

Abstract
Understanding how cells spatially organize signaling events is important in normal biology and pathological conditions such as cancer. Here, we uncover a membraneless, protein granule-based subcellular structure that can organize receptor tyrosine kinase (RTK)-mediated RAS/MAPK pathway signaling, which is thought to occur exclusively from lipid-membrane compartments in mammalian cells. De-novo assembly of cytoplasmic protein granules by certain RTKs, including oncogenic gene fusions involving ALK and RET, is dependent on multimerization domains in the RTK fusion partners. Protein granule formation is both necessary and sufficient to locally concentrate the RAS activating complex GRB2/SOS1 to initiate MAPK pathway signaling. Our findings reveal membraneless, higher-order protein assembly as a principle by which cells can organize kinase-mediated proliferative signals.
One Sentence Summary Kinase/RAS signaling via protein granules
Main Text
The organization of cellular signaling events is enabled by subcellular compartments. For example, the plasma membrane (PM) serves as a scaffold to spatially concentrate signaling molecules in receptor tyrosine kinase (RTK)-mediated RAS GTPase/MAPK (mitogen activated protein kinase) pathway signaling [1]. Recent reports also demonstrate that the PM resident T-cell receptor and associated proteins undergo phase separation in the presence of lipid bilayers to physically compartmentalize signaling events [2, 3]. On the other hand, non-lipid-membrane structures represent a field of emerging complexity, with recent studies highlighting alternative means of subcellular compartmentalization through primarily protein-based membraneless organelles such as P-bodies, nucleoli and stress granules [4, 5]. These subcellular structures, which we generally refer to as protein granules, exhibit a spectrum of liquid-to gel-like properties which impact the dynamics and enrichment of granule constituents [6]. The full extent and mechanistic underpinnings of protein granules in organizing cell signaling pathways, particularly in diseases such as cancer, remain to be elucidated.
RTK/RAS/MAPK signaling is broadly important in regulating the proliferation and survival of normal human cells and is often hyper-activated through various mechanisms in human cancer [7]. Prominent examples are chromosomal rearrangements involving RTKs such as anaplastic lymphoma kinase (ALK) or RET, which generate chimeric (fusion) oncoproteins that are validated therapeutic targets across multiple cancer subtypes [8, 9]. Virtually all oncogenic ALK fusion proteins retain the intracellular domain of ALK but lack the native transmembrane domain [8]. We discovered that the echinoderm microtubule-associated protein-like 4 (EML4)-ALK fusion oncoprotein present in lung and other cancer subtypes is exquisitely dependent upon RAS GTPase activation and downstream RAF/MEK/ERK (MAPK pathway) signaling for its oncogenic output [10]. We and other groups showed that EML4-ALK is not localized to the PM, but instead to intracellular, punctate cytoplasmic structures of unknown identity [10, 11]. This specific intracellular localization is essential for EML4-ALK to activate RAS and downstream MAPK signaling [10]. Neither the biophysical and biochemical nature of these cytoplasmic structures nor the mechanism through which they promote oncogenic signaling is clear.
To answer these questions, we focused our initial study on EML4-ALK variant 1, the most common form in human cancers [12]. Given the well-established requirement for lipid membranes in mediating RAS GTPase activation [13, 14], we first tested whether EML4-ALK localizes to an intracellular lipid-membrane containing structure. Live-cell microscopy in a nontransformed human lung epithelial cell line (Beas2B) expressing fluorescent-protein-tagged EML4-ALK showed no significant colocalization of EML4-ALK cytoplasmic puncta with plasma or intracellular membranes as marked by a membrane intercalating dye, nor with a panel of established protein markers labeling canonical intracellular lipid-containing organelles [15] (Fig. S1). Additionally, subcellular fractionation in patient-derived cancer cell lines expressing endogenous EML4-ALK protein produced an EML4-ALK fractionation pattern unaffected by membrane-solubilizing detergents, which was distinct from the pattern of PM (epidermal growth factor receptor, EGFR) or internal membrane proteins (calnexin and early endosome antigen 1, EEA1) but similar to that of well-known cytoplasmic protein granule constituents (the P-body protein decapping mRNA 1B, DCP1B [16]) (Fig. 1, A and B, and Fig. S2). We confirmed by fluorescence microscopy that EML4-ALK puncta do not co-localize with P-bodies, suggesting they form distinct protein-based cytoplasmic structures consistent with protein granules (Fig. S1). We validated by immunofluorescence (IF) the similar presence of EML4-ALK puncta in patient-derived cancer cells (H3122) that endogenously express this variant and in Beas2B cells expressing FLAG-tagged EML4-ALK (Fig. S3, A and B), verifying that these puncta were not the result of artificial expression or fluorescent protein-mediated multimerization [17].

(A, B) Subcellular fractionation by ultracentrifugation +/− detergent (1% Triton X-100) in EML4-ALK expressing cancer cell line H3122, followed by Western blotting. EML4-ALK and DCP1B are statistically distinct (p < 0.01) from the lipid membrane-associated proteins, which shift from the insoluble fraction (pellet) to the supernatant (sup) with detergent. Percent in pellet calculated as ratio of the insoluble to supernatant fractions as assessed by Western blotting, N=3. (C) SIM images of 2 distinct YFP::EML4-ALK puncta in Beas2B cells. SIM box size: 2 μm × 2 μm × 2 μm. (D) FRAP analysis of YFP::EML4-ALK expressed in human epithelial cell line Beas2B. Each curve represents photobleaching and recovery of fluorescence intensity for an individual EML4-ALK puncta. N = 30 cells. (E) Live-cell confocal imaging in Beas2B with endogenous mNG2-tagging of GRB2, GAB1, and SOS1 in the presence or absence of mTagBFP2::EML4-ALK. White arrows indicate a representative EML4-ALK cytoplasmic protein granule with local enrichment of respective signaling proteins (multiple non-highlighted granules also show colocalization between EML4-ALK and signaling proteins). (F) Quantification of colocalization between EML4-ALK granules and relevant signaling proteins, at least 100 total cells were scored in each condition over 3 independent experiments. (G) Fold-enrichment of signaling proteins at EML4-ALK granules +/− 24 hour treatment with 1 μM Crizotinib (criz). For all panels, error bars represent ± SEM, ** p < 0.01.
We further investigated the biophysical nature of EML4-ALK cytoplasmic structures using a suite of established cellular assays for protein granules [18, 19]. Live-cell microscopy of EML-ALK puncta rarely showed fission or fusion events. Higher resolution images by Structured Illumination Microscopy (SIM) revealed that these puncta exhibit porous and curvilinear features corresponding to gel-like granules, in contrast to the more uniform appearance characteristic of liquid-like granules (Fig. 1C) [4, 18]. Consistent with a partially gel-like state, fluorescence recovery after photo-bleaching (FRAP) showed an overall low fraction of exchange of EML4-ALK between the puncta and the surrounding cytosol (Fig. 1D). This recovery fraction was heterogeneous, varying from negligible to 40% in a few minutes among puncta within the same cell, possibly reflecting an on-going aging process from more liquid-like to more gel-like on the experimental timescale [19]. We also found that EML4-ALK puncta mostly persist after hexanediol treatment, which potently disrupts many liquid-like protein granules (Fig. S4) [20]. These data suggest that oncogenic EML4-ALK mostly exists in gel-like cytoplasmic protein granules.
To uncover the connection between EML4-ALK granules and RAS activation, we created a library of gene-edited Beas2B cell lines by introducing a split mNeonGreen21-10/11 tag (mNG2) at the endogenous locus of canonical adaptor and effector proteins in the RTK/RAS/MAPK signaling pathway, including GRB2, GAB1, SOS1, RAS GTPases (H/N/K isoforms) and RAF (A/B/C isoforms) [21]. This suite of isogenic cell lines avoids potential biases that can arise when overexpressing labeled proteins or fixing and permeabilizing cells for immunofluorescence. In this set of cell lines, we found that expression of EML4-ALK specifically re-localized key upstream RAS pathway proteins, including GRB2, GAB1, and SOS1, from a diffuse cytosolic pattern to the discrete EML4-ALK granules (Fig. 1, E and F) but not to the PM. This is distinct from the pattern of PM re-localization seen in the control case of expressing an oncogenic form of the transmembrane RTK EGFR (Fig. S5). Treatment with the ALK kinase inhibitor crizotinib for 24 hours substantially reduced the recruitment of these adaptor proteins, indicating that this process requires ALK kinase activation (Fig. 1G). We orthogonally confirmed recruitment of the key adaptor, GRB2, both in patient-derived cancer cells and through dual overexpression of EML4-ALK and GRB2 (Fig. S3, A and C). Additionally, we observed a low but heterogeneous FRAP recovery behavior for GRB2 at the granules similar to that of EML4-ALK (Fig. S6).
Our imaging and biochemical data led to the unanticipated hypothesis that RAS GTPase activation may occur via a non-lipid-membrane containing structure (e.g. EML4-ALK membraneless protein granules), potentially through a cytosolic pool of RAS that is known to exist but with unclear functional significance [22] (see Supplementary Note 1). We directly tested whether cytosolic RAS can become activated in a lipid-membrane-independent manner by EML4-ALK protein granules. While the expression of either EML4-ALK or the PM-localized oncogenic EGFR increased RAS-GTP levels (Fig. 2A and Fig. S7), only the former increased RAS-GTP levels of established, mutant forms of RAS (KRAS-C185S, H/NRAS-C186S) that abrogate membrane targeting and are retained exclusively in the cytosol [14] (Fig. 2B and Fig. S7, see Supplementary Note 1 on RAS prenylation). Furthermore, inhibition of EML4-ALK with crizotinib in H3122 patient-derived cancer cells suppressed not only wild-type RAS-GTP levels, but also the levels of GTP-bound, cytosolic KRAS-C185S (Fig. 2, C and D). Control experiments treating a distinct patient-derived cancer cell line HCC827 expressing endogenous oncogenic EGFR (PM-localized) with an established EGFR inhibitor [23] confirmed suppression of wild-type RAS-GTP levels but showed no effect on KRAS-C185S RAS-GTP levels (Fig. S8). These findings demonstrate the specificity of cytosolic RAS activation by membraneless EML4-ALK cytoplasmic protein granules.

(A, B) Stable expression of KRAS wild-type or C185S cytosolic mutant in 293T cells, followed by transfection of empty vector (EV), EML4-ALK, or oncogenic EGFR. RAS-GTP levels normalized to relevant total RAS species (KRAS wild-type or C185S) and then standardized against EV, N=3. (C, D) EML4-ALK expressing H3122 cancer cell line with stable expression of KRAS wild-type or cytosolic KRAS C185S mutant +/− two hours of 100 nM crizotinib. RAS-GTP levels normalized to relevant total RAS species (KRAS wild-type or C185S) and then standardized against DMSO treated H3122 cells (no drug), N=3. (E) Live-cell confocal imaging of mTagBFP2::EML4-ALK expressing human epithelial cell line Beas2B with mEGFP::ARAF or mEGFP::ARAF-R52L. White arrows indicate a representative EML4-ALK cytoplasmic protein granule with local enrichment of ARAF (multiple non-highlighted granules also show colocalization between EML4-ALK and ARAF). (F) Quantification of colocalization between EML4-ALK granules and ARAF wild-type or R52L mutant, N = 3 with at least 30 cells per replicate. (G, H) Quantification of western blotting results for EML4-ALK cancer cell line (H3122) or oncogenic EGFR cancer cell line (HCC827) treated with siRNAs against A/B/C-RAF or all three for 5 days. pERK levels normalized to total ERK protein levels and then displayed relative to control scramble (SCR) siRNA sample. N = 4, knockdown confirmed by Western blotting (Fig. S13). For all panels, error bars represent ± SEM, * denotes p < 0.05, ** p < 0.01, n.s. denotes non-significant comparison.
Consistent with previous observations that RAS proteins are not substantially recruited to RTK-containing structures upon RTK activation [24–26] (see Supplementary Note 1), we did not observe enrichment of either endogenously-tagged H/N/KRAS or overexpressed H/N/KRAS at EML4-ALK granules; however, we did confirm presence of RAS proteins in the cytosol in addition to lipid-membrane subcellular compartments (Fig. S9). To test whether EML4-ALK granules display evidence of activated RAS (i.e. RAS-GTP), we used overexpressed fluorescently tagged A/B/CRAF proteins (see Supplementary Note 2, Figs. S10 and S11) as livecell reporters given their specificity and high affinity binding to RAS-GTP [25, 27]. We validated this approach using cells expressing oncogenic KRAS as a positive control (Fig. S10). In EML4-ALK expressing cells, we observed enrichment of ARAF, but not BRAF or CRAF, at EML4-ALK cytoplasmic protein granules (Fig. 2, E and F, and Fig. S12). Importantly, an established mutant form of ARAF that impairs the interaction with RAS-GTP, ARAF R52L [28] (Fig. S10), showed diminished localization at EML4-ALK granules (Fig. 2, E and F). These findings support a model of RAS activation locally by membraneless EML4-ALK cytoplasmic protein granules. The data also reveal an unanticipated isoform specificity of RAF interaction, as has been described in other contexts [28, 29]. Consistent with these findings, knockdown of ARAF in H3122 patient-derived lung cancer cells with endogenous EML4-ALK substantially reduced MAPK signaling (Fig. 2G and Fig. S13A), contrasting with a distinct patient-derived cancer cell line expressing endogenous oncogenic EGFR (HCC827) wherein BRAF was the major isoform critical for MAPK signaling (Fig. 2H and Fig. S13B). Altogether, the data reveal that oncogenic RTK/RAS/MAPK signaling can occur via membraneless cytoplasmic protein granules and that cytosolic RAS that is not lipid-membrane associated can undergo RTK-mediated activation in mammalian cells.
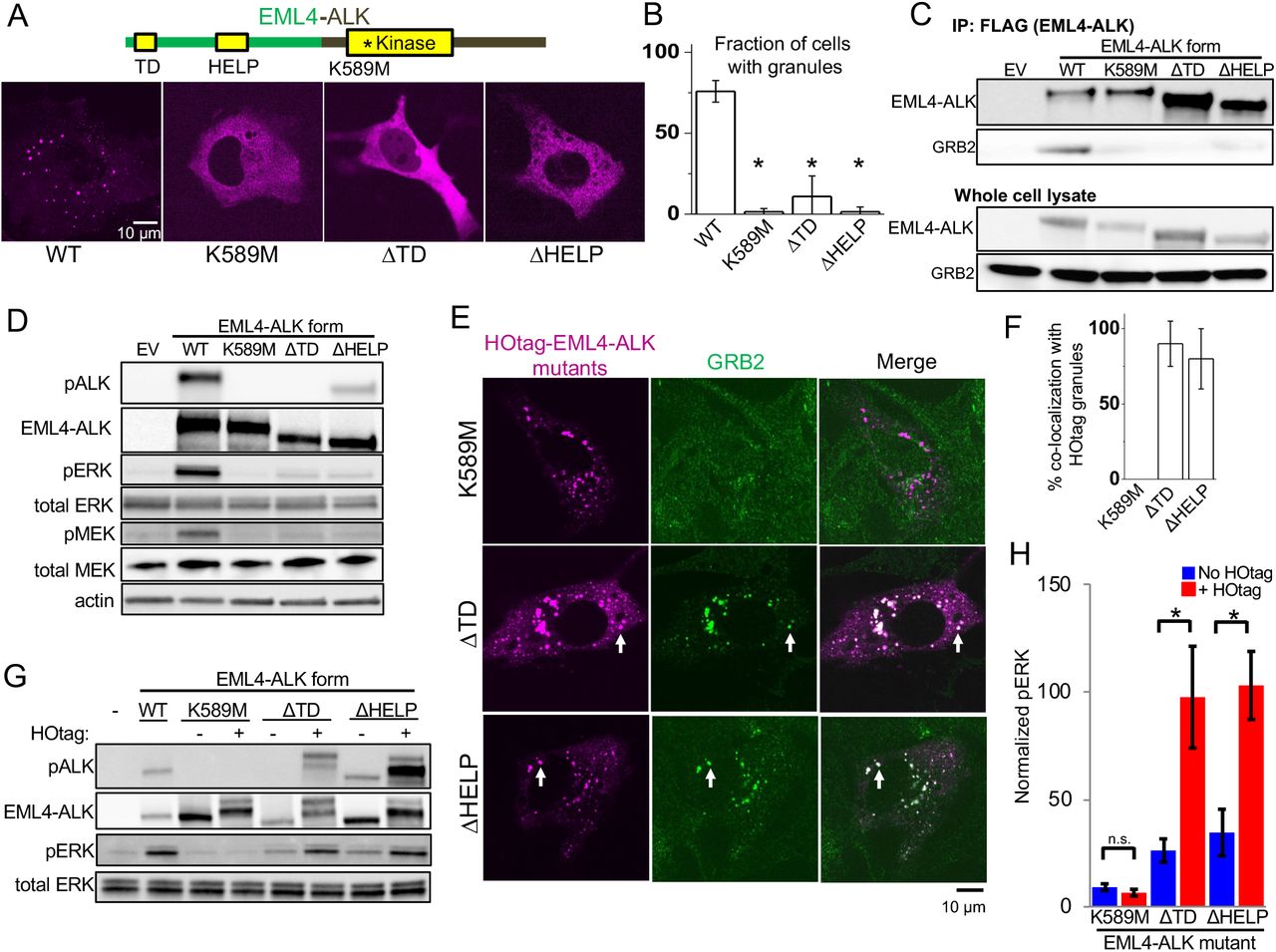
We next investigated the molecular determinants of RTK-mediated de-novo cytoplasmic protein granule formation and productive RAS/MAPK signaling. We first determined that the EML4-ALK protein itself is sufficient to spontaneously form granules in the absence of GRB2 or SOS1 (Fig. S14). The EML4 portion of the chimeric EML4-ALK oncoprotein contains an N-terminal trimerization domain (TD) and a truncated WD-propeller domain [12]. Deletion of the TD or the hydrophobic EML protein (HELP) motif in the propeller domain disrupted protein granule formation, resulting instead in a diffuse cytoplasmic distribution of EML4-ALK labeled by either fluorescent protein or FLAG-tag (Fig. 3, A and B, and Fig. S15). ΔTD or ΔHELP mutants of EML4-ALK demonstrated loss of ALK trans-phosphorylation and GRB2 interaction (Fig. 3, C and D) and impaired RAS/MAPK activation (Fig. 3D and Fig. S16). These data implicate de-novo protein granule formation mediated by the EML4 portion of the fusion protein as critical for productive RAS/MAPK signaling. We also observed disrupted granule formation and absent RAS/MAPK signaling with a kinase-deficient mutant (K589M) form of EML4-ALK (Fig. 3, A to D, and Figs. S15 and S16), which may be due to phosphorylation events regulating EML4-ALK protein granule formation, as shown in other protein granule systems [30, 31].

(A) Structure schematic of the EML4-ALK fusion protein with highlighted trimerization domain (TD), HELP motif, and ALK kinase domain. Live-cell confocal imaging of mTagBFP2::EML4-ALK (denoted as WT for wild-type EML4-ALK) or kinase-deficient (K589M), ΔTD or ΔHELP mutants in human epithelial cell line Beas2B. (B) Quantification of fraction of cells with granules. At least 75 cells were scored over 3 independent replicates. (C) Co-immunoprecipitation of FLAG-tagged wild-type (WT) EML4-ALK or respective mutants expressed in 293T cells, followed by Western blotting to assess GRB2 binding. EV denotes empty vector control, images representative of at least 3 independent experiments. (D) Western blotting upon expression of EML4-ALK or respective mutants in 293T cells. Images representative of 5 independent replicates. (E) Live-cell confocal imaging of HOtag-mTagBFP2::EML4-ALK ΔTD, ΔHELP, and K589M mutants in Beas2B cells with endogenous mNG2-tagging of GRB2. White arrows indicate representative HOtag EML4-ALK ΔTD or ΔHELP protein granules with local enrichment of GRB2 (multiple non-highlighted granules also show colocalization between HOtag EML4-ALK mutants and GRB2). (F) Quantification of percent colocalization between HOtag protein granules of EML4-ALK mutants and GRB2. N = 130 total cells for each condition over 3 independent experiments. (G, H) Western blotting upon expression of wild-type EML4-ALK or respective mutants +/− HOtag in 293T cells. For quantification, pERK levels were normalized to total ERK and then displayed relative to wild-type EML4-ALK sample which was set to 100, N = 5. For all panels, error bars represent ± SEM, * denotes p<0.05, n.s. denotes non-significant comparison.
Our findings prompted a new model in which clustering of an RTK in membraneless cytoplasmic protein granules is sufficient to organize activation of RAS/MAPK signaling. To directly test this hypothesis, we utilized the HOtag method recently developed for forced protein granule formation through multivalent interactions [32] (Fig. S17). HOtag-forced cytoplasmic granule formation of either the ΔTD or ΔHELP mutants of EML4-ALK locally recruited GRB2 (Fig. 3, E and F), increased RAS-GTP levels (Fig. S16) and restored RAS/MAPK signaling (Fig. 3, G and H). As an important negative control, HOtag-forced clustering of the kinase-deficient EML4-ALK did not promote GRB2 recruitment or RAS/MAPK signaling (Fig. 3, E to H, and Fig. S16), highlighting the dual importance of cytoplasmic protein granule formation and intact kinase activity for productive signaling. Compared to wild-type EML4-ALK, the ΔTD mutant that is expressed diffusely in the cytoplasm also demonstrated substantially reduced levels of activated (GTP-bound) cytosolic KRAS-C185S, which could be restored through HOtag-forced clustering (Fig. S18). Collectively, our data show that membraneless EML4-ALK protein granules can spatially concentrate, organize, and initiate RAS/MAPK pathway signaling events.
We tested the generality of this model. First, multiple variants of EML4-ALK have been described in cancer patients [12], all comprising the intracellular domain of ALK (but not its transmembrane domain) fused to N-terminal fragments of EML4 of varying lengths. We demonstrated that another recurrent form of oncogenic EML4-ALK (variant 3), which contains a further truncation of the propeller domain but retains the TD [12], also formed cytoplasmic granules that locally recruited GRB2 and increased RAS/MAPK signaling (Fig. 4A and Fig. S19). Second, we engineered an intracellular EGFR (iEGFR) protein that lacked the native extracellular and transmembrane domains. This iEGFR is similar to naturally-occurring truncated forms of this RTK and others [33, 34] and is distributed in the cytoplasm when expressed alone (Fig. 4B). HO-tag forced clustering of iEGFR recruited GRB2 and increased RAS/MAPK signaling in a kinase-dependent manner, analogous to oncogenic ALK (Fig. 4, B and C).
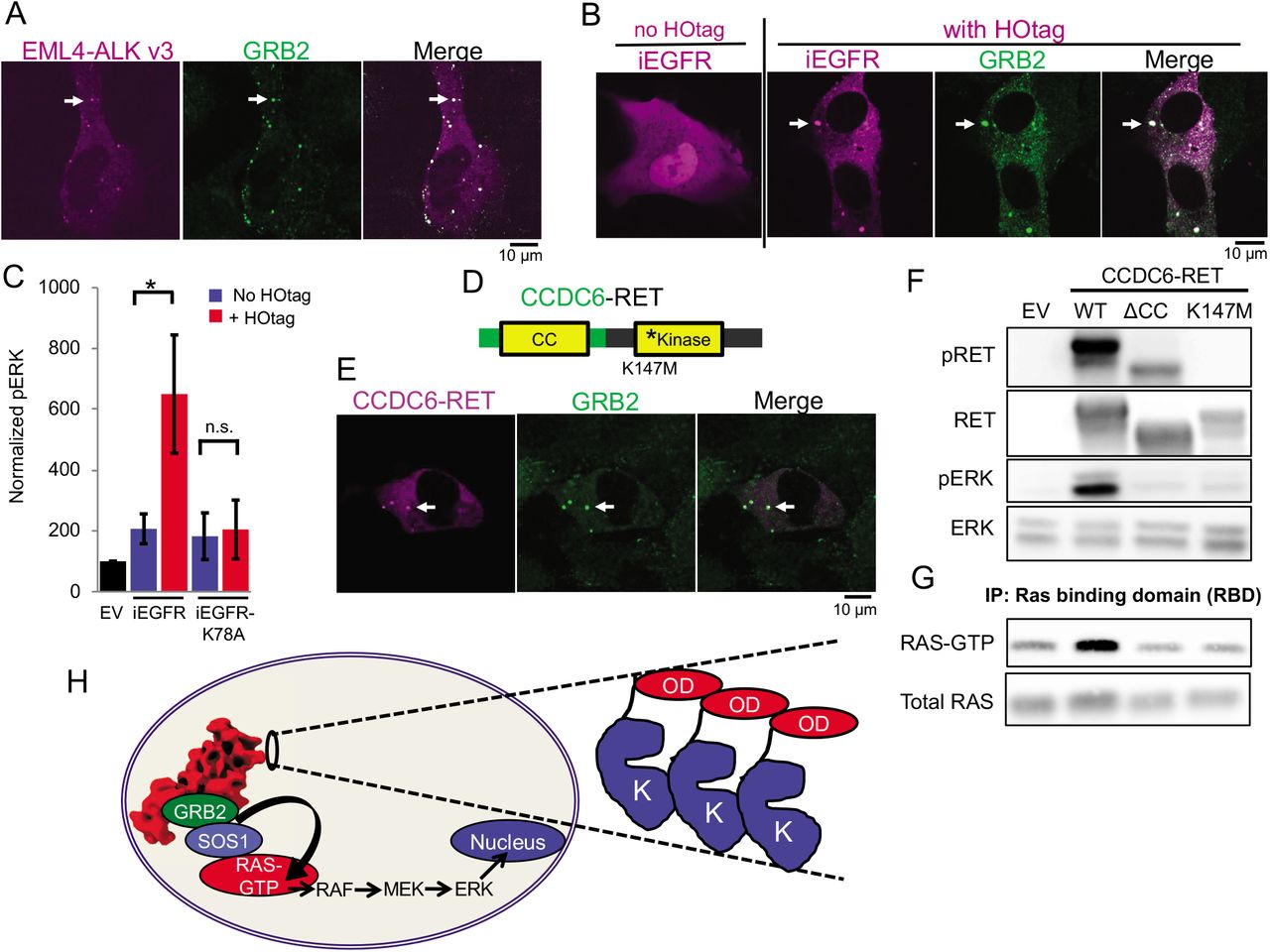
(A) Live-cell confocal imaging of mTagBFP2::EML4-ALK variant 3 in Beas2B cells with endogenous mNG2-tagging of GRB2. White arrows indicate a representative EML4-ALK variant 3 cytoplasmic protein granule with local enrichment of GRB2 (multiple non-highlighted granules also show colocalization between EML4-ALK variant 3 and GRB2). (B) Live-cell confocal imaging of mTagBFP2::iEGFR +/− forced clustering (HOtag) in Beas2B cells. White arrows indicate a representative HOtag iEGFR protein granule with local enrichment of GRB2 (multiple non-highlighted granules also show colocalization between HOtag iEGFR and GRB2). (C) Quantification of Western blotting results upon expression of empty vector (EV), iEGFR or iEGFR kinase-deficient mutant (iEGFR-K78A) +/− HOtag in 293T cells. pERK levels normalized to total ERK, N = 6, error bars represent ± SEM, * denotes p < 0.05, n.s. denotes non-significant comparison. (D) Schematic structure of the CCDC6-RET fusion protein with CCDC6 coiled-coiled domain (CC) and RET kinase domain. (E) Live-cell confocal imaging of mTagBFP2::CCDC6-RET in Beas2B cells with endogenous mNG2-tagging of GRB2. White arrows indicate a representative CCDC6-RET cytoplasmic protein granule with local enrichment of GRB2 (multiple non-highlighted granules also show colocalization between CCDC6-RET and GRB2). (F, G) Western blotting and RAS-activation assay upon expression of empty vector (EV), wild-type CCDC6-RET (WT), and CCDC6-RET ΔCC and kinase-deficient (K147M) mutants. Quantification of at least 4 replicates shown in Figure S22. (H) Model for cytoplasmic protein granule-mediated RTK/RAS/MAPK signaling. Inset highlights common structural features of oncogenic RTK fusion proteins with a cytoplasmic kinase domain (K) fused to a partner with an oligomerization domain (OD). All images in this figure are representative of at least 25 analyzed cells in 3 independent experiments.
Next, we studied another oncogenic RTK, RET, that also undergoes multiple distinct gene rearrangements in human cancer, leading to the elimination of the extracellular and transmembrane domains from the fusion oncoprotein [9]. The recurrent fusion oncoprotein CCDC6-RET formed de-novo cytoplasmic protein granules (Fig. 4E) and did not demonstrate PM localization or co-localize with intracellular lipid-containing organelles or a lipid-intercalating dye (Fig. S20). CCDC6-RET cytoplasmic protein granules recruited GRB2 (Fig. 4E) and locally enriched RAS-GTP as measured by the RAF reporter assay (Fig. S21), resulting in increased RAS activation and downstream MAPK signaling (Fig. 4, F and G, and Fig. S22). Interestingly, our findings indicated that CCDC6-RET protein granules recruited both ARAF and CRAF, but not BRAF (Fig. S21), reflecting different isoform specificity in RAF interaction compared to EML4-ALK. Structure-function studies showed that a CCDC6-RET mutant lacking the coiled-coil oligomerization domain in the CCDC6 component abrogated granule formation (Fig. S23) and reduced RAS/MAPK activation (Fig. 4, F and G, and Fig. S22). A kinase-deficient (K147M) mutant form of CCDC6-RET still formed cytoplasmic protein granules but was unable to recruit GRB2 or activate RAS/MAPK signaling (Fig. 4, F and G, and Figs. S22 and S24). These results reinforce the dual importance of cytoplasmic protein granules and kinase activity in driving oncogenic RTK/RAS/MAPK signaling. The data also reveal differences between EML4-ALK and CCDC6-RET in the dependence on kinase activity for granule formation that may relate to structural differences in either the kinase or the fusion partner. Altogether, these cases show that RTK/RAS/MAPK signaling emanating from membraneless cytoplasmic protein granules may represent a general mechanism of organizing signaling events in cancer.
Our findings reveal that kinase-mediated proliferative signaling can occur via membraneless cytoplasmic protein granules. The data establish initial examples of this previously unrecognized mode of cell signaling. While RTK/RAS/MAPK signaling from cytoplasmic protein granules represents a new paradigm, our results and model are not discordant with the well-established importance of lipid membranes in organizing and regulating RTK/RAS/MAPK signaling cascades [1, 2, 13]. Our findings provide a new conceptual framework that is distinct from, yet analogous to prior work showing the importance of lipid-membrane-based micro-domain regulation as a means to concentrate RAS/MAPK signaling components to promote signaling [1, 35]. Cytoplasmic protein granule assembly by certain RTK forms acts to increase the local concentration of RAS-activating proteins (i.e. GRB2/SOS1) and the levels of GTP-bound RAS, which in turn initiates downstream MAPK signaling (Fig. 4H). Our studies do not exclude the possibility that after activation at the cytoplasmic protein granule, RAS GTPases might traffic to and back from the PM and engage with RAF. In this sense, PM-based and cytoplasmic protein granule-based RTK/RAS/MAPK signaling could co-exist. The differential localization of RTKs, distinct cellular pools of RAS and MAPK pathway proteins, and the known dynamic nature of RAS subcellular localization may create multiple subcellular platforms to regulate signaling [22, 25, 36]. Such a model of spatial regulation across different subcellular compartments could further explain how RAS GTPases as binary molecular switches are subject to exquisite control and mediate pleiotropic and diverse signaling outputs in mammalian cells and cancer.
Our work defines the first naturally occurring example, to our knowledge, of a lipid-membrane independent, protein granule-based structure than can directly organize proliferative and oncogenic signaling events in mammalian cells. Deciphering the rules governing formation and stability of protein granules presents significant new opportunities for molecular diagnostics and targeted drug development to identify and disrupt protein granules that drive disease pathogenesis. Many RTKs that undergo gene rearrangements in cancer share a similar structure that we describe as a key and potentially conserved feature of this mode of cell signaling organization: loss of the extracellular and transmembrane domain, resulting in chimeric fusion of the kinase domain to a protein partner with a multimerization domain [8, 9] (Fig. 4H). We show that the multimerization domains of RTK gene fusion partners are essential for oncogenic ALK and RET to form cytoplasmic protein granules and activate RAS/MAPK signaling. Our findings reveal a general principle in which the organization of kinase-mediated proliferative cell signaling is enabled by membraneless, higher-order structural assembly of signaling proteins that we propose may exist more broadly in normal cells, cancer, and other diseases.
Funding
This research project was conducted with support from the National Institutes of Health (R01CA231300 to T.G.B and B.H., U54CA224081, R01CA204302, R01CA211052 and R01CA169338 to T.G.B, R21GM129652, R01GM124334 and U19CA179512 to B.H), Pew and Stewart Foundations (to T.G.B), the UCSF Marcus Program in Precision Medicine Innovation (to B.H. and T.G.B.), the UCSF Byers Award for Basic Science (to B.H.) and the UCSF Physician-Scientist Scholar Program (to A.T.). B.H. is a Chan Zuckerberg Biohub investigator. A.T. also received financial support from Alex’s Lemonade Stand, the St. Baldrick’s Foundation, the A.P. Giannini Foundation, the Campini Family Foundation, and the Posey Family Foundation. D.N received support from F30 CA210444-04.
Author Contributions
A.T., J.G., D.N., B.H. and T.G.B. designed the study. A.T., J.G., D.N., H.L., A.H., H.A., S.P., A.R., X.S., B.Y., S.F. performed experiments, collected and analyzed data. A.T., J.G., D.N., B.H. and T.G.B wrote the manuscript. B.H. and T.G.B oversaw the study. All authors have approved the manuscript.
Competing interests
T.G.B. is an advisor to Array Biopharma, Revolution Medicines, Novartis, Astrazeneca, Takeda, Springworks, Jazz Pharmaceuticals, and receives research funding from Novartis and Revolution Medicines.
Data and Materials Availability
All data is available in the main text or the supplementary materials.
Acknowledgments
The authors would like to acknowledge Amit Sabnis, Franziska Haderk and Zoji Bomya for experimental help and manuscript review, Mark Philips for generously providing plasmids and manuscript review, and Michael Rosen for scientific input and manuscript review.





{kind=link}
{kind=link}
{kind=link}
{kind=link}