

ABSTRACT
Marine microbial eukaryotes underpin the largest food web on the planet and influence global biogeochemical cycles that maintain habitability. They are also remarkably diverse and provide insights into evolution, including the origins of complex life forms, as revealed through genome analyses. However, their genetic tractability has been limited to a few species that do not represent the broader diversity of eukaryotic life or some of the most environmentally relevant taxa. Here, we report on genetic systems developed as a community resource for experimental cell biology of marine protists from across the eukaryotic tree of life. We outline DNA delivery methods, expression constructs, and genome editing approaches that proved successful, as well as results from taxa where a working system was not achieved. The reported breakthroughs on genetic manipulation position the community to dissect cellular mechanisms from a breadth of protists, which will collectively provide insights into ancestral eukaryotic lifeforms, protein diversification and evolution of cellular pathways.
INTRODUCTION
The ocean represents the largest continuous planetary ecosystem, hosting an enormous variety of organisms. These range from some of the largest creatures on Earth to a vast microscopic biota including unicellular eukaryotes (protists). Despite their small size, protists play key roles in marine biogeochemical cycles and harbor tremendous evolutionary diversity1–3. Notwithstanding their significance for understanding the evolution of life on Earth and their role in marine food webs, as well as driving biogeochemical cycles to maintain habitability, little is known about their cell biology including reproduction, metabolism, and signalling4. Most of the biological information available is based on comparison of proteins from cultured genome-sequenced species to homologs in genetically tractable model taxa, such as yeast5–9. A major impediment to understanding the cell biology of these diverse eukaryotes is that protocols for genetic modification are only available for a small number of species that represent neither the most ecologically relevant marine protistan species nor the breadth of eukaryotic diversity.
The development of genetic tools requires reliable information about gene organization and regulation of the emergent model species. Over the last decade, some of this information has become available through genome5,6,8,10 and transcriptome sequencing initiatives7,9,11,12 resulting in nearly 120 million unigenes from protists13. Insights from these projects have enabled the phylogenetically-informed approach7 used herein for selecting and developing key marine protists into model systems. Forty-one scientific groups took part in a community-based effort resulting in the development of genetic tools that significantly expand the number of eukaryotic lineages, which can be manipulated, and which encompass multiple ecologically important marine protists. These genetic tools transform our ability to address questions about the evolution and cell biology of these microorganisms and, by extension, other eukaryotes.
Here, we summarize detailed methodological achievements by this collaborative effort and analyse results to provide a synthetic ‘Transformation Roadmap’ for creating new microeukaryotic model systems. Although the organisms reported here are diverse, the paths to overcome difficulties share similarities, highlighting the importance of building a well-connected community to overcome technical challenges and accelerate the development of genetic tools. The new model species presented herein will not only extend our knowledge of cell biology and functional biodiversity, but also serve as platforms to advance microbial biotechnology.
RESULTS
Overview of studied organisms
Taxa were selected from multiple eukaryotic supergroups1,7 to maximize the potential to compare fundamental aspects of cellular biology and to evaluate the numerous “hypothetical” unigenes found in marine protists (Fig. 1). Previously, reproducible transformation of marine protists was limited to only a few species such as Thalassiosira pseudonana, Phaeodactylum tricornutum, and Ostreococcus tauri (Supplementary Table 1). Our initiative included 40 species, specifically, 6 Archaeplastida, 2 Haptophyta, 2 Rhizaria, 10 Stramenopila, 12 Alveolata, 4 Discoba, and 4 Opisthokonta (Fig. 1). Most of them were isolated from coastal habitats, the focus area of several major culture collections7. More than 50% of the selected species are considered photoautotrophs, with another 35% divided between heterotrophic osmotrophs and phagotrophs, the remainder being predatory mixotrophs. Almost 20% of the chosen species are symbionts and/or parasites of marine plants or animals, 5% are associated with detritus, and several are responsible for harmful algal blooms (Supplementary Table 2). The main challenge of this initiative was to develop reverse genetics tools applicable to transforming all of these species, which not only require different cultivation conditions but are also phenotypically extremely diverse.

A phylogenetically-informed approach was used to select protists for concerted genetic manipulation efforts. A schematic view of the eukaryotic tree of life with effigies of main representatives based on Keeling (2019). Colour-coordinated species we have attempted to genetically modify, are listed below. Current transformability status is schematized in circles indicating: DNA delivered and shown to be expressed (yellow); DNA delivered, but no expression seen (grey); no successful transformation achieved despite efforts (blue). Delivery methods and construct types are shown pictorially. Overall, protocols and specific reagents are available to transfect 23 protist species belonging to 7 eukaryotic supergroups.
Roadmap for establishing new model organisms
The research teams met and conversed over a three-year period to identify and optimize the steps required to create new model systems (Fig. 2). Selectable markers, transformation conditions, and reporters were compared across species (Supplementary Tables 3, 4 and 5), and efforts were detailed using consistent terminology (Table 1; Fig. 3; Fig. 4). Information on partially successful or failed approaches was also summarized (see Supplementary Results) to facilitate future research efforts.
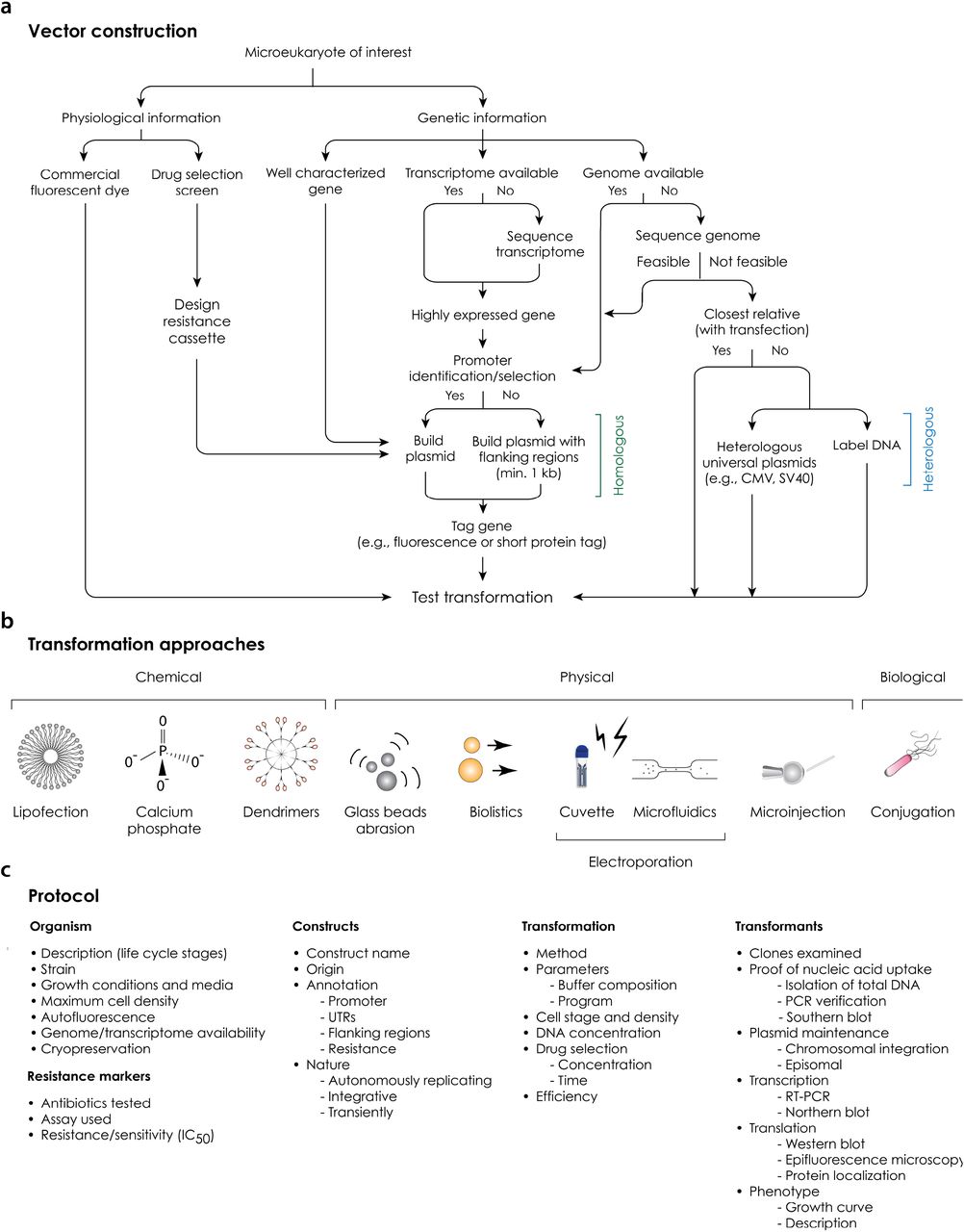
(A) Vector design and construction. Alternative routes employing a range of information and strategies are shown. (B) Transformation approaches. All methods (chemical, physical or biological) for introducing DNA/RNA/protein into a living cell are shown using icons. We highly recommend testing several of them, since successful DNA delivery depends on a number of specific features of each organism. (C) Protocol. Key features needed to obtain and demonstrate a successful transformation are listed in an abbreviated form.

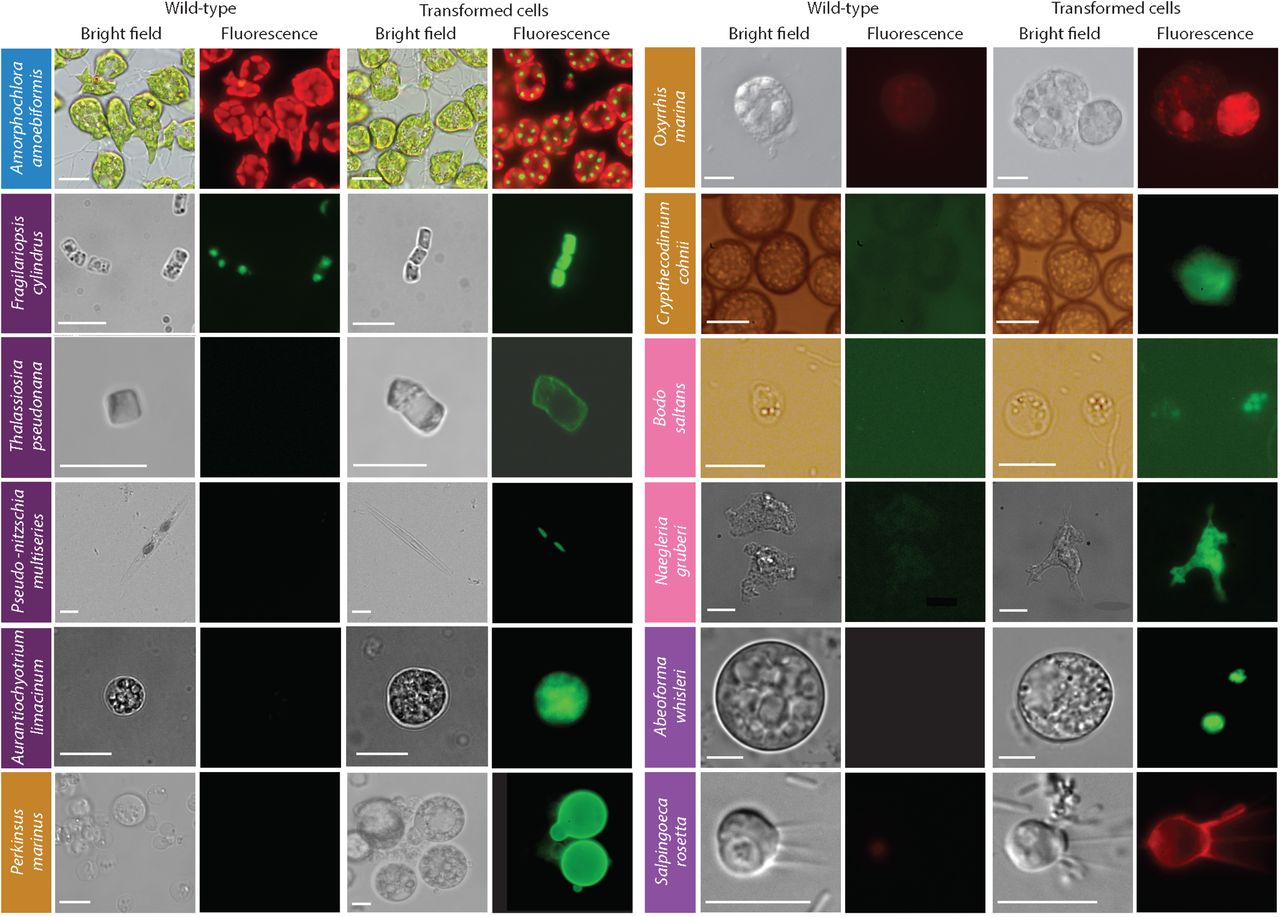
Fluorescent microscopy images showing the transformants and wild type cells of 11 protist species.Colored boxes behind species names reflect supergroup assignments in Fig. 1. Scale bars are as follows: 10 μm for Thalassiosira pseudonana, Amorphochlora (Lotharella) amoebiformis, Bodo saltans, Naegleria gruberi, Abeoforma whisleri, and Salpingoeca rosetta; 11 μm for Crypthecodinium cohnii; 15 μm for Perkinsus marinus; 20 μm for Fragilariopsis cylindrus and Oxyrrhis marina; 100 μm for Pseudo-nitzschia multiseries.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
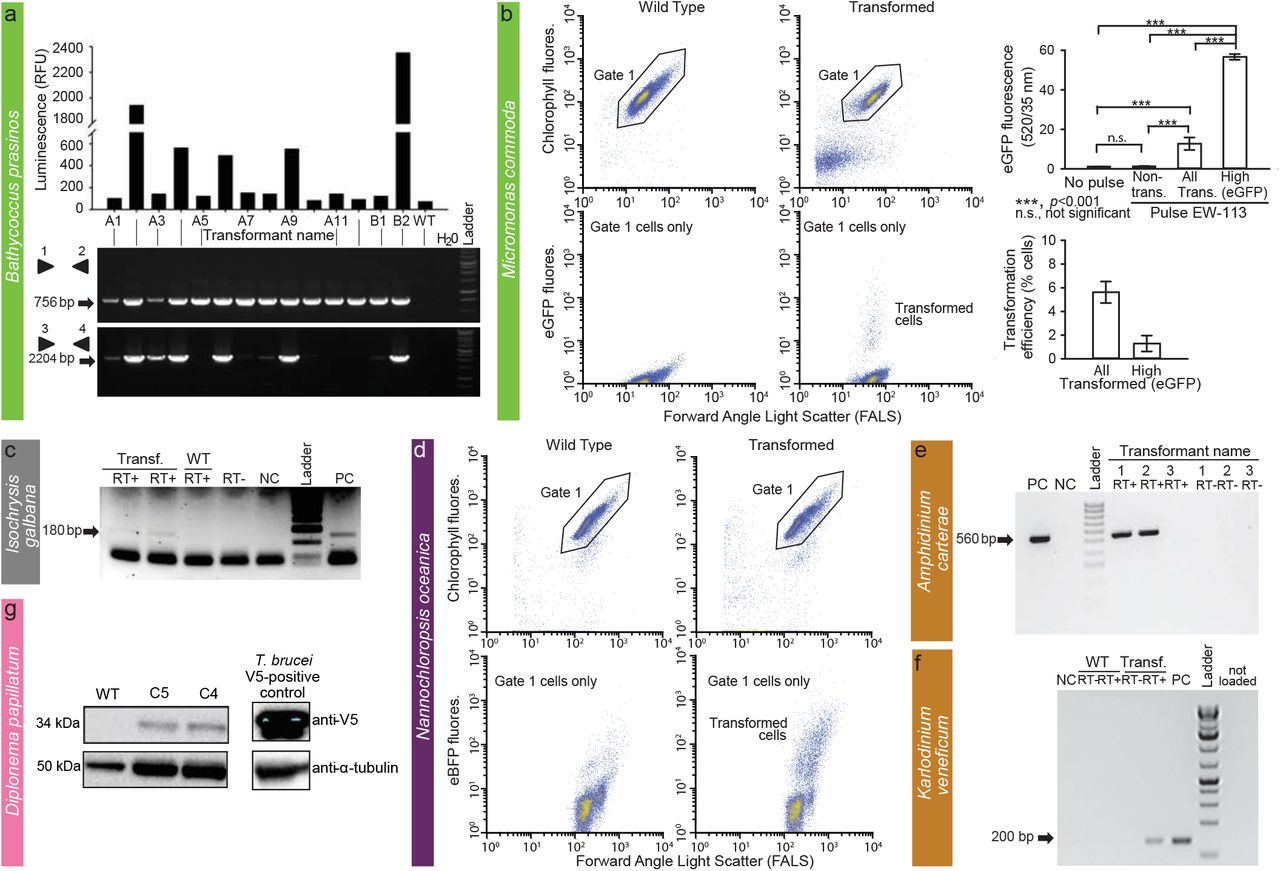
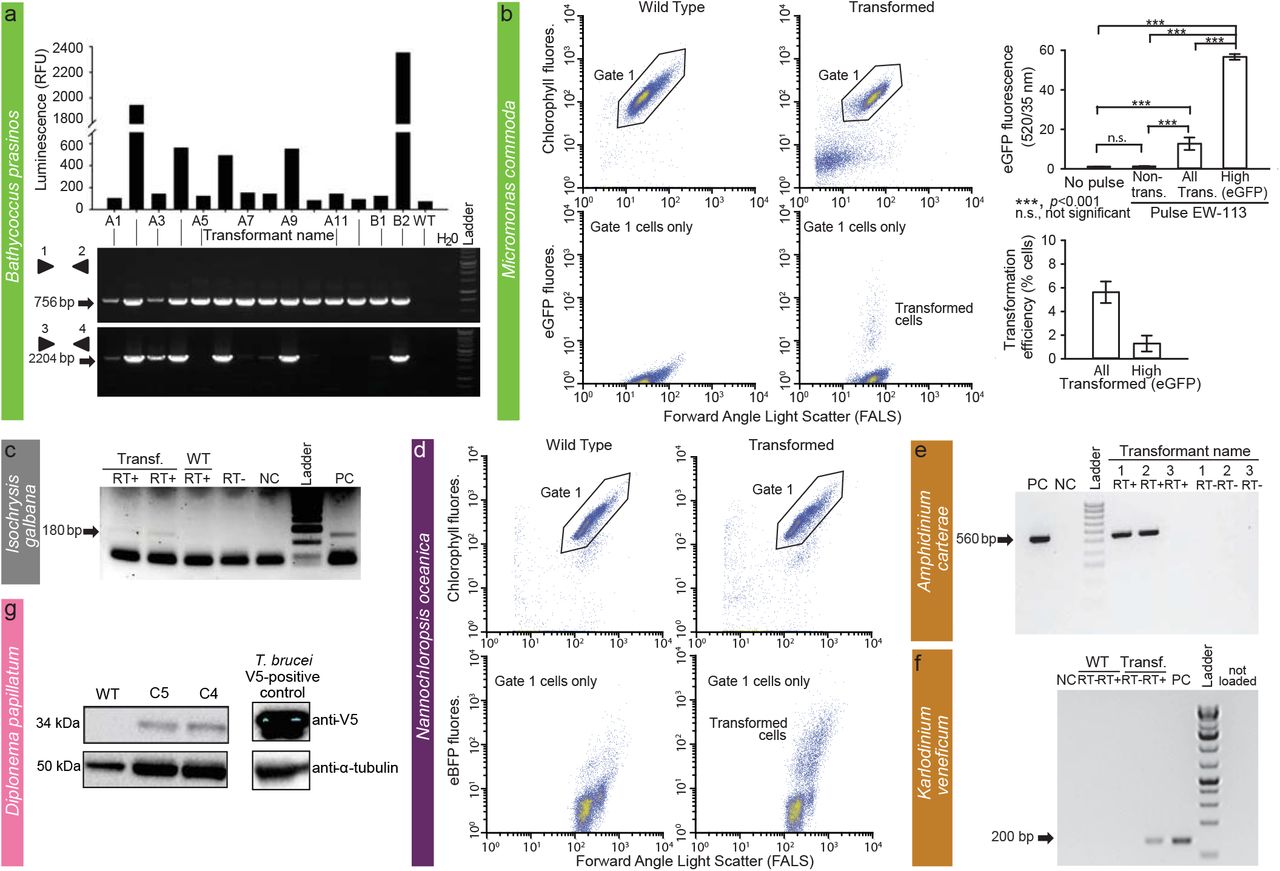
FACS and other methods were used to confirm transformation in two prasinophytes (A, B), one haptophyte (C), one stramenopile (D), two dinoflagellates (E, F) and one diplonemid (G). (A) In vivo luminescence of 14 B. prasinos transformants resistant to G418 depicted as relative luminescence unit (RLU) per 5 s and a corresponding gel showing PCR amplification from DNA of transformants of pH4:KanMx (primers 1 and 2) and pHAPT: luciferase sequences (primers 3 and 4). (B) FACS analysis of M. commoda cells in two treatments: controls (wild type [WT] or “no-pulse”), in which constructs were added but no electroporation pulse applied, and in the treatment to which a pulse was applied. Note that lower panels include only the population of healthy cells selected using the depicted gate in upper panels. Bar graphs show the mean and standard deviation of eGFP fluorescence from biological triplicates in the same experiment, analyzed as control (WT or “non-pulse”) and pulsed (EW-113 treatment, with cells in the latter treatment analyzed as non-transformed, all eGFP and high GFP (cells for which eGFP fluorescence was an order of magnitude higher than in controls). Transformation efficiencies also reflect mean and standard deviations of biological triplicates. (C) Expression of NAT in pIgNAT transformed I. galbana. RT-PCR products from transformed cells resistant to nourseothricin. (D) FACS analysis of N. oceanica control cells (no plasmid added but pulse applied) and cells subjected to a pulse with a plasmid, after which a subset of cells appeared to have been transformed at 24 h. Product size (arrow) is 180 bp. (E) Transformation of A. carterae chloroplast genome with pAmpChl. RT-PCR with or without reverse transcriptase (RT), as indicated. Size of band (arrow) is 560 bp. (F) RT-PCR of the Neo gene in Karlodinium transformant “N6” under selection with kanamycin and wild type cells that were not grown under antibiotic selection. “-” and “+” depict negative (no template) and positive (neo gene as template) PCR controls, respectively. “RNA–” depicts no reverse transcriptase control, which gave negative results, and “-N” depicts nested PCR negative control. cDNA libraries were made with 200 ng of RNA from wild type and CA-137 N6 cells. (G) Western blot of D. papillatum wild type (WT) and C4 and C5 transformants that express the V5-tagged aminoglycoside 3’-phosphotransferase (Neo). Monoclonal mouse α-V5 antibodies (1:2,000) and secondary α-mouse antibodies coupled to horseradish peroxidase (1:1,000) were used for visualisation. V5 tagged-mNeonGreen cell line of T. brucei cells served as a positive control and mouse anti-α-tubulin antibodies (1:5,000) were used as a loading control.
For some of the selected species, the first step was to identify cultivation conditions for robust growth in the laboratory and compatibility with transformation protocols. The aim was to generate axenic cultures and establish conditions supporting either high cell densities or large culture volumes to obtain sufficient amounts of biomass for use with a variety of molecular biology methods. Unlike established microbial model species, cultivation of marine protists can be challenging, especially for predatory taxa that require co-cultivation with their prey. Subsequent steps included the identification of suitable antibiotics and their corresponding selectable markers (Supplementary Table 3), conditions for introducing exogenous DNA (Supplementary Table 4), and selection of promoter and terminator sequences for designing transformation vectors (Fig. 2; Supplementary Table 5).
A variety of methods were used to test whether exogenous DNA was integrated into the genome or maintained as a plasmid and whether the introduced genes were expressed. Approaches to show the former included inverse PCR, Southern blots and whole genome sequencing, whereas approaches to demonstrate the latter included RT-PCR, epifluorescence microscopy, fluorescence-activated cell sorting (FACS), antibody-based methods, and growth assays in the presence of antibiotics to confirm transcription and translation of introduced selection and reporter genes (e.g., eGFP, YFP, mCherry). Transformation outcomes for each species were parsed into three groups according to the level of success or lack thereof and are discussed below according to their phylogenetic position (Fig. 1).
Archaeplastids
Prasinophytes are important marine green algae distributed from polar to tropical regions. They form a sister group to chlorophyte algae, and together, these two groups branch adjacent to land plants, collectively comprising the Viridiplantae, which are part of the Archaeplastida1,8 (Fig. 1). Genome sequences are available for the picoprasinophytes (<3 μm cell diameter) tested herein, specifically, Micromonas commoda, Micromonas pusilla, Ostreococcus lucimarinus and Bathycoccus prasinos. A homologous recombination system exists for Ostreococcus tauri14, which we extend here to O. lucimarinus. Additionally, we introduced the first genetic system(s) for Bathycoccus, a scaled, non-motile genus, and Micromonas, a motile, naked genus with larger genomes than Bathycoccus and Ostreococcus8.
O. lucimarinus (RCC802) and B. prasinos (RCC4222) were transformed using protocols adapted from O. tauri15. Briefly, using electroporation (Supplementary Table 4) for transfer of exogenous genes, O. lucimarinus was transformed using a DNA fragment encoding the O. tauri high-affinity phosphate transporter (HAPT) gene fused to a luciferase gene and a KanMX selection marker, which resulted in transient luciferase expression 24 h after electroporation (Table 1; Supplementary Fig. 1). After 2 weeks of growth in low melting agarose plates containing neomycin (1 mg/ml), 480 colonies were obtained, picked, and grown in artificial seawater with neomycin. Of these, 76 displayed luminescence ≥ 2.5 fold above background (80 Relative Luminescence Units, RLU), with widely variable levels (200 to 29100 RLU), likely reflecting either the site of integration and/or the number of integrated genes (Supplementary Fig. 1). The O. tauri construct did not work in B. prasinos, while the use of the B. prasinos histone H4 and high affinity phosphate transporter sequences in an otherwise identical construct and conditions was successful. Although luciferase expression was not detected 24 h after electroporation, 48 neomycin-resistant colonies were obtained 2 weeks later, 20 being luminescent when grown in liquid medium. Analysis of 14 resistant transformants revealed that the luciferase sequence was integrated into the genome of 5 clones that were luminescent, and one non-luminescent clone, suggesting that the chromatin context at integration sites in the latter was not favourable to luciferase expression (Fig. 4A).
The above methods for Bathycoccus and Ostreococcus failed in Micromonas. However, Lonza nucleofection was successful with M. commoda (CCMP2709) (Table 1; Fig. 4B) using 2 codon-optimized plasmids, one encoding the luciferase gene (NanoLuc, Promega) flanked by an exogenous promoter and terminator sequence from the 5′- and 3′-untranslated regions (UTRs) of histone H3 in M. polaris8, and the other encoding an eGFP gene flanked by endogenous promoter and terminator sequences from the ribosomal protein S9. Acclimated mid-exponential M. commoda cells grown in L1 medium at 21 °C were spun at 5000 × g for 10 min, the pellet was resuspended in Buffer SF (Lonza) premixed with carrier DNA (pUC19) and the plasmid, and 3 × 107 cells were used per reaction. After applying the EW-113 pulse, 100 μl of ice-cold recovery buffer (10 mM HEPES-KOH, pH 7.5; 530 mM sorbitol; 4.7% [w/v] PEG 8000) was added to each well and incubated for 5 min at room temperature. Each reaction was then transferred into 2 ml L1, placed at 21 °C and light was increased stepwise over 72 h. Sensitivities to antibiotics were established (Supplementary Table 3). Constructs did not include a selectable marker, as we aimed to introduce and express foreign DNA while developing conditions suitable for transfection that supported robust growth in this cell walllacking protist. Transformants revealed a significantly higher level of eGFP fluorescence than wild type cells, with 1.3% of the population showing fluorescence per cell 45-fold higher than both the non-transformed portion of the culture and the wild type cells (Fig. 4B). Additionally, the RLU was 1500-times higher than controls when using the luciferase-bearing construct, such that multiple experiments with both plasmids confirmed expression of exogenous genes in M. commoda.
Haptophytes (incertae sedis)
Haptophytes are a group of photosynthetic protists that are abundant in marine environments and include the major calcifying lineage, the coccolithophores. Genome sequences are available for Emiliania huxleyi6 and Chrysochromulina tobin16, and there are few reports of nuclear transformation of haptohytes17. Here, a stable nuclear transformation system was developed for Isochrysis galbana, a species that lacks coccoliths, but represents an important feedstock for shellfish aquaculture18.
I. galbana (CCMP1323) was transformed by biolistic bombardment with the pIgNAT vector, which contains nourseothricin N-acetyltransferase (NAT, for nourseothricin resistance) driven by the promoter and terminator of Hsp70 from E. huxleyi (CCMP1516). Twenty four hours after bombardment, cells were transferred to liquid f/2 medium at 50% salinity containing 80 μg/ml nourseothricin (NTC) and left to grow for 2-3 weeks to select for transformants (Table 1). The presence of NAT in NTC-resistant cells was verified by PCR and RT-PCR (Fig. 4C) and the sequence verified. To confirm NTC resistance was a stable phenotype, cells were subcultured every 2-4 weeks at progressively higher NTC concentrations (up to 150 μg/ml NTC) in the above-mentioned media. Cells remained resistant to NTC for approximately 6 months, as confirmed by PCR screening to identify the presence of the NAT gene.
Rhizarians
Rhizarians include diverse non-photosynthetic protists, as well as the photosynthetic chlorarachniophytes that acquired a plastid via secondary endosymbiosis of a green alga. Uniquely, they represent an intermediate stage of the endosymbiotic process, since their plastids still harbor a relict nucleus (nucleomorph)5. Here, we have advanced a transformation protocol for the chlorarachniophyte Amorphochlora (Lotharella) amoebiformis for which low-efficiency transient transformation has previously been achieved using particle bombardment19.
A. amoebiformis (CCMP2058) cells (1 × 107) were resuspended in 100 μl of Gene Pulse Electroporation Buffer (BioRad) with 20 to 50 μg of the reporter plasmid encoding GFP-RubisCO fusion protein under the control of the native rbcS1 promoter and subjected to electroporation (Supplementary Table 4). Cells were immediately transferred to fresh ESM medium and incubated for 24 h. Transformation efficiency was estimated by the fraction of cells expressing eGFP, resulting in 0.03-0.1% efficiency, as enumerated by microscopy, showing an efficiency up to 1,000-fold higher than the previous study19 (Table 1). Stable transformants were generated by manual isolation using a micropipette, and a transformed line has maintained eGFP fluorescence for at least 10 months without antibiotic selection (Fig. 3).
Stramenopiles
Stramenopiles are a diverse lineage with many important photoautotrophic, mixotrophic and heterotrophic taxa. As the most studied class in this lineage, diatoms (Bacillariophyceae) were early targets20,21 for the development of reverse genetics tools. Diatoms alone are estimated to contribute approximately 20% of annual carbon fixation22 and, like several other algal lineages, are used in bioengineering applications, such as the production of high-value end products and biofuels23. The work presented here builds on established reverse genetics tool for diatoms21 and recently developed for conjugation-based DNA delivery24 and genome editing25,26. An improved method for episome-based and CRISPR/Cas-driven gene knockout is presented for P. tricornutum (see Supplementary Results) More efficient T. pseudonana conjugation-based DNA delivery is also described. New transformation protocols are presented for Fragilariopsis cylindrus, the first cold-adapted alga to have such a system, as well as the coastal species Pseudo-nitzschia multiseries (producer of the neurotoxin domoic acid) and Pseudo-nitzschia australis. We also present the first successful transformations for other marine stramenopile groups. Specifically, for Nannochloropsis oceanica, a photosynthetic eustigmatophyte and for the non-photosynthetic labyrinthulomycete Aurantiochytrium limacinum. All of these stramenopiles except P. australis have a sequenced genome27.
Microparticle bombardment was used on F. cylindrus (CCMP1102), grown and maintained at 4 °C during the protocol. Exponential phase cells (5 × 107) were harvested onto a 1.2 μm membrane filter (Millipore) which was then placed on an 1.5% agar Aquil plate for bombardment with beads coated with a plasmid containing zeocin resistance and eGFP, both controlled by an endogenous fucoxanthin chlorophyll a/c binding protein (FCP) promoter and terminator (Table 1; Supplementary Tables 3 and 4)28. Transformation was performed using 0.7 μm tungsten particles and the biolistic particle delivery system PDS1000/He (BioRad).
Rupture discs for 1,350 and 1,550 pounds per square inch (psi) gave the highest colony numbers (efficiencies of 20.7 colony forming units (cfu)/108 cells and 30 cfu/108 cells, respectively. Following bombardment, the filter was turned upside down and left to recover (24 h) on the plate, then cells were rinsed from the plate/filter and spread across five 0.8% agar Aquil plates with 100 μg/ml zeocin. Colonies appeared 3 to 5 weeks later. PCR on genomic DNA showed that 100% and 60% of colonies screened positive for the zeocin resistance and eGFP genes, respectively. Confirmed by FACS and microscopy, eGFP was localized to the cytosol and was distinguishable from plastid autofluorescence (Fig. 3). Both genes were present in transformants after multiple transfers (> 10) 2 years later, indicating long-term stability.
We selected the silaffin precursor TpSil3p (Table 1) as the target gene for improving bacterial conjugation in T. pseudonana (CCMP1335). TpSil3p was fused to eGFP flanked by an FCP promoter and terminator, cloned into pTpPuc3 episomal backbone, and transformed into mobilization plasmid-containing EPI300 E. coli cells (Lucigen). The donor cells were grown in SOC medium at 37 °C until OD600 of 0.3–0.4, centrifuged and resuspended in 267 μl SOC medium. Next, 200 μl donor cells were mixed with 4 × 107 T. pseudonana cells, co-cultured on pre-dried 1% agar plates, dark incubated at 30 °C for 90 min, then at 18 °C in constant light for 4 h, followed by selection in 0.25% agar pour plates containing 100 μg/ml nourseothricin. Colonies were observed after 2 weeks, inoculated into 300 μl L1 medium and supplemented with 200 μg/ml nourseothricin to reduce the number of false positives. Positive transformants were identified by colony PCR screening (Supplementary Fig. 2) and epifluorescence microscopy (Fig. 3).
The diatoms Pseudo-nitzschia multiseries (15093C) and P. australis and other members of this genus form buoyant linear chains with overlapping cell tips during active growth, and were unconducive to punctate colony formation on agar, where their growth is generally poor. A low-gelation-temperature agarose seawater medium (LGTA) was developed to facilitate growth, antibiotic selection, and cell recovery. Both diatoms exhibited growth inhibition at relatively low concentrations under nourseothricin, formaldehyde, and zeocin (Supplementary Table 3). Biolistic transformation of two other Pseudo-nitzschia species had been demonstrated at low efficiency29. To complement this approach and explore potentially higher efficiency methods for transformation with diatom episomal plasmids, we modified the existing conjugation-based method24. The published conjugation protocol was modified to enhance P. multiseries post-conjugation viability by reducing SOC content. An episomal version of the Pm_actP_egfp_actT expression cassette was transfected into E. coli EPI300+pTAMOB and used for conjugation. After 48 h in L1 medium, cells were plated in LGTA and eGFP-positive cells were observed 7 days later (Fig. 3). PCR revealed the presence of plasmids in all eGFP positive colonies. Similarly, conjugation with the episome pPtPUC3 (bleomycin selection marker)-containing bacterial donors was followed under zeocin selection (200 μg/ml). After 7 days, only viable cells (based on bright chlorophyll fluorescence) contained the episome, as confirmation by PCR. Propagation of transformants after the first medium transfer (under selection) has so far been unsuccessful.
The electroporation of N. oceanica (CCMP1779) was optimized based on the ability to treat cells with fluorescein-conjugated 2000 kDa dextran and their subsequent survival (Supplementary Table 4). Increasing the sorbitol concentration to 800 mM and electroporating at between 5 and 9 kV/cm resulted in highest cell recovery. This protocol was used to introduce plasmids containing the gene for the blue fluorescent reporter mTagBFP2 under control of the cytomegalovirus (CMV), the cauliflower Mosaic Virus 35S, or the VCP1 promoter previously described from Nannochloropsis sp.30. Transient expression of blue fluorescence (compared to cells electroporated simultaneously with the same protocol without plasmid) appeared within 2 h, lasted for at least 24 h, and disappeared by 48 h in subsets of cells electroporated with mTagBFP2 under the control of CMV (Fig. 4D). The effectivness of transient transformation was much higher when a linearized plasmid was used compared to a cirular plasmid (Table 1). VCP1 did not induce blue fluorescence with a circular plasmid, while 35S gave inconsistent results when both circularized and linearized plasmids were used.
Stable transformation of A. limacinum (ATCC MYA-1381 or NIBH SR21) was achieved by knock-in of a resistance cassette composed of the bleomycin-resistance gene (shble) driven by 1.3 kb promoter and 1.0 kb terminator regions of the endogenous glyceraldehyde-3-phosphate dehydrogenase gene carried in a pUC19-based plasmid (18GZG) along with the native 18S rRNA gene, and by knock-in of a similar construct containing a yeGFP:shble fusion (18GeZG) (Supplementary Fig. 3). Approximately 1 × 108 cells were electroporated (Supplementary Table 4), adapting the electroporation protocol used for Schizochytrium31. The highest transformation efficiency was achieved using 1 μg of linearized 18GZG plasmid with 2 pulses, resulting in a time constant of ~5 ms (Supplementary Table 4). Expression of the fusion protein was confirmed by both the zeocin-resistance phenotype and the detection of eGFP (Fig. 3). Six 18GZG transformants derived from uncut and linearized plasmids were examined in detail. All maintained antibiotic resistance throughout 13 serial transfers, first in selective, and subsequently in non-selective media, and then again in selective medium. Integration of the plasmid into the genome was confirmed by PCR as well as by Southern blots using a digoxigenin-labeled shble gene probe, showing that 4 transformants had integrations by single homologous recombination, while in 2 transformants, additional copies of the antibiotic resistance cassette were integrated by non-homologous recombination elsewhere in the genome (Supplementary Fig. 3).
Alveolates
This species-rich and diverse group is subdivided into ciliates, apicomplexans, and dinoflagellates (Fig. 1). As a link between apicomplexan parasites and dinoflagellate algae, perkinsids are key for understanding the evolution of parasitism, and also have potential biomedical applications. Techniques currently exist for transformation of only a small number of ciliates, perkinsids and apicomplexans32,33. Here, we present results for the perkinsid Perkinsus marinus, a major pathogen of marine mollusks, fish, and amphibians34. Additionally, advances in transformation methods were made for 4 dinoflagellate species: Oxyrrhis marina, a basal-branching phagotroph that lacks photosynthetic plastids, Crypthecodinium cohnii, a heterotroph used in food supplements, Amphidinium carterae with a highly reduced plastid genome containing only a small number of genes encoding proteins for photosynthetic electron transport, rRNAs and one tRNA, and Karlodinium veneficum, a mixotroph (combining photosynthetic and phagotrophic nutrition) that produces fish-killing karlotoxins35.
We advanced the P. marinus (PRA240) transformation system36,37 using a newly formulated transformation 3R buffer (200 mM Na2HPO4; 70 mM NaH2PO4; 15 mM KCl; 1.5 mM CaCl2; 150 mM HEPES-KOH, pH 7.3) that makes the electroporation reactions cheaper and therefore more accessible. We were able to co-express 2 genes and efficiently select transient and stable transformants using FACS (Fig. 3; Table 1). In addition, we established the integration profile of ectopic DNA once introduced into the P. marinus genome. We saw no evidence of integration through homologous recombination, and a propensity for plasmid fragmentation and integration within transposable elements sites. Furthermore, an optimized alternative protocol for transformation using glass bead abrasion was developed. In brief, 5 × 107 cells were resuspended in 330 μl of fresh ATCC Medium 1886 and were mixed with 5.0 μg of linearized and circular [1:1] plasmid and 300 μl of glass beads (Sigma) in a 1.5 ml tube, vortexed for 30 s at maximum speed, and the cells in 500 μl of culture medium were transferred to 6-well plates in a final volume of 3 ml. Two versions of the previously published Moe gene promoter were tested36. Whereas the 1.0 kb promoter version induced expression after 2 or 3 days, the truncated version (0.5 kb) took 7 days for expression to be detected. Resistance genes to bleomycin, blasticidin and puromycin have all been shown to confer resistence to transformed P. marinus; however, selection regimes are still relatively slow and inefficient, indicating further room for improvement37.
O. marina (CCMP1788) was cultured in f/2 medium with a diverse bacterial community and fed weekly with heat-killed E. coli. Selection trials revealed that 5 different antibiotics led to 100% mortality of O. marina in 6 days (Supplementary Table 3). Fluorescently-labelled DNA or DNA analogs such as FITC dextran were used to test the efficiency of delivery using a variety of chemical or electrical methods. For instance, incubation with CaCl2 allowed introduction of Alexa 488-labeled DNA (Molecular Probes) with 20% efficiency. Briefly, a mix of 1 to 8 μg of DNA and CaCl2 (f.c. 0.25 M) was combined with an equal volume of HeBS (274 mM NaCl; 10 mM KCl; 1.4 mM Na2HPO4; 15 mM D-glucose; 42 mM HEPES, pH 7.1) and incubated with 1 ml of O. marina culture. FITC-dextran incorporation was achieved when using Gene Pulser Electroporation Buffer in combination with three 5 ms square-wave pulses (0.1 ms pause between pulses) using a field strength of 0.5 kV/cm (Supplementary Table 4). Transient expression of mCherry was observed after introducing a plasmid encoding mCherry gene with a flanking sequence of the hsp90 gene from O. marina using the transformation protocol from above (Fig. 3; Table 1).
The cell cycle of C. cohnii consists of motile G1 cells, which encyst when they shed their flagella completing the remaining cell-cycle phases38. After cytokinesis, the daughter cells remain inside the mother cell, and in this stage, treatment with PEG results in the release of the non-motile spheroplasts39. Transfection of C. cohnii (CCMP316) was attempted using physical (electroporation, microfluidics, particle bombardment) and chemical (lipofection) methods in motile daughter cells and spheroplasts with both electroporation and lipofection resulting in DNA delivery. C. cohnii spheroplasts were electroporated with 5 μg of plasmid using Amaxa Cell Line Optimization Solution V or 3R buffer and program D-023 in Nucleofector (Lonza). The swimming cells and spheroplasts were resuspended in 1 mg of FITC-Dextran and 300 μl of glass beads, vortexed at maximum speed for 15-30 s and subsequently recovered in fresh medium. Biolistics was also tried on 2.5 × 107 swimming cells and spheroplast using 7.5 μg of HEM plasmid, FITC-dextran, 3 μg of Cas9/sgRNA and 7.5 μg of GFP-carrying plasmid. Cells were precipitated in the presence of gold beads and shooting was carried out in a vacuum at >25 Hg using a 1550 psi rupture disk. Plasmids from P. marinus (PmMOE:GFP-11)36, Hematodinium sp. (UB-GFP and EF-GFP) under human ubiquitin promoters, PAY and PAYCO using E. huxleyi ubiquitin promotors, purified Cas9 protein with synthetic sgRNA and donor DNA template with the GFP gene and site-directing homology flanking sequences resulted in no fluorescent cells independently of the delivery method attempted. We also tested transformation using chemically labeled DNA (Alexa Fluor 488) and FITC-labeled 150 kDa dextran. C. cohnii withstands electroporation with low or no damage, and its swimming cells were difficult to disrupt with glass-bead abrasion and particle bombardment, whereas spheroplasts were much more sensitive. Electroporation of spheroplasts was successful using the Lonza program X-001 or lipofection, both with labeled DNA (Fig. 3), but expression of encoded genes was not confirmed and the number of transfected cells was very low. We also attempted electroporation and microfluidics (5 square waves and 6 exponential decays)40 with plasmids PmMOE:GFP-11, UB-GFP, EF-GFP, PAY and PAYCO but with no positive outcome.
For A. carterae (CCMP1314), we successfully conducted transformation of plastid DNA. In brief, we selected psbA (encodes D1 of photosystem II), which is a target of the herbicide atrazine (Supplementary Table 3). Two shuttle vectors using an E. coli plasmid backbone, both based on the psbA minicircle, were constructed. One introduced a mutation in the psbA gene to confer atrazine resistance (pAmpPSBA) and in the other vector psbA was replaced by chloramphenicol acetyl transferase (CAT; pAmpChl). While no transformants were seen for glass-bead agitation, NEPA electroporation or Lonza nucleofection, biolistic transformation with DNA-coated gold microparticles rendered transformed cells. Those with pAmpPSBA were not stable long-term; however, the pAmpChl transformants were stable for at least 12 months, and RT-PCR (Fig. 4E) showed that the CAT gene was transcribed41, albeit missing the polyU tail which is normally added post-transcriptionally to all A. carterae plastid transcripts. Attempts to transform Symbiodinium microadriaticum using the same approach were unsuccessful42.
Since K. veneficum (CCMP1975) is sensitive to kanamycin (Supplementary Table 3), a neomycin resistance gene (Neo) was added to the backbone of a dinoflagellate-specific expression vector43, named DinoIII-Neo. After linearization, the vector was successfully electroporated by Nucleofector (Lonza). The preprogrammed Nucleofector optimization pulse codes, buffer SF/Solution I (Lonza), and 2 μg/μl of linearized DinoIII-Neo were used. Electroporated cells were selected under 150 μg/ml kanamycin 3 days post-electroporation. New seawater with kanamycin was added every 2 weeks to the cultures and new subcultures were inoculated monthly. Total RNA was isolated and cDNA synthesized as previously reported44 using random hexamer as the primer. Out of 16 pulse codes tested, CA-137 and DS-138 resulted in long-term survival of K. veneficum under kanamycin selection. CA-137 resulted in high cell densities for several months, with the resistance gene successfully detected by PCR (Fig. 4F).
Discobans
This diverse group, recently split into Discoba and Metamonada45, includes heterotrophs, photoautotrophs, predatory mixotrophs, as well parasites. The Discoba include parasitic kinetoplastids with clinical significance, such as Trypanosoma brucei, T. cruzi and Leishmania spp., for which efficient transformation protocols are available46. However, such protocols are missing for marine species. Here, we describe transformation protocols for the kinetoplastid Bodo saltans, the diplonemid Diplonema papillatum, and the heterolobosean Naegleria gruberi.
B. saltans (‘Lake Konstanz’ strain) was transformed with a plasmid containing a cassette designed to fuse an endogenous EF-1 α gene with GFP for C-terminal tagging. This cassette includes downstream GFP, a B. saltans tubulin intergenic region followed by the aminoglycoside 3’-phosphotransferase gene (Neo), conferring resistance to neomycin. EF-1α genes exist in tandem repeats; however, the homologous regions that flank the cassette to induce homology-directed repair were chosen to target only one copy of the gene. As transcription in B. saltans is polycistronic47, insertion of the tubulin intergenic region into the plasmid is essential for polyadenylation of the EF1-α/GFP fusion and trans-splicing of the Neo gene. Square-wave electroporation (Nepa21) was used with a poring pulse of 250V (25 ms) and 5 transfer pulses of 60V (99 ms) in the presence of Cytomix buffer (120 mM KCl; 0.15 mM CaCl2; 10 mM KH2PO4; 2 mM EGTA; 5 mM MgCl2; 25 mM HEPES-KOH, pH 7.6). Selection of transfected cells began with 2 μg/ml of neomycin added 24 h after electroporation,and gradually increased over 2 weeks to 5 μg/ml (Table 1). Cells were washed and subcultured into fresh selection medium every 4 days, and neomycin-resistant cells emerged 7 to 9 days post-electroporation. The GFP signal was detected 2 days post-electroporation, albeit with low intensity. This may be due to the inefficient translation of eGFP since it has not been codon-optimized for B. saltans (Fig. 3). Genotyping analysis 9 months post-transfection confirmed the presence of the Neo gene and at least partial plasmid sequence. However, plasmid integration into the B. saltans genome through homologous recombination is still unconfirmed. This suggests either off-target plasmid integration or that the plasmid is maintained episomally.
D. papillatum (ATCC 50162) was transformed by electroporation using 3 μg of SwaI-linearised fragment (cut from p57-V5+NeoR plasmid) containing V5-tagged Neo gene flanked by partial regulatory sequences derived from the hexokinase gene of the kinetoplastid Blastocrithidia (strain p57) (Table 1) using a published protocol48. About 18 h after electroporation, 75 μg/ml neomycin was added to the medium and after 2 weeks 7 neomycin-resistant clones were recovered. Transcription of Neo was verified in 2 clones by RT-PCR, and the expression of the tagged Neo protein was confirmed by Western blots using α-V5 antibody (Fig. 4G). As preliminary data indicate that the homologous recombination machinery is present and active in D. papillatum, extending the 5’and 3’ homologous regions (> 1 kb) may increase integration efficiency.
For N. gruberi (ATCC 30224) two plasmids were designed. The first one carried the hygromycin-resistance gene with an actin promoter and terminator, along with an HA-tagged eGFP driven by the ubiquitin promoter and terminator. The second plasmid carried the Neo gene instead. For each individual circular plasmid, 4 μg was electroporated (Supplementary Table 4). About 48 h after electroporation, dead cells were removed from the suspension and viable cells were washed with PBS. Afterwards, 300 μg/ml of hygromycin B or 700 μg/ml of neomycin was added to the fresh media (Table 1). One to 4 weeks later several resistant clones were recovered and expression of eGFP and/or hygromycin was confirmed by Western blots. Expression of eGFP was observed by epifluorescence microscopy (Fig. 3) with ~80% of transformants maintaining hygromycin or neomycin resistance in addition to expressing eGFP.
Opisthokonts
The opisthokont clade Holozoa includes animals and their closest unicellular relatives Choanoflagellata, Filasterea, Ichthyosporea, and Corallochytrea. The establishment of molecular genetic tools in non-metazoan holozoans promises to help illuminate the cellular and genetic foundations of animal multicellularity9. Genomic and transcriptomic data are available for multiple representatives characterized by diverse cell morphologies, some of which can even form multicellular structures9,12. Here, we show that transient transformations have been achieved for the filasterean Capsaspora owczarzaki49, the ichthyosporean Creolimax fragrantissima50 and the choanoflagellate Salpingoeca rosetta51. A novel protocol is presented for transforming the ichthyosporean Abeoforma whisleri, isolated from the digestive tract of mussels, and we have improved a transformation protocol for S. rosetta51.
All A. whisleri life stages are highly sensitive to a variety of methods for transformation. We developed a Lonza 4D-nucleofection-based protocol using 16-well strips, wherein PBS-washed cells were resuspended in 20 μl of buffer P3 (Lonza) containing 40 μg of carrier plasmid (empty pUC19) and 1-5 μg of the reporter plasmid (A. whisleri H2B fused to mVenus fluorescent protein, mVFP) (Table 1), and subjected to code EN-138 (Lonza). Immediately after the pulse, cells were recovered by adding 80 μl of marine broth (Gibco) prior to plating in 12-well culture plates previously filled with 1 ml marine broth. After 24 h, ~1% of the culture was transformed based on the fraction of cells expressing mVFP in the nucleus (Fig. 3).
As part of this initiative, we also developed a method for transiently transfecting the choanoflagellate S. rosetta using a modified protocol for Lonza nucleofection51 (Fig. 3). Improvement to this protocol include the use of selection for expression of a puromycin resistance gene that enables stable transformation of S. rosetta and allows for genetic complementation (Table 1). Lessons from these efforts, including the use of the highly sensitive reporter nanoluc gene, a hypertonic recovery buffer and large amounts of carrier DNA helped with the establishment and improvement of transformation conditions for Micromonas sp. and A. whisleri, respectively.
DISCUSSION
Marine organisms play essential roles in global biogeochemical cycles and produce approximately half of the Earth’s oxygen1,22. Decades of research by marine biologists, ecologists, protistologists, and oceanographers have contributed to an increasingly coherent picture of the oceanic ecosystem. These studies highlight the diversity of ocean life, including the protistan component2,3. Remarkable strides have also been made in developing an overview of the genomes and predicted proteomes of these protists13. However, without genetic manipulation systems, these taxa remain an untapped resource for providing deeper insights into their cell biology, with potentially valuable outcomes for evolutionary studies, nanotechnology, biotechnology, medicine, and pharmacology.
Global synthesis of the approaches developed herein provides a Transformation Roadmap that will guide future efforts to establish new and emergent model organisms (Fig. 2). Notably, our studies did not result in a universally applicable protocol, likely because transformability and a range of other key conditions varied greatly across taxa and approaches. Factors influencing outcomes include intrinsic features of the genome (e.g., presence/absence of homologous recombination, extrachromosomal elements, genome size), as well as morphology and structural features of the cell. In general, electroporation proved the most common method for introducing exogenous DNA stably into the cell. This approach was utilized for naked cells and protoplasts, yet frequently also worked, albeit with lower efficiency, on cells protected by cell walls. Linearized plasmids were most effective for delivery, and 5’ and 3’ UTRs-containing promotors of highly expressed endogenous genes provided the strongest expression of selective reporters and markers.
Our results significantly expand the segment of extant eukaryotic diversity amenable to reverse genetics approaches. Out of the 40 microbial eukaryotes selected, we were able to deliver and express exogenous DNA in ca. 50% of them. This high rate of success testifies to the benefits of taking on high-risk research in a large, open, and collaborative context52. The new systems reported herein open a wealth of opportunities for exploring functional differences between members of relatively conserved protein families shared across eukaryotes, or even domains of life. These novel transformation systems also enable us for the first time to shed light on the function of species-specific genes which likely reflect key adaptations to specific niches in dynamic ocean habitats.
AUTHOR CONTRIBUTIONS
The project was conceived and designed by A.C.J., J.Z.K., S.B., D.F., J.L., R.E.R.N., J.A.F.R., E.C., L.S., A.Z.W., T.M., A.E.A., F.Y.B, C.B, Ch.B., H.C., T.C., J.L.C., K.C., C.L.D., V.E., V.H., Y.H., C.J.H., P.J.K., N.K., S.L., C.M., J.M., I.R.T., P.A.S., C.H.S., G.J.S., A.T., P.V.D., A.T. and R.F.W. Data analysis was carried out by M.A.J., C.A., C.B., A.C.B., P.B., D.S.B., S.A.B., A.B., G.B., R.C., M.A.C., D.B.C., L.C., R.D., E.E., P.A.E., F.F., V.F.B., N.J.F., K.F., P.A.G., P.R.G., F.G., S.G., J.G., Y.H., E.R.H.C., E.H., A.Hi., A.Ho., I.H., J.I., N.A.T.I., Y.I., N.E.J., A.K., K.F.K., B.K., E.K., L.A.K., N.L., I.L., Z.L., J.C.L., F.L., S.M., T.M., M.M., S.R.N., D.N., I.C.N., L.N., A.M.G.N.V., M.N., I.N., A.Pa., A.Pi., S.P., J.P., J.S.R., M.R., D.R., A.R., M.A.S., E.S., B.S., R.S.,T.V.H., L.T., J.T., M.V., V.V., L.W., X.W., G.W., A.W. and H.Z. The manuscript was written by D.F., R.E.R.N., J.A.F.R., E.C., L.S., T.M., A.Z.W. and J.L. with input from all authors.
COMPETING INTERESTS
The authors declare no competing interests.
ACKNOWLEDGEMENTS
We thank M. Salisbury for assistance, C. Poirier and M. Hamilton (Monterey Bay Aquarium Research Institute) for FACS analysis; and Vinay K. Nagarajan, Monica Accerbi, and Pamela J. Green (University of Delaware) who carried out the Agrobacterium studies. We are grateful to L. Teytelman from protocols.io. This collaborative effort was supported by the Gordon and Betty Moore Foundation EMS Program of the Marine Microbiology Initiative and other forms of grant support to the participating laboratories.
REFERENCES




