

Abstract
Over 70% of breast cancers express the estrogen receptor (ER) and depend on ER activity for survival and proliferation. While hormone therapies that target receptor activity are initially effective, patients invariably develop resistance which is often associated with activation of the PI3K/Akt/mTOR pathway. While the mechanism by which estrogen regulates proliferation is not fully understood, one gene target of ER, growth regulation by estrogen in breast cancer 1 (GREB1), is required for hormone-dependent proliferation. However, the molecular function by which GREB1 regulates proliferation is unknown. Herein, we validate that knockdown of GREB1 results in growth arrest and that exogenous GREB1 expression initiates oncogene-induced senescence, suggesting that an optimal level of GREB1 expression is necessary for proliferation of breast cancer cells. Under both of these conditions, GREB1 is able to regulate signaling through the PI3K/Akt/mTOR pathway. GREB1 acts intrinsically through PI3K to regulate PIP3 levels and Akt activity. Critically, growth suppression of estrogen-dependent breast cancer cells by GREB1 knockdown is rescued by expression of constitutively activated Akt. Together, these data identify a novel molecular function by which GREB1 regulates breast cancer proliferation through Akt activation and provides a mechanistic link between estrogen signaling and the PI3K pathway.
Introduction
Breast cancer is the most frequently diagnosed malignancy in women (1). Over 70% of breast cancer patients are diagnosed with the estrogen receptor-positive (ER+) subtype, which is characterized by the expression of the transcription factor ER and dependence on ER activity for tumor cell growth and survival (2–6). Patients diagnosed with the ER+ subtype of breast cancer are typically prescribed endocrine therapies that target ER activity (2, 3, 6). However, resistance to endocrine therapies invariably occurs, leading to re-activation of the ER, expression of ER-target genes, and ultimately, patient relapse (2, 3, 6). Treatment options for patients that are resistant to endocrine therapies are limited, highlighting the need for innovative therapies that target downstream of the ER (2, 3, 6).
Crosstalk between the ER and the PI3K/Akt/mTOR pathway has been implicated in ER+ breast cancer progression and resistance to endocrine therapies (7–10). The PIK3CA gene, which encodes the catalytic subunit of the PI3K enzyme, is the most commonly mutated gene in ER+ breast cancer patients with over 40% incidence (11). This mutation is speculated to be a causal event in breast cancer progression, suggesting there is some need to upregulate this pathway in the development of ER+ breast cancer (11). Studies have indicated the ER can interact with PI3K to regulate its kinase activity in an estrogen-dependent manner (12). Further, inhibition of the PI3K/Akt/mTOR pathway increases ER expression and sensitivity of breast cancer cells to tamoxifen treatment, suggesting that activation of this pathway is associated with resistance to endocrine therapies through downregulation of ER (13). Together, these data indicate interdependence between ER and PI3K/Akt/mTOR signaling, however, the molecular basis and clinical relevance for this cooperation remains unclear.
Herein, we show that the ER gene target, growth regulation by estrogen in breast cancer 1 (GREB1), is a regulator of the PI3K/Akt/mTOR pathway, linking ER activation to this critical signaling pathway. Expression of GREB1 has been highly correlated to ER-positivity in breast cancer cell lines and patient samples (14–17). Previous studies have shown that knockdown of GREB1 results in significantly reduced proliferation and colony formation of ER+ breast cancer cell lines indicating, a required role for GREB1 in regulation of estrogen-dependent proliferation (16–18). However, it appears that an optimal level of GREB1 expression is necessary for proliferation of breast cancer cell lines as exogenous expression of GREB1 inhibits growth (18). Interestingly, growth repression by exogenous expression of GREB1 was also observed in ER-negative cell lines, indicating the ability of GREB1 to regulate proliferation of breast cancer cells is independent of ER activity (18). Despite the clear association of GREB1 and proliferation of ER+ breast cancer, the molecular function of the protein and the mechanism by which it regulates proliferation remain largely unknown. In this study, we characterize a novel mechanism by which GREB1 regulates proliferation of ER+ breast cancer cell lines through activation of Akt.
Materials and methods
Cell lines and reagents
MCF7, T47D, ZR-75-1, and HEK-293T cells were validated using Short Tandem Repeat analysis by the Genomics Core in the Research Technology Support Facility (Michigan State University, East Lansing, MI 48824). HCC1500 cells were purchased from the American Type Culture Collection. Cells were maintained as previously described (18). For experiments with EGF stimulation, cells were cultured in serum-free media for 16 hours before being stimulated with 1 ng/mL recombinant human EGF (Thermo) for the indicated time. The inhibitors GDC-0941 was obtained from Cayman Chemicals and were used at the indicated concentration for 24 hours prior to harvest of the cells.
Plasmids
3XFLAG PCDNA and H2BGFP have been described previously (18–20). GIPZ lentiviral non-specific shRNA (# RHS4346) and lentiviral GREB1-targeted shRNA plasmids (V2LHS_139192 and V3LHS_372339) were obtained from Open Biosystems. MISSION shRNA constructs targeted to PIK3CA (TRCN0000196 582, TRCN0000195 203, TRCN0000010 406), PTEN (TRCN0000002 745, TRCN0000002 747, TRCN0000002 749), and PDK1 (TRCN0000001 476, TRCN0000039 778, TRCN0000039 782, TRCN0000010 413) were purchased from Sigma Aldrich. Myristoylated (Myr) AKT1 from pBabe-Puro-Myr-Flag-AKT1 (21) was cloned into a pLenti-hygro backbone to create pLenti hygro Myr FLAG AKT1 (CA AKT) using standard Gibson cloning (NEB).
Immunoblot analysis and antibodies
Cell lysates were prepared, subjected to immunoblot analysis, and visualized on a LI-COR Odyssey system as previously described (18, 22). Immunoblots were probed with the following antibodies: GREB1 (abcam; ab72999 or CST; P76195 Clone 9C1), β-actin (Cell Signaling Technologies (CST); 3700), phosphor-p38 (CST; 9211S), p38 (CST; 9212), phosphor-MEK1/2 (CST; 9121), MEK1/2 (CST; 9122), phosphor-ERK1/2 (CST; 4370); ERK1/2 (CST; 4695), phospho-MKK3/6 (CST; 12280), phospho-MSK1 (CST; 9595), phospho-ATF2 (CST; 5112), phospho-HSP27 (CST; 9709), phospho-MAPKAPK2 (CST; 3007), phospho-PTEN (CST; 9551), PTEN (CST; 9556), phospho-PDK1 (CST; 3438), PDK1 (CST; 5662), phospho-Akt Thr308 (CST; 13038), phospho-Akt Ser473 (CST; 9271S), Akt (CST; 2920S), PP2A (CST; 2041T), phospho-GSK3β (CST; 5558), mTOR (CST; 2983), Rictor (CST; 2114), Raptor (CST; 2280), and p110α (CST; 4249T).
Adenovirus
GREB1 adenovirus was purified as previously described (18). Ad5-CMV-eGFP adenovirus (Baylor College of Medicine Vector Development Labs, Houston, TX 77030) was used as a control.
Alamar blue assay
Cells were treated with 0.04 g/L resazurin sodium salt in phosphate buffered saline (PBS) at 37°C for 1 hour. Fluorescence was measured on a BioTek Synergy microplate reader using a 540/35 excitation filter and a 590/20 emission filter. Data are depicted as mean fluorescence normalized to day 0 ± SD for each condition from 3 biological replicates. Statistical significance for alamar blue assays was determined using either a two-tailed Student’s t-test (exogenous GREB1 expression) or a one-way ANOVA with post-hoc Tukey’s HSD test (shRNA experiments).
SA-β-gal staining
Cells transduced with GFP or GREB1 adenovirus were plated on poly-L-lysine coated coverslips. Cells were fixed and stained for SA-β-gal activity using the Senescence β-Galactosidase Staining Kit (CST #9860) as previously described (23).
Conditioned media assay
MCF7 cells were transduced with either GFP or GREB1 adenovirus. After 24 hours, transduced cells were washed in PBS and fresh media added. The following day, media was collected from the transduced cells and centrifuged at 500 × g for 5 minutes to pellet any cellular debris. Target cells were washed twice with PBS prior to the addition of conditioned media. After 24 hours, all cells were harvested by scraping in cold PBS containing 10 nM calyculin A (Cell Signaling).
Co-culture assay
MCF7 cells were transduced with adenovirus expressing either GFP or GREB1 (both adenovirus vectors express GFP). The following day, transduced cells were trypsinized and replated at a 1:1 ratio with un-transduced MCF7 cells. Cells were harvested after 24 hours by trypsinization and washed twice with cold PBS containing 10 nM calyculin A (Cell Signaling). Cells were sorted from both the adGFP and adGREB1 co-cultures using a Becton Dickinson FACSAria II cell sorter into GFP-positive (transduced) and GFP-negative (un-transduced) populations. Cell lysates were prepared from the sorted populations and immunoblot analysis was performed as described above.
Immunofluorescence microscopy
Cells were plated on poly-L-lysine-coated coverslips. Following treatment, cells were fixed in 4% methanol-free formaldehyde (Thermo) diluted in PBS for 15 minutes at room temperature. Cells were then washed three times in PBS before permeabilization with 0.5% saponin, 1% BSA PBS solution at room temperature for 15 minutes. The cells were labeled with the indicated primary antibodies for 2 hours at room temperature in a humidified chamber. Coverslips were then washed three times in PBS before incubation with secondary antibodies (Alexa Fluor 555 goat anti-mouse and Alexa Fluor 555 goat anti-rabbit; Invitrogen) at room temperature for 1 hour in a humidified chamber, protected from light. Coverslips were mounted on microscope slides with VECTASHIELD Hard Set Mounting Medium with DAPI (Vector Laboratories). Images were obtained using a spinning disk confocal microscope (Ultra-VIEW VoX CSU-X1 system; Perkin Elmer) and analyzed using Velocity (Perkin Elmer).
PIP3 quantification
MCF7 cells were transduced with either GFP or GREB1 adenovirus. 24 hours after transduction, cells were placed in serum-free, phenol-red-free DMEM for 16 hours. Lipids were extracted and PIP3 levels measured using the PIP3 Mass ELISA kit (Echelon K-2500s) according to the manufacturer’s instructions.
Results
GREB1 initiates cellular senescence
Our previous work has suggested that an optimal level of GREB1 expression is necessary for proliferation of breast cancer cell lines. This work showed that both GREB1 knockdown (18) and exogenous expression of GREB1 results in growth arrest (Fig. 1A-B)(18). We previously reported that exogenous expression of GREB1 did not induce apoptosis (18), thus we investigated the ability of GREB1 overexpression to initiate oncogene-induced cellular senescence. Two ER+ breast cancer cell lines, MCF7 and ZR-75-1 cells, were transduced with adenovirus expressing either GFP or GREB1. Following 7 days of exogenous GREB1 expression, the cells were fixed and stained for SA-β-galactosidase activity, a marker of cellular senescence (23, 24). Compared to GFP control cells, cells overexpressing GREB1 had a large, flattened morphology and characteristic blue staining associated with SA-β-galactosidase activity (Fig. 1C). These data suggest that exogenous GREB1 expression is able to induce cellular senescence to inhibit proliferation of breast cancer cell lines.

MCF7 or ZR-75-1 cells were transduced with adenovirus expressing GFP or GREB1. A) Immunoblot depicting the relative overexpression of GREB1 in cells transduced with GREB1 adenovirus compared to GFP control. B) Proliferation of transduced cells was measured by alamar blue assay. Data are plotted as mean fluorescence normalized to Day 0 ± SD; n=3 for each cell line. *p≤0.05, ** p≤0.005. C) Cells were fixed and stained for SA-β-galactosidase activity 7 days post-transduction.
Exogenous GREB1 expression induces hyperactivation of the PI3K/Akt/mTOR pathway
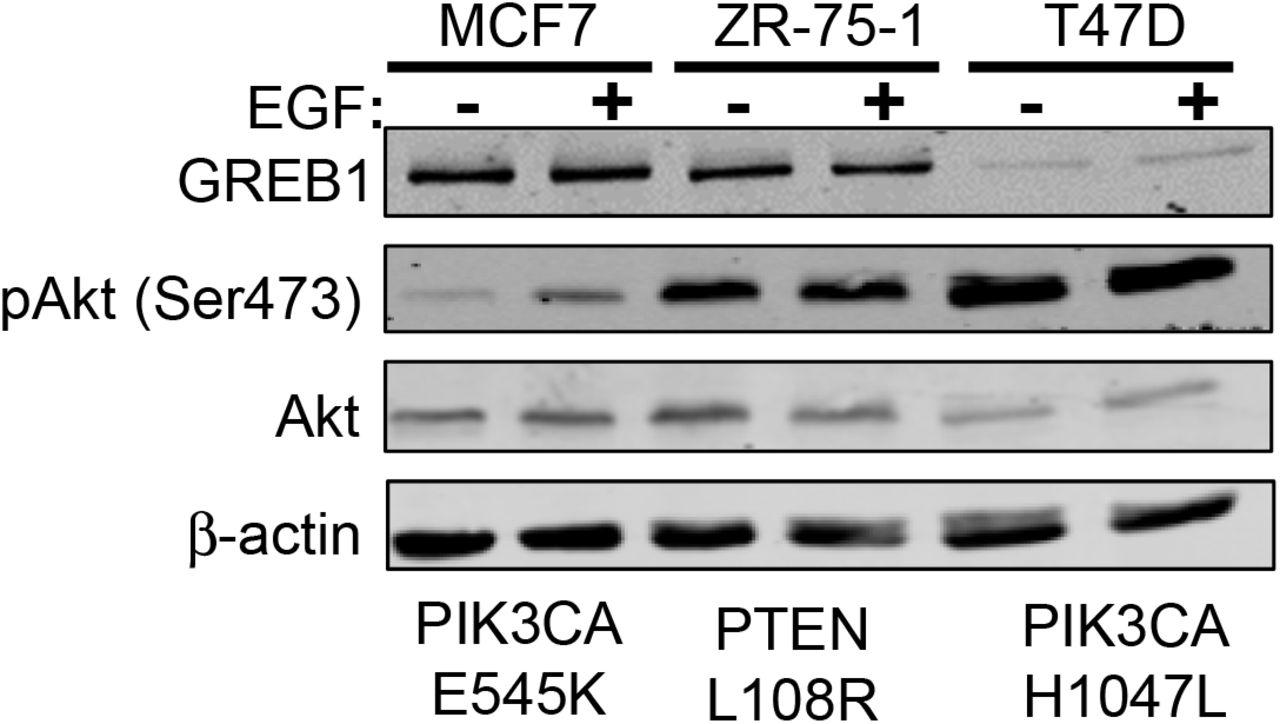
In order to delineate the mechanism by which GREB1 regulates proliferation, we chose to focus our attention on two signaling pathways thought to play a critical role in regulating both senescence and proliferation phenotypes: the p38 MAPK pathway and the PI3K/Akt/mTOR pathway (25–27). MCF7 cells were transduced with adenovirus expressing GFP or GREB1 and after 48 hours, cell lysates were analyzed by immunoblot for activation of various nodes in the p38 MAPK pathway (Fig. 2A) and the PI3K/Akt/mTOR pathway (Fig. 2B). Our data indicate that exogenous GREB1 expression induces an increase in the activation and phosphorylation of p38 and its downstream effector, MAPKAPK2, however, the activation and/or expression of upstream regulators of p38 (MEK1/2, ERK1/2, MKK3/6) and other downstream effectors (MSK1, ATF2, and HSP27) were largely unaffected (Fig. 2A). Analysis of the PI3K/Akt/mTOR pathway revealed hyperactivation of Akt, as well as increased phosphorylation of GSK3β, a downstream effector of Akt, when GREB1 was exogenously expressed in MCF7 cells (Fig. 2B). As the PI3K/Akt/mTOR pathway is frequently altered in breast cancer, we analyzed the effect of GREB1 overexpression on Akt activation in a panel of ER+ breast cancer cell lines with wildtype or differing alterations to the PI3K/Akt/mTOR pathway (28). While MCF7, ZR-75-1, and T47D cells harbor mutations in this pathway, each cell line has varying levels of Akt activation with MCF7 cells demonstrating the least constitutive activity (Supplemental Fig. 1). In cells lines without constitutive maximal activity of this pathway, GREB1 induces a strong activation of Akt (Fig. 2C). In contrast, T47D cells, which have a PIK3CA mutation resulting in constitutively active Akt that does not respond to serum deprivation (Supplemental Fig. 1), are unaffected by GREB1 expression (Fig. 2C).

MCF7, ZR-75-1, and T47D cells were serum starved for 16 hours before stimulation with 1 ng/mL of EGF for 1 hour. Cell lysates were harvested and analyzed by immunoblot for the indicated proteins.

A) MCF7 cells were transduced with adenovirus expressing GFP or GREB1. Cell lysates were harvested 2 days post-transduction and analyzed by immunoblot for indicated proteins in the p38 MAPK pathway. B) MCF7 cells were transduced with adenovirus expressing GFP or GREB1. Cell lysates were harvested 2 days post-transduction and analyzed by immunoblot for indicated proteins in the PI3K/Akt pathway. C) HCC1500, ZR-75-1, MCF7, and T47D cells were transduced with adenovirus expressing GFP or GREB1. Cell lysates were harvested 2 days post-transduction and analyzed by immunoblot for indicated proteins.
GREB1-induced hyperactivation of Akt is PI3K-dependent
Akt requires phosphorylation at two sites, Thr308 and Ser473, by PDK1 and mTORC2 respectively, for maximal activation (29–31). Both phosphorylation events are dependent on PI3K and occur downstream of PI3K conversion of PIP2 to PIP3 (29, 31). Thus, we sought to determine if GREB1 was acting to regulate Akt activation upstream or downstream of PI3K. To this end, we targeted PI3K activity with the pharmaceutical inhibitor GDC0941. MCF7 cells were simultaneously treated with DMSO or GDC0941 and transduced with adenovirus expressing GFP or GREB1. After 24 hours, cell lysates were harvested and activation of Akt at Thr308 and Ser473 were evaluated by immunoblot analysis. The expected hyperactivation of Akt was observed in DMSO-treated cells that were transduced with GREB1 adenovirus (Fig. 3A). PI3K inhibition by GDC0941 demonstrated a marked decrease in basal Akt activation in the control GFP-transduced cells (Fig. 3A). A decrease in GREB1-mediated activation was also observed, particularly at Ser473 (Fig. 3A), suggesting that GREB1 is activating the canonical PI3K/Akt axis.

A) MCF7 cells were transduced with adenovirus expressing GFP or GREB1 and treated with DMSO or 40 nM GDC0941 simultaneously. Cell lysates were harvested 24 hours post-transduction and immunoblot analysis was performed with the indicated antibodies. B) MCF7 cells were transduced with lentivirus targeted to a nonspecific control (shNS), PIK3CA, PTEN, or PDK1. Following selection, cells were transduced with GFP or GREB1 adenovirus. Cell lysates were harvested 24 hours post adenovirus transduction. Following SDS-PAGE, immunoblot analysis was performed with indicated antibodies.
To confirm this result and further probe other nodes in the pathway, MCF7 cells were transduced with lentivirus expressing shRNA targeted to a non-specific control (shNS), PIK3CA, PTEN, or PDK1. Following selection, cells were transduced with adenovirus expressing GFP or GREB1. After 24 hours, cell lysates were harvested and analyzed via immunoblot. Control cells transduced with non-specific shRNA demonstrated the expected increase in phosphorylation of Akt (Thr308 and Ser473) and GSK3β when GREB1 was exogenously expressed (Fig. 3B). Knockdown of PIK3CA expression reduced Akt activation in both control and GREB1-expressing cells (Fig. 3B). GREB1-induced hyper-phosphorylation of GSK3β was also reduced when PIK3CA was knocked-down (Fig. 3B). Knockdown of PTEN enhanced GREB1-induced hyperactivation of Akt at Thr308, suggesting the mechanism of GREB1 action is not through phosphatase inhibition (Fig. 3B). In contrast to our pharmacological approach (Fig. 2B), GREB1-induced hyperactivation of Akt at Thr308 was completely blocked by knockdown of PDK1, the primary kinase for this site (29–32)(Fig. 3B). As expected, PDK1 knockdown had no effect on GREB1-induced hyperactivation of Akt at Ser473, as this is not the primary kinase for this site (29–31) (Fig. 3B). Knockdown of PDK1 diminished GREB1-induced phosphorylation of GSK3β (Fig. 3B). These data further demonstrate that GREB1-induced hyperactivation of Akt is dependent on signaling through the canonical PI3K pathway.
GREB1 activates Akt through intracellular mechanisms
Canonical activation of PI3K and Akt occurs through activation of receptor tyrosine kinases (RTK) or G-protein-coupled receptors (GPCR) by external stimuli (29, 33). We first sought to determine if GREB1-mediated Akt regulation is dependent upon induction and secretion of a signaling molecule that activates the PI3K/Akt/mTOR pathway. To this end, MCF7 cells were transduced with adenovirus expressing GFP or GREB1. Media from transduced cells was transferred to un-transduced MCF7 cells. After 24 hours, cell lysates were harvested and activation of Akt was analyzed by immunoblot. As expected, exogenous expression of GREB1 induced hyperactivation of Akt at Ser473 when compared to GFP-transduced cells (Fig, 4A). However, conditioned media was unable to induce hyperactivation of Akt in un-transduced cells (Fig. 4A).

A) Conditioned media from MCF7 cells transduced with GFP or GREB1 adenovirus was added to un-transduced cells. Cell lysates from transduced cells (GFP or GREB1) and un-transduced cells cultured in conditioned media (GFP CM or GREB1 CM) were harvested 24 hours later. Lysates were analyzed by immunoblot for indicated proteins. B) MCF7 cells were transduced with adenovirus expressing GFP or GREB1. Transduced cells were then cultured with un-transduced cells at a 1:1 ratio for 24 hours. Cells were harvested and sorted for GFP. Cell lysates were analyzed via immunoblot for expression of the indicated proteins.
Alternatively, exogenous GREB1 may induce the expression of a membrane-bound signaling molecule that could activate the PI3K/Akt/mTOR pathway. To investigate this possibility, MCF7 cells were transduced with adenovirus expressing GFP or GREB1 and then co-cultured at a 1:1 ratio with un-transduced MCF7 cells. After 24 hours, the cells were harvested and GFP-positive, adenovirus-transduced cells (GFP or GREB1), were sorted from GFP-negative, un-transduced cells. All populations were then analyzed for activation of Akt by immunoblot. Both GFP-transduced cells and cells co-cultured with the GFP-transduced cells had similar levels of Akt activation (Fig. 4B). Exogenous expression of GREB1 induced the expected hyperactivation of Akt at Ser473 within the transduced cells; however, the untransduced co-culture cells did not demonstrate Akt hyperactivation (Fig. 4B). While these data are negative, they clearly demonstrate that GREB1 regulates Akt activation through in an intracellular mechanism that does not require extracellular activation of RTKs.
Exogenous GREB1 promotes recruitment of Akt to the plasma membrane
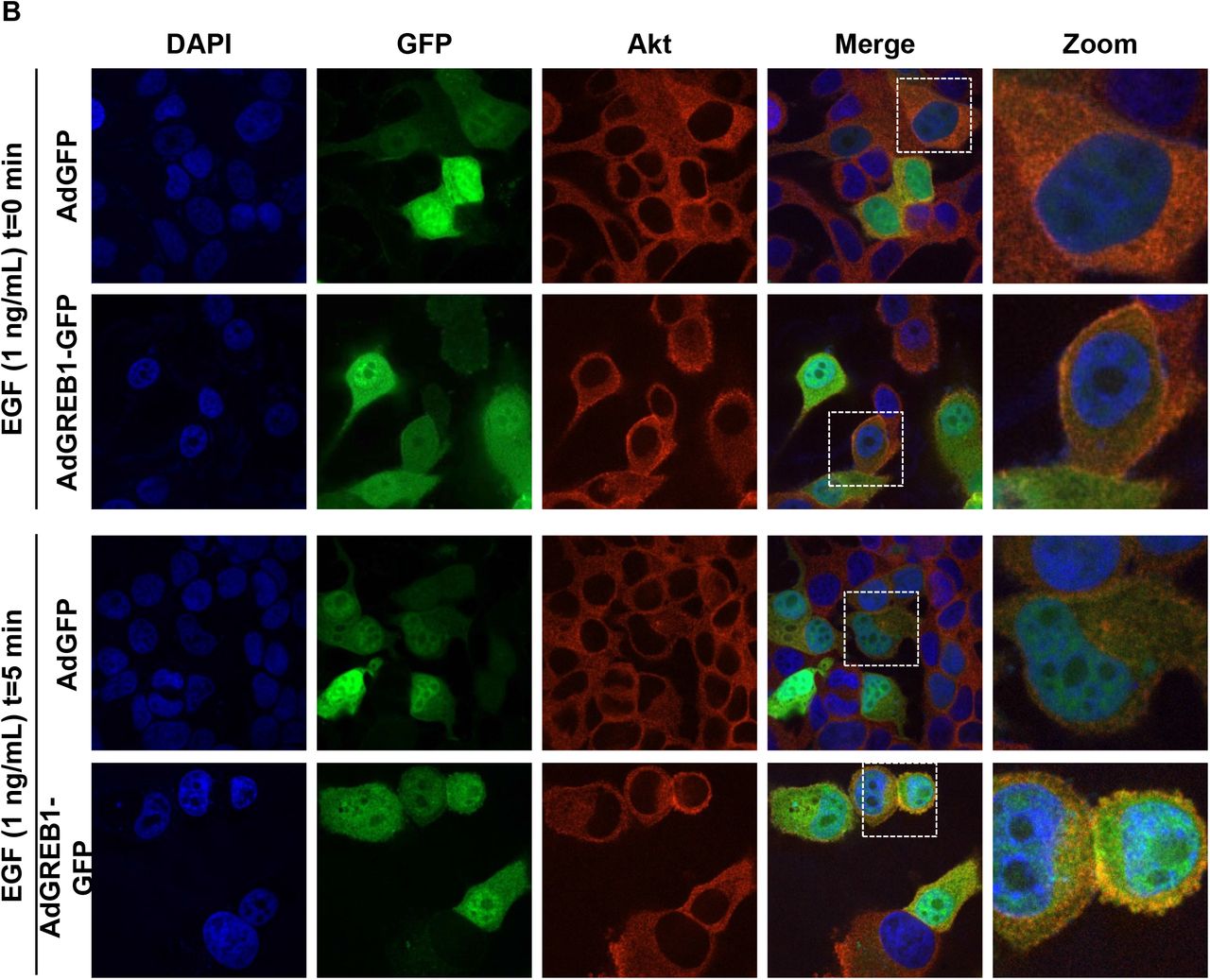
Akt is typically activated via recruitment to the plasma membrane by interaction with PIP3, however, it is believed that there are other pools of activated Akt on endomembrane surfaces and within the nucleus (29). To determine the localization of activated Akt induced by exogenous GREB1 expression, MCF7 cells were transduced with either GFP or GREB1 adenovirus. Following transduction, the cells were serum-starved for 16 hours to reduce basal Akt activation before stimulation of the pathway with EGF. Cells were then fixed and stained with DAPI and the indicated Akt antibodies. As both adenoviral vectors expressed GFP, we focused our imaging on transduced cell populations. In serum-starved, GFP-transduced cells, staining for activated Akt was minimal and staining for total Akt resulted in diffuse staining throughout the cytoplasm (Fig. 5A, Supplemental Fig. 2A-B). In contrast, cells transduced with GREB1 adenovirus under serum-starved conditions had distinct staining for activated and total Akt, primarily localized to the plasma membrane (Fig. 5A, Supplemental Fig. 2A-B). When stimulated with EGF for 5 minutes, both GFP- and GREB1-transduced cells demonstrated activated Akt and total Akt at the plasma membrane (Fig. 5A, Supplemental Fig. 2A-B). Activation of Akt and focal localization of total Akt at the plasma membrane was noticeably stronger in GREB1-transduced cells when compared to GFP-transduced cells in the presence of EGF (Fig. 5A, Supplemental Fig. 2A-B). These data further suggest that GREB1 may act through PI3K to increase PIP3 levels and Akt re-localization to the plasma membrane.

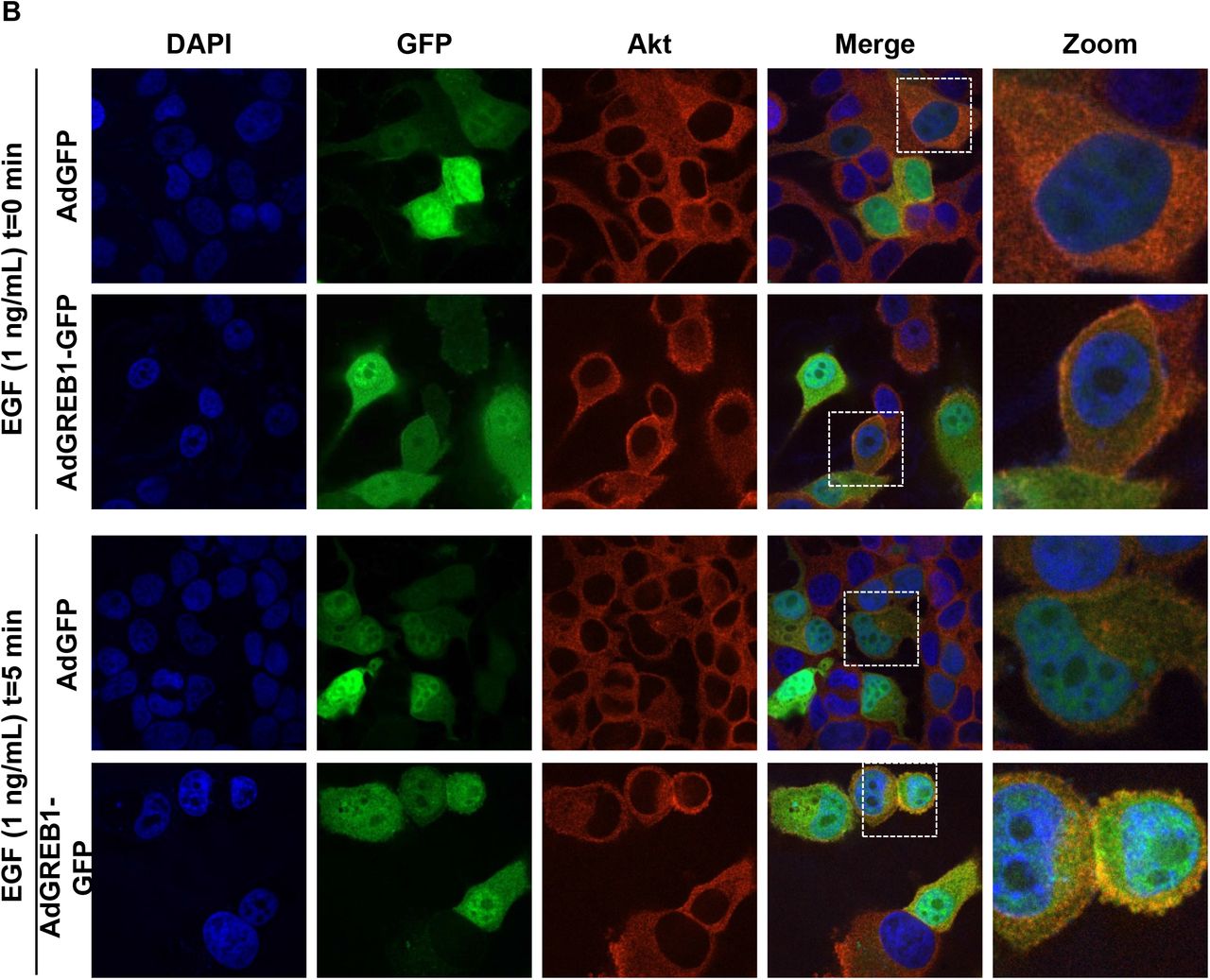
MCF7 cells were transduced with adenovirus expressing GFP or GREB1. The cells were then cultured in serum-free media for 16 hours before being stimulated with 1 ng/mL EGF for 0 or 5 minutes. Cells were fixed and stained for DAPI and A) Akt or B) p-Akt (Thr308). Immunofluorescence microscopy was used to visualize the activation and localization of Akt.

A) MCF7 cells were transduced with adenovirus expressing GFP or GREB1. The cells were then cultured in serum-free media for 16 hours before being stimulated with 1 ng/mL EGF for 0 or 5 minutes. Cells were fixed and stained for DAPI or p-Akt (Ser473). Immunofluorescence microscopy was used to visualize the activation and localization of Akt. B) MCF7 cells were transduced with adenovirus to express exogenous GFP or GREB1 and serum starved for 16 hours. Lipids were extracted from all samples and levels of PIP3 were measured via ELISA. Graphs represent mean PIP3 (pmol) + SD (n=3).
We sought to directly test if GREB1 expression influenced the conversion of PIP2 to PIP3. MCF7 cells were transduced with GFP and GREB1 adenovirus and then placed in serum-free media as depicted in Figure 5A. Following serum starvation for 16 hours, lipids were extracted and PIP3 levels measured by ELISA. Expression of exogenous GREB1 induced a significant increase in PIP3 levels, further indicating that GREB1 augments PI3K activity.
Previous studies have suggested that GREB1 is primarily localized to the nucleus in patient samples and breast cancer cell lines (15, 16), thus, it remained unclear how GREB1 was able to regulate signaling through a primarily cytoplasmic pathway. Interestingly, we discovered that under serum-starved conditions, endogenous GREB1 is diffuse throughout the cytoplasm and nucleus in MCF7 cells, but upon stimulation with EGF, the vast majority of GREB1 rapidly re-localizes to the cytoplasm (Supplemental Fig. 3A). As this is contradictory to previously published reports, we performed nuclear/cytoplasmic fractionation to verify cytoplasmic expression of GREB1. Under normal growth conditions (i.e. media containing FBS), GREB1 is primarily located within the cytoplasm of MCF7 cells (Supplemental Fig. 3B-C). Thus, in response to growth factor activation, GREB1 localizes to the cytoplasm wherein it can modulate the PI3K pathway.

A) MCF7 cells were serum starved for 4 hours and stimulated with 1 ng/mL EGF for 0, 5, or 15 minutes. Cells were fixed and stained for DAPI and endogenous GREB1. Immunofluorescence microscopy was used to visualize GREB1 localization. B) Cytoplasmic and nuclear fractions were extracted from MCF7 whole cell lysate using high-speed centrifugation. Fractionated cell lysates were subjected to SDS-PAGE and analyzed via immunoblot for indicated proteins. C) MCF7 cells cultured in full serum media were fixed and stained for DAPI and endogenous GREB1. Immunofluorescence microscopy was used to visualize GREB1 localization under normal growth conditions.
GREB1 regulates breast cancer proliferation through activation of the PI3K/Akt/mTOR pathway
In order to determine if GREB1-mediated Akt activation is imperative for proliferation of ER+ breast cancer cells, we tested whether constitutively activated Akt can rescue proliferation loss in GREB1 depleted cells. Thus, we made use of T47D cells which are ER+ and GREB1-expressing, but harbor a PI3KCAH1047L mutation rendering this pathway constitutively active and unresponsive to typical PI3K/Akt/mTOR-activating stimuli, including GREB1 exogenous expression (Fig. 2C and Supplemental Fig. 1). T47D cells were transduced with shRNA targeting a nonspecific control (shNS) or shRNA targeting GREB1 (shGREB1 #1 or shGREB1 #2). Immunoblot analysis confirmed knockdown of GREB1, as well as hyperactivation of Akt in T47D cells (Fig. 6A). Proliferation of these cells was then monitored via alamar blue assay. GREB1 knockdown had no effect on the proliferation of T47D cells (Fig. 6B), suggesting constitutive Akt activation abrogates the need for GREB1 expression.

A) T47D cells were transduced with lentivirus expressing non-specific shRNA or shRNA targeted to GREB1 (shGREB1 #1 or shGREB1 #2). Immunoblot depicting the expression of indicated proteins. B) Proliferation was measured via alamar blue assay. Data are plotted as mean fluorescence normalized to Day 0 ± SD; n=3. C) MCF7 cells were transduced with lentivirus expressing empty vector (EV) and either non-specific shRNA or shRNA targeted to GREB1 (shGREB1 #1 or shGREB1 #2). Immunoblot showing the expression of labeled proteins. D) Proliferation was measured via almar blue assay. Data are plotted as mean fluorescence normalized to Day 0 ± SD; n=3. E) MCF7 cells were transduced with lentivirus expressing myristoylated Akt (CA AKT) and either non-specific shRNA or shRNA targeted to GREB1 (shGREB1 #1 or shGREB1 #2). Immunoblot demonstrating the expression of indicated protein. F) Proliferation was measured via alamar blue assay. Data are plotted as mean fluorescence normalized to Day 0 ± SD; n=3.
The proliferation of ER+ and GREB1-expressing MCF7 cells has previously been shown to be dependent on expression of GREB1 (16–18). While MCF7 cells also harbor a mutation in PIK3CAE545K, which is thought to be an activating mutation (34), these cells are still responsive to typical PI3K/Akt/mTOR-activating stimuli (Supplemental Fig. 1). Thus, we sought to determine if constitutively activated Akt (myristoylated-Akt) would rescue proliferation in GREB1-depleted MCF7 cells. MCF7 cells were transduced with empty vector lentivirus (EV) or lentivirus expressing constitutively activated Akt (CA AKT) in combination with lentivirus expressing shRNA targeted to a non-specific control (shNS) or to GREB1 (shGREB1 #1 or shGREB1 #2). Knockdown of endogenous GREB1 resulted in decreased Akt activation in cells co-transduced with GREB1-targeted shRNA and empty vector lentivirus (Fig. 6C) as well as parental cells infected with control lentivirus (Supplemental Fig. 4). The knockdown of GREB1 significantly impaired the growth of MCF7 cells co-transduced with empty vector lentivirus (Fig. 6D). However, expression of constitutively active Akt (Fig. 6E), rescues the proliferation phenotype caused by GREB1 knockdown to that of control transduced cells (Fig. 6F). Together, these data demonstrate that the primary mechanism by which GREB1 drives estrogen-dependent proliferation is through modulation of Akt activity. Interestingly, stable expression of constitutively active Akt results in long-term silencing of GREB1 (Supplemental Fig. 5), suggesting a feedback loop and potentially explaining the observation that GREB1 expression is reduced in hormone-refractory disease {Mohammed, 2013 #169}.

MCF7 cells transduced with lentivirus expressing non-specific shRNA (shNS) or one of two shRNAs targeted to GREB1 (shGREB1 #1 or shGREB1 #2) were placed in serum/phenol red free media for 16 hours followed by 1 hour of activation with 1ng/ml EGF. Cells were harvested and lysates analyzed via immunoblot for Akt activation pathway.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Following transduction with control (EV) or constitutively active Akt (CA) expressing lentivirus, MCF7 stable lines were generated by placing cells on selection. Lysates from different passages were probed for GREB1 expression and Akt. activation Two distinct stable lines are depicted (top panel and bottom panel, respectively).
Discussion
Despite extensive research on hormone signaling in breast cancer, the explicit mechanism by which ER drives proliferation remains largely undefined. In order to delineate this mechanism, concerted efforts have been made to identify ER-target genes involved in estrogen-induced proliferation of breast cancer cells. Several of these studies have identified GREB1 as a gene that is required for estrogen-stimulated proliferation of breast cancer cell lines (14, 16, 17). Previous studies have suggested that GREB1 regulates proliferation through modulation of ER activity (16). However, our findings show that GREB1 is not a potent regulator of ER activity and has the ability to affect the proliferation of breast cancer cell lines independent of ER expression and action (18). Here, we suggest a novel mechanism by which GREB1 regulates proliferation through fine-tuning of PI3K/Akt/mTOR signaling.
GREB1 regulates proliferation of ER+ breast cancer cells through modulation of Akt activity
Several studies have indicated complex crosstalk between ER signaling and PI3K/Akt/mTOR pathway activation and implications for this crosstalk in resistance to endocrine therapy in breast cancer patients (7–9, 35–37). However, no studies have described a comprehensive connection between activation of these signaling pathways and proliferation of breast cancer cells. The difficulty to assess this connection is compounded by the fact that the vast majority of available breast cancer cell lines contain mutations involved in the PI3K/Akt/mTOR pathway (38). Although most ER+ breast cancer cell lines contain mutations within this pathway, the specific mutations have distinctly different effects on the activation of the PI3K/Akt/mTOR pathway. Specifically, breast cancer cell lines harboring PIK3CAH1047R mutations (ex. T47D) have significantly higher intrinsic PI3K activity compared to breast cancer cell lines harboring PIK3CAE545Kmutations (ex. MCF7), which have subtle effects on activation of the PI3K/Akt/mTOR pathway (11, 39). Using this to our advantage we show that in T47D cells, which harbor a constitutively active PI3K/Akt/mTOR pathway, GREB1 is no longer required for proliferation (Fig. 6A-B). However, in MCF7 cells, which harbor a PIK3CA mutation but still respond to pathway-activating stimuli, GREB1 is still required but knockdown of GREB1 can be rescued by constitutively active Akt (Fig. 6C-F). These data demonstrate that the requirement of GREB1 for hormone-responsive proliferation is dependent upon this novel function to alter Akt activity. Furthermore, the activation of the PI3K/Akt by GREB1 occurs through intracellular mechainisms (Fig. 4), highlighting a potentially new mode of modulating this signaling pathway without the need for RTK activation.
GREB1 and endocrine resistance
Despite the clear association between GREB1 and proliferation of breast cancer cells, expression of GREB1 has been correlated to better prognosis in ER+ breast cancer patients (16, 40). In a study that included only patients that received adjuvant tamoxifen monotherapy, higher GREB1 expression correlated with both prolonged disease-free survival and sensitivity to tamoxifen treatment (40). Similarly, in an in vitro model of tamoxifen resistance, MCF7 cells that were resistant to tamoxifen treatment had significantly less GREB1 expression compared to the parental line, suggesting GREB1 expression is lost in hormone-refractory breast cancer cells (16). These data have implicated loss of GREB1 as a causal event for therapy-resistance. Here, we show that proliferation of breast cancer cells with constitutively active PI3K/Akt/mTOR signaling no longer require GREB1 expression (Fig. 6). Constitutive activation of the PI3K/Akt/mTOR pathway is frequently associated with resistance to endocrine therapies and is the basis for numerous clinical trials investigating PI3K/Akt/mTOR pathway inhibitors in endocrine-resistant patient populations (7–10). In patients with hormone-refractory disease with hyperactivation of the PI3K/Akt/mTOR pathway, the pressure to express GREB1 is lost. Thus, decreased GREB1 expression in advanced disease may be the result of constitutive PI3K/Akt/mTOR activity rather than a cause of therapeutic bypass. In support of this notion, stable expression of constitutively active Akt resulted in silencing of GREB1 expression after several passages (Supplemental Fig. 5). These findings warrant further research into the use of GREB1 as a clinical biomarker for treatment selection.
Declaration of interest
The authors have no conflicts of interest to disclose.
Funding
This research was funded in part by Pelotonia (CNH).
Acknowledgements
We thank Paul Herman, Clarissa Wormsbaecher, Weiwei Liu, and Makanko Komara for critical reading of this manuscript prior to submission.
References




