increasing our understanding of the compositional makeup of these structures[8].
Systematic experiments posit that PB assembly is a two-step process requiring the generation of a pool of non-translating mRNP complexes that then interact via proteins that harbor self-interaction and low complexity domains (LCDs). First, supporting the role of non-translating mRNPs in PB assembly, treatment of cells with cycloheximide, which traps mRNAs in polysomes, inhibits PB assembly[5]. Conversely, inhibition of translation initiation[5] or treatment with puromycin[9], all increase the amount of non-translating mRNPs, and result in an increase in PB assembly. Furthermore, in vitro RNase treatment of semi-purified PBs result in their disassembly[5]. Second, supporting the role of specific protein interactions in PB assembly, deletion of genes including Edc3, Pat1 and Lsm4 lead to defects in PB assembly [10][11]. Edc3 has a self-interaction domain (Yjef-N) and Lsm4 has a glutamine/asparagine (Q/N) rich low complexity domain that are both implicated in PB assembly[10]. Furthermore, Edc3 directly interacts with multiple PB proteins like Dcp2 and Dhh1, whereas Pat1 can additionally bind to Xrn1, Dcp2, the Lsm1-7 and Ccr4-Not complexes[12,13][14]. By virtue of these multivalent interactions with numerous PB proteins, Edc3 and Pat1 act as protein scaffolds in PB assembly. Thus, like other mRNP granules, PB assembly is driven by multiple proteins, with no single protein seemingly essential for PB formation[15].
To date, knowledge about PB assembly has largely been gleaned from candidate gene analyses. However, an unbiased genetic screen coupled to live cell yeast microscopy to identify genes that alter PB assembly identified rps28bΔ as having a severe defect in PB formation under normal growth conditions[16]. Rps28 is a protein of the 40S ribosomal subunit and binds near the mRNA exit tunnel. In yeast it is encoded by paralogous genes RPS28A and RPS28B. Rps28a and Rps28b are identical except for an S3N change. However, they differ significantly with respect to their mRNA 3’UTRs. RPS28B mRNA possesses an unusually long 3’ UTR of ~643nts. The 3’UTR also harbors a stem loop structure, thought to bind Edc3[17], that has been extensively studied for enabling an auto-regulatory circuit that regulates RPS28B mRNA and protein levels [18]. Specifically, it is proposed that high Rps28 protein levels, generated from either RPS28A or RPS28B mRNA, leads to Rps28-Edc3 binding. This is thought to promote recruitment of decapping proteins to the mRNA, and drive deadenylation-independent decapping and decay of RPS28B mRNA[17–19]. Thus, Rps28 protein levels are thought to regulate RPSB28B mRNA abundance, thus helping maintain a homeostatic balance of Rps28 protein. Interestingly, in normal growth conditions at mid-log, Rps28a protein is reportedly 11 fold more abundant than Rps28b, but RPS28A mRNA levels are only 50% greater than that of RPS28B [18,20], suggesting that RPS28B mRNA is less translationally active, for reasons that remain unknown.
While deletion of RPS28B could cause a decrease in Rps28 protein levels resulting in PB assembly defects, we hypothesized that the mRNA itself might also have a role as a novel mRNA scaffold driving PB assembly for two reasons. 1) RPS28B mRNA 3’UTR interacts with Edc3, a major PB assembly factor[17,18]. 2) The RPS28B 3’UTR is unusually long. Recent studies have shown that mRNA lengths correlate with their enrichment in RNA granules[21,22]. Furthermore, RNAs themselves can drive liquid-liquid phase separation (LLPS)[23,24]. LLPS is a process thought to facilitate granule formation wherein biomolecules with high valency phase separate once critical local concentrations are attained. While RNAs can accelerate this process in vitro[23] (though not always [25]), importantly, no specific mRNA has been identified to uniquely and potently facilitate PB (or stress granule; SG) assembly in vivo.
In this work, our data suggests that the RPS28B mRNA 3’UTR acts as a novel PB nucleating mRNA scaffold. However, Rps28 protein, translated in cis from the RPS28B mRNA, is also important for PB assembly. Strikingly, the cis translation of Rps28 from the RPS28B mRNA with its native 3’UTR is necessary for efficient Rps28-Edc3 protein interaction, which in turn is necessary for PB assembly under normal growth conditions. This work suggests that mRNA scaffolds might be a common theme in RNA granule assembly. More broadly, and consistent with recent work [26][27,28], an under-appreciated role of mRNA 3’UTRs may be enhancement of protein-protein interactions involving nascently encoded proteins and previously 3’UTR-bound binding partners.
Materials and methods
Yeast strains and growth conditions
The strains used in this study are described in Supplemental Table 1. The strains knocked out for specific genes were obtained from the Yeast Knockout Collection. Strains were grown on YPD or synthetic media (VWR Glucose 2%, Difco Yeast Nitrogen Base 0.17%, Fisher Ammonium sulphate 5 g/L, appropriate amino acids and nucleotides). All strains were grown at 30°C in shaking water baths. A standard lithium acetate technique was used for yeast transformations. Glucose deprivation stress was applied for 10 min as previously described[11].
Plasmids
The plasmids used in this study are described in Supplementary Table 1. To construct plasmid pRB224, GFP was amplified from plasmid pRB001 using oligos oRB446 and oRB447. This PCR product along with XhoI digested pRB011 was recombined in yeast via homologous recombination. To make plasmids pRB378, pRB229 and pRB230 (RPS28B 3’UTR truncation constructs1, 2 and 3), oligos oRB396 and oRB445, oRB398 and oRB399, oRB400 and oRB401 were used to carry out linear amplification of the plasmids with deletion of the respective regions. This was followed by PNK treatment and ligation to circularize the plasmids. To make plasmids pRB379, pRB380, pRB381 and pRB382, plasmids pRB225, pRB227, pRB228 and pRB226 were digested with NotI and SalI. Required gel extracted fragments were then ligated into NotI/SalI cut pRS315.
Microscopy
The screen that identified rps28bΔ as having a severe defect in PB formation has been previously described[16]. Analyses of PB assembly was conducted as previously described[29], using a Deltavision Elite (100x objective) followed by image analysis using Fiji software[30]. For every strain, a minimum of 100 cells/per replicate were quantified, with a minimum of 3 biological replicates examined for each microscopy dataset.
Polysome analysis
Polysome analyses were conducted as previously described[31] with the following minor differences. Cells were either harvested in midlog (OD600 0.3-0.6) or after being subject to 15 min glucose deprivation stress.
Western Blots
Western blotting was carried out by standard protocols. Protein extracts were loaded onto SDS-polyacrylamide gels. Rps28 was detected by using Rabbit anti-Rps28 (Thermo Fisher Scientific) at a 1:2500 dilution. GAPDH was detected by using Mouse anti-GAPDH (Thermo Scientific) at a 1:25000 dilution. Detection was carried out by using Li-Cor fluorescently labelled secondary antibodies and the Li-Cor Odyssey Imaging system. Secondary antibodies used were IRDye 800CW Goat anti-Rabbit IgG (H + L) and IRDye 800CW Goat anti-Mouse IgG (H + L) to detect anti-Rps28 and anti-GAPDH antibodies respectively.
Northern Blots
5 ml yeast cultures were grown to midlog (OD600 ~0.3-0.6) after which RNA was extracted using a hot phenol extraction protocol described previously[32]. Northern blots were carried out using previously established protocols[33]. 30μg of total RNA was subject to agarose gel electrophoresis (1.25% agarose). mRNAs were visualized by PhosphorImager analysis (Typhoon) of nylon membranes that were probed with 5’ end [32P]-radiolabeled DNA probes. RPS28B was also specifically visualized with a probe complementary to the RPS28B 3’UTR (See supplemental table 1). Image analysis was carried out using Fiji[30].
qRT-PCR
2 ml yeast cultures were grown to midlog (OD600 ~0.3-0.6) after which RNA was extracted using the Trizol RNA extraction method. Briefly, cells were centrifuged at 4000rpm for 15 min followed by resuspension in 1 ml of Trizol reagent. Glass beads were added to the microfuge tubes and cells were disrupted using the Fisher Vortex Genie 2 for 5 min at 4°C. 200μl chloroform was added, vortexed for 15 sec followed by a 5 min incubation at RT. Tubes were centrifuged at 13,300rpm for 5 min at 4°C. Aqueous layer was recovered followed by another chloroform extraction. RNA was precipitated by adding 500μl of isopropanol and incubating on ice for 15 min. Tubes were centrifuged at 13,300rpm for 15 min at 4°C. Pellets were washed with 70% ethanol and re-dissolved in 50μl RNase free distilled water. qPCR was carried out using the SuperScript III Platinum SYBR Green One-Step qRT-PCR Kit as per manufacturer’s instructions using oligos mentioned in supplemental table 1. Control experiments were carried out to assess the specificity of the probes used.
FISH
FISH was carried out using Stellaris FISH probes for RPS28B designed using the Stellaris Probe designer. The FISH experiment was carried out using the manufacturers protocol for S. cerevisae with the following modifications. Spheroblasting buffer used had components as mentioned in the immunofluorescence protocol above but dissolved in 1 ml Fixation Buffer. Hybridization was carried out at 32°C. Probes are listed in Supplemental Table 1.
Immunoprecipitation
400ml culture of OD600~0.6 was centrifuged at 3000rpm for 2min followed by transfer to a microfuge tubes, centrifugation at 3000rpm for 1min. Pellets were frozen in liquid nitrogen and stored at −80°C until used. Tubes were thawed on ice followed by addition of 300μl lysis buffer (50 mM Tris, pH 7.4, 1 mM EDTA, 150 mM NaCl, 0.5% NP-40) with fungal-specific protease inhibitor (1Ul/100Ul; Sigma) and 1mM PMSF. Acid washed glass beads were added to 500μl and cells were disrupted by using the Fisher Vortex Genie 2 for 3 min at 4°C followed by resting on ice for 2 min. This was repeated twice. Holes were created at the base of microtubes harboring lysed cells and glass beads using a hot syringe needle, and then placed in 15ml tubes. After centrifugation at 2000g for 2 min at 4°C, supernatant was added to equilibrated Chromotek GFP-Trap-MA beads and rotated on a nutator for 1h. This was followed by 4 washes in the abovementioned buffer (excluding NP-40 from this wash buffer). SDS Sample loading buffer was added, samples were heat at 95°C for 10 min followed by western blotting as mentioned above. Rps28 was detected by using Rabbit anti-Rps28 (Thermo Fisher Scientific) at a 1:500 dilution and GFP was detected by using Rabbit anti-GFP (Abcam) at a 1:5000 dilution.
EMSA assay
Electrophoretic mobility shift assay was carried out as previously described[34], specifically the protocol for simple interactions. The reaction mixture of 32P end-labelled RNA and Edc3 was incubated for 1 h at 30°C. Edc3 (N-terminal SNAP and C-terminal His6 tagged) was a gift from the Parker lab.
Results
RPS28A and RPS28B are both important for PB assembly
A previous genome-wide microscopy screen to identify genes that affected PB and stress granule assembly in yeast identified RPS28B as a gene that, when deleted, conferred a severe defect in PB assembly under normal growth conditions[16]. However, deletion of its paralog RPS28A was not tested. We found that in addition to rps28bΔ, rps28aΔ strains are also severely defective in PB assembly under normal growth conditions, with rps28bΔ showing ~100% and rps28aΔ showing ~70% reduction in number of PBs per cell respectively as assessed by Edc3 foci (Figure 1A-B). We also examined PB assembly in the above strains during glucose deprivation stress. As expected, PBs increased in size and number in WT cells under stress as previously observed, and although rps28aΔ and rps28bΔ strains did not completely block PB formation, a significant inhibitory effect was still observed, similar to that of an edc3Δ strain in magnitude[11] (Figure 1C-D). Impaired PB assembly in rps28aΔ and rps28bΔ strains was also observed when other PB markers like Dhh1 and Dcp2 were used to assess PB formation (Figure S1A-B). The PB assembly defect is also unlikely due to general ribosome impairment caused by absence of any given ribosomal protein, as strong PB assembly defects were not seen in multiple other viable ribosomal protein deletion mutants in the original screen[16], or mutants re-assessed here under normal growth conditions (Figure S1C-D). Edc3-GFP protein levels in WT, rps28aΔ and rps28bΔ strains also showed no differences, thus arguing impaired PB assembly is not an artefact of altered PB marker expression (Figure S1E). Finally, WT and rps28aΔ growth rates are almost equal, with rps28bΔ strains showing only a minor growth defect (Figure S1F). In summary, deletion of RPS28A and RPS28B genes results in severe impairment in PB formation under normal growth and stress conditions.

A,B) The PB defect in the rps28aΔ and rps28bΔ strains is consistent when analyzed by other PB markers. Logphase WT, rps28aΔ and rps28bΔ strains transformed with plasmids expressing Dcp2-GFP (pRB038) or Dhh1-GFP (pRB038) and Edc3-mCherry (PB markers) were examined for the presence of PB foci. C,D) Other ribosomal protein deletion mutants do not have a severe defect in PB assembly. Log-phase WT, rps10aΔ, rps10bΔ, rps21aΔ, rps21bΔ and rps24bΔ strains were assessed for their ability to form PBs as in A. Average number of PBs per cell. Data generated from 3 biological replicates with mean ± s.d shown. Paired 2-tailed student T-tests were used to assess significance. E) Levels of the PB marker Edc3-GFP do not differ significantly in WT, rps28aΔ and rps28bΔ strains. Log-phase WT, rps28aΔ and rps28bΔ strains transformed with Edc3-GFP (pRB224) were assessed for Edc3-GFP levels by western blotting. F) A growth curve was carried out to assess growth of WT, rps28aΔ and rps28bΔ strains. Data generated from 3 biological replicates with mean ± s.d shown.
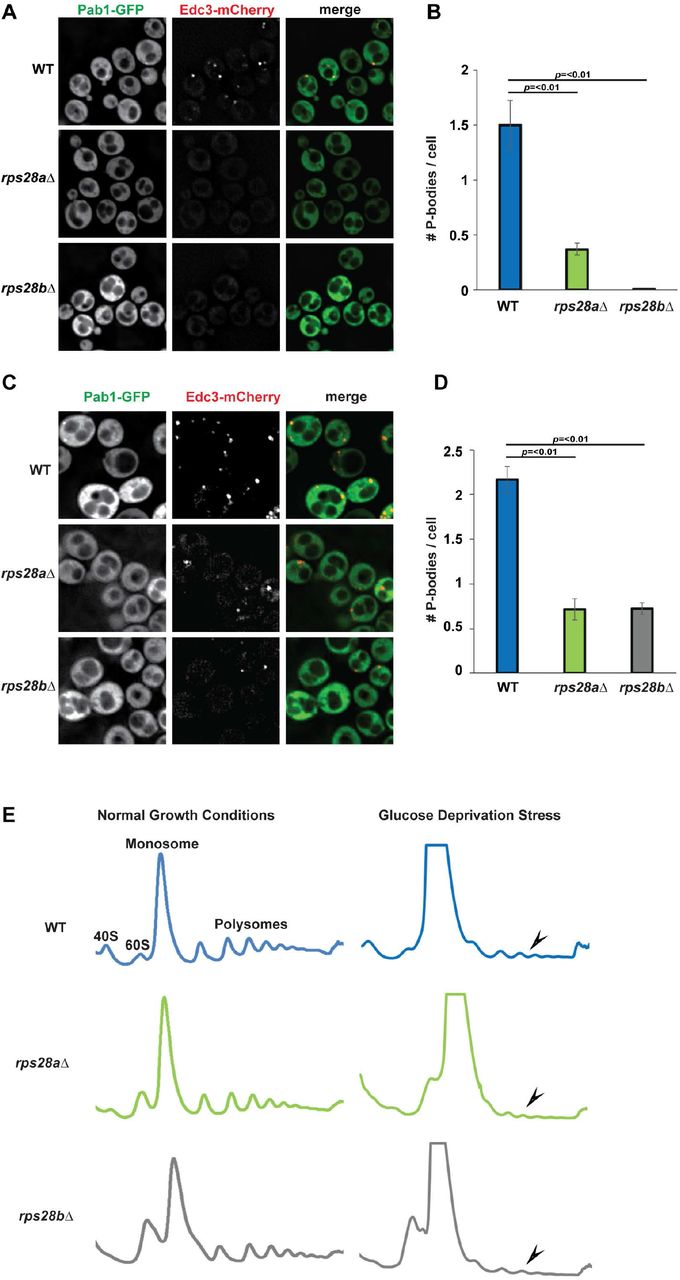
A) Log-phase wild-type S.cerevisiae BY4741 (WT), rps28aΔ and rps28bΔ strains were transformed with pRB001 expressing both Pab1-GFP (SG marker) and Edc3-mCh (PB marker) and examined for the presence of PB foci. B) Quantification of A; average number of PBs per cell. Data generated from 3 biological replicates with mean ± s.d shown. An ANOVA with Dunnetts post-hoc test was used to assess significance. C) As in A, except cells were subject to 10minute glucose deprivation stress. D) Quantification of C; average number of PBs per cell. Data generated from 3 biological replicates with mean ± s.d shown. An ANOVA with Dunnetts post-hoc test was used to assess significance. E) rps28aΔ and rps28bΔ strains are not defective in global translation repression. Log-phase WT, rps28aΔ and rps28bΔ strains growing under normal growth conditions and under 10 min of glucose deprivation stress were subject to polysome analysis. Data is representative of observations from three biological replicates.
rps28aΔ and rps28bΔ strains are not defective in global translation repression
Prior literature suggests that PB assembly defects can be due to either loss of assembly factors (e.g. Edc3, Lsm4[10]), or a defect in translation repression or polysome dissociation, which hampers the accumulation of non-translating mRNPs that are necessary for PB assembly[5,35]. To address this second possibility, we subjected rps28aΔ and rps28bΔ strains to polysome analysis under normal growth and glucose deprivation stress, which elicits a strong translational repression response (Figure 1E); this was previously used to identify Pat1 and Dhh1 as factors affecting translational repression, the first event in PB assembly[35]. In normal growth conditions, polysome abundance in WT, rps28aΔ and rps28bΔ strains is approximately equal. Following glucose deprivation, WT, rps28aΔ and rps28bΔ strains all exhibited a strong polysome collapse, indicating no significant defects in global translation repression under stress. Of note, relative to WT, the rps28aΔ and rps28bΔ strains show a small 40S peak and a large 60S peak, both in normal and stress conditions. This is in keeping with earlier studies that have shown that ribosomal proteins gene deletions lead to aberrancies in 40S and 60S peaks[36,37]. In summary, PB assembly defects in the rps28aΔ and rps28bΔ strains are unlikely to be due to a general defect in translation or translation repression and suggests a defect at a later step of PB assembly.
Rps28 protein levels do not account for PB assembly defects in rps28aΔ and rps28bΔ strains
In principle, rps28aΔ and rps28bΔ strains could affect PB assembly due to decreased levels of Rps28 protein. Indeed, Rps28 interacts with Edc3, Pat1, Lsm2/4/8 and Pby1 PB proteins as assessed by yeast two hybrid assays[38,39]. However, Rps28b protein reportedly only accounts for ~8% of the Rps28 protein population[20,40,41], and yet rps28bΔ strains exhibit a striking PB assembly defect, greater than that seen with rps28aΔ strains. An alternative hypothesis is that the RPS28B mRNA, whose 3’UTR has the ability to recruit Edc3, may act as an mRNA scaffold driving PB assembly.
To assess the relative roles of Rps28 protein and RPS28B mRNA, we assessed PB assembly in WT, rps28aΔ and rps28bΔ strains transformed with WT RPS28B (RPS28B+3’UTR) or empty control vectors (Figure 2A). PBs form normally in the WT strain with the empty vector, with a modest increase (not significant) when a second copy of RPS28B is expressed from a plasmid (Figure 2A, B). In the rps28bΔ strain, WT RPS28B+3’UTR plasmid expression rescues PB assembly whereas empty vector expression does not, as expected (Figure 2A,B). Since Rps28 protein levels do not differ significantly between rps28bΔ strains transformed with the two constructs (Figure 2D), this argues that Rps28 protein levels are not responsible for PB assembly phenotypes in these strain backgrounds.

A) WT, rps28aΔ and rps28bΔ strains were transformed with empty vector, pRS315 or pRS315 expressing RPS28B + 3′UTR and assessed for the presence of PB foci. B) Quantitation of data in panel A. Data generated from 3 biological replicates with mean ± s.d shown. An ANOVA with Tukey’s post-hoc test was used to asses significance. C) Northern analysis of above strains with probe for RPS28B 3’UTR sequence. D) Western analysis of above strains to assess total Rps28 protein levels.
Notably, in the rps28aΔ strain, which already possess an endogenous RPS28B gene, only expression of a second copy of RPS28B from a plasmid rescues PB assembly while empty vector expression does not. Again, Rps28 protein levels show no discernible difference in either of the transformed rps28aΔ strains, whereas RPS28B mRNA levels are clearly increased in both rps28aΔ transformed strains relative to WT cells, but more so in those expressing the RPS28B plasmid (compare Figure 2C, lanes 3-4 versus 1). Thus, while RPS28B mRNA levels do alter in correlation with PB formation, suggesting a role for the RPS28B mRNA, there is not a correlation between overall abundance and PB assembly (i.e. no constant threshold mRNA level to induce PB formation).
One explanation to reconcile PB phenotypes with these observations of RPS28B mRNA and Rps28 protein abundance, and prior data in the field, is that increased abundance and/or translation of RPS28B mRNAs in a rps28aΔ strain may be compensating for the absence of RPS28A mRNA (compare 2C lanes 1 v 3-4). This ultimately results in normal Rps28 protein levels in both cases (Figure 2D). Importantly, given there are no obvious changes in Rps28 protein stability in WT, rps28aΔ or rps28bΔ strains (Figure S2), the differences in RPS28B mRNA abundance in rps28aΔ cells expressing either an empty vector or second RPS28B gene copy logically suggests that RPS28B mRNA must be more heavily translated in rps28aΔ empty vector cells. Sequestration in heavier polysomes may thus prevent effective PB scaffolding by RPS28B mRNA in empty vector expressing rps28aΔ strains, whereas non-translating RPS28B mRNA is more likely to exist in rps28aΔ cells with higher RPS28B mRNA abundance owing to a second RPS28B gene copy. Thus, the critical threshold amount of RPS28B mRNA necessary to form PBs may not relate to abundance, but rather the amount of RPS28B mRNA in a non-translating state, capable of entering into and scaffolding PBs (see discussion).
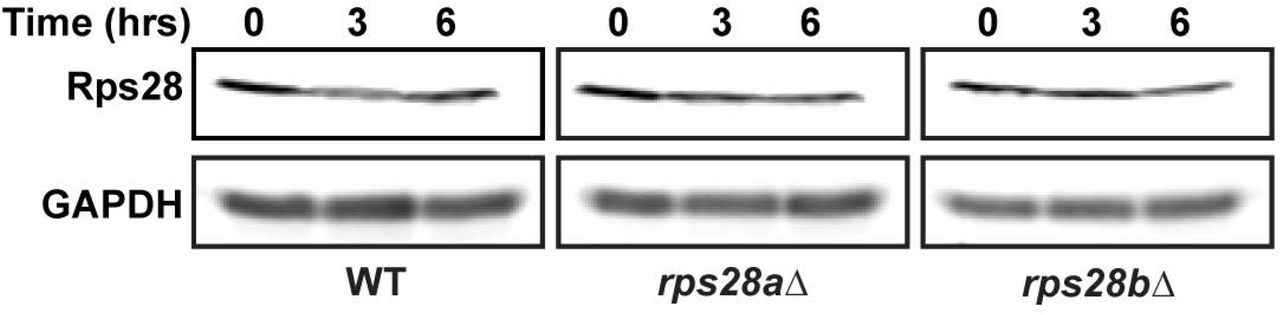
Turnover of Rps28 protein in the indicated strains was carried out by using a cycloheximide (100μg/ml) shut-off experiment.
In summary, our results suggest that total Rps28 protein levels do not account for PB assembly defects in the rps28aΔ and rps28bΔ strains and suggests that RPS28B mRNA might be important for PB assembly.
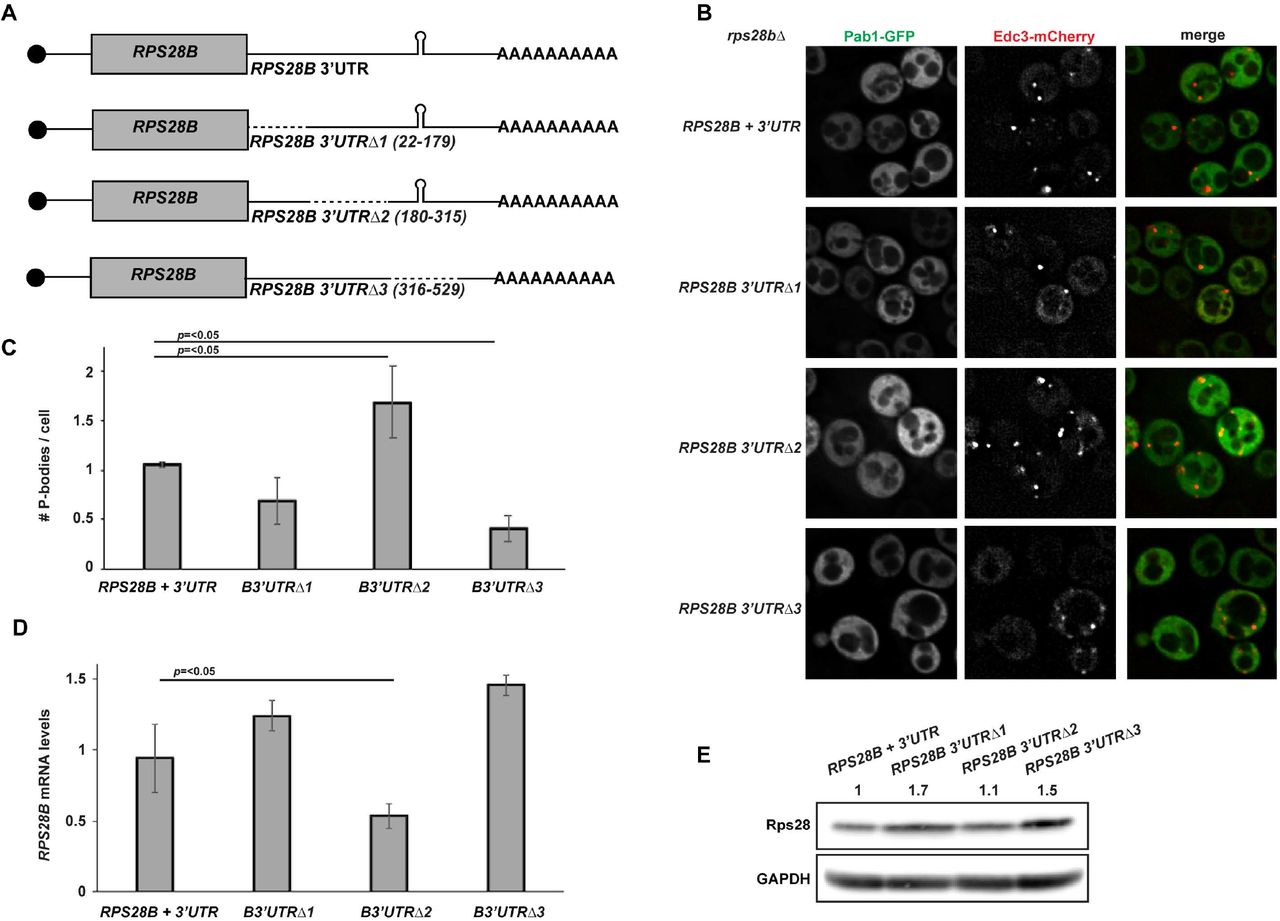
Truncations of the RPS28B 3’UTR can negatively and positively affect PB assembly
To more directly assess the importance of the RPS28B 3’UTR in PB assembly, we created 3 truncations of RPS28B 3’UTR and assessed their effects on PB assembly in a rps28bΔ strain background (Figure 3). The distal region of RPS28B 3’UTR (Region 3, Δ316-529) harboring the stem-loop previously reported to interact with Edc3, significantly impaired PB assembly. Deletion of the ORF-proximal region (Region 1, Δ22-179) modestly impaired PBs also, thought this effect was not significant. Nonetheless, it is formally possible that Edc3 or other PB-stimulating proteins may bind the RPS28B 3’UTR at more than one site (see discussion). Interestingly, deletion of region 2 (Δ180-315) significantly stimulates PB assembly. Importantly, the PB assembly defect in region 3 and 1 deletion mutants is not a result of lower RPS28B mRNA and protein levels; in fact both are slightly higher in these mutants than WT cells (Figure 3D,E). In summary, PB assembly can be negatively and positively modulated by truncations of the RPS28B 3’UTR, bolstering the argument that the RPS28B 3’UTR is important for PB assembly.

A) Schematic of RPS28B 3’UTR truncations generated. B) Log-phase rps28bΔ strains were transformed with plasmids harboring different RPS28B 3’UTR truncations and pRB001 expressing both Pab1-GFP (SG marker) and Edc3-mCh (PB marker) and assessed for their effects on PB assembly by fluorescence microscopy. C) Quantitation of data in panel B. Data generated from 3 biological replicates with mean ± s.d shown. An ANOVA with Dunnetts post-hoc test was used to assess significance. D) RT-qPCR analysis to assess RPS28B mRNA levels of above strains. An ANOVA with Dunnetts post-hoc test was used to assess significance. E) Western analysis of above strains to assess total Rps28 protein levels.
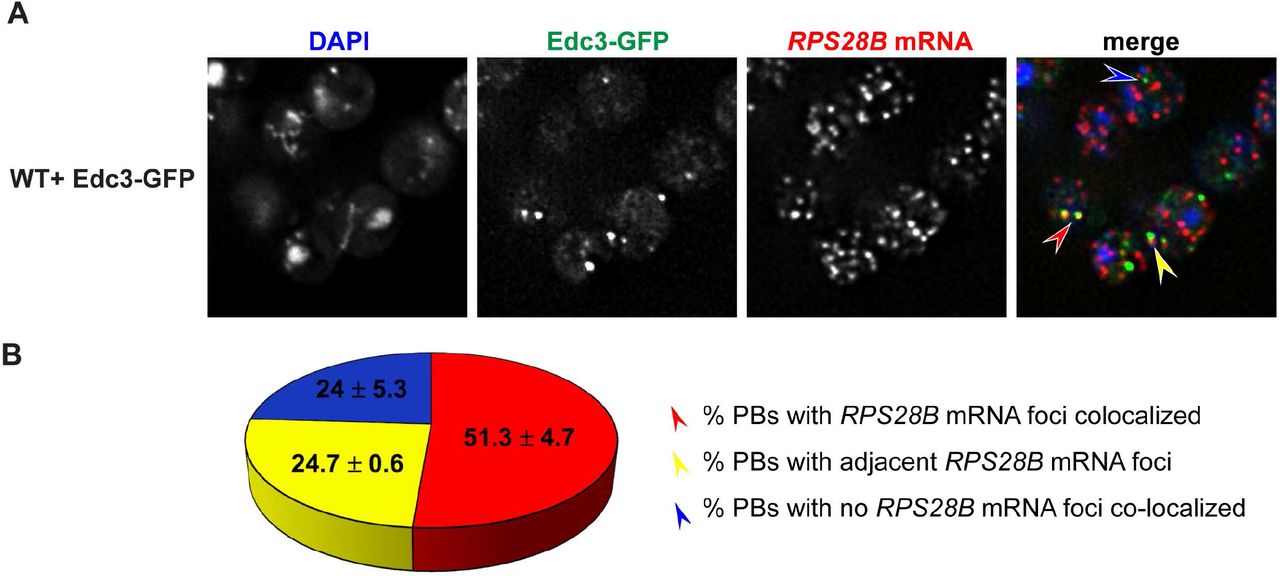
RPS28B mRNA localizes to PBs
If RPS28B mRNA directly facilitates PB assembly, perhaps by scaffolding interactions of key PB assembly proteins, one would expect RPS28B mRNA to localize to PBs. To assess this, we carried out single molecule FISH. ~51% of PBs have RPS28B mRNA perfectly co-localized, ~25% of PBs have RPS28B mRNAs adjacent to them while another 24% do not have visible RPS28B mRNA co-localized (Figure 4). The high percent of PBs with RPS28B mRNA associated with them is consistent with the idea that RPS28B mRNA might aid PB assembly by acting as an mRNA scaffold. The 25% PBs without visible RPS28B mRNA might be explained by limitations of single molecule FISH (e.g. limited probe access to dense mRNP granule structures[42]; loss of mRNA from PBs during hybridization process), RPS28B mRNAs being in the process of degradation or that RPS28B mRNA is only transiently required to localize within PBs in order to stimulate PB assembly.

A) WT transformed with Edc3-GFP was assessed for localization of RPS28B mRNA to PBs by single molecule FISH. B) Quantitation of data in A. Arrow colors in A correspond to phenotypes mentioned in B.
Rps28 protein expressed in cis from an RPS28B 3’UTR-containing mRNA is required for PB assembly
The data presented above argues that the 3’UTR of the RPS28B mRNA has a significant role to play in PB assembly. However, although Rps28 protein levels did not correlate with PB assembly phenotypes (Figure 2B, D), this did not conclusively prove that RPS28B mRNA-based effects operated independently of the presence of Rps28 protein. Thus, to further test if the RPS28B 3’UTR alone could drive PB formation, chimeric constructs (Figure 5A) featuring either the PGK1 ORF, RPS28A or B ORFs, or the reverse complement of the RPS28B ORF, fused to WT RPS28B 5’ and 3’UTRs, were expressed in rps28bΔ strains; these constructs were then assessed for their ability to rescue PB assembly.

A) Schematic of chimeric constructs used. B) Log-phase rps28bΔ strains were transformed with chimeric constructs and pRB001 and assessed for presence of PB foci. C) Data generated from 3 biological replicates with mean ± s.d shown. An ANOVA with Dunnetts post-hoc test was used to assess significance. D) RT-qPCR using RPS28B 3’UTR-specific probe(s) was used to assess chimeric mRNA levels. An ANOVA with Dunnetts post-hoc test was used to assess significance.
Surprisingly, we found that only the chimeric mRNA expressing RPS28A or RPS28B ORFs fused to RPS28B 3’UTR were sufficient to fully rescue PB assembly in rps28bΔ strains (Figure 5B-C). PGK1 or reverse complemented RPS28B ORFs fused to the RPS28B 3’UTR failed to stimulate PB assembly in the rps28bΔ background. These results were not artefacts of altered expression levels of the chimeric mRNAs; in fact, strains lacking PBs typically expressed higher levels of their RPS28B+3’UTR containing chimeric mRNAs than cells expressing fully WT RPS28B mRNA, where PBs did form (Figure 5D). Interestingly, the RPS28A+RPS28B 3’UTR construct was previously shown to undergo decay similar to WT RPS28B+RPS28B 3’UTR while the other chimeric mRNAs fail to be degraded as efficiently[17], suggesting a possible relationship between RPS28B mRNA decay and PB assembly (see discussion). Additionally, expressing start codon mutants of otherwise WT RPS28B+3’UTR constructs in a rps28bΔ background failed to rescue PBs (data not shown). The simplest explanation given these results is that translation of Rps28a or b protein in cis from an mRNA harboring the RPS28B 3’UTR is critical for PB assembly.
Rps28 protein translated in cis from an RPS28B 3’UTR containing mRNA facilitates interaction of nascent Rps28 protein with Edc3
Based on prior Edc3-Rps28 and Edc3-RPS28B 3’UTR interaction studies[17–19] and the above data, we hypothesized that “cis-translation” of Rps28 protein from an RPS28B 3’UTR-harboring mRNA facilitates interaction of nascent Rps28 with 3’ UTR-bound Edc3 (Figure 6A), which in turn enhances PB assembly via an unclear mechanism.
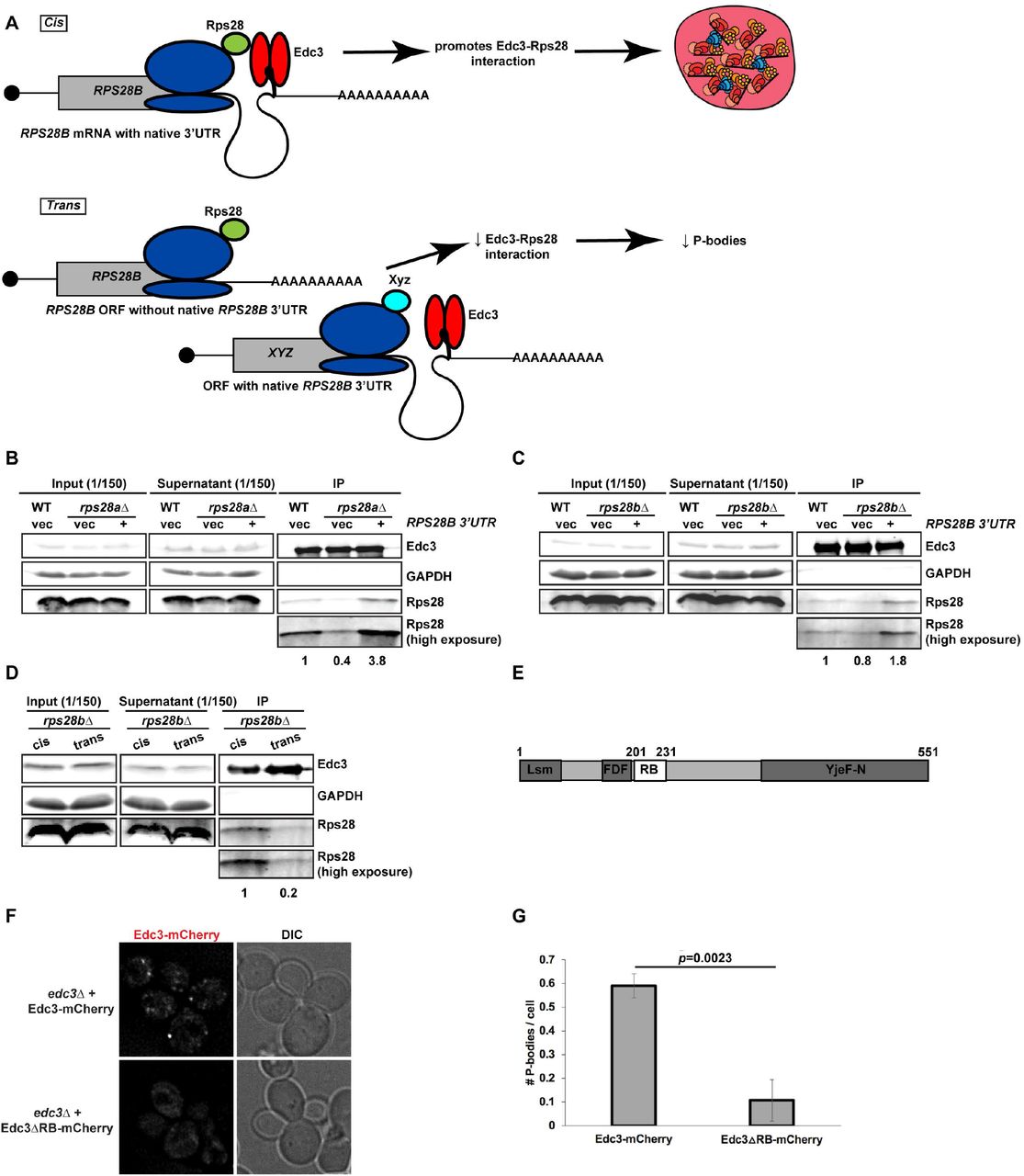
A) Working model of nascently synthesized Rps28 interacting better with Edc3 if Rps28 ORF lies immediately upstream of Edc3-bound RPS28B 3’UTR, which in turn aids PB assembly. B,C) Log-phase WT, rps28aΔ and rps28bΔ strains transformed with empty vector and RPS28B were assessed for Rps28-Edc3 interaction by carrying out an IP for Edc3-GFP and probing for Rps28. Rps28 IP exposure is same duration as in input/supernatant lanes; longer Rps28 exposures are also shown to better quantify relative differences in Edc3-Rps28 interaction. Quantification of Rps28 band intensity is normalized to Edc3 IP band intensity. Images/quantification representative of 2 biological replicates. D) “cis” rps28bΔ yeast transformed with RPS28B ORF-RPS28B 3’UTR and “empty vector” (tTA plasmid - pRB079); “trans” rps28bΔ yeast transformed with PGK1 ORF-RPS28B 3’UTR and RPS28 ORF-CYC1 3’UTR, tTA (pRB388). Images, quantification and biological replicates as in B-C. E,F) Log-phase edc3Δ RP840 strains transformed with Edc3-mCherry/Edc3ΔRB-mCherry were assessed for Edc3-mCherry PB foci by microscopy. G.) Quantification of data in F; data was analyzed via a 2-tailed student’s T-test.
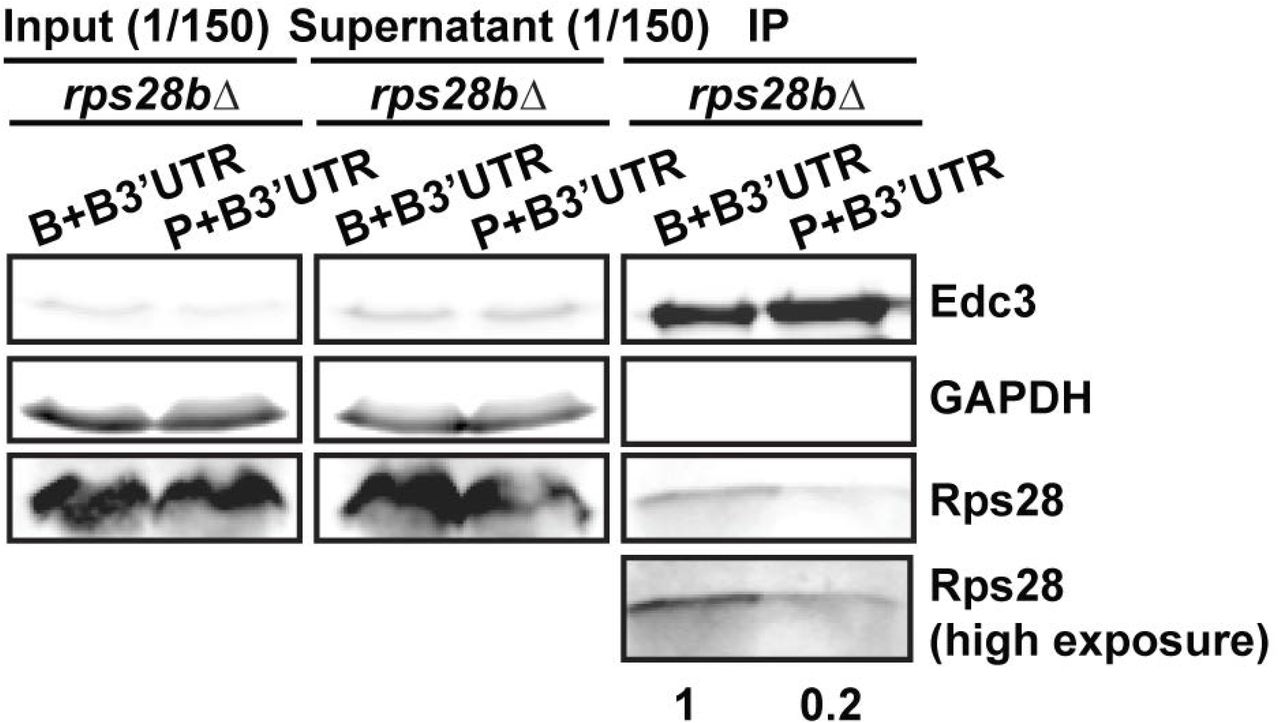
To test this hypothesis, we first determined if Edc3 can bind the RPS28B 3’UTR directly, as previous Yeast-3-hybrid (Y3H) data[17] did not fully rule out the possibility of a bridging protein being involved. We conducted an Electrophoretic Mobility shift assay (EMSA) using purified Edc3 and a 60mer oligo encapsulating the RPS28B 3’UTR stem loop region and confirmed that Edc3 can indeed bind the RPS28B 3’UTR directly (Figure S3); interestingly, a stepped pattern suggests multiple Edc3 binding events at higher concentrations may occur. Next, we carried out an immunoprecipitation of Edc3-GFP and assessed its ability to interact with Rps28 protein in a WT strain expressing an empty vector, and an rps28aΔ or rps28bΔ strain expressing either an empty vector or a WT RPS28B construct (Figure 6B,C), which our prior data indicated rescued PBs (Figure 2). Interestingly, Rps28 protein interaction with Edc3 was barely detectable in rps28aΔ or rps28bΔ strains expressing empty vectors but rose significantly in WT cells and rps28aΔ or rps28bΔ strains expressing WT RPS28B constructs. Thus, increased Rps28-Edc3 interaction correlates with PB assembly. This suggests that the RPS28B 3’UTR facilitates interaction of nascently-translated Rps28 protein with Edc3. A simple model is that this in cis 3’UTR-driven protein-protein interaction reflects the closer spatial proximity of Rps28 synthesis to 3’UTR-bound Edc3 than in the scenario of an RPS28 ORF being separated from its endogenous 3’UTR (Figure 6A). To directly test this, we examined the effect of supplying rps28bΔ cells with a transcript bearing the RPS28B ORF and RPS28B 3’UTR in cis, or where the RPS28B ORF and 3’UTR were supplied in trans on separate transcripts (Figure 6D). Strikingly, despite equal Edc3 and Rps28 protein levels, we again saw a stronger interaction of Edc3 and Rps28 protein in cells bearing the cis RPS28B ORF and 3’UTR transcript. Similar results were observed in rps28bΔ strains using the WT and PGK1 chimeric plasmids (B+B3’UTR and P+B3’UTR) outlined in Figure 5A (Figure S4). Collectively, this data strongly argues that the presence of RPS28B ORF and 3’UTR in an rps28bΔ strain only facilitates a robust Edc3-Rps28 protein interaction when both these elements are on the same transcript.

EMSA was carried out using Edc3 protein and a synthesized 60nt RPS28B RNA oligo containing the predicted RPS28B stem-loop structure. The indicated concentration of Edc3 was mixed with 32P end-labeled RNA (0.5μM). Following incubation at 30°C for 60 min, the reaction mixtures were resolved on 6% native PAGE gel.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Log-phase rps28bΔ strains transformed with RPS28B ORF-RPS28B 3’UTR and PGK1 ORF-RPS28B 3’UTR chimera and assessed for Rps28-Edc3 interaction as in Figure 6B and C
Interestingly, if proximity of Edc3 to nascently synthesized Rps28 is indeed key to their interaction, this might also explain the increase in PBs with truncation 2 that deletes a region in the middle of the RPS28B 3’UTR (Figure 3B,C). Deletion of this region would result in bringing the RPS28B 3’UTR region 3 with the stem-loop (Edc3 binding site) closer to the RPS28B ORF, possibly facilitating the Rps28-Edc3 interaction.
In summary, in both rps28bΔ and rps28aΔ backgrounds, the degree to which PBs form correlates with the ability of Edc3 and Rps28 protein to immunoprecipitated, which occurs most strongly when the RPS28B ORF and 3’UTR are in cis.
The Rps28-Edc3 protein interaction facilitates PB assembly
To determine if the Rps28-Edc3 protein interaction indeed facilitates PB assembly, we turned to a previously described mutant of Edc3 impaired in this interaction[19]. Using edc3Δ cells expressing plasmid-borne WT Edc3-mCherry or the Rps28 binding mutant (Edc3ΔRB-mCherry; Figure 6E)[19], we observed that PB assembly is strongly inhibited in Edc3ΔRB-mCherry expressing cells compared to cells expressing WT Edc3-mCherry (Figure 6F-G). Thus, the Edc3-Rps28 protein interaction is indeed important for PB assembly.
Discussion
Previous work has emphasized protein-protein interactions as key drivers of PB and RNA granule assembly in general[10,43,44][45]. However, given that RNAs can base pair with other RNA molecules and bind to numerous RNA-binding proteins, they too are clearly multivalent and thus, perhaps unsurprisingly, also play a key role in RNA granule assembly. Supporting this, treating cells with cycloheximide completely inhibits PB and SG assembly due to sequestration of mRNA in polysomes, whereas puromycin treatment stimulates PBs and SGs due to release of mRNAs from polysomes [9,46,47]. Additionally, biochemical evidence indicates that RNAs can drive liquid-liquid phase separation (LLPS) of granule proteins in vitro [24]. In vivo, RNA promotes phase separation of Meg proteins in P-granules Caenorhabditis elegans [48]. Finally, yeast total RNA can self-assemble in vitro, in the absence of any proteins, into phase separated granules whose RNA content closely resembles the transcriptome of in vivo purified SGs; this suggests that RNA-RNA interactions are often likely to be sufficient to recruit RNAs to SGs and drive their assembly[24]. While RNA is clearly a driver of RNP granule assembly, specific RNAs (with the exception of the lncRNA NEAT1 in paraspeckles[49]) have not been identified as scaffolding the assembly of RNP granules. This study is the first to our knowledge showing that a specific yeast mRNA, RPS28B, drives PB assembly.
Several features of RPS28B mRNA make it a good candidate for scaffolding PBs. First, the RPS28B mRNA 3’UTR is one of the longest 3’UTRs in yeast, about 643 nucleotides long with the median yeast 3’UTR being ~120 nucleotides long[50]. Second, in WT cells, RPS28B mRNA is seemingly translated weakly compared to its paralog RPS28A, making it more available to scaffold PBs which only harbor non-translating mRNAs[51]. Supporting this, previous data and our data (not shown) suggests that while RPS28A mRNA is only 50% more abundant than RPS28B at steady state in WT cells[18], Rps28a protein is ~11 fold higher than Rps28b protein in WT cells[20], indicating a large difference in translational efficiency. Furthermore, ribosome profiling data indicates that RPS28A is translated more efficiently than RPS28B[51]. Third, the RPS28B 3’UTR directly binds (Figure S3; [17]) to Edc3, one of the major yeast PB assembly proteins. Given that our RPS28B truncation data (Figure 3A-C) hints that more than 1 site in the 3’UTR may contribute to PB assembly, it is also quite possible that the RPS28B 3’UTR might be a binding site for other protein interactions that drive PB assembly. Indeed CLIP data previously showed that Dhh1 binds to RPS28B mRNA but not RPS28A mRNA[52]. Dhh1 is a core PB component previously implicated in repression of translation initiation, elongation and stimulating mRNA decapping [35,53]. Alternatively, Edc3 may also bind at more than 1 RPSB28B 3’UTR site. Regardless, the ability of RPS28B mRNA to drive PB assembly in yeast raises an intriguing question; do other specific RNAs, mRNA or otherwise, drive the formation of other RNA granules? In addition to genetic and phenotypic screening, recent advances in purifying RNA granules, coupled with sequencing of their RNA content, may help reveal the answer to this question.
RPS28B mRNA is a well-studied example of autoregulatory control of ribosomal protein production which our study adds new insight to and raises new questions. Work by the Jaquier, Seraphin and Jacobson labs suggests that when Rps28 protein (A or B) levels are in excess, an Edc3-dependent but deadenylation-independent rapid decay of RPS28B mRNA occurs. A stem loop structure in the RPS28B 3’UTR appears critical to this Edc3-facilitated mRNA turnover[18]. The importance of the Edc3-Rps28 binding interaction to RPS28B mRNA turnover is less clear, with the Seraphin lab describing a necessity for this interaction for Edc3-facilitated RPS28B mRNA decay[19], whereas no effect was seen by the Jacobson lab on steady state RPS28B mRNA levels[17]. In our study, we find that overall Rps28 protein levels are remarkably well controlled, with essentially no variation in overall abundance in WT, rps28aΔ or rps28bΔ strains that do or do not express a WT copy of RPS28B on a plasmid (Figure 2), nor an alteration in Rps28 protein stability in WT, rps28aΔ or rps28bΔ strains (Figure S2). However, RPS28B mRNA levels do vary significantly, with RPS28B steady state levels strongly increasing in rps28aΔ nulls (Figure 2) as expected. Determining whether RPS28A mRNA is subject to autoregulation is an area of future interest, especially given that this transcript seemingly accounts for the majority of total Rps28 protein in WT cells. It is also intriguing that rps28bΔ, but not rps28aΔ strains exhibit a modest growth defect (Figure S1F), suggesting a possible functional importance for RPS28B beyond simply production of Rps28 protein.
The ability to form RPS28B-stimulated PBs seems to rely on similar elements reported as being required for forming an Edc3-decay competent RPS28B mRNP. An RPS28 ORF needs to be upstream of the RPS28B 3’UTR in a rps28bΔ background ([17]; and Figure 1), and an interaction between Rps28 and Edc3 is required ([19]; and Figure 6). Intuitively, it may seem odd that an mRNA subject to accelerated decay could also facilitate assembly of PBs. However, it is possible that the RPS28B mRNA merely plays a transitory role in stimulating the assembly of a Rps28-Edc3 protein complex, and that this complex, and PBs themselves may persist after the RPS28B mRNA itself is degraded. This would be consistent with our smFISH data (Figure 4).
A simple model for the RPS28B 3’UTR scaffolding PB assembly predicts that it’s presence alone in cells would be sufficient to stimulate RNA and/or protein interactions that drive PB formation; to our surprise, this was not the case. PB assembly requires an RPS28 ORF (A or B) upstream of the RPS28B 3’UTR (Figure 5A-B). Additionally, a previously described Rps28-Edc3 protein interaction[17,19] is strengthened when the RPS28B ORF is upstream of the RPS28B 3’UTR (Figure 6B-D). Finally, impairing Rps28-Edc3 protein interaction directly with an Edc3 Rps28 binding mutant also impairs PB assembly (Figure 6E-G). Note, this differs from findings by the Seraphin lab[19], who reported no effects on PB assembly in cells expressing WT or Rps28 binding-mutant Edc3; however quantitative data was not presented, and a second copy of Dcp2 was also expressed in their system which may conceivably alter PB assembly thresholds given that Dcp2 interacts with Edc3 and other PB proteins. Regardless, the above observations suggest that translation of Rps28 protein directly upstream of the RPS28B 3’UTR increases the probability of binding Edc3 (a cis-translational interaction), and that this interaction in turn stimulated PB assembly. How Rps28-Edc3 protein interaction achieves this is unclear; indeed, different evidence has been presented for existence of heterodimeric and trimeric (2 Edc3:1 Rps28) protein complexes in vitro and in vivo respectively[17,19]. This remains an area of future study but nonetheless represents another example of a ribosomal protein with an intriguing extra-ribosomal function. The fact that this function happens to be facilitating the assembly of PBs, whose assembly is anti-correlated with bulk translation levels[5,35], suggests a possible control point for balancing general translation activity with translation repression and decay in yeast.
A role for 3’UTRs driving formation of functionally important protein interactions, in which the cis-translated protein is one of the protein interacting partners has recently been described for two mRNA 3’UTRs in human cells by the Mayr lab[26–28]. In one example, an extended 3’UTR isoform of the CD47 gene (a cell-surface ‘marker of self’ protein), specifically recruits the RNA binding protein HuR which in turn recruits SET. CD47 translated in cis in turn interacts with SET, which ultimately enhances CD47 localization to the plasma membrane [26]. In contrast, a short 3’UTR isoform of the CD47 transcript fails to recruit HuR and SET, causing CD47 to preferentially localize to the ER, where CD47 translation occurs. In the other example, mass spectrometry analyses revealed that the BIRC3 gene, an E3 Ubiquitin ligase, when encoded from a long 3’UTR isoform (but not a short 3’UTR isoform), leads to formation of many distinct BIRC3-containing protein complexes[28]. These included BIRC3 forming a complex with protein trafficking factors IQGAP and RALA, which are themselves recruited to the BIRC3 long 3’UTR by RNA binding proteins Staufen and HuR. Ultimately the BIRC3 long 3’UTR, and the resulting BIRC3-IQGAP-RALA protein complex facilitates recycling to the cell surface of receptor proteins CXCR4 and CD27. In a BIRC3 long 3’UTR isoform null context, impaired CXCR4 membrane localization likely underpins an observed cell migration defect in a malignant B cell model system [28]. Thus, the Mayr lab studies and our own illustrate two key principles. First, 3’UTRs may not just serve as regulators of mRNA function, but also as facilitators of protein-protein interactions with broadreaching functional consequences for the protein interactors. Secondly, the effects of 3’UTRs on promoting consequential protein-protein interactions seem to depend on cis-translation for one of the protein interactors, and recruitment of the other protein interactor(s) to the 3’UTR. Our work demonstrates this phenomenon extends from yeast to mammals and can happen in very different biological contexts (cell surface protein localization versus PB assembly), and as a result of differing gene expression regulatory mechanisms (alternatively spliced 3’UTRs versus differentially expressed gene paralogs). A key open question is how widespread the role of 3’UTRs and cis-translation is in facilitating functional protein-protein interactions.
Acknowledgements
We are grateful to the Jacquier, Badis, Jacobson and Séraphin Lab for plasmids, and the Parker Lab for plasmids and purified Edc3 protein; we particularly acknowledge Bhalchandra Rao and Laura Mizoue for their help here. This work was supported by startup funds to J.R.B. from the University of Arizona and support from NIGMS (NIH RO1-GM1145664).
Footnotes
Two datasets added; demonstration that RPS28B (or A) deletion also impairs stress-induced PBs (Fig1 C-D), and a cis and trans experiment (Fig 6D) demonstrating Edc3-Rps28 interaction is increased when RPS28B ORF and 3'UTR are on the same transcript versus different transcripts. Other general fixes (typos etc).
References




