

SUMMARY
FSHD is caused by the loss of repression at the D4Z4 locus leading to DUX4 expression in skeletal muscle, activation of its early embryonic transcriptional program and muscle fiber death. While progress toward understanding the signals driving DUX4 expression has been made, the factors and pathways involved in the transcriptional activation of this gene remain largely unknown. Here, we describe the identification and characterization of p38α as a novel regulator of DUX4 expression in FSHD myotubes. By using multiple highly characterized, potent and specific inhibitors of p38α/β, we show a robust reduction of DUX4 expression, activity and cell death across FSHD1 and FSHD2 patient-derived lines. RNA-seq profiling reveals that a small number of genes are differentially expressed upon p38α/β inhibition, the vast majority of which are DUX4 target genes. Our results reveal a novel and apparently critical role for p38a in the aberrant activation of DUX4 in FSHD and support the potential of p38α/β inhibitors as effective therapeutics to treat FSHD at its root cause.

INTRODUCTION
Facioscapulohumeral muscular dystrophy (FSHD) is a rare and disabling disease with an estimated worldwide population prevalence of between 1 in 8,000-20,000 (Deenen et al., 2014; Statland and Tawil, 2014). Most cases are familial and inherited in an autosomal dominant fashion and about 30% of cases are known to be sporadic. FSHD is characterized by progressive skeletal muscle weakness affecting the face, shoulders, arms, and trunk, followed by weakness of the distal lower extremities and pelvic girdle. Initial symptoms typically appear in the second decade of life but can occur at any age resulting in significant physical disability in later decades (Tawil et al., 2015). There are currently no approved treatments for this disease.
FSHD is caused by aberrant expression of the DUX4 gene, a homeobox transcription factor in the skeletal muscle of patients. This gene is located within the D4Z4 macrosatellite repeats on chromosome 4q35. DUX4 is not expressed in adult skeletal muscle when the number of repeats is >10 and the locus is properly silenced (Lemmers et al., 2010). In the majority of patients with FSHD (FSHD1), the D4Z4 array is contracted to 1–9 repeat units on one allele. FSHD1 patients carrying a smaller number of repeats (1–3 units) are on average more severely affected than those with a higher number of repeats (8-9) (Tawil et al., 1996). Loss of these repetitive elements (referred to as contraction) leads to de-repression of the D4Z4 locus and ensuing aberrant DUX4 expression activation in skeletal muscle (de Greef et al., 2009; Wang et al., 2018). In FSHD2, patients manifest similar signs and symptoms as described above but genetically differ from FSHD1. These patients have longer D4Z4 repeats but exhibit similar derepression of the D4Z4 locus with low levels of DNA methylation (Calandra et al., 2016; Jones et al., 2014; 2015). This loss of chromatin repression is caused by mutations in SMCHD1, an important factor in the proper deposition of DNA methylation across the genome (Dion et al., 2019; Jansz et al., 2017). SMCHD1 has also been identified as the cause of Bosma arhinia microphthalmia syndrome (BAMS), a rare condition characterized by the lack of an external nose (Gordon et al., 2017; Mul et al., 2018; Shaw et al., 2017). Similarly, modifiers of the disease, such as DNMT3B, are thought to participate in the establishment of silencing (van den Boogaard et al., 2016).
DUX4 expression in skeletal muscle as a result of the D4Z4 repeat contraction or SMCHD1 mutations leads to activation of a downstream transcriptional program that causes FSHD (Bosnakovski et al., 2014; Homma et al., 2015; Jagannathan et al., 2016; Shadle et al., 2017; Yao et al., 2014). Major target genes of DUX4 are members of the DUX family itself and other homeobox transcription factors. Additional target genes include highly homologous gene families that are clustered on chromosomes, including the preferentially expressed in melanoma (PRAMEF), tripartite motif-containing (TRIM), methyl-CpG binding protein-like (MBDL), zinc finger and SCAN domain containing (ZSCAN) and ret-finger protein-like (RFPL) families (Geng et al., 2011; Shadle et al., 2017; Tawil et al., 2014; Yao et al., 2014). Expression of DUX4 and its downstream transcriptional program in skeletal muscle cells is toxic, leading to oxidative stress, interference with sarcomere organization, impairment of contractile function and cell death (Bosnakovski et al., 2014; Himeda et al., 2015; Homma et al., 2015; Rickard et al., 2015; Statland et al., 2015; Tawil et al., 2014).
Several groups have made progress towards understanding the molecular mechanisms regulating DUX4 expression (van den Boogaard et al., 2015; Campbell et al., 2018; van den Boogaard et al., 2016). However, factors that drive transcriptional activation of DUX4 in the skeletal muscle of FSHD patients are still largely unknown. By screening our annotated chemical probe library to identify disease-modifying small molecule drug targets that reduce DUX4 expression in FSHD myotubes, we have identified multiple chemical scaffolds that inhibit p38 α and β mitogen-activated protein kinase (MAPK). We found that inhibitors of p38α kinase or its genetic knockdown, reduce DUX4 and its downstream gene expression program in FSHD myotubes, thereby impacting the core pathophysiology of FSHD.
Members of the p38 MAPK family, composed of α, β, γ and δ, isoforms are encoded on separate genes and play a critical role in cellular responses needed for adaptation to stress and survival (Krementsov et al., 2013; Martin et al., 2015; Whitmarsh, 2010). In many inflammatory, cardiovascular and chronic disease states, p38 MAPK stress-induced signals can trigger maladaptive responses that aggravate, rather than alleviate, the disease process (Martin et al., 2015; Whitmarsh, 2010). Similarly, in skeletal muscle, a variety of cellular stresses including chronic exercise, insulin exposure and altered endocrine states, myoblast differentiation, reactive oxygen species as well as apoptosis have all been shown to induce the p38 kinase pathways (Keren et al., 2006; Zarubin and Han, 2005). Moreover, these pathways can be activated by a number of external stimuli, including pro-inflammatory cytokines and cellular stress environments, that lead to activation of the dual-specificity MAPK kinases MKK3 and MKK6. Activation of MKK3 and MKK6, which in turn phosphorylate p38 in its activation loop, trigger downstream phosphorylation events. These include phosphorylation of other kinases, downstream effectors like HSP27 and transcription factors culminating in gene expression changes in the nucleus (Cuenda and Rousseau, 2007; Kyriakis and Avruch, 2001; Viemann et al., 2004).
P38α is the most abundantly expressed isoform in skeletal muscle and it has an important role in the development of skeletal muscle tissue, controlling the activity of transcription factors that drive myogenesis (Knight et al., 2012; Segalés et al., 2016; Simone et al., 2004). P38α abrogation in mouse myoblasts inhibits fusion and myotube formation in vitro (Perdiguero et al., 2007; Zetser et al., 1999). However, conditional ablation of p38α in the adult mouse skeletal muscle tissue appears to be well-tolerated and alleviates some of the phenotypes observed in models of other muscular dystrophies (Wissing et al., 2014).
Here, we show that selective p38α/β inhibitors potently decrease the expression of DUX4, its downstream gene program and cell death in FSHD myotubes across a variety of FSHD1 and FSHD2 genotypes. Using RNA-seq and high content image analysis we also demonstrated that myogenesis is not affected at concentrations that result in downregulation of DUX4.
RESULTS
Identification of inhibitors of DUX4 expression
To model FSHD in vitro, we differentiated FSHD1 patient-derived immortalized myoblasts into skeletal muscle myotubes. We allowed myoblasts to reach >70% confluency and added differentiation medium lacking growth factors (Figure 1A) (Brewer et al., 2008; Krom et al., 2012; Thorley et al., 2016). After one day of differentiation, we detected DUX4 expression by RT-qPCR and its expression increased throughout the course of myogenic fusion and formation of post-mitotic, multinucleated FSHD myotubes (Figure 1B). Because of the stochastic and low expression levels of DUX4 in FSHD cells, we measured DUX4-regulated genes as an amplified readout of the expression and activity of DUX4. These include ZSCAN4, MBD3L2, TRIM43, LEUTX and KHDC1L which are among the most commonly described DUX4 targets (Chen et al., 2016; Geng et al., 2011; Jagannathan et al., 2016; Tasca et al., 2012; Wang et al., 2018; Whiddon et al., 2017; Yao et al., 2014). These genes were downregulated after DUX4 antisense oligonucleotide treatment of FSHD myotubes and were nearly undetectable or completely absent in FSHD myoblasts or wild-type myotubes (Figure 1C). We concluded that these transcripts were solely dependent on DUX4 expression in differentiating myotubes. Although a number of DUX4-dependent transcripts have been previously described, we selected an assay to specifically detect MBD3L2 for high-throughput screening because it displayed the best signal window of differential expression in our in vitro system comparing FSHD to healthy wildtype myotubes (Figure 1D). With this assay, we identified several small molecules that reduced MBD3L2 expression after 5 days of differentiation and treatment and showed good reproducibility across replicates (Figure 1E). Validating our results, we found several molecules identified previously to reduce DUX4 expression, including BET inhibitors and β-adrenergic agonists exemplified in Figure S1 (Campbell et al., 2017; Cruz et al., 2018). However, when treating differentiating FSHD myotubes in our assay, we observed a reduction in fusion as indicated by visual inspection and by the reduction of MYOG expression with BET inhibitors. Importantly, we identified multiple scaffolds that inhibit p38 α and β and strongly inhibit the expression of MBD3L2 without affecting differentiation.

Bromodomain containing proteins inhibitors (A) and β-adrenergic agonist reduced the expression of MBD3L2 in a concentration dependent manner as previously described (Campbell et al., 2017). Arrow indicates concentration at which effects in differentiation started to be observed by visual inspection.

(A) Schematic describing the cellular assay used to identify small molecules that result in the inhibition of DUX4 expression and activity. In short, immortalized FSHD myoblasts (C6, 6.5 D4Z4 RUs) were seeded in 96-well plates 2 days before differentiation was induced. After myoblasts reached confluence, media was replaced and compounds for treatment were added. At day 2, fusion was observed and at day 5, differentiated myotubes were harvested for gene expression analysis or fixed for immunostaining. Representative image of the alpha-actinin staining in differentiated myotubes. (B) DUX4 expression is rapidly induced after differentiation of immortalized FSHD myotubes in vitro. To measure DUX4 transcript, C6 FSHD myotubes were grown in 12-well plates similarly to A, cells were harvest on day 5 for RNA extraction. RT-qPCR was used to determine expression of DUX4 mRNA and its downstream gene MBD3L2 (normalized using HMBS as housekeeping). These transcripts were not detected in wild-type immortalized myotubes derived from healthy volunteers. (C) Canonical DUX4 target genes are specifically detected in FSHD myotubes and are downregulated when DUX4 is knocked down using a specific antisense oligonucleotide (ASO). RT-qPCR analysis was used to detect expression in immortalized myoblasts/myotubes. ASO knockdown in FSHD myotubes (mt) was carried out during the 5 days of differentiation. Bars indicate mean±SD. (D) A 96-well plate cell-based assay was optimized to screen for inhibitors of DUX4 expression. An assay measuring MBD3L2 by RT-qPCR was selected because of robust separation and specificity reporting DUX4 activity. MBD3L2 signal was normalized using POLR2A as a housekeeping gene. Bars indicate mean±SD. (E) Hits identified in small molecule screen potently reduced the activity of DUX4. X and Y axis show the normalized MBD3L2 signal obtained from the two replicate wells analyzed.
p38α signaling participates in the activation of DUX4 expression in FSHD myotubes
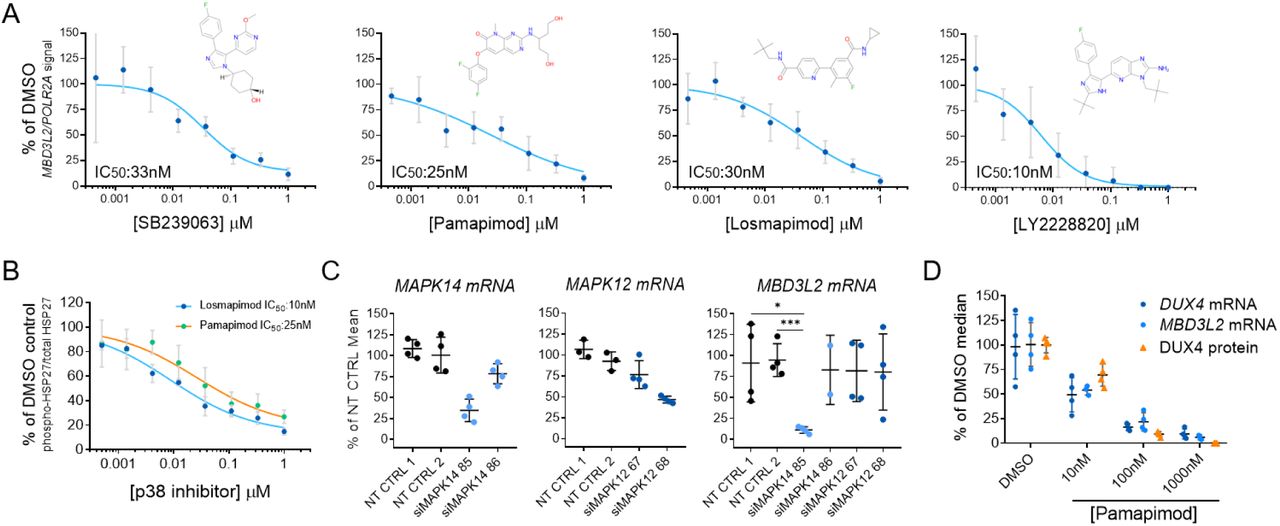
Potent and selective inhibitors of p38α/β have been previously explored in multiple clinical studies for indications associated with the role of p38α in the regulation of the expression of inflammatory cytokines and cancer (Coulthard et al., 2009). We tested several p38α/β inhibitors of different chemical scaffolds in our assays which showed significant inhibition of MBD3L2 expression (Figure 2A). Importantly, half maximal inhibitory concentrations (IC50) obtained for MBD3L2 reduction were comparable to reported values by other groups in unrelated cell-based assays that measured p38 α/β inhibition, suggesting the specificity for the assigned target (Campbell et al., 2014; Fehr et al., 2015; Underwood et al., 2000). P38α and β kinases phosphorylate a myriad of substrates, including downstream kinases like MAPKAPK2 (also known as MK2) which phosphorylates effector molecules such as heat shock protein 27 (HSP27), as well as a variety of transcription factors including myogenic transcription factors like MEF2C (Knight et al., 2012; Segalés et al., 2016a; Simone et al., 2004). To determine p38α/β signaling activity in differentiating myoblasts, we measured the levels of phosphorylation of HSP27. As reported previously, we observed increased p38 signaling rapidly upon addition of differentiation media (Figure S2) (Perdiguero et al., 2007). We observed P38α/β inhibitors reduced phosphorylated HSP27 levels with similar IC50 values to that of MBD3L2 (Figure 2B). To further validate our findings, we electroporated FSHD myoblasts with siRNAs against p38 α and β. After 3 days of differentiation, transient knockdown of p38α showed robust inhibition of expression of MBD3L2 in FSHD myotubes (Figure 2C) and no significant effects in fusion were observed (Figure S3). We observed that close to 50% reduction of MAPK14 (p38α) mRNA was sufficient to inhibit MBD3L2 expression without impacting myogenesis and this level of reduction may account for the differences on myogenesis observed between this study and those previously reported using p38 mouse knockout myoblasts (Perdiguero et al., 2007).
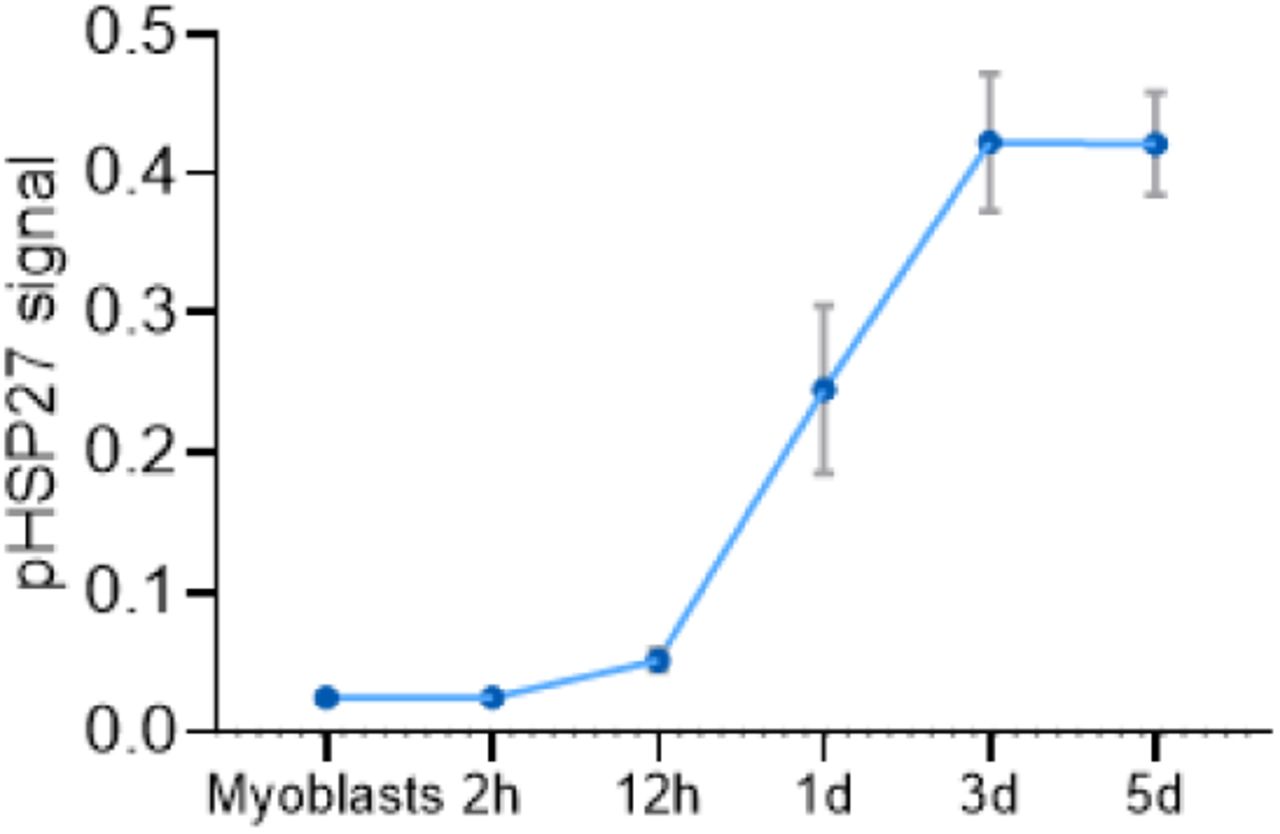
Levels of phosphorylated-HSP27 increase during myogenic differentiation in C6 FSHD myotubes.

Differentiation of C6 FSHD myotubes was not affected by MAPK12 and MAPK14 partial knockdown that resulted in MBD3L2 level reduction.
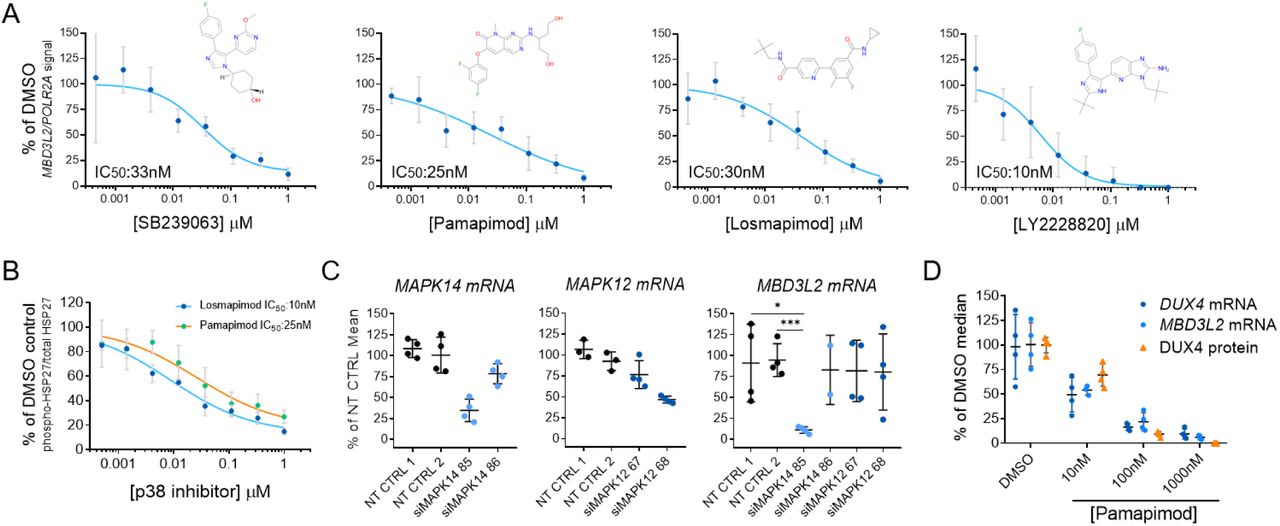
(A) Diverse inhibitors of p38α/β reduce the expression of MBD3L2 in differentiating FSHD myotubes. Concentration-dependent responses were observed with all tested inhibitors. Four replicates per concentration were tested to measure reduction of MBD3L2 in immortalized C6 FSHD myotubes and bars indicate mean±SD. (B) P38α/β pathway inhibition in C6 FSHD myotubes. The ratio between phosphorylated HSP27 to total HSP27 was measured by an immunoassay (MSD) after 12h of treatment of C6 FSHD myotubes with the indicated inhibitors. Half maximal inhibitory concentrations (IC50) observed for p-HSP27 were comparable to those obtained for reduction of MBD3L2 expression. Bars indicate mean±SD for four replicate wells. (C) Knockdown of p38α (MAPK14) results in reduction of MBD3L2 expression. Immortalized C6 myoblasts were electroporated with siRNAs specific for MAPK14 (p38α) and MAPK12 (p38β) plated and differentiated for 3 days. Expression of the indicated transcripts was measured using RT-qPCR and normalized against POLR2A. Reduction of MBD3L2 expression was observed when >50% knockdown of MAPK14 was achieved. Bars indicate mean±SD. (D) P38α/β inhibition results in the reduction of DUX4 expression. After inhibition, correlated reduction of DUX4 mRNA, protein and downstream gene MBD3L2 was observed. To measure DUX4 protein a novel immunoassay was developed using previously described antibodies (see methods and Figure S4). Bars indicate mean±SD, t-test p value * <0.01, *** 0.0002
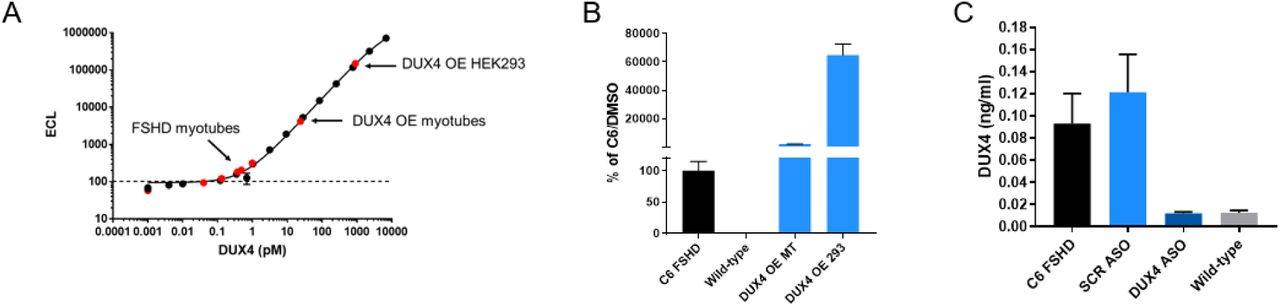
Specific detection of DUX4 protein in mesoscale electro-chemiluminescent immunoassay (A) Recombinant GST-DUX4 calibrator curve. (B) C6 FSHD or wild type 5-day differentiated myotubes, DUX4 overexpressed 1-day differentiated myotubes infected with DUX4 bacmam, DUX4 overexpressed in 293 cells transfected with CMV-DUX4 plasmid. (C) C6 FSHD myotubes treated with scrambled or DUX4 anti-sense oligonucleotide or wild type control.
Our results suggest the p38α pathway is an activator of DUX4 expression in FSHD muscle cells undergoing differentiation. To further understand the reduction in DUX4 expression, we measured the expression of DUX4 transcript and protein upon inhibition of p38 α and β. To measure protein, we developed a highly sensitive assay based on the electrochemiluminescent detection of DUX4 on the Mesoscale Diagnostics (MSD) platform using two previously generated antibodies (Figure S4). We observed that p38α/β inhibition resulted in a highly correlated reduction of DUX4 transcript and protein (Figure 2D). We concluded this led to the reduction in the expression of DUX4 target gene, MBD3L2.
p38 α and β inhibition normalizes gene expression of FSHD myotubes without impacting the myogenic differentiation program
We further examined the effect of p38 α and β selective inhibition on myotube formation because this pathway has been linked to muscle cell differentiation (Perdiguero et al., 2007; Segalés et al., 2016b; 2016a; Simone et al., 2004; Wissing et al., 2014). We developed a quantitative assay to measure cell fusion and myotube formation to assess skeletal muscle differentiation in vitro. In this assay, we stained immortalized FSHD myotubes cells using antibodies against Myosin Heavy Chains (MHC) and quantified the number of nuclei detected inside MHC-stained region. This provided a way quantitate the number of cells that successfully underwent the process of in vitro myogenesis. P38α/β inhibition by LY2228820 and GW856553X (losmapimod) did not impact differentiation of myoblasts into skeletal muscle myotubes. Treated cells fused properly at all tested drug concentrations to levels comparable to the DMSO control (Figure 3A).

(A) Quantification of myotube differentiation after p38α/β inhibition. Two inhibitors were used to demonstrate the effects of p38α/β inhibition in a high-content imaging assay to quantify the number of nuclei that properly underwent differentiation by activation of expression of myofiber specific proteins (i.e. MHC). No changes were observed in the morphology of C6 myotubes treated for 5 days. Bars indicate mean±SD. (B) Heat map representing fold change of expression levels of differentially expressed genes after p38α/β inhibition in FSHD myotubes for 5 days. 86 genes showed significant changes in expression after treatment with two different inhibitors (abs(FC)>4; FDR<0.001). Each condition was tested in triplicate represented as rows in the heatmap (C) DUX4 target genes are specifically downregulated by p38 inhibition. X-axis indicates the fold changes observed in members of the gene families indicated. Diameter of dots represent p-value.
We also further assessed gene expression changes in FSHD myotubes upon p38α/β inhibition. We performed RNA-seq analysis of FSHD and WT myotubes after four days of treatment with vehicle or p38α/β inhibitors. Inhibition of the p38 signaling pathway during differentiation did not induce significant transcriptome changes, and resulted in less than 100 differentially expressed genes (abs(FC)>4; FDR<0.001). Around 80% of these differentially expressed genes were known DUX4-regulated transcripts and were all downregulated after p38 α and β inhibition (Figure 3B). This set of DUX4-regulated genes overlapped significantly with genes upregulated in FSHD patient muscle biopsies (Wang et al., 2018). Moreover, key driver genes of myogenic programs such as MYOG, MEF and PAX genes and markers of differentiation such as myosin subunits and sarcomere proteins were not affected by p38 inhibition (Figure 3C).
Inhibition of DUX4 expression results in the reduction of cell death in FSHD myotubes
DUX4 activation and downstream DUX4-regulated target gene expression in muscle cells is toxic, leading to oxidative stress, changes in sarcomere organization, and apoptosis, culminating in reduced contractility, and muscle tissue replacement by fat (Block et al., 2013; Bosnakovski et al., 2014; Choi et al., 2016; Homma et al., 2015; Rickard et al., 2015; Tawil et al., 2014). In particular, apoptotic cells have been detected in skeletal muscle of FSHD patients supporting the hypothesis that programmed cell death is caused by aberrant DUX4 expression and contributes to FSHD pathology (Sandri et al., 2001; Statland et al., 2015). To test this hypothesis in vitro, we evaluated the effect of p38α/β inhibition on apoptosis in FSHD myotubes. We used an antibody recognizing caspase-3 cleavage products by immunofluorescence to quantify changes in the activation of programmed cell death. Cleavage of caspase-3 is a major step in the execution of the apoptosis signaling pathway, leading to the final proteolytic steps that result in cell death (Dix et al., 2008; Fuentes-Prior and Salvesen, 2004; Mahrus et al., 2008). We detected activated caspase-3 in FSHD but not in wild-type myotubes and observed a stochastic pattern of expression of DUX4 in FSHD as previously reported (Figure 4A) (van den Heuvel et al., 2018; Jones et al., 2012; Snider et al., 2010). Levels of cleaved caspase-3 were reduced in a concentration-dependent manner with an IC50 similar to what we observed for inhibition of the p38 pathway and DUX4 expression (Figure 4B). Moreover, we measured SLC34A2, a DUX4 target gene product using a similar immunofluorescence assay (Figure 3B). This protein was expressed in a similar stochastic pattern observed for active caspase-3 and its expression was also reduced by p38α/β inhibition (Figure 4B and C). Our results demonstrate that DUX4 inhibition in FSHD myotubes results in a significant reduction of apoptosis.

(A) A high-content imaging assay was developed to measure cleaved caspase-3 in differentiating myotubes. C6 FSHD myotubes were differentiated and treated for 5 days as indicated above and stained to measure MHC, cleaved-caspase-3 and nuclei. Representative images show that cleaved caspase-3 was only detected in FSHD myotubes, not in wild-type controls or after inhibition of the p38 pathway. Six replicates were imaged and cleaved caspase-3 signal under MHC staining was quantified. (B) Stochastic expression of DUX4 target gene, SLC34A2, in C6 FSHD myotubes. Expression of SLC34A2 was measured by immunostaining in similar conditions as image above. No expression was detected in wild-type control or p38 inhibitor-treated myotubes. Signal of SLC34A2 under MHC staining was quantified in two replicates (C) Concentration-dependent inhibition of the expression of DUX4 target genes is highly correlated to the inhibition of programmed cell death in C6 myotubes. Bars indicate mean±SD.
p38 α and β inhibition results in downregulation of DUX4 expression and suppression of cell death across multiple FSHD1 and FSHD2 genotypes
FSHD is caused by the loss of repression at the D4Z4 locus leading to DUX4 expression in skeletal muscle due to the contraction in the D4Z4 repeat arrays in chromosome 4 or by mutations in SMCHD1 and other modifiers such as DNMT3B. Primary FSHD myotubes were used to study the in vitro efficacy of p38α/β inhibitors across different genotypes. We tested eight FSHD1 primary myoblasts with 2-7 D4Z4 repeat units and three FSHD2 cell lines with characterized SMCHD1 mutations. Upon differentiation, the primary cells tested expressed a wide range of MBD3L2 levels (Figure 5A, number of D4Z4 repeat units or SMCHD1 mutation indicated in parenthesis), comparable to what we and others have observed in other FSHD myotubes (Jones et al., 2012). However, we observed significant inhibition of the DUX4 program expression following treatment with multiple p38α/β inhibitors in all primary myotubes tested from FSHD1 and FSHD2 patients (Figure 5B). Furthermore, this reduction in the DUX4 program resulted in concomitant reduction of cleaved caspase-3 (Figure 5C) without any measurable effects on myotube differentiation (Figure 5D). Our results suggest that the p38α/β pathway critically regulates the activation of DUX4 independently of the mutation driving its expression in FSHD muscle cells.

(A) Levels of MBD3L2 expression across different primary and immortalized myotubes determined RT-qPCR. DUX4 activity is only detected in FSHD1/2 lines after 4 days of differentiation. Bars indicate mean±SD and repeat number is indicated in parenthesis in FSHD1 lines and SMCHD1 mutation for FSHD2 lines used. (B) Inhibition of the p38α/β pathway results in potent reduction of MBD3L2 expression activation across the entire set of FSHD primary cells tested. Three different inhibitors were used, and each circle indicates a different FSHD cell line tested. FSHD1 in blue and FSHD2 in green. Expression levels were measured by RT-qPCR in six replicates. (C and D) p38α/β pathway inhibition reduces activation of programmed cell death across primary FSHD cell lines with different genotypes. Stochastic activation of caspase-3 in a small number of FSHD myotubes was detected by immunostaining and quantified in all lines. Six replicates were used to quantify signal of cleaved caspase-3 under MHC stained myotubes. Wilconox test, P value **0.002, ***0.0002.
DISCUSSION
Recent studies have advanced our understanding of the mechanisms that normally lead to the establishment and maintenance of repressive chromatin at the D4Z4 repeats. Similar to other repetitive elements in somatic cells, chromatin at this locus is decorated by DNA methylation and other histone modifications associated with gene silencing, such as H3K27me3 and H3K9me3 (Cabianca et al., 2011; Huichalaf et al., 2014; van Overveld et al., 2003; van den Boogaard et al., 2016; Zeng et al., 2009). Factors involved in the deposition of these modifications like SMCHD1 and DNMT3B have been identified by genetic analysis of affected FSHD populations (Calandra et al., 2016; Lemmers et al., 2012; van den Boogaard et al., 2016). Other factors like NuRD and CAF1 have been identified by biochemical approaches isolating proteins that associate with the D4Z4 locus (Campbell et al., 2018). However, sequence-specific transcriptional activators of DUX4 have remained elusive not only in skeletal muscle but also in the regulation of DUX4 in the developing embryo, where this factor is normally expressed. Because of the effects of expression of DUX4 in FSHD and the apparent tissue specific expression of DUX4 in skeletal muscle, it has been hypothesized that myogenic regulatory elements upstream of the D4Z4 repeats regulate the expression of DUX4 in FSHD (Himeda et al., 2014), yet this finding has not led to the identification of other factors that can specifically activate DUX4.
In this study, by modelling FSHD in vitro and screening a library of probe molecules, we identified p38α as a novel activator of DUX4 expression in patient-derived FSHD cells. This signaling kinase directly phosphorylates transcription factors involved in myogenesis and may signal directly to activate DUX4 expression in differentiating myoblasts. Using highly selective and potent small molecules extensively characterized previously, we have studied the pharmacological relationships between the inhibition of this signaling pathway and the inhibition of the expression of DUX4, its downstream gene program expression and its consequences in muscle cells from FSHD patients. These relationships are maintained across multiple FSHD genotypes, including FSHD1 and FSHD2, indicating that this mechanism acts independent of the genetic lesion present in these patients. Our studies show a specific effect of p38 α and β inhibition in downregulation of the DUX4 program and normalization of gene expression compared to cells from healthy donors. Notably, no effects in differentiation were detected at the tested concentrations of p38 inhibitor.
Other recent efforts to identify targets for the treatment of FSHD have reported similar studies in which the investigators followed the expression of MBD3L2 as a readout for DUX4 expression or by using a reporter driven by the activity of DUX4 in immortalized FSHD myotubes in vitro (Campbell et al., 2017; Cruz et al., 2018). Our results have reproduced their identification of β-adrenergic agonists and BET inhibitors as inhibitors of DUX4 expression. However, these molecules also caused downregulation of the transcription factor MYOG expression or affected myoblasts fusion at concentrations similar to the half maximal inhibitory concentration for DUX4 expression inhibition in our model (Figure S1B, lack of fusion indicated by arrow).
In previous clinical studies in non-FSHD indications under an anti-inflammatory therapeutic hypothesis, many p38 α/β inhibitors were tested extensively and shown to be safe and tolerable, however they never met efficacy endpoints including in diseases such as rheumatoid arthritis, chronic obstructive pulmonary disease and acute coronary syndrome (Barbour et al., 2013; Damjanov et al., 2009; Hammaker and Firestein, 2010; Hill et al., 2008; MacNee et al., 2013; Norman, 2015; Patnaik et al., 2016). Here, we present evidence from in vitro studies that support the therapeutic hypothesis of treatment of FSHD at its root cause, prevention or reduction of aberrant expression of DUX4, via inhibition of p38 α/β.
MATERIALS AND METHODS
Cell lines and cell culture
Immortalized myoblasts from FSHD (AB1080FSHD26 C6) and healthy individuals (AB1167C20FL) were generated and obtained from the Institut Myologie, France. In short, primary myoblast cultures were obtained from patient samples and immortalized by overexpression of TERT and CDK4 (Krom et al., 2012). Primary myoblasts were isolated from FSHD muscle biopsies and were obtained from University of Rochester.
Immortalized myoblasts were expanded on gelatin-coated dishes (EMD Millipore, #ES-006-B) using Skeletal muscle cell growth media (Promocell, #C-23060) supplemented with 15% FBS (ThermoFisher, #16000044). Primary myoblasts were also expanded on gelatin-coated plates but using media containing Ham’s F10 Nutrient Mix (ThermoFisher, #11550043), 20% FBS and 0.5% Chicken embryo extract (Gemini Bio-product, #100-163P). For differentiation, immortalized or primary myoblasts were grown to confluency in matrigel-coated plates (Corning, #356234) and growth media was exchanged for differentiation media (Brainbits, #Nb4-500) after a PBS wash. DMSO (vehicle) or compounds (previously dissolved in DMSO at 10 mM stock concentrations) were added at the desired concentration at the time differentiation media was exchanged and maintained in the plates until harvesting or analysis.
Small molecule compounds and antisense oligonucleotides
SB239063, Pamapimod, LY2228820 and Losmapimod were purchased from Selleck Chem (#S7741, S8125, S1494 and S7215). 10 mM stock solutions in DMSO were maintained at room temperature away from light. DUX4 antisense oligonucleotides (gapmer) were purchased from QIAGEN and were designed to target exon 3 of DUX4. The lyophilized oligos were resuspended in PBS at 25 mM final concentration and kept frozen at −20°C until used. This antisense oligonucleotide was added to cells in growth media 2 days before differentiation and maintained during the differentiation process until harvesting.
Detection of DUX4 and target gene expression by RT-qPCR
RNA from myotubes was isolated from C6 FSHD cells differentiated in 6-well plates using 400 μl of tri-reagent and transfer to Qiagen qiashredder column (cat#79656). An equal amount of 100% Ethanol was added to flow through and transferred to a Direct-zol micro column (Zymo research cat# 2061) and the manufacturers protocol including on-column DNA digestion was followed. RNA (1 μg) was converted to cDNA using Superscript IV priming with oligo-dT (Thermofisher cat# 18091050). Pre-amplication of DUX4 and housekeeping gene HMBS was performed using preamp master mix (Thermofisher cat#4384267) as well as 0.2X diluted taqman assays (IDT DUX4 custom; forward Forward: 5’-GCCGGCCCAGGTACCA-3’, Reverse: 5’-CAGCGAGCTCCCTTGCA-3’, and Probe: 5’-/56-FAM/CAGTGCGCA/ZEN/CCCCG/3IABkFQ/-3’; and HMBS HS00609297m1-VIC). After 10 cycles of pre-amplification, reactions were diluted 5-fold in nuclease-free water and qPCR was performed using taqman multiplex master mix (Thermofisher cat#4461882).
To measure DUX4 target gene expression in a 96-well plate format, cells were lysed into 25 μL Realtime Ready lysis buffer (Roche, #07248431001) containing 1% RNAse inhibitor (Roche, #03335399001) and 1% DNAse I (ThermoFisher, #AM2222) for 10 min while shaking on a vibration platform shaker (Titramax 1000) at 1200 rpm. After homogenization, lysates were frozen at −80°C for at least 30 min and thawed on ice. Lysates were diluted to 100 μL using RNase-free water. 1 μL of this reaction was used for reverse transcription and preamplification of cDNA in a 5 μL one-step reaction using the RT enzyme from Taqman RNA-to-Ct (ThermoFisher, #4392938) and the Taqman Preamp Master Mix (ThermoFisher, #4391128) according to manufacturer’s specifications. This preamplification reaction was diluted 1:4 using nuclease-free water, 1μL of this reaction was used as input for a 5 μL qPCR reaction using the Taqman Multiplex Master Mix (ThermoFisher, #4484262). Amplification was detected in a Quantstudio 7 Flex instrument from ThermoFisher. The following Taqman probes were purchased from ThermoFisher; MBD3L2 Taqman Assay (ThermoFisher, Hs00544743_m1, FAM-MGB). ZSCAN4 Taqman Assay (ThermoFisher, Hs00537549_m1, FAM-MGB). LEUTX Taqman Assay (Thermo Fisher, Hs01028718_m1, FAM-MGB). TRIM43 Taqman Assay (ThermoFisher, Hs00299174_m1, FAM-MGB). KHDC1L Taqman Assay (ThermoFisher, Hs01024323_g1, FAM-MGB). POLR2A Taqman Assay (ThermoFisher, Hs00172187_m1, VIC-MGB).
Detection of HSP27 by Electrochemiluminescence
Total and phosphorylated HSP27 was measured using a commercial MesoScale Discovery assay, Phospho (Ser82)/Total HSP27 Whole Cell Lysate Kit (MesoScale Discovery, # K15144D). Myotubes were grown in 96-well plates using conditions described above and were lysed using 25 μL of 1X MSD lysis buffer with protease and phosphatase inhibitors. The lysates were incubated at room temp for 10 minutes with shaking at 1200 rpm using Titramax 1000. Lysates were stored at −80 °C until all timepoints were collected. Lysates were then thawed on ice and 2 μL were used to perform a BCA protein assay (ThermoFisher, # 23225). 10 μL of lysate were diluted 1:1 in 1X MSD lysis buffer and added to the 96-well Mesoscale assay plate. Manufacturer instructions were followed, and data was obtained using a MesoScale Discovery SECTOR S 600 instrument.
Myotube nuclei isolation and detection of DUX4 by Electrochemiluminescence
DUX4 was measured using a novel MesoScale Discovery assay developed at Fulcrum Therapeutics. Anti-DUX4 monoclonal capture antibody (clone P2B1) was coated overnight at 5 μg/ml in 0.1 M sodium Bicarbonate pH=8.4 onto a Mesoscale 384 well plate (L21XA). The plate was blocked with 5% BSA/PBS for at least 2 hours. Human FSHD myotubes grown in 100 mm plates in the conditions described above were harvested 4 days post differentiation using TrypLE express solution (Gibco, #12605-010), neutralized with growth media and the myotubes were pelleted by centrifugation. Myotubes were resuspended in ice cold nuclei extraction buffer (320 mM Sucrose, 5 mM MgCl2, 10 mM HEPES, 1% Triton X-100 at pH=7.4). Nuclei were pelleted by centrifugation at 2000 xg for 4 minutes at 4°C. Nuclei were resuspended in ice cold wash buffer (320 mM Sucrose, 5 mM MgCl2, 10 mM HEPES at pH=7.4) and pelleted by centrifugation at 2000 xg for 4 minutes at 4°C. Nuclei were suspended in 150 μl of RIPA buffer at 4°C (+150 mM NaCl). Extracts were diluted 1:1 with assay buffer and 10 μl per well was added to 384 well pre-coated/blocked MSD plate and incubated for 2 hours. Anti-DUX4-Sulfo Conjugate (clone E5-5) was added to each well and incubated for two hours. Plates were washed and 40 μl per well of 1X Read T buffer was added. Data was obtained using a MesoScale Discovery SECTOR S 600 instrument.
Quantitative Immunofluorescent detection of Myosin Heavy Chain, SLC34A2 and cleaved Caspase-3
Myotubes were grown and treated as described above. At day 5 after differentiation was induced, cells were fixed using 4% paraformaldehyde in PBS during 10 min at room temperature. Fixative was washed, and cells were permeabilized using 0.5% Triton X-100 during 10 min at room temperature. After washing, fixing and permeabilizing, the cells were blocked using 5% donkey serum in PBS/0.05% Tween 20 during 1 h at room temperature. Primary antibodies against MHC (MF20, R&D systems, #MAB4470), SLC34A2 (Cell signaling, #66445) and active Caspase-3 (Cell signaling, #9661) were diluted 1:500 in PBS containing 0.1% Triton X-100 and 5% donkey serum and incubated with cells for 1 h at room temperature. After 4 washes, secondary antibodies were added (ThermoFisher, #A32723 and # R37117) in a 1:2000 dilution and incubated during 1 h at room temperature. During the last 5 min of incubation a 1:2000 dilution of DAPI was added before proceeding with final washes and imaging. Images were collected using the CellInsight CX7 (ThermoFisher). Images were quantified using HCS Studio Software. Differentiation was quantified by counting the percentage of nuclei in cells expressing MHC from the total of the well. SLC34A2 and active Caspase-3 signal was quantified by colocalization of cytoplasmic cleaved Caspase-3 within MHC expressing cells.
Knockdown of MAPK12 and MAPK14 in FSHD myotubes
Exponentially dividing immortalized C6 FSHD myoblasts were harvested and counted. 50000 myoblasts were electroporated using a 10 μL tip in a Neon electroporation system (ThermoFisher). Conditions used were determined to preserve viability and achieved maximal electroporation (Pulse V=1100V, pulse width=40 and pulse #=1). After electroporation, cells were plated in growth media and media was changed for differentiation 24h after. 3 days after differentiation, cells were harvested and analyzed for KD and effects in MBD3L2 using the RT-qPCR assay described before. siRNAs used were obtained from ThermoFisher (4390843, 4390846, s3585, s3586, s12467, s12468).
Gene expression analysis by RNA-seq
RNA from myotubes grown in 6-well plates in conditions described above was isolated using the RNeasy Micro Kit from Qiagen (#74004). Quality of RNA was assessed by using a Bioanalyzer 2100 and samples were submitted for library preparation and deep sequencing to the Molecular biology core facility at the Dana Farber Cancer Institute. After sequencing, raw reads of fastq files from all samples were mapped to hg38 genome assemblies using ArrayStudio aligner. Raw read count and FPKM were calculated for all the genes, and DESeq2 was applied to calculate differentially expressed genes using general linear model (GLM). Statistical cutoff of absolute fold change (abs(FC) > 4, FDR < 0.001) were applied to identify differentially expressed protein coding genes. (DATA DEPOSITION INFO TBD)
ACKNOWLEDGEMENTS
We thank Peter Jones, Takako Jones and Charis Himeda from the University of Reno for technical advice and guidance during the development of assays in this manuscript and insightful discussions about the regulation of DUX4 expression. Peter Jones for providing us with constructs for DUX4 overexpression used to validate our DUX4 protein detection assay. Vincent Mouly (Institut Myologie) and Silvère Van der Maarel (LUMC) for providing access to immortalized myoblasts lines. In addition, the authors would like to thank members of Fulcrum Therapeutics for helpful discussions throughout the project. We would also like to thank patients participating in previous studies that have provided tissues to generate cell lines used in this manuscript.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}