

ABSTRACT
The cis-polyisoprenoid lipids namely polyprenols (POH), dolichols (DOH) and their derivatives are linear polymers of several isoprene units and have recently gained special attention due to several breakthroughs in the field including improvements in the analytical techniques for their analysis, partial identification of their enzymatic machinery, and the discovery of new biological functions beyond glycosylation. In eukaryotes, POH and DOH are synthesized as a mixture of four or more homologues of different length with one or two predominant species with sizes varying among organisms. Interestingly, POH are hardly detectable in eukaryotic cells under normal conditions with the exception of plants and sporulating yeast. Our metabolomics studies revealed that cis-polyisoprenoids are more prevalent and diverse in the protozoan parasite Plasmodium falciparum than previously postulated as we uncovered the active biosynthesis of medium-long POH and DOH (15 to 19 isoprene units). Moreover, a distinctive POH and DOH profile both within the intraerythrocytic asexual cycle and between asexual and gametocyte stages was observed. These results suggest that cis-polyisoprenoid biosynthesis is developmentally regulated. In addition, we confirmed that DOH biosynthesis occurs via reduction of the POH to DOH by an active polyprenol reductase (PfPPRD) and metabolomics analyses of a PfPPRD conditional mutant suggest that a salvage mechanism of DOH may exist.
Malaria is caused by protozoan parasites of the genus Plasmodium and most cases of life-threatening malaria are attributable to infection with Plasmodium falciparum. The parasite has a complex life cycle that involves the human host and its vector, the Anopheles mosquitoes. All clinical features are caused during the asexual intraerythrocytic life cycle due to the repeated invasion of human red blood cells (RBCs). The intraerythrocytic cycle of P. falciparum lasts around 48 h, during which the parasite progresses through four morphologically different stages: ring, trophozoite, and schizont stages, ending with the rupture of the erythrocyte and release of merozoites that will invade new erythrocytes. Transmission of the malaria parasite requires the development of male and female gametocytes (gametocytogenesis), which are ingested by female mosquitoes during a blood meal and undergo sexual reproduction in the mosquito’s midgut. Nondividing P. falciparum gametocytes take between 10 and 12 days to fully mature and progress through five morphologically distinct forms (stages I to V), which are different from other Plasmodium species.
Over one billion people are at risk of contracting malaria. In 2016, an increase of 5 million malaria cases were reported, mainly in the Americas, and 98 million additional treatment courses of artemisinin-based combination therapy (ACT) were obtained by countries as compared to 2015 (1). ACT combines two or more drugs with different modes of action with artemisinin and is the first-line treatment of P. falciparum infection. Unfortunately, the rapid development of resistance to both the partner drug and now to artemisinin supports the urgent need for identifying different therapeutic targets to expand our repertoire of antimalarials (2).
Historically, metabolic pathways have been a source of therapeutic targets especially for infectious diseases when pathways or their enzymes are sufficiently different or absent in the human host. The mature RBCs are capable of only a few metabolic functions since they do not contain organelles. Among the metabolic pathways that are not active in mature RBCs is the mevalonate pathway which synthesizes the isoprenoid building blocks isopentenyl diphosphate (IPP) and dimethylallyl diphosphate (DMAPP). The downstream cis-polyisoprenoid biosynthesis for dolichols that occurs mainly in the endoplasmic reticulum (ER) is also inactive in mature RBCs (3). On the other hand, P. falciparum has active isoprenoid biosynthesis during the asexual intraerythrocytic cycle as well as during gametocytogenesis where the isoprenoid precursors are synthesized through the methylerythritol phosphate (MEP) pathway (4–6).
The MEP pathway is localized in the apicoplast (7,8), a unique chloroplast-like organelle essential for growth and pathogenesis of the malaria parasite (9) (Fig.1). Moreover, supply of the isoprenoid precursor IPP is the sole metabolic function of the apicoplast in asexual intraerythrocytic and gametocyte stages (6,10). The fact that IPP alone allows parasites lacking the apicoplast to normally grow and develop indicates that IPP is transported out of the apicoplast where the synthesis of cis-polyisoprenoid products is predicted to occur (Fig. 1). cis-Polyisoprenoids (polyprenols (POH), dolichols (DOH) and their derivatives) are linear polymers of several isoprene units. These lipids are present in all membrane systems (11,12) but their biological functions besides protein glycosylation in the ER remains largely unknown. Recently, cis-polyisoprenoids gained special attention due to several breakthroughs in the field including improvements in the analytical techniques for their analysis (13), partial identification of their enzymatic machinery (14), and the discovery of new biological functions beyond glycosylation (15,16). Among some of these biological functions are the regulation of the membrane fluidity (16), stimulation of spore wall formation in yeast (15), and scavenging free radicals in cell membranes (17–19). However, cis-polyisoprenoid metabolism and its biological functions in P. falciparum remain poorly understood.

Phosphoenolpyruvate (PEP); pyruvate (PYR); glyceraldehyde 3-phosphate (G3P); 1-deoxy-D-xylulose-5-phosphate (DOXP); DOXP reductoisomerase (DXR); fosmidomycin (FOS); 2-C-methyl-D-erythritol-4-phosphate (MEP); hydroxy-3-methyl-but-2-enyl diphosphate (HMBPP); dimethylallyl diphosphate (DMAPP); isopentenyl diphosphate (IPP); geranyl diphosphate (GPP); farnesyl diphosphate (FPP); farnesyl-geranylgeranyl diphosphate synthase (FGDPS); cis-prenyltransferase (CPT); polyprenyl-diphosphate (PDP); polyprenyl diphosphate phosphatase (PPP); polyprenol reductase (PPRD); dolichol kinase (DK); dolichyl phosphate (Dol-P). The α-saturated isoprene unit of DOH is indicated by a red circle. The genes encoding enzymes depicted with question mark (?) have not been identified in the Plasmodium genome.
The initial step of cis-polyisoprenoid biosynthesis (POH and DOH) in the malaria parasite is common to all isoprenoid products (Fig. 1) where a multifunctional enzyme synthesizes farnesyl diphosphate (FPP) and geranylgeranyl diphosphate (GGPP) (20). Synthesis of cis-polyprenyl-diphosphate (PDP) is then catalyzed by a cis-prenyltransferase (CPT) which adds several IPP molecules in cis-configuration to FPP or GGPP. This step is called elongation and in eukaryotes occurs in the ER where their downstream products dolichyl phosphates (Dol-P) serve as lipid linked oligosaccharide (LLO) carrier for protein glycosylation and glycosylphosphatidylinositol (GPI) synthesis. In other organisms, cis-polyisoprenoid synthesis also occurs in peroxisomes (rat (21), yeast (22)), lipid droplets (yeast (15,23)) and chloroplasts (plants, cyanobacteria (16,24)). The postulated final steps of cis-polyisoprenoid biosynthesis involve dephosphorylation of PDP to POH followed by reduction of the α-isoprene unit by polyprenol reductase (PPRD), also called SRD5A3 or DFG10 in mammals and yeast, respectively. Whether dephosphorylation of PDP is catalyzed by one or two enzymes (25) has not been elucidated in any organism. Moreover, it has also been suggested that an isopentenol or isopentanol, instead of IPP, is added at the last step, which would avoid the need of dephosphorylation or dephosphorylation and reduction, respectively, but it remains to be addressed (11,26).
In eukaryotes, DOH and POH are synthesized as a mixture of four or more different lengths (indicated by the total number of carbons or IPP units, e.g. POH/DOH 11 or C55) with one or two predominant species where the size varies among organisms. Interestingly, POH are hardly detectable in eukaryotic cells under normal conditions with the exception of plant roots and sporulating yeast. Previously, it was reported that the malaria parasite synthesizes DOH of 11 and 12 isoprene units (27) and their biosynthesis peaks at the schizont stage (4,5). Here, our metabolomic studies uncovered active biosynthesis of medium-long POH and DOH (15 to 19 isoprene units) in the malaria parasite as well as a distinctive POH and DOH profile within the intraerythrocytic asexual cycle and between asexual and gametocyte stages. In addition, we confirmed that DOH biosynthesis occurs via reduction of the POH to DOH by an active PfPPRD (PF3D7_1455900) and metabolic analyses in a conditional knockdown of PfPPRD suggest the presence of a salvage mechanism of DOH.
Results and Discussion
Medium-long polyprenols and dolichols are present in the asexual and sexual intraerythrocytic stages of P. falciparum
Dolichol biosynthesis in the malaria parasites has been assessed previously using radiolabeled metabolic precursors followed by TLC or HPLC analysis. Thus, in order to expand our knowledge of cis-polyisoprenoids present in the malaria parasite especially in gametocyte stages, we standardized an untargeted metabolomics approach using ion-key separation, a high-pressure microfluidic device that provides sub-2-µm Ultra-Performance Liquid Chromatography (UPLC) separations with the SYNAPT G2-Si mass spectrometer, enabling highly sensitive and efficient microflow LC-HRMS (High Resolution Mass Spectrometry) separations that enhance detection of metabolites (see experimental procedures). Surprisingly, we identified a distinctive POH and DOH profile between asexual and gametocyte stages that also differs from the human erythrocyte cell host (Fig. 2). To our knowledge, this is the first time that detectable levels of POH are reported to be present in another eukaryotic cell besides plants and sporulating yeast.

Metabolites from P. falciparum schizont and gametocyte stages as well as uninfected red blood cells (RBCs) were analyzed by LC-HRMS. The area of the signal corresponding to each metabolite was normalized to the cell number. Values are average ± S.E.M. from 3 independent experiments. The structures of polyprenol (POH) and dolichol (DOH) are illustrated where (n) indicates the number of internal cis-isoprene units. The α-isoprene unit of POH (unsaturated) and DOH (saturated) is indicated by a red circle.
Our studies revealed that cis-polyisoprenoids are more prevalent and diverse in the malaria parasite than previously postulated (27,28). In the asexual intraerythrocytic stages, we detected the presence of both POH and DOH of 15 to 19 isoprene units, with 15-, 16-and 17-isoprene units being the predominant species (Fig. 2). Interestingly, ring and trophozoite stages presented higher DOH/POH ratios than schizonts (Table 1). In stage IV gametocytes, DOH of 15 to 20 isoprene units were also present, but the predominant species are 17-, 18-, 19-isoprene units (Fig. 2). Interestingly, POH were difficult to detect in gametocytes under our experimental conditions. As expected, human RBCs contain low levels of only DOH of 18 to 20 isoprene units. This is the first time that the cis-polyisoprenoid profile is determined by LC-HRMS in P. falciparum, and under our experimental conditions DOH and POH of 11 and 12 were not detected probably due to their low abundance.
The area of the signal corresponding to each metabolite was normalized to the cell number and used to calculate the DOH/POH ratios at each stage of the P. falciparum asexual intraerythrocytic cycle. Values represent the mean ± S.E.M. from 3 independent experiments. POH-ND, not detected under the experimental conditions used in this study.
The cis-polyisoprenoid lipids are present in all membrane systems (11,12) but their biological functions besides protein glycosylation in the ER remain largely unknown. A recent fluorescence anisotropy analysis performed in the protein-dense thylakoid membranes of the chloroplast, revealed that membrane areas with less POH had greater membrane fluidity and photosynthesis was affected (16). These analyses suggest that POH have an effect on the biophysical characteristics of biological membranes. Moreover, it was recently demonstrated in yeast that lipid droplets of vegetative cells contain mainly DOH of 14-17 isoprene units while sporulating cells not only shift toward longer species (19 to 24 isoprene units) but also accumulate similar levels of long POH (19 to 24 isoprene units). Long-chain POH synthetized by a developmentally regulated CPT present in lipid droplets (Srt1) promote spore wall formation by serving as a signal that activates chitin synthase (directly or indirectly which remains to be determined) possibly through changes in the membrane structure introduced by POH (15). Therefore, the presence of a distinctive POH and DOH profile within the intraerythrocytic asexual cycle and between asexual and gametocyte stages suggests that cis-polyisoprenoid biosynthesis is developmentally regulated and that these lipids may be involved in other cellular processes besides glycosylation.
Medium-long polyprenols and dolichols are synthesized de novo by P. falciparum
In order to confirm that the medium-long POH and DOH detected were in fact synthesized by the parasite, de novo biosynthesis was assessed by metabolic labeling using [1-13C]glucose and [3-13C]IPP (Fig. 3A). Glucose was selected as metabolic precursor for cis-polyisoprenoid biosynthesis because the mevalonate and MEP pathway-specific labeling patterns of isoprene units resulting from glucose metabolism via glycolysis are well established (13,29). In addition, the mevalonate pathway is inactive in the host cell, the human RBCs, thus, only MEP pathway-specific labeling patterns are observed.

A) Scheme used for metabolic labeling with [1-13C]glucose or [3-13C]IPP in highly synchronous ring stage cultures. Parasites were recovered at schizont stage for LC-HRMS analysis. A representative Giemsa-stained smear is shown and scale bar indicates 2 μm. The fate of 13C through the MEP pathway for [1-13C]glucose is showed as a half-black circle to indicate 13C abundance, which is 50% of the initial one. B) Mass spectra of POH-17 ([M+NH4]=1193.1145) and DOH-17 ([M+NH4]=1195.1208) detected in P. falciparum schizont stage are shown. Natural isotopic distribution for POH and DOH is indicated for the standard. Arrows indicate absence of unlabeled POH. (*) Indicates presence of unlabeled DOH. C) Mass spectra of DOH-18 ([M+NH4]=1263.1868) from uninfected human RBCs cultured in the absence or presence of [1- 13C]glucose under the same conditions as P. falciparum showing lack of de novo DOH biosynthesis.
Highly synchronous infected RBC in ring stage were first incubated in glucose-free media for 2 h and then supplemented with [1-13C]glucose (4 g/L) for 24 h which is metabolized into [1,5-13C]IPP (Fig. 3A). A similar scheme was used for the metabolic precursor [3-13C]IPP, but in this case cultures were treated for 2 h with 10 μM fosmidomycin (FOS), an inhibitor of 1-deoxy-D-xylulose 5-phosphate reductoisomerase in the MEP pathway (DOXP →MEP) (4,8), to reduce >75% the endogenous IPP pool as we reported previously (30), and then supplemented with [3-13C]IPP:12C5-IPP (1:1, 200 μM final concentration) in the presence of FOS. As previously described (13,29), we observed 13C-enrichment as a Gaussian distribution for both 13C-precursors (Fig. 3B and S1), where m/z shift is determined by the remaining of unlabeled metabolic precursor pools in the cell and the number of 13C-atoms incorporated into IPP based on the isotopic dilution due to metabolism (13,31). As expected, only unlabeled DOH was detected in uninfected RBCs since there is no active de novo biosynthesis of isoprenoids (Fig. 3C). Thus, these results confirmed that the detected POH and DOH of 15 to 19 isoprene units are biosynthesized by the malaria parasite.
During optimization of the 13C-biolabeling experimental design, we established that in order to observe 13C-enrichment in POH and DOH, parasites required at least 20-24 h of growth in the presence of the 13C-precursor starting at ring stage. Interestingly, independently of the metabolic 13C-precursor used, unlabeled DOH was always detected while unlabeled POH was indistinguishable from noise in the presence of a 13C-precursor (Fig. 3B). In addition, we showed that POH and DOH achieved similar levels as the parasite progress from ring to the schizont stage. Altogether, these results suggest that synthesis of POH and DOH is developmentally regulated and that a reserve of cis-polyisoprenoids may be carried over to the next asexual cycle.
It has been demonstrated that stage III gametocytes incorporate and metabolize 13C-glucose (32). In addition, we previously reported that the MEP pathway intermediates are present in late-stage gametocytes and that IPP synthesis is essential for normal gametocytogenesis since gametocytes lacking the apicoplast require exogenous IPP supplementation until they reach stage IV (6). Surprisingly, 13C-enrichment in DOH was not detected in stage IV gametocytes when [1-13C]glucose was supplemented starting at stage III (Fig. 4). Therefore, these results suggest that synthesis of DOH occurs earlier than stage III in the gametocytogenesis or potentially these lipids are carried over from the previous asexual cycle and that IPP is required for other isoprenoid products than DOH in gametocyte stages III to IV (6).
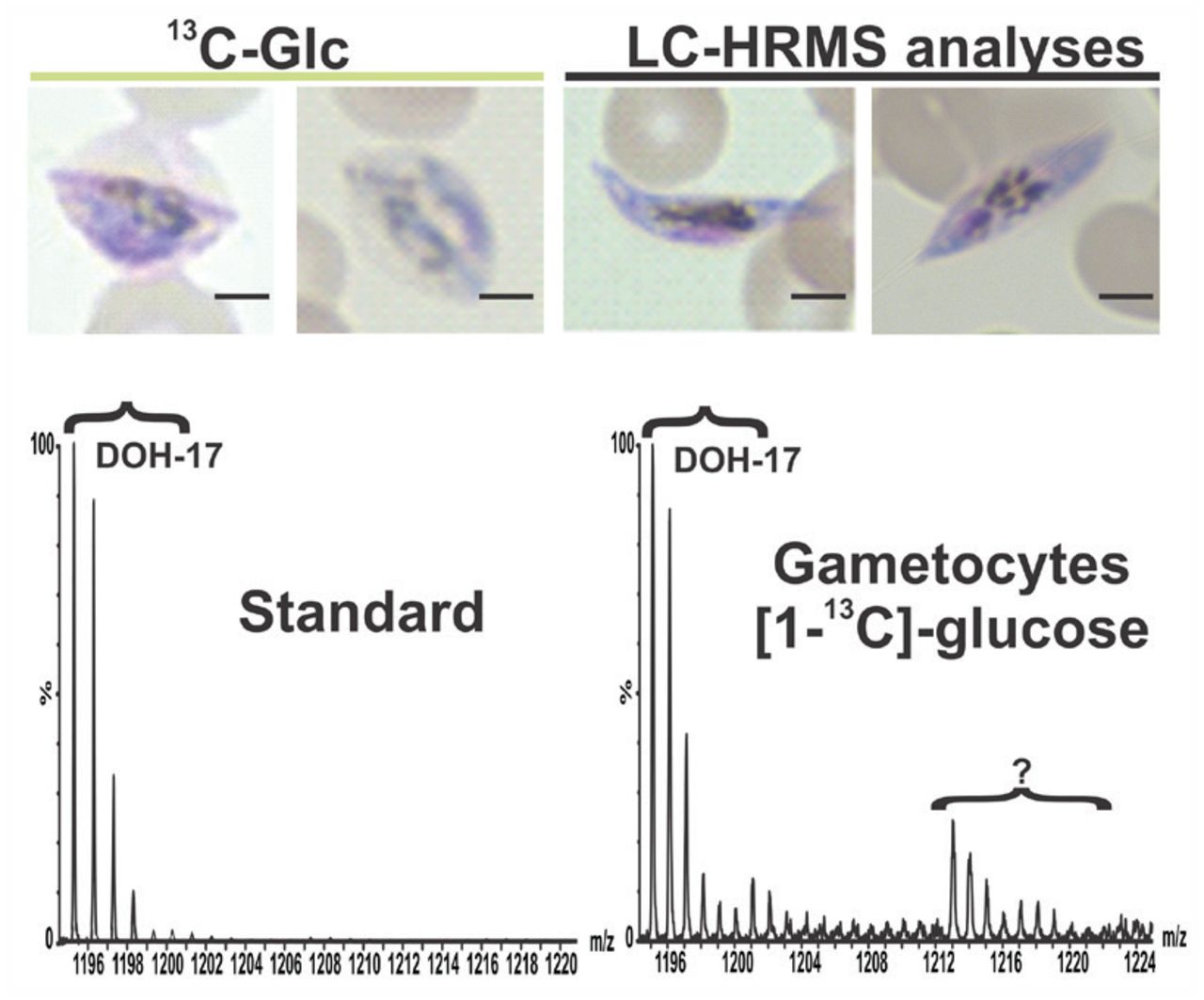
Representative mass spectrum of DOH-17 ([M+NH4]=1195.1208) detected in P. falciparum gametocyte cultures that were metabolically labeled with [1-13C]glucose starting at stage III of gametocytogenesis and recovered at stage IV for LC-HRMS analyses as indicated in the scheme. (?) Indicates unidentified m/z features. Scale bar indicates 2 μm.
P. falciparum polyprenol reductase restored glycosylation of mCPY and reduced POH levels in yeast dfg10 Δ mutant cells
We hypothesized that the presence of POH in P. falciparum indicates that DOH biosynthesis may occur via reduction of the POH to DOH by an active PfPPRD (PF3D7_1455900) (Fig. 1). Therefore, the coding sequence of the putative PfPPRD was optimized for codon usage in yeast and its enzymatic function was confirmed by complementation assays in Saccharomyces cerevisiae. It was previously reported by Cantagrel and colleagues (33) that human PPRD (SRD5A3) can complement the N-glycosylation defects caused by an inactive yeast DFG10, which is the PPRD in this organism for DOH synthesis. As mentioned above, their downstream products Dol-P serve as lipid linked oligosaccharide carrier for protein glycosylation (Fig. 1). In this work, we used the dfg10Δ mutant cells where the DFG10 gene is deleted from the chromosome and displays a lack of N-glycosylation. Mature carboxypeptidase Y (mCPY) has four N-glycosylation sites which are all occupied under normal growth conditions in yeast (34) and depend on a functional PPRD (Fig. 5A) (33). A single band is observed in wild-type (WT) cells when mCPY is fully glycosylated while four extra bands were detected in dfg10Δ mutant cells representing the different glycoforms (−1, −2, −3, −4) (Fig. 5A, lane 2). The mutant transformed with yeast DGF10 or PfPPRD under the control of the endogenous DFG10 promoter showed full correction of the CPY underglycosylation. Some underglycosylated CPY was still detected in cells expressing PfPPRD suggesting that this enzyme was not as active as yeast DFG10.

A) Glycosylation status of CPY in dfg10Δ mutant cells expressing yeast DFG10 and PfPPRD. Positions of mature CPY (mCPY) and its glycoforms (−1, −2, −3, −4) are indicated. Empty vector (vector) was used as control. B) Extracted ion chromatogram (EIC) from LC-HRMS analyses of yeast strains showing that for WT only DOH (15–16) are detected while in dfg10Δ mutant strain DOH is accompanied by their respective POH (15–16). Dfg10Δ mutant strain transformed with PfPPRD showed considerable reduction of POH 15 and 16.
In order to confirm the biochemical effects underlying the yeast complementation, we assessed the POH and DOH profile by LC-HRMS (Fig. 5B). As described previously (3,33), only DOH 15 and 16 were detected in wildtype (WT) yeast while POH along with DOH were detected in the dfg10Δ mutant cells. As expected, dfg10Δ mutant cells expressing PfPPRD showed reduction of the POH levels confirming its predicted enzymatic function (POH →DOH).
Changes in the POH and DOH profiles without major growth defects suggests the presence of a salvage pathway
Two recent functional profiling studies of the Plasmodium genome indicate that Plasmodium PPRD is predicted to be essential (35,36). Thus, we used the CRISPR/Cas9 and the TetR-DOZI conditional system (37) to generate a transgenic strain for inducible knockdown of PfPPRD to assess if changes in the DOH/POH ratios may affect the in vitro growth of P. falciparum (Fig. 6A). Synthesis of DOH is predicted to occur in the ER, thus, a triple-HA tag was added to the C-terminus of PfPPRD to assess its localization. However, multiple attempts to tag PfPPRD with the TetR-DOZI-HA construct failed (Fig. S2), suggesting that PfPPRD tagged at the C-terminus is not efficiently translated or the protein is unstable. It is also possible that the highly conserved catalytic domain of PfPPRD is located in the C-terminal as predicted for the human and plant orthologs (33,38) and tagging interferes with proper enzymatic function. Therefore, we designed a similar construct (PfPPRD-TetR-DOZI) lacking a C-terminal tag. Using the untagged construct, we successfully modified the PfPPRD genomic locus and confirmed correct integration by PCR (Fig. 6B).

A) Schematic of plasmids used and integration of the linearized TetR-DOZI plasmid with 10x aptamer system into PfPPRD gene. The endonuclease cas9 promotes a double-stranded break in the genomic locus while the repair plasmid provides a homology region at the C-terminus of PfPPRD (PfPPRD-C-t) for double crossover homologous recombination to introduce a 3′-untranslated region (UTR) RNA aptamer sequence that binds a tetracycline repressor (TetR) and development of zygote inhibited (DOZI) fusion protein to generate TetR-DOZI conditional system. B) PCR analysis confirming integration at the recombinant locus. Wild type parasites were detected using primers P8 and P2 (Table S1) after 4 days of transfection (A’). Conditional knockdown parasites were detected after 30 days post-transfection (B’) using primers P8 and P9.
The conditional knockdown system was induced by removing anhydrotetracycline (aTc), a stabilizer of the TetR-DOZI complex (37). The absence of aTc allows the TetR-DOZI complex to bind at the repetitive aptamer region reducing the translation of PfPPRD enzyme (Fig. 7A). Interestingly, a slight but significant growth defect was observed only after 8 days (5th intraerythrocytic life cycle) without aTc. At this point certain morphological defects started to be observed in parasites (Fig. 7B). In the absence of PfPPRD tagging, it was not possible to determine whether the observed growth and morphological defects were attributed to a gradual or incomplete reduction in PfPPRD protein levels. Instead, the levels of POH and DOH were determined by LC-HRMS to assess whether PfPPRD enzymatic activity was reduced in the absence of aTc confirming that PfPPRD synthesizes DOH in vivo. These analyses were performed in trophozoite stage parasites where the highest DOH/POH ratios are observed (Table 1). Parasites grown in the absence of aTc for 3 days displayed a pronounced accumulation of POH 15 to 18 and a slight reduction of DOH 15 to 17 without changes in the levels of DOH 18 to 20 suggesting that some residual PfPPRD activity may still be present (Fig. 7C). Surprisingly, LC-HRMS analyses performed after at least 10 days in the absence of aTc revealed similar pronounced accumulation of POH 15 to 18 but DOH 15, 16 and 17 were reduced to undetectable levels while DOH 18, 19 and 20 were not significantly reduced under our experimental conditions (Fig. 7D). The observed reduction of DOH 15, 16 and 17 after 3 days of aTc removal was not statistically significant, but these lipids were undetectable after 10 days in the absence of aTc. Metabolic labeling experiments using [1-13C]glucose in cultures after 10 days of aTc removal revealed that while 13C-enrichment was observed in POH 18 supporting that PfCPT is active, 13C-enrichment in DOH 18 was reduced to undetectable levels. Therefore, the detected DOH 18 was not biosynthesized de novo by parasites under our experimental conditions (Fig. S3). Moreover, 13C-enrichment in DOH 19 was not detected in cultures where aTc was not removed. This result was not unexpected since low levels of 13C-enrichment using [1-13C]glucose were also observed using wild type parasites (Fig. S1). Since DOH 18, 19 and 20 are the species found in uninfected RBCs (Fig. 2), it is possible that the malaria parasite is able to uptake or sequester DOH species from the erythrocyte host but it becomes detectable, or probably intensified, when de novo DOH biosynthesis is impaired such as is the case of reduced PfPPRD activity. However, we cannot rule out that the parasite accumulated or salvaged these DOH species from its own biosynthesis as a result of reduced PfPPRD activity overtime which was negligible when 13C-biolabeling was performed.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A) Schematic of the PfPPRD conditional knockdown mechanism. The modified PfPPRD locus contains a 3′-UTR RNA aptamer sequence that binds to TetR and DOZI fusion protein. In the presence of anhydrotetracycline (aTc), the TetR-DOZI complex is stabilized and the 3′-UTR aptamer is unbound. Removal of aTc causes binding of the TetR+DOZI repressor to the aptamer region which results in reduced protein levels. B) Time-course of growth of PfPPRD-TetR-DOZI knockdown parasites treated with BSD only or BSD+aTc. Data are shown as mean ± SD (n = 3) with error bars smaller than the dots. Individual values are shown in Table S2. (*) Indicates p < 0.0001. A representative Giemsa-stained thin smear of parasites cultured in the presence or absence of aTc showing the morphological defects observed in some parasites after 8 days of aTc removal. The levels of POH and DOH species present in P. falciparum PfPPRD-TetR-DOZI were analyzed by LC-HRMS in trophozoite stage after aTc was removed from cultures for 3 days (panel C) and 10 days (panel D). The area of each metabolite was normalized to the cell number. Values are average ± S.E.M. from 3 independent experiments.
Since the biological role of these lipids remains largely unknown, it cannot be ruled out that POH may be able to perform some of the functions of DOH when their synthesis is compromised. Moreover, as mentioned above, areas of thylakoid membranes of a chloroplast with less POH had greater membrane fluidity and photosynthesis was affected (16). Therefore, it is also possible that the higher levels of POH detected in the absence of aTc may had affected membranes and their functions, thus, contributing to the growth and morphological defects observed.
Conclusion
Our studies using 13C-profiling by LC-HRMS uncovered for the first time in P. falciparum co-occurrence of POH and DOH as well as synthesis of longer DOH species than previously reported. The presence of a distinctive POH and DOH profile within the intraerythrocytic asexual cycle and between asexual and gametocyte stages suggest that cis-polyisoprenoid biosynthesis is developmentally regulated. Interestingly, in mammals and plants increased level of POH results in aberrant glycosylation (39). However, it is difficult to assess if a similar response occurs in the malaria parasite since N-glycosylation in Plasmodium has been controversial despite the presence of very short N-glycans was previously reported in this parasite (40,41). Nevertheless, it is possible that the co-occurrence of POH and DOH suggest that these lipids may be involved in other cellular processes beside N-glycosylation as has been recently demonstrated in yeast (15) and plants (16) which guarantees further investigation.
The presence of POH in P. falciparum supports that DOH biosynthesis occurs via reduction of the POH to DOH and this step is catalyzed by PfPPRD (PF3D7_1455900). Interestingly, a knockdown of PfPPRD revealed that after aTc has been removed for several days, shorter DOH were undetectable while unlabeled DOH of 18 to 19 isoprene units were still present at which point a slight but significant growth defect as well as the observation of abnormal parasites were observed. Altogether, these results suggest that a salvage mechanism for DOH may be present and further studies are needed to establish their source (endogenous vs the host cell). Interestingly, P. falciparum cannot synthesize cholesterol (42), another isoprenoid product. Instead, the malaria parasite salvages cholesterol from its mammalian host (43,44) supporting the hypothesis that DOH salvage from RBCs may also occur.
As a result of several breakthroughs in the field of cis-polyisoprenoids (13–15), new biological functions of these lipids besides their known role in protein glycosylation are starting to be unveiled. Further analyses are still needed to answer why detectable levels of POH are present in the malaria parasite as well as why cis-polyisoprenoid species change during the parasite’s life cycle which may reveal new biological functions of these unique lipids.
Experimental procedures
Parasite cultures
P. falciparum 3D7 and NF54 strains (sulfadoxine-resistant) were obtained through the MR4 Malaria Reagent Repository (ATCC, Manassas, VA) as part of the BEI Resources Repository, NIAID, NIH. Parasites were maintained at 5% hematocrit in O+ human erythrocytes (The Interstate Companies, TN) in RPMI 1640 (GIBCO Life Technologies) supplemented with 5 g/L Albumax I (GIBCO Life Technologies), 2 g/L glucose (Sigma-Aldrich), 2.3 g/L sodium bicarbonate (Sigma-Aldrich), 370 µM hypoxanthine (Sigma-Aldrich), 25 mM HEPES, and 20 mg/L gentamycin (GIBCO Life Technologies). Cultures were maintained at 37°C under reduced oxygen conditions (5% CO2, 5% O2, and 90% N2).
Stable isotope labeling and metabolite extraction of P. falciparum-infected and uninfected RBCs for polyisoprenoids profiling
Mycoplasma-free parasite cultures were tightly synchronized by 5% sorbitol treatment. Each biological replicate was obtained from 75 mL of P. falciparum-infected RBC cultures at 5% hematocrit and 7% parasitemia. For stage-specific polyisoprenoid profiling, parasites in trophozoite and schizont stages were obtained from P. falciparum-infected RBC cultures starting with early ring stage at 7% parasitemia and harvesting parasites at each indicated stage. Parasites at ring stage were obtained from synchronized cultures starting with early rings of P. falciparum at 3% parasitemia and recovering parasites after 48 h incubation to avoid collecting cells after sorbitol treatment.
For metabolic labeling experiments using [1-13C]glucose, cultures in ring stage (5% hematocrit and 7% parasitemia) were first incubated in glucose-free medium for 2 h and then supplemented with 4 g/L of [1-13C]glucose (99% isotopic abundance, Cambridge Isotope Laboratories, CIL) for 22-24 h. For metabolic labeling experiments using [3-13C]IPP (99% isotopic abundance), cultures in ring stage were first treated with 10 μM fosmidomycin for 2 h and then supplemented with [3-13C]IPP:IPP (1:1, 200 μM) in the presence of fosmidomycin. Each biological replicate obtained for infected RBCs was accompanied by its corresponding uninfected human RBCs cultures as controls.
In order to assess the DOH/POH profile in early trophozoite culture of the PfPPRD conditional knockdown, parasites were grown in media supplemented either with 2.5 µg/mL Blasticidin S and 0.5 μM aTc, or only 2.5 µg/mL Blasticidin S for the indicated times (Fig. 7). 13C-Biolabeling experiments were performed under similar conditions to induce the knockdown and then metabolically labeled as described above.
In all cases, infected RBCs were released from the host cell by lysis with 0.03% saponin in cold phosphate-buffered saline (PBS) solution containing 2 g/L glucose and washed three times with PBS/glucose by centrifuging 7 min at 10,000 × g at 4°C and 10 μL were collected in the last wash to count parasites using a Countess cell counter (Thermo Fisher). Cell pellets were flash-frozen and stored at −80°C until metabolite extractions were performed.
Gametocyte stages were obtained using P. falciparum NF54 strain. Briefly, highly synchronous cultures at early ring stage were obtained by sorbitol treatment and purified using a MACS® magnetic affinity column (day 0) and set at 8-12% parasitemia and 5% hematocrit. On day 0 and 1, gametocytogenesis was induced by incubating cultures with 50% spent media and 50% fresh media for 40-48 h, as described previously (45). After reinvasion, asexual parasites were depleted from culture by addition of fresh media containing 20 U/mL of heparin for 4 days (day 2 to 5) and sorbitol treatment (46). After day 5, the media was replaced every two days until gametocytes reached stage IV. Development of parasites was monitored by microscopic evaluation of Giemsa-stained thin smears. For metabolic labeling experiments, stage III gametocyte cultures where obtained as described above (day 6 after induction). Then, parasites were incubated in glucose-free media for 2 h before adding [1- 13C]glucose (4 g/L) and incubated for 24 h when media was changed to add fresh [1-13C]glucose (4 g/L) and to complete a total of 48 h labeling until gametocytes reached stage IV.
Extraction of metabolites was performed by treating samples with 1 mL cold methanol and three consecutive extractions with 2 mL of hexane followed by pulse-vortex for 1 min and 10 min sonication in an ultrasonic bath. The upper phases were combined, dried under nitrogen and stored at −80°C until LC-HRMS analysis. For LC-HRMS analysis, samples were resuspended in 100 µL of methanol/acetonitrile/2-propanol (60:25:15, v/v) pulse vortex 5 times and sonicate for 10 min.
Ultra-performance liquid chromatography and high-resolution mass spectrometry analysis
Analysis of polyisoprenoids was performed on an ionKey/MS system composed of an ACQUITY UPLC M-Class, the ionKey source, and an iKey HSS T3, 100 Å, 1.8 µm (particle size), 150 µm × 100 mm column coupled to a SYNAPT G2-Si mass spectrometer (Waters Corporation, Milford, MA, USA). Isoprenoid separation was accomplished using a binary gradient system consisting of methanol/acetonitrile (3:1, v/v), with 10 mM ammonium formate + 0.1% formic acid (mobile phase A) and 2-propanol/acetonitrile (3:1, v/v) containing 10 mM ammonium formate + 0.1% formic acid (mobile phase B). Sample analysis was performed using a linear gradient over a total run time of 35 min. The gradient was programmed as follows: 0.0-2.0 min 18% B, 2-16 min from 18% B to 89% B, 16-20 min at 89 % B, 20-25 min from 89% B to 18% B, and re-equilibrated for 10 min. The flow rate was 3 µL/min, the injection volume was 5 µL in full loop mode, and the ionKey temperature was set at 40°C. MS analyses were performed acquiring in MSE mode from 80 to 1500 m/z in positive electrospray ionization mode with a capillary voltage of 3 kV. Data were collected in two channels all of the time: low collision energy (6.0 V), for the molecular ions, and high collision energy (15-40 V), for product ions. The source temperature was set at 110°C. Leucine enkephalin (50 pg/mL) was used as the lock mass (m/z 556.2771) with parameters set to 1 s scan at 20 s intervals. Infusion flow rate for lock mass was 1 µL/min. For unlabeled samples, peak identification, alignment, normalization, and statistical treatment of the data was performed using Progenesis QI software (Nonlinear Dynamics, Waters Corporation, Milford, MA, USA). For 13C-biolabeled samples, MassLynx software (Waters Corporation, Milford, MA, USA) was used for data processing and isotopologue labeling distribution analysis.
Heterologous expression of PfPPRD and analysis of CPY glycosylation in Saccharomyces cerevisiae transformants
HA-tagged DFG10 and PfPPRD (optimized for codon usage in yeast) were expressed under the control of the DFG10 endogenous promoter from a centromeric plasmid, pRS416. Empty vector pRS416 was included as a negative control. Saccharomyces cerevisiae strain BY4741 (MATa his3Δ1 leu2Δ met15Δ ura3Δ) and BY4741-based dfg10Δ strains were grown at 30°C in synthetic defined (SD) medium containing 2% galactose as the carbon source. After two passages (36 to 48 h) in SD medium, cells were harvested when the optical density at 600 nm (OD600) reached between 0.4 and 1.0. Two OD600 units of cells were harvest for protein analysis and 25 OD600 units of cells were harvested for analysis of polyisoprenoids by LC-HRMS.
Total proteins were extracted as described previously (47). Equal amount of proteins was analyzed using a 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a polyvinylidene difluoride (PVDF) membrane. Mouse anti-CPY (1:5000; Life Technologies) was used as the primary antibody; horseradish peroxidase (HRP)-conjugated anti-mouse antibody (1:5,000 dilution; Thermo Scientific) together with the Supersignal West Femto substrate (Thermo Scientific) were used to detect the target protein.
Generation of a P. falciparum strain with an inducible knockdown of PfPPRD
Genomic DNA was isolated from P. falciparum strain 3D7 and homology regions were PCR amplified using PrimeStar GXL DNA polymerase (Takara). The 3’-untranslated homology region (3’-UTR) was amplified using forward P1 and reverse P2 (Table S1). The C-terminal homology region was amplified using forward P3 and reverse primer with triple-hemagglutinin (HA) tag P4 or without HA tag P5. The 3’-UTR and C-terminal homology regions were combined using forward P1, reverse P4 or P5 and PrimeStar GXL DNA polymerase (Takara). The resulted PCR product was cloned into TetR-DOZI plasmid (37) by sequence-and ligation-independent cloning (SLIC) using T4 DNA polymerase (Thermo Scientific™) as previously described (48). Before transfection, the constructs PfPPRD-TetR-DOZI+HA and PfPPRD-TetR-DOZI were linearized with EcoRV (New England BioLabs). The RNA guide (gRNA) sequence was chosen using ChopChop online platform (http://chopchop.cbu.uib.no/). A set of primers forward P6 and reverse P7 were annealed and inserted into pUF1-Cas9-guide (49) as previously described (50). Briefly, pUF1-Cas9-guide was digested with BtgZI and annealed oligos were inserted using the SLIC method.
For transfection, 20 µg of PfPPRD-TetR-DOZI+HA or PfPPRD-TetR-DOZI plasmids were linearized with EcoRV (NEB), mixed with 60 µg of pUF1-Cas9-guide and precipitated. DNA pellet was dissolved in Cytomix Buffer (37) and combined with fresh red blood cells (RBCs). The DNA/RBC mixture was electroporated using BioRad Gene Pulser Xcell™ system at 0.32 kV, 925 μF. A culture of P. falciparum in schizont stage at high parasitemia was added to the RBCs and parasites were grown in the presence of 0.5 μM aTc (Cayman Chemical). Drug pressure was applied 48 hours post-transfection using 2.5 µg/mL Blasticidin S (BSD) (Sigma). Media was changed daily for the first 7 days and every other day thereafter. P. falciparum cultures were split 1:1 every 7 days to provide fresh uninfected RBCs and to perform DNA extraction for integration analysis.
Growth and morphological assessment of the PfPPRD-TetR-DOZI
In order to assess potential morphological defects due to the reduced expression of PfPPRD, P. falciparum cultures treated with BSD only or BSD+aTc as indicated above were monitored by Giemsa-stained smears every 24 h during 8 days. Potential growth defects were assessed by seeding cultures in a 96 well plate (0.5% early trophozoite stages at 4% hematocrit) and treated with BSD only or BSD+aTc with each condition tested in triplicate. Throughout the course of the experiment, parasites were sub-cultured to maintain the parasitemia between 1-5%. Parasitemia was monitored every 24 hours by flow cytometry using a CytoFLEX S (Beckman Coulter, Hialeah, Florida). A sample of 0.5 µL from each well was stained with Hoechst (Thermo Scientific) at 0.8 µg/mL in PBS and data were analyzed using Prism (GraphPad Software, Inc.). Relative parasitemia at each time point was back calculated based on actual parasitemia multiplied by the relevant dilution factors.
Statistical analysis
The t-test and Benjamini and Hochberg procedure were used to analyze differences between different conditions, and a false discovery rate of 0.01 was used to identify statistically significant differences.
Conflict of interest
The authors declare that they have no conflicts of interest with the contents of this article. “The content is solely the responsibility of the authors and does not necessarily represents the official views of the National Institutes of Health.”
Acknowledgements
This work was supported by the National Institutes of Health (AI108819 to M.B.C.) and American Heart Association Postdoctoral Fellowship (18POST34080315 to A.F.). The following reagents were obtained through MR4 as part of the BEI Resources Repository, NIAID, NIH: P. falciparum, strain NF54 (MRA-1000), contributed by M. Dowler, Walter Reed Army Institute of research and strain 3D7 (MRA-102) contributed by Daniel J. Carucci. We thank Julie Nelson and the CTEGD core facility for providing access to flow cytometry equipment. We thank Dennis Kyle for the gift of [3-13C]IPP. The yeast strains were kindly supplied to E.S. by Vincent Cantagrel. We thank Grant Butschek for comments and corrections.
References




