

Abstract
Phosphorylated derivatives of phosphatidylinositol (PIPs), are key membrane lipid residues involved in clathrin-mediated endocytosis (CME). CME relies on PI(4,5)P2 to mark endocytic sites at the plasma membrane (PM) associated to clathrin-coated vesicle (CCV) formation. The highly diverged parasitic protist Giardia lamblia presents disordered and static clathrin assemblies at PM invaginations, contacting specialized endocytic organelles called peripheral vacuoles (PVs). The role for clathrin assemblies in fluid phase uptake and their link to internal membranes via PIP-binding adaptors is unknown.
Here we provide evidence for a robust link between clathrin assemblies and fluid-phase uptake in G. lamblia mediated by proteins carrying predicted PX, FYVE and NECAP1 PIP-binding modules. We show that chemical and genetic perturbation of PIP-residue binding and turnover elicits novel uptake and organelle-morphology phenotypes. A combination of co-immunoprecipitation and in silico annotation techniques expands the initial PIP-binding network with addition of new members. Our data indicate that, despite the partial conservation of lipid markers and protein cohorts known to play important roles in dynamic endocytic events in well-characterized model systems, the Giardia lineage presents a strikingly divergent clathrin-centered network. This includes several PIP-binding modules, often associated to domains of currently unknown function that shape and modulate fluid-phase uptake at PVs.
Introduction
Phosphorylated derivatives of the minor membrane phospholipid phosphatidylinositols (PIPs) are surface molecules of most eukaryotic endomembrane compartments [1–3]. PIPs play important roles in diverse pathways including signaling cascades, autophagy and membrane remodelling [2, 4–8]. Their diverse functions are reflected in their distinct subcellular distribution. PI(4,5)P2 is highly enriched at the plasma membrane (PM) with PI(3,4,5)P3 [4, 5]. PtdIns(4)P’s largest pool is at Golgi membranes, with smaller amounts found at the the PM. PI(3)P is converted into PI(3,5)P2 on early endosomes during transition to multivesicular bodies and then late endosomes [6, 7]. PI(3)P is also a marker of phagosomes [8] while PI(5)P marks both the PM and endomembranes [9]. At least 14 distinct PIP-binding modules haven been identified in eukaryotes, demonstrating a wide range of selective protein-lipid interactions associated with the PM and internal membranes [10].
In addition to their structural functions in membranes, PIPs are involved in spatiotemporal organization of membrane remodeling processes such as clathrin-coated vesicles (CCV) formation during clathrin-mediated endocytosis (CME). In particular, PI(4,5)P2 marks sites of endocytosis at the PM and recruits proteins involved in the formation of CCVs [11]. The protein interactomes of mammalian PI(4,5)P2-binding proteins include the early-acting clathrin interacting partners AP2 [12–15], AP180/CALM [16, 17] and epsin [17, 18]. These factors carry specific PIP-binding domains that can discriminate between PIP variants to achieve membrane targeting specificity.
Giardia lamblia (syn. intestinalis, duodenalis) is a widespread parasitic protist that colonizes the upper small intestine of vertebrate hosts. Its life cycle is marked by the alternation between an environmentally-resistant, infectious cyst stage responsible for parasite transmission, and a trophozoite stage proliferating by binary fission. Nutrient uptake of trophozoites in the lumen of the small intestine is almost entirely routed through peripheral vacuoles (PVs). These organelles are positioned just beneath the PM and are contacted by funnel-shaped invaginations of the PM that are likely conduits for uptake of fluid-phase extracellular material [19].
A recent characterization of the PV protein interactome using the highly conserved G. lamblia clathrin heavy chain (GlCHC) as affinity handle confirmed the endocytic nature of these organelles by highlighting the presence of giardial AP2 (GlAP2) subunits, the single dynamin-like protein GlDRP and a putative clathrin light chain Gl4259 (GlCLC; [19]). Notably absent were components for CCV uncoating and disassembly, consistent with a lack of measurable clathrin assembly turnover and in line with the observations that CCVs are missing in G. lamblia and clathrin assemblies are static and long-lived. Therefore, G. lamblia presents an unusual endocytic system, characterized by divergent endocytic compartments (PVs) associated to static clathrin assemblies that are not predicted to form ordered arrays or higher-order structures such as CCVs yet are closely membrane-associated.
Included in the giardial CHC interactome were three proteins with predicted PIP-binding domains: FYVE domain protein Gl16653 and two PX-domain proteins (Gl7723 and Gl16595), the latter part of a six-member protein family (Table 1; [19, 20]). In a previous study, we hypothesized that Gl16653 (GlFYVE), Gl7723 (GlPXD1) and Gl16595 (GlPXD2) act as PIP-binding adaptors to link and maintain static clathrin assemblies at the PM and PV membrane interface in G. lamblia [19]. We further hypothesized that a perturbation of PIP-binding protein levels and/or function would lead to impaired fluid-phase uptake by affecting PV functionality. To test these hypotheses, we performed an in-depth functional characterization of all previously-identified PIP-binding proteins associated to clathrin at PVs. We assessed their lipid-binding preferences and visualized their subcellular localizations using electron microscopy and both conventional and super resolution light microscopy. By manipulating protein levels and/or function we could elicit novel fluid-phase uptake and PV morphology-related phenotypes, thereby establishing PIPs as a link between the role of clathrin as a membrane remodeling proteins and PV-based endocytosis in G. lamblia. Furthermore, we used a combination of co-immunoprecipitation and in silico annotation techniques to expand protein interactomes established previously, thereby discovering a new set of PIP-binding proteins with roles likely reaching beyond the PV compartment. Lastly, we propose an updated working model summarizing the complex networks between PIP-binding proteins and clathrin assemblies at PVs.
G. lamblia PIP-binding proteins.
A compilation of all PIP-binding domains identified in the Giardia Genome Database (www.giardiadb.org; GDB) using previously characterized domains [24] as baits for HMM-based homology searches (column 1). Predicted Giardial orthologs are present for PIP-binding domains ENTH, PH, FYVE, PX, BAR, FERM and PROPPINS (column 2) and mostly retrieve the correct domains when used as a baits for reverse HHpred searches (column 4). Except for Glepsin, GlPXD2 and GlPROP1 and 2, all others are currently annotated on GDB as generically “hypothetical”, i.e. of unknown function (column 6). Each orthologue was assigned a name used throughout this report (column 7). Functional domain predictions using SMART (http://smart.embl-heidelberg.de/; column 8) and subcellular localization data (column 9) either previously reported or acquired in this study (column 10), are also included.
Results
The G. lamblia genome encodes at least seven distinct PIP-binding modules
Given that several types of PIP-binding modules have been identified in eukaryotes, we determined how many endocytosis-associated module types were actually represented in the Giardia genome, in addition to the known G. lamblia epsin, FYVE and PXD variants [19–23]. For this reason, we selected a total of 14 protein types from various organisms known to harbor PIP-binding domains, some of them involved in endocytosis. These are: ANTH (AP180 N-terminal homology), ENTH (epsin N-terminal homology), PH (Pleckstrin homology domain), FYVE (Fab1, YOTB, Vac1 and EEA1), PX (Phox homology), BAR (bin, amphiphysin and Rvs), FERM (4.1, ezrin, radixin, moiesin), PROPPINs (β-propellers that bind PIs), C2 (conserved region-2 of protein kinase C), GOLPH3 (Golgi phosphoprotein 3), PDZ (postsynaptic density 95, disk large, zonula occludens), PTB (phosphotyrosine binding), Tubby modules and the PH-like module of the endocytosis-associated NECAP1 protein [24]. Representatives for each module were used as bait for the HMM-based tool HHpred [25] for protein structure prediction and the detection of remotely related sequences in the G. lamblia predicted proteome (Table 1). Putative Giardia protein homologs (Table 1) were then subjected to the online tools SMART [26, 27] and InterProScan [28] to identify, conserved structural domains and sequence motifs within a query sequence (Fig 1A).

(A) Predicted functional domains for all identified PIP-binding proteins including positions of repetitive motifs and putative lipid and Zn -binding residues using HHPRED, HMMER and InterProScan. Ptd – Phosphatidylinositol. (B) Conventional confocal light-microscopy analysis of representative non-transgenic trophozoites labelled with Dextran-OG (first panel) to mark PV lumina and of immune-labelled trophozoites expressing HA-tagged PIP-binding protein reporters. and. Except for Glepsin and GlFERM, all tested reporter proteins localise in close proximity to peripheral vacuoles (PVs) at the cell cortex. Epitope-tagged GlNECAP1, GlPXD5 and GlPROP1 additionally show signal distribution throughout the cell. Cells were imaged at maximum width, where nuclei and the bare-zone are at maximum diameter. Epitope-tagged Glepsin-expressing cells were imaged at maximum width of the ventral side. Insets: DIC images. Scale bar: 1 µm (C) Confocal STED microscopy analysis of trophozoites expressing clathrin assemblies-associated epitope-tagged PIP-binding reporter proteins for GlPXD1-6, GlFYVE and GlNECAP1 (red channel) co-labelled with Dextran-OG as a marker for PV lumina (green channel). As shown in the merged insets, although all reporters are clearly PV-associated, reporters for proteins GlFYVE and GlPXD1 and 2 are proximal to the PM with respect to Dextran-OG, indicating they reside at the PV-PM interface. In contrast, reporters for GlPXD3 and GlNECAP1 appear to intercalate PVs. Scale bars: 1 µm for full cell image, 1 µm for insets.
This data mining approach detected high-confidence homologs for hitherto undiscovered G. lamblia proteins containing PH-like, FERM, BAR, FYVE and Proppin PIP-binding domains (Table 1, Fig 1A). No homologs could be found for the ANTH, C2, GOLPH3, PDZ, PTB, Tubby and PH PIP-binding module types.
Protein GL50803_17195 (GlNECAP1) is a predicted NECAP1 homolog containing a PH-like domain. Similarly, a conserved PH-like domain found at the C-terminus of FERM proteins was correlated with high confidence to protein GL50803_115468 (GlFERM). Immunofluorescence assays (IFAs) and confocal microscopy imaging of an epitope-tagged GlNECAP1 reporter expressed as an extra copy under its own promoter showed localization in close proximity to PVs and in the cytosol (Fig 1B). Similar to GlNECAP1, BAR domain-containing proteins GL50803_15847 and GL50803_14045 (GlBAR1 and 2) localize in close proximity to PVs (Fig 1B). Tagged reporters for new FYVE and PROPPIN members GL50803_16801 (Gl16801) and GL50803_16957 (GlPROP2), respectively, both localise in close proximity to PVs (Fig 1B). In contrast, a tagged GlFERM reporter presents a diffused cytosolic subcellular distribution (Fig 1B).
To extend the initial annotation of giardial PIP-binding proteins we performed multiple sequence alignment (MSA) analyses for each giardial PIP-binding module with selected orthologs to delineate lipid-binding motifs and residues critical for PIP recognition (Fig S1). In silico structural analyses of the lipid-binding domains of giardial proteins and their closest homologs were performed ab initio using the online tool I-TASSER [29–31]. Comparative analysis of structure models generated with I-TASSER clearly demonstrated positional conservation of residues critical for PIP binding (Fig S1). Since GlPXD1-2, GlFYVE and GlNECAP1 were experimentally shown to be associated to giardial clathrin assemblies [19], we selected these proteins and GlPXD3-6 for more detailed subcellular localization experiments. Stimulated emission-depletion (STED) microscopy in co-labelling experiments with Dextran-OG as a marker for fluid-phase endocytosis, unequivocally confirmed accumulation for GlPXD1-4 and 6, GlFYVE and GlNECAP1 epitope-tagged reporters at PVs (Fig 1C). The signal generated by GlPXD5 reporters was insufficient for a conclusive localization.
Taken together, in silico analysis identifies seven distinct PIP-binding module types encoded in the G. lamblia genome, conserved on both sequence and structural levels. Subcellular localization of epitope-tagged variants by fluorescence microscopy indicates clear association to PVs with the exception of Glepsin.
PIP-binding proteins associated with clathrin assemblies present distinct lipid-binding profiles in vitro
PX domains [32] and FYVE [33–35] preferentially bind PI(3)P. Even though PH domains have rather promiscuous binding preferences, a subset of PH domains binds strongly to PtdIns(3,4,5)P3 and PtdIns(4,5)P2, as well as PtdIns(3,4)P2 [36–38]. Based on the presence of conserved residues for lipid-binding in the giardial PXD1-6, FYVE and NECAP1 proteins (Fig S1), we hypothesized that their lipid-binding preferences would also be conserved. We tested this experimentally by expressing MBP-fused, epitope-tagged GlPXD1-6, GlFYVE and GlNECAP1 lipid-binding domains (Fig S2A and B). The recombinant fusion proteins were affinity-purified and used in lipid binding assays either for commercially-available PIP gradients as membrane-supported arrays (1.56-100 pmol/spot) (Fig 2A) or membrane strips spotted with defined amounts (100 pmol/spot) of PIPs (Fig S2C). The negative control for binding consisted of a PIP array probed with purified epitope-tagged MBP alone, whereas the positive control consisted of a PIP array probed with a commercially-available anti-PI(4,5)P2 antibody (Fig 2A). Quantification of the chemiluminescence signals shows a marked preference of MBP-GlPXD1 for PI(4,5)P2 in PIP gradients (Fig 2B) which was corroborated by experiments using PIP strips (Fig S2C). Under these conditions, GlPXD2, 3 and 6 show unexpectedly promiscuous binding preferences, with GlPXD2 presenting a marked affinity for PI(3)P and PI(4,5)P2, GlPXD3 for PI(3)P and to a lesser extent PI(5)P, and GlPXD6 for PI(3)P, PI(4)P and PI(5)P (Fig 2B). These data were in line with results from independent PIP strip experiments (Fig S2C and D). MBP-GlPXD4 and MBP-GlPXD5 binding preferences could only be probed using PIP strips (Fig S2C), showing in both cases a marked affinity for PI(3,5)P2 and PI(4,5)P2 (Fig S2C and D). Binding preferences for MBP-GlFYVE could not be determined, given that no signal was ever obtained on both PIP arrays and strips (Fig 2A, Figs S2C and E). Surprisingly, testing of GlNECAP1 consistently detected cardiolipin as the preferred lipid moiety (Fig 2C; Fig S2E), with no detectable preference for PIP residues (Fig S2C). Taken together, our data shows clearly distinguishable lipid binding profiles in vitro, with varying degrees of promiscuity for different PIP-binding domains.

(A) Membrane-supported lipid arrays spotted with gradients of different phosphorylated variants of phosphatidylinositol (PtdIns), from 100pmol (A) to 1.56pmol/spot (G), were probed with fixed amounts (2.5 µg) of clathrin assemblies-associated epitope-tagged PIP-binding domains from GlPXD1-6, GlNECAP1 and GlFYVE, followed by immunodetection of the epitope tag. Lipid-binding preferences for the protein fusion partner MBP (MBP alone) and for antibodies raised against PI(4,5)P2 (anti-PI(4,5)P2) were included as negative and positive controls for binding, respectively. No signal using arrays was obtained for MBP::GlPXD4 and MBP::GlPXD5, however, binding preferences for these fusions were determined using lipid strips (Figs S2A and B). (B) Plots of densitometric analyses using FIJI for each MBP-fused PIP-binding domain and each spotted PI/PIP residue based on array data presented in (A). (C) Testing of the binding affinity of the MBP-fused PIP-binding domain from GlNECAP1 on a wider range of lipid residues detects cardiolipin as the preferred substrate.
Saturation of PI(3)P, PI(4,5)P2 and PI(3,4,5)P3, but not PI(4)P binding sites in vivo inhibits PV-mediated uptake of a fluid-phase marker
The marked preference of GlPXD1-6 for PIP residues PI(3)P and PI(4,5)P2 raised the question whether their saturation of in vivo would elicit loss of function phenotypes in fluid phase uptake by Giardia trophozoites. Using a combination of commercially available antibodies, heterologous reporter constructs and chemical treatment we saturated sites of PI3P, PI(4,5)P2, and in addition PI(3,4,5)P3 and PI(4)P.
Detection of PI(3)P, PI(4,5)P2 and PI(3,4,5)P3 in chemically fixed trophozoites by immunofluorescence microscopy with primary PIP-targeted antibodies highlights enrichment for all PIP moieties in the cortical region containing PVs (Fig S3).
Ectopic expression of fluorescent high-affinity reporters for PI(3)P and PI(4)P 2xFYVE::GFP and GFP::P4C [39], respectively, in transgenic G. lamblia trophozoites was used to both identify as well as saturate membranes enriched for PI(3)P and PI(4)P deposition (Figs 3A-D). Live microscopy of cells expressing 2xFYVE::GFP shows distinct reporter accumulation in cortical areas consistent with binding to PV membranes (Fig 3B, green panels), whereas representative cells from line GFP::P4C show a more diffused cytosolic staining pattern, with some accumulation at PVs (Fig 3D, green panels). Fluid-phase uptake of Dextran-R was assessed in cells from both transgenic lines and compared to wild-type cells using quantification of signal intensity. Wild-type control cells and transgenic cells expressing small amounts of 2xFYVE::GFP (Fig 3A) incorporated large amounts of Dextran-R (Fig 3E). Conversely, a strong 2xFYVE::GFP signal correlated with low amounts of endocytosed Dextran-R detected at the cell periphery and with noticeably enlarged cells (Fig 3B). In contrast, there was no detectable difference in either Dextran-R uptake efficiency (based on fluorescent signal intensity) or cell width between weak (Fig 3C) and strong expressors (Fig 3D) of the GFP::P4C line. Cell width (Fig 3F) and fluid-phase uptake (Fig 3G) aberrant phenotypes in 2xFYVE::GFP cells were recorded with respect to wild-type control and GFP::P4C cells and tested for significance (p>0.05) on 100 cells/line selected in an unbiased fashion. These data translate into a significant negative correlation between expression of the PI(3)P-binding 2xFYVE::GFP reporter and fluid-phase uptake (Fig 3H) whereas only a slight albeit insignificant correlation was found between Dextran uptake and GFP::P4C expression (Fig 3I).

Light microscopy-based immunofluorescence analysis of representative transgenic trophozoites expressing Legionella-derived PIP-binding constructs. (A-B) Compared to low 2xFYVE::GFP-expressing cells from the same population, saturation of PI(3)P binding sites in cells highly overexpressing a regulated encystation-dependent epitope-tagged construct 2xFYVE::GFP (anti-HA) inhibits uptake of fluid-phase marker Dextran-R. Scale bars: 1 µm. (C-D) Expression levels of PI(4)P-binding epitope-tagged construct GFP::P4C expression (anti-HA) have no visible impact on Dextran-R signal at PVs of transfected cells. Scale bars: 1 µm. (E) Dextran-R uptake in non-transgenic wild-type cells as negative controls for construct-induced uptake phenotypes. Scale bars: 1 µm (F) Box-plot representing the distribution of cell width (in µm) across at least 100 wild-type, 2xFYVE::GFP- and GFP::P4C-expressing cells selected in an unbiased fashion. A statistically significant (two-sided t-test assuming unequal variances, p<0.05) increase in median cell width with respect to non-transgenic cells is detected for 2xFYVE::GFP-but not GFP::P4C-expressing cells. Asterisks indicate statistical significance. n.s.: not significant. (G) Box-plot representing the distribution of measured Dextran-R signal intensity across at least 100 wild-type, 2xFYVE::GFP- and GFP::P4C-expressing cells selected in an unbiased fashion. A statistically significant (two-sided t-test assuming unequal variances, p<0.05) decrease in Dextran-R signal intensity, normalized to wild-type cells (100%), is detected for 2xFYVE::GFP-but not for GFP::P4C-expressing cells. Asterisks indicate statistical significance. n.s.: not significant. (H) A statistically significant (p<0.5) linear correlation exists between Dextran-R signal (x-axis, intensity_red channel [%]) and 2xFYVE::GFP expression (y-axis, intensity_green channel [%]) measured across 100 cells. (I) The apparent linear correlation between GFP::P4C expression (y-axis, intensity_green channel [%]) and Dextran-R signal (x-axis, intensity_red channel [%]) is not statistically significant (p<0.5). (J) Wide-field microscopy-based immunofluorescence analysis of the impact of Neomycin treatment on Dextran-R uptake to deplete PI(4,5)P2 and PI(3,4,5)P3 binding sites in non-transgenic wild-type cells. With respect to non-treated cells (WT; left panel), Dextran-R signal at PVs is visibly impacted in neomycin-treated cells (WT_Neo; right panel). Scale bars: 10 µm for full wide-field image, 1 µm for a single cell. (K) Box-plot representing the distribution of measured Dextran-R signal intensity across 100 wild-type cells, either untreated (WT) or treated with neomycin (WT_Neo). Neomycin treatment causes a statistically significant (two-sided t-test assuming unequal variances, p<0.05) decrease in Dextran-R signal. Scale bars: wide-field: 10 µm; single cells:: 1 µm. For all images, nuclei are labelled with DAPI (blue). Insets: DIC images.
The cationic antibiotic neomycin binds tightly to the headgroup of phosphoinositides with a marked preference for PI(4,5)P2 and PI(3,4,5)P3 [40, 41]. As a means to saturate PI(4,5)P2 and PI(3,4,5)P3 binding in Giardia trophozoites, we tested its effect on fluid-phase uptake by treating wild-type trophozoites with 2mM neomycin followed by uptake of Dextran-R. Quantitative light microscopy image analysis revealed a significantly lower level of Dextran-R in treated trophozoites (p<0.05). (Fig 3J, K). Taken together, the data indicate that saturation of PI(3)P, PI(4,5)P2, and PI(3,4,5)P3, but not PI(4)P binding significantly impacts fluid-phase endocytosis through G. lamblia PVs.
Functional characterization of GlPXD1-4 and 6, GlFYVE and GlNECAP1
Manipulation of PIP residue homeostasis elicited PV-dependent fluid-phase uptake phenotypes. We hypothesized that changing expression levels of giardial PIP-binding proteins previously identified in clathrin interactomes would elicit aberrant uptake phenotypes in Giardia trophozoites. In addition we explored the functional boundaries of each PIP-binding module by defining their protein interactomes. To test this, we used the previously-generated epitope-tagged reporter lines for full-length GlPXD1-4 and 6, GlFYVE and GlNECAP1 (Fig 1C) for assessing the effects of ectopic expression on fluid-phase uptake phenotypes. Furthermore, we used the same lines as baits in antibody-based affinity co-immunoprecipitation (co-IP) and identification of reporter-associated protein complexes. Further investigation of GlPXD5 was abandoned at this stage due to its intractably low levels of expression.
The extended interactomes of GlPXD1, GlPXD4 and GlPXD6
Epitope-tagged, full-length GlPXD1 is a validated GlCHC interaction partner; its extended interactome confirms association to all core clathrin assembly components (GlCHC, GlCLC, GlDRP, GlAP2) (Fig 4 and Table S1) [19]. A weaker interaction with GlPXD2 was also found. The GlPXD4 interactome includes GlCHC and GlDRP and, uniquely for the GlPXD protein family, a previously confirmed interaction with GlPXD2 [19] albeit detected at lower stringencies (95_2_95, 2 hits) (Fig 4; Table S4). A putative SNARE protein GL50803_5785, previously identified in the GlTom40 interactome [42], was detected at lower stringencies (95_2_95, 2 hits). Similar to GlPXD1, GlPXD6 showed strong interaction with the β subunit of GlAP2 and GlCHC (Fig 4), although the reverse interaction was not detected in the previously-published clathrin-centered interactome [19]. Using lower stringency parameters (95_2_50, 3 hits), revealed interaction with GlFYVE, GlPXD3 and GlDRP (Fig 4; Table S). The GlPXD6 interactome includes Gl16717, a protein of unknown function predicted to carry a StAR-related lipid-transfer domain (Steroidogenic Acute Regulatory protein, START) domain [43]. Ectopic expression of epitope-tagged GlPXD1, 4 and 6 elicited no discernible PV-related phenotypes.

Curated core interactomes for GlPXD1, GlPXD4 and GlPXD6. All three epitope-tagged variants used as affinity handles in co-immunoprecipitation experiments identify GlCHC as a strong interaction partner for GlPXD1, 4 and 6. GlPXD1 and 4 further interact with other known clathrin assembly components such as GlCLC, GlAP2 subunits α, β and µ, and GlDRP. GlPXD2, albeit at low stringency, is the only other PXD protein found in all three interactomes. The GlPXD4 interactome includes a putative SNARE protein (5785; [42] while GlPXD6 as an affinity handle pulled down another PIP residue binder, GlFYVE, known to be associated to clathrin assemblies in G. lamblia [19]. Solid lines: interactions detected at high stringency. Dashed lines: interactions detected at low stringency. Yellow partners are currently annotated on GDB as “hypothetical protein” i.e. proteins of unknown function.
Ectopic expression of tagged GlPXD2 severely perturbs PV organisation
Mining the GlPXD2 protein interactome dataset with high stringency parameters confirmed interactions with GlCHC, GlAP2 and GlPXD4 (Fig 5A; Table S2). Furthermore, we identified three predicted SNARE proteins: GL5785, GL50803_14469 (Gl14469; at lower stringencies 95_2_50, 9 hits) and GL50803_10013 (Gl10013; Fig 5A) [44]. The SNARE GL5785 was detected also in the interactomes of GlPXD4 GlTOM40 [42]. GlNECAP1 was also identified as a GlPXD2 interacting partner, albeit only by applying low stringency parameters (95_2_50, represented by a dashed line, Fig 5A).

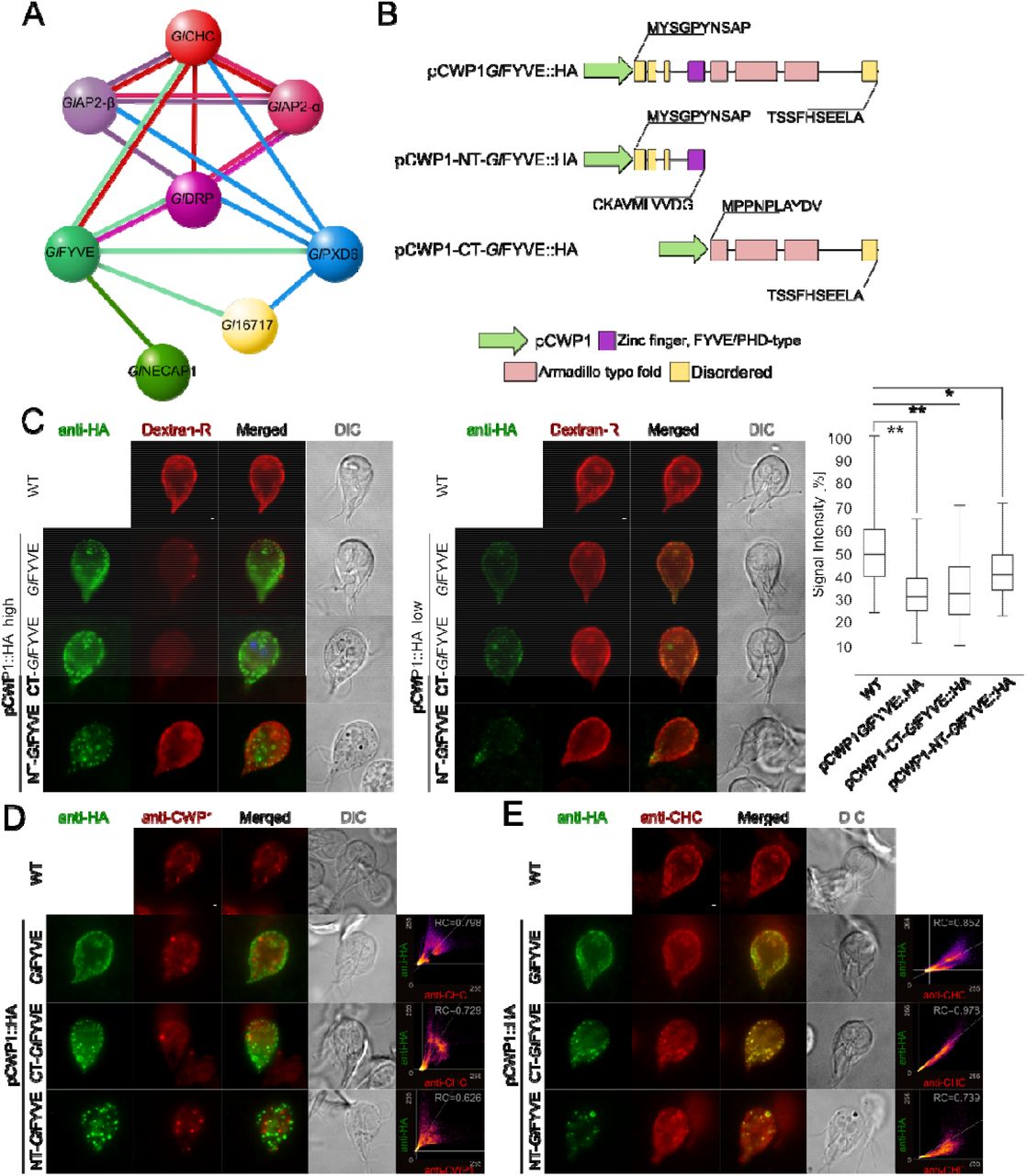
(A) The extended interactome analysis of epitope-tagged GlFYVE confirms confirms tight association to GlCHC, GlDRP and GlPXD6. GlNECAP1 as an alternative PIP-binding module was also detected. (B) C-terminally epitope-tagged full-length (top; pCWP1-GlFYVE::HA), C-terminal truncated (middle; pCWP1-NT-GlFYVE::HA, residues 1-300) and N-terminal truncated (bottom; pCWP1-CT-GlFYVE::HA, 301-990 residues) constructs were generated for regulated expression and phenotype testing. (C) Confocal imaging and immunofluorescence analysis of non-transgenic wild-type cells and in cells overexpressing constructs GlFYVE::HA, NT-GlFYVE::HA or pCWP1-CT-GlFYVE::HA (anti-HA) shows statistically significant (two-sided t-test assuming unequal variances, p<0.05) differences in their ability to take up Dextran-R. Cells overexpressing construct pCWP1-NT-GlFYVE::HA present additional membrane-bound structures that are not detected in other lines and do not associate with Dextran-R labelling. Asterisks indicate statistical significance: * p<0.05; ** p<0.005. n.s.: not significant. DIC: differential interference contrast. Scale bars: 1 µm. (D) Confocal imaging and immunofluorescence analysis of non-transgenic wild-type cells and cells overexpressing constructs GlFYVE::HA, NT-GlFYVE::HA or pCWP1-CT-GlFYVE::HA (anti-HA) using anti-CWP1-TxRed antibody (anti-CWP1) shows that the membrane compartments found in NT-GlFYVE::HA-expressing cells are not related to ESVs. Scale bars: 1 µm. (E) Antibody-based detection and immunofluorescence analysis of GlCHC deposition (anti-CHC) in non-transgenic wild-type cells and in cells overexpressing constructs GlFYVE::HA, NT-GlFYVE::HA or pCWP1-CT-GlFYVE::HA (anti-HA) detects a significant degree of GlCHC association to the CT-GlFYVE::HA variant, with only partial association to NT-GlFYVE::HA and GlFYVE::HA constructs. Scale bars: 1 µm.
In contrast to ectopic expression of tagged GlPXD1, 4, and 6, expression of an epitope-tagged reporter HA::GlPXD2 elicited a distinct phenotype. In contrast to non-transgenic wild-type cells (Fig 5C) and weakly-expressing HA::GlPXD2 cells (Figs 5D and E upper panels), gated STED imaging of trophozoites strongly expressing HA::GlPXD2 showed large membranous clusters which also accumulated Dextran-R (Fig 5D) and were bound by both anti-GlCHC (Fig 5E) and anti-PI(3)P (Fig 5F) antibodies. Transmission electron microscopy (tEM) analysis confirmed the presence of randomly distributed peripheral PV clusters in cells expressing HA::GlPXD2 (Fig 5G; left panel) which were not present in representative wild-type control cells (Fig 5G; right panel).
The GlPXD3 interactome is connected to clathrin assemblies and includes a novel dynamin-like protein
GlDRP, GlCHC, and GlAP2 (α/β subunits) were detected in the GlPXD3 interactome, thereby establishing the association of this PX domain protein with clathrin assembly structures at the PV/PM interface (Fig 6A; Table S3). A pseudokinase (Gl15411 [45]) previously identified in GlCHC assemblies was also found in the GlPXD3 interactome (Fig 6A; [19]). Furthermore, the GlPXD3 and Gl15411 interactomes share proteins GL50803_16811 (Gl16811) tentatively annotated as a ZipA protein in GDB, and proteins GL50803_87677 (Gl87677) and GL50803_17060 (Gl17060), annotated as a NEK kinase and an ankyrin-domain carrying protein, respectively (Fig 6A). Unique interaction partners for GlPXD3 include the SNARE protein Gl7309 [44] and GlNSF (GL50803_114776) [46]. In addition, protein GL50803_103709 carrying a predicted N-terminal BRO domain and protein GL50803_9605 were identified as unique GlPXD3 interaction partners (Fig 6A). Furthermore, the StAR-related lipid-transfer protein Gl16717, already found in the GlPXD6 interactome, was also found to be a low-stringency interaction partner for GlPXD3 and Gl15411, thereby connecting the GlPXD3 and GlPXD6-GlFYVE circuits. IFA analysis for Gl15411, Gl16811, Gl103709, and Gl7309 shows PV-associated labelling profiles for all corresponding epitope-tagged reporters whereas GlNSF presents a diffused localization pattern and Gl9605 shows a diffused yet punctate deposition pattern (Fig 6B).

(A) Left panel: Analysis of the extended GlPXD3 interactome using an epitope-tagged variant as affinity handle reveals robust interactions with clathrin assembly components GlCHC, α and β GlAP2 subunits, and GlDRP. Predicted inactive inactive NEK kinase 15411 [45] is similarly associated to clathrin assemblies [19] and further shares proteins Gl16811, Gl87677 and Gl17060 as interaction partners with GlPXD3. Predicted SNARE protein Gl7309, GlNSF (GL50803_1154776) and proteins Gl103709 and Gl9605 are unique GlPXD3 interaction partners. The GlPXD3 interactome is connected to the GlPXD6 circuit both directly and through Gl16717. Solid lines: interactions detected at high stringency. Dashed lines: interactions detected at low stringency. Yellow partners are currently annotated on GDB as “hypothetical protein” i.e. proteins of unknown function. Right panel: Total spectral counts as a measure of relative abundance for interaction candidates in the interactomes of GlPXD3 and Gl15411 epitope-tagged variants. Hashtag: detection at low stringency (95_2_50) and visualised with a dashed line in the interactome. (B) Light-microscopy-based immunofluorescence analysis of representative transgenic trophozoites expressing epitope-tagged reporter variants for proteins Gl15411, Gl16811, Gl103709, GlNSF, Gl9605, Gl7309 and Gl16717. Cells were imaged at maximum width, where nuclei and the bare-zone are at maximum diameter. Nuclei are labelled with DAPI (blue). Insets: DIC images. Scale bars: 1 µm (C) MSA analysis G1-Ploop, G2 switch1, G3 switch 2 and G4 regions of the conserved GTPase domains of Gl9605, GlDRP, Campylobacter jejuni DLP1 (Uniprot accession CJ0411) and DLP2 (CJ0412), Nostoc punctiforme BDLP1 (B2IZD3), Bacillus subtilis DynAD1 (P54159), Bacillus cereus DynAD2 (CUB17917), and Escherichia coli LeoA (E3PN25) bacterial dynamin-like proteins (BDLPs), Homo sapiens MFN1 (Q8IWA4), MFN2 (O95140), OPA1 (O60313) and DYN1 (Q05193), and Saccharomyces cerevisiae Fzo1p (P38297). Conserved positions are highlighted in grey. (D) I-TASSER de novo predicted 3D structure for Gl9605 (blue) and its closest known structural homologue, C. jejuni DLP2 (5owvC; green) indicating the GTPAse, neck and trunk regions that characterize BDLPs. (E) A close-up view of the overlapping structures in the GTPase domains of Gl9605 (blue) and C. jejuni DLP2 (5owvC; green) marking specific residues important for GTP binding and catalytic activity. (F) Quantitative microscopy-based immunofluorescence analysis of Dextran-R signal in cells overexpressing either a non-mutated full length epitope-tagged Gl9605 or mutated Gl9605 K73E and S74N variants. In contrast to non-transgenic wild-type controls and Gl9605::HA overexpressing cells, expression of Gl9605 K73E and S74N variants inhibited Dextran-TxR uptake in a statistically significant fashion (box-plot). Asterisks indicate statistical significance. n.s.: not significant.
Protein Gl9605, the sixth most abundant hit in the GlPXD3 interactome (Fig 6A), and currently annotated as having an unknown function, was identified as a highly-diverged dynamin-like protein (Fig 6C). In support of this, the predicted GTPase domain in Gl9605 contains signature motifs in the P-loop (G1), switch 1 (G2) and switch 2 (G3) regions [47–49]. Conserved motifs in the G4 region are only partially maintained (Fig 6D). To test residue conservation on a structural level, Gl9605 was subjected to ab initio modelling using I-TASSER and the resulting tertiary structure was superimposed on that of a dynamin-like 2 (DLP2 Cj:5ovW) [50], Gl9605’s closest structural homologue (Fig 6D). A structural overlap TM-score of 0.913 suggests an almostperfect structural match, with clear chemical and positional conservation of key residues involved in GTPase activity (Fig 6E). We sought to elicit a dominant-negative phenotype by engineering Gl9605 K73E and S74N mutants [51]. In contrast to either wild-type cells or cells overexpressing a wild-type epitope-tagged Gl9605 control, expression of Gl9605 K73E and S74N mutant reporters inhibited fluid-phase uptake of Dextran-R in a statistically significant manner (p<0.05; Fig 6F).
Regulated ectopic expression of GlFYVE variants inhibits fluid-phase uptake and induces the emergence of novel membrane-bound compartments
GlFYVE is a confirmed interactor of clathrin assemblies (Zumthor et al., 2016) through specific association to GlCHC and GlDRP (Fig 7A) Table S6). GlFYVE’s extended interactome includes GlPXD6, GlNECAP1 and protein GL50803_16717 (Gl16717). The latter was also found in the GlPXD6 interactome (Fig 4) and partially localizes to PVs as an epitope-tagged reporter (Fig 7A).

(A) The extended interactome analysis of epitope-tagged GlFYVE confirms confirms tight association to GlCHC, GlDRP and GlPXD6. GlNECAP1 as an alternative PIP-binding module was also detected. (B) C-terminally epitope-tagged full-length (top; pCWP1-GlFYVE::HA), C-terminal truncated (middle; pCWP1-NT-GlFYVE::HA, residues 1-300) and N-terminal truncated (bottom; pCWP1-CT-GlFYVE::HA, 301-990 residues) constructs were generated for regulated expression and phenotype testing. (C) Confocal imaging and immunofluorescence analysis of non-transgenic wild-type cells and in cells overexpressing constructs GlFYVE::HA, NT-GlFYVE::HA or pCWP1-CT-GlFYVE::HA (anti-HA) shows statistically significant (two-sided t-test assuming unequal variances, p<0.05) differences in their ability to take up Dextran-R. Cells overexpressing construct pCWP1-NT-GlFYVE::HA present additional membrane-bound structures that are not detected in other lines and do not associate with Dextran-R labelling. Asterisks indicate statistical significance: * p<0.05; ** p<0.005. n.s.: not significant. DIC: differential interference contrast. Scale bars: 1 µm. (D) Confocal imaging and immunofluorescence analysis of non-transgenic wild-type cells and cells overexpressing constructs GlFYVE::HA, NT-GlFYVE::HA or pCWP1-CT-GlFYVE::HA (anti-HA) using anti-CWP1-TxRed antibody (anti-CWP1) shows that the membrane compartments found in NT-GlFYVE::HA-expressing cells are not related to ESVs. Scale bars: 1 µm. (E) Antibody-based detection and immunofluorescence analysis of GlCHC deposition (anti-CHC) in non-transgenic wild-type cells and in cells overexpressing constructs GlFYVE::HA, NT-GlFYVE::HA or pCWP1-CT-GlFYVE::HA (anti-HA) detects a significant degree of GlCHC association to the CT-GlFYVE::HA variant, with only partial association to NT-GlFYVE::HA and GlFYVE::HA constructs. Scale bars: 1 µm.
To characterize the function of GlFYVE and to test whether a dominant-negative effect on uptake could be elicited, we performed a deletion analysis by generating epitope-tagged C-terminal (pCWP1-NT-GlFYVE::HA) and N-terminal (pCWP1-CT-GlFYVE::HA) truncation constructs, consisting of either the disordered region followed by the FYVE domain (Fig 7B), residues 1-300) or the armadillo repeat-rich (ARM repeats) domains (Fig 7B), residues 301-990), respectively. Expression of both constructs is regulated by an inducible promoter which is de-repressed during induction of encystation [52]. After a short (6hrs) induction pulse, transfected cells were subjected to Dextran-R uptake. Both in cells expressing the full-length pCWP1-GlFYVE::HA and truncated variants the amount of Dextran-R accumulated in PVs was significantly (p<0,05) lower (Fig 7C, box plot). Furthermore, IFA analysis of pCWP1-NT-GlFYVE::HA cells revealed the presence of membrane-bound compartments which overlapped neither with Dextran-R-labelled PVs (Fig 7C) nor with encystation specific vesicles (ESVs) labeled with the anti-CWP1 antibody (Fig 7D). In contrast, CT-GlFYVE::HA and full length GlFYVE::HA localized predominantly to PVs as (Fig 7C and D). The subcellular localization of GlCHC in these lines and in a wild-type control overlapped with the truncated CT-GlFYVE::HA variant, but only partially with NT-GlFYVE::HA and GlFYVE::HA (Fig 7E).
Ectopic expression of GlNECAP1 significantly impairs fluorescent Dextran uptake
Co-IP using epitope-tagged GlNECAP1 (Figs 1B and C) confirmed interaction with clathrin assembly components GlAP2-β, µ and α subunits, GlCHC and GlDRP (Table S7). Interaction with GlFYVE (Fig 7A) and, at lower stringency also for GlPXD2 (Fig 5A) could be confirmed (Fig 8A). Three putative conserved AP2-interacting motifs were identified using multi-sequence alignment; the high affinity WxxF motif at the N-terminus, two residues being invariant throughout evolution, K147 and G149, and AP2-beta linker interacting residues binding sites (Fig 8B) [53]. De novo 3D modelling confirms overall structural conservation of all key residues in GlNECAP1 (Fig 8B) when compared with mammalian NECAP1 (Fig 8C). Furthermore, the interacting interface of NECAP1 with the β-linker region of AP2 was also identified in the structural model for GlNECAP1 (Fig 8C).

(A) A GlNECAP1-centered interactome highlights association to clathrin assembly components and to additional PIP-residue binders GlFYVE and GlPXD2. (B) Multiple sequence alignment analysis of GlNECAP1 and NECAP1 orthologues from Arabidopsis thaliana (Uniprot accession Q84WV7), Trichinella pseudospiralis (A0A0V1JQ20), Caenorhabditis elegans (Q9N489), Echinococcus multilocularis (A0A087VZS0), Ceratitis capitata (W8CD89), Homo sapiens (Q8NC96) and Mus musculus (Q9CR95) identifies conserved motifs and residues for interaction with AP2. GlNECAP1 presents partial conservation, with a WXXF motif (orange) shifted to the N-terminus with respect to other orthologues. (C) Ab initio template-based 3D modelling of G. lamblia and H. sapiens NECAP1 (1tqz) homologues predicts similar structures, with conservation of key residues involved in the interaction between NECAP1 proteins and AP2 complexes (shaded in blue and green). (D) Wild-type non-transgenic control cells (WT) and cells overexpressing either epitope-tagged GlNECAP1 reporters GlNECAP1::HA, GlNECAP1::APEX2-2HA or the ΔWVIF deletion construct GlNECAP1∆WVIF::HA (green) were tested for Dextran-R (red). Dextran-R signal intensity was significantly (p<0.05) decreased in GlNECAP1::HA- and GlNECAP1::APEX2-2HA-expressing cells compared to wild-type controls and GlNECAP1∆WVIF::HA-expressing cells (box-plot). (E) Quantitative tEM analysis of GlNECAP1::APEX2-2HA-expressing cells (upper panels) and wild-type non-transgenic cells (WT; lower panels) shows visibly enlarged PVs in GlNECAP1::APEX2-2HA-expressing cells, with a statistically significant (p<0,05) increase in median PV area (in µm2; box-plot).
To test whether expression of a GlNECAP1 variant lacking the putative high-affinity motif WVIF could elicit a dominant-negative uptake effect, a deletion construct GlNECAP1∆WVIF::HA lacking this motif (Fig 8B) for conditional expression in induced trophozoites. Accumulation of Dextran-R into PVs detected by microscopy was significantly lower (p<0.05) in induced cells expressing GlNECAP1::HA or an APEX- and epitope-tagged variant GlNECAP1::APEX2-2HA compared with wild type controls (Fig 8D, box plot). Conversely, inducible expression of a deletion construct GlNECAP1∆WVIF::HA (Fig 8D, GlNECAP1∆WVIF::HA) had no discernible effect on accumulation of Dextran-R in PVs (Fig 8D, box plot). Inducible expression of the genetically encoded enzymatic reporter [54, 55] GlNECAP1::APEX2-2HA showed subcellular distribution of APEX-derived deposits around significantly enlarged PVs in tEM compared to wild type controls (Fig 8E; Fig S4).
GlPXD3 associates specifically with PVs as membrane coat
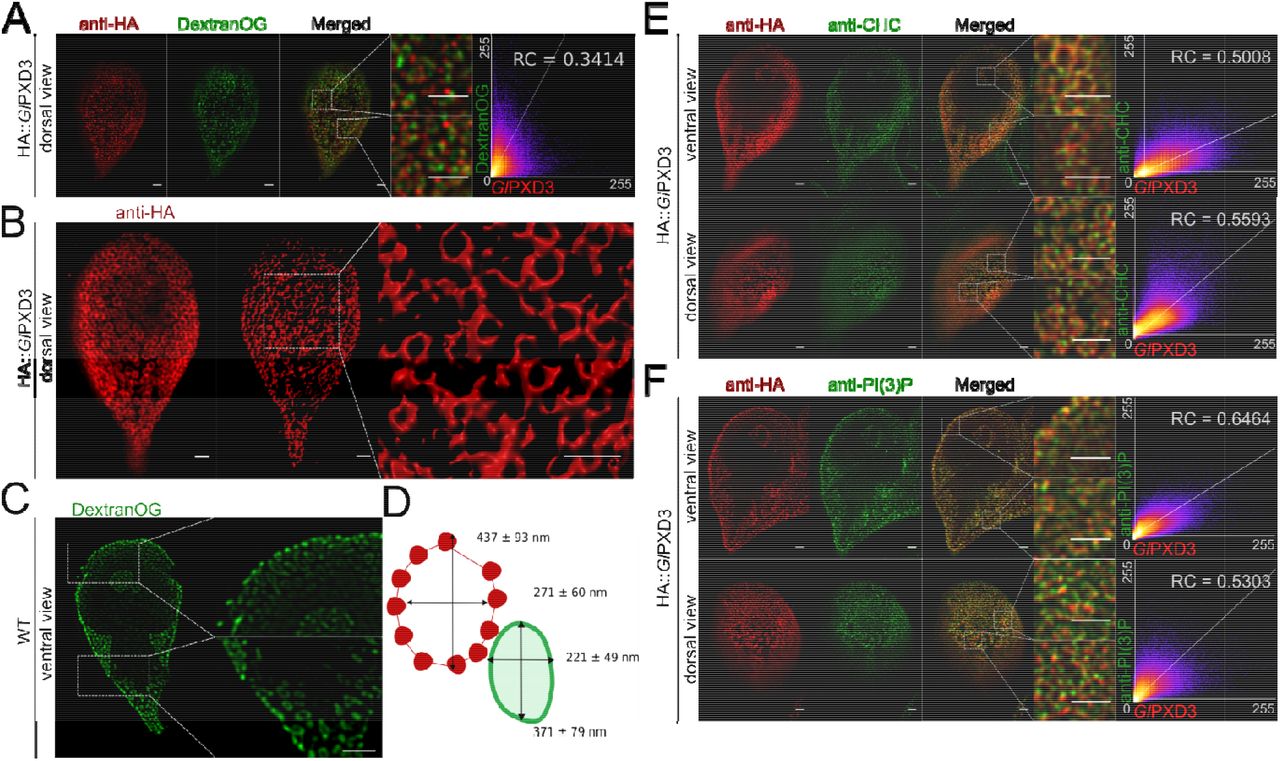
Co-localization studies with Dextran-OG and ectopically expressed HA:: GlPXD3 show apparent coating of the entire PV membrane on the cytoplasmic side by the reporter construct (Fig 9A; Fig S2C) This provided us with an opportunity to generate measurements of PV organelles in optical sections using 3D STED microscopy followed by reconstruction and rendering with IMARIS. Rendered images show hive-like GlPXD3-labelled structures predominantly in the cortical area of the cell underneath the PM that clearly surround the entire PV membrane (Fig 9B). The major and minor principal axes of these structures measured 437 +/− 93 nm and 271 +/− 60 nm. Consistent with the subcellular localization of this marker on the cytoplasmic side of PV membranes, these values were significantly higher (p≤ 0.05) than those obtained from PVs labeled with Dextran-OG (371 +-79 nm and 221 +/− 49 nm) (Fig 9C, D). Signal overlap of epitope-tagged GlPXD3 with endogenous GlCHC as a marker for the PM-PV interface [19] in fluorescence microscopy is low. The image data indicate that both labels have distinct distributions but may spatially overlap at focal clathrin assemblies in small areas at the PV-PM interface (Fig 9E). Similarly, labelling for both PI(3)P and a reporter GlPXD3 variant showed minimal signal overlap (Fig 9F), despite the strong affinity of the latter for this lipid in in vitro lipid-array binding experiments (Fig 2A and B).

(A) A dorsal view of representative cells expressing an epitope-tagged GlPXD3 reporter (red) and co-labelled for Dextran-OG (green). STED confocal imaging followed by signal overlap analysis (scatter plot) shows proximal yet distinct deposition patterns, with GlPXD3 reporters closely associated to Dextran-OG-illuminated PVs. Scale bars: whole cell 1 µm; close-ups 1 µm. (B) 3D STED microscopy (left panel) followed by reconstruction using IMARIS (middle panel) of a representative cell expressing an epitope-tagged GlPXD3 reporter reveals fenestrated GlPXD3-delimited areas distributed under the PM and throughout the whole cell (close-up view of inset in the right panel). Scale bars: whole cell 1 µm; close-ups 1 µm. (C) STED microscopy analysis of PVs in a representative non-transgenic wild-type cell labelled with Dextran-OG. Scale bars: whole cell 1 µm; close-ups 1 µm. (D) Average length of the major and minor principle axes of GlPXD3-delimited fenestrated structures (in red) and Dextran-labelled PV organelles in wild-type non-transgenic cells (in green) measured across at least 100 structures/organelles. (E) STED confocal microscopy analysis of ventral and dorsal views of a representative cell expressing an epitope-tagged GlPXD3 reporter (anti-HA) and co-labelled for GlCHC (anti-CHC) shows how fenestrated GlPXD3-delimited structures are decorated with GlCHC foci. Scatter plots are included for signal overlap analysis. Scale bars: whole cell 1 µm; close-ups 1 µm. (F) Similar to GlCHC, anti-PI(3)P antibodies (anti-PI(3)P) detect foci of PI(3)P accumulation in close proximity to GlPXD3 epitope-tagged reporters (anti-HA) in HA::GlPXD3-expressing cells analysed with STED microscopy. Scatter plots are included for signal overlap analysis. Scale bars: whole cell 1 µm; close-ups 1 µm.
Discussion
PIPs and PIP binders in G. lamblia
PIPs are recognized spatiotemporal organizers and decorate the surface of the eukaryotic cell’s plasma and endo –membrane system [1–3]. G. lamblia is no exception; despite its significant reduction in endomembrane complexity, this species maintains a variety of PIP residues, mostly located at the cell periphery. We identified 13 novel proteins, in most cases of unknown function, that carry predicted PIP-binding modules and primarily localize in close proximity to PVs.
All hitherto identified PIP-binding proteins in G. lamblia can be loosely grouped in two categories; they are either relatively small proteins (up to 400 amino acid residues) consisting almost entirely of the PIP-binding module (e.g. GlPXD6 and GlNECAP1) or they are large proteins consisting of domains of unknown function associated to a single predicted domain for PIP-binding (e.g. GlPXD2 and GlFYVE). A full functional characterization of the latter is a challenge given the level of genomic sequence divergence in G. lamblia. This makes it currently difficult to determine whether sequences are lineage-specific or so diverged as to be unrecognizable orthologues of previously-characterized proteins. Hence, structural annotation of large G. lamblia proteins carrying PIP-binding modules such as GlPXD2 or GlFYVE is limited to the lipid binding domain.
Eight out of 14 identified PIP-binding modules are either directly or indirectly associated to clathrin assemblies. Their PIP binding preferences, as measured using in vitro lipid-binding assays, are clearly distinct despite showing a varying degree of promiscuity, consistent with previously published data [20]. In contrast to previous reports, we could not measure PIP residue binding activity for GlFYVE using in vitro lipid-binding assays [22]. Furthermore, GlNECAP1 showed a distinctive and highly-specific binding preference for cardiolipin. This is a surprising finding since cardiolipin is an abundant phospholipid of the inner mitochondrial membrane [56] whose presence in Giardia is controversial [57, 58]. Although GlNECAP1 lacks canonical motifs for cardiolipin binding [59], previous reports on the identification of cardiolipin-binding PH domains [60, 61] lend support to the observation that the PH-like domain in GlNECAP1 could bind cardiolipin, at least in vitro. The evolutionary implications for the presence of cardiolipin in an organism with “bare-bones” mitochondrial remnants i.e. mitosomes, with no maintenance of membrane potential nor ATP synthesis activity [62], provide for an exciting research direction worth pursuing.
An interactome-based model for PIP-binding proteins and clathrin assemblies at PVs
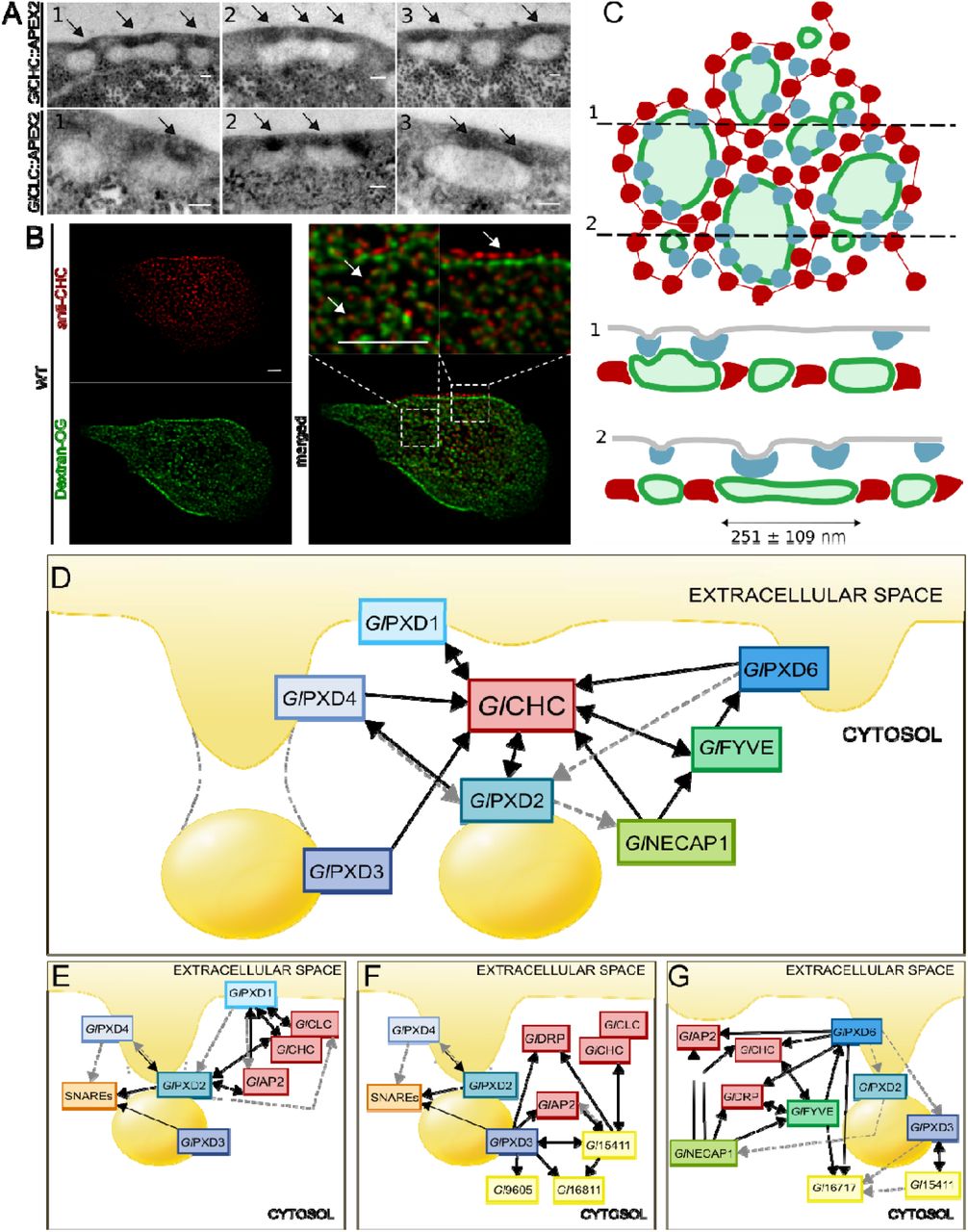
Unpublished data derived from APEX-mediated tEM experiments on transgenic trophozoites expressing APEX-tagged clathrin assembly components (GlCHC and GlCLC; [19]) show how larger PVs are associated to more than one PM-derived clathrin-marked invagination (Fig. 10A). This is supported by data from IFA and STED microscopy analysis of trophozoites loaded with Dextran-OG and labelled with anti-GlCHC antibodies (Fig. 10B). By combining APEX-derived tEM data with STED microscopy data for both Dextran-OG and GlPXD3 labelling, a quantified suborganellar model for PV organization can be built which takes into account organelle size and relative distribution of clathrin assemblies (Fig. 10C). In this model, GlPXD3 clearly emerges as a membrane coat that surrounds individual PV organelles (Fig 10C, upper panel) on the cytoplasmic side of clathrin assemblies at the PV-PM interface (Fig 10C, lower panel).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Electron microscopy images of G. lamblia cells expressing an APEX2-tagged GlCHC (upper panels) or GlCLC (lower panels) reporter show darker APEX2-derived deposits at the PM-PV interface (arrows). Scale bar: 0.1 µm. (B) IFA analysis of a representative non-transgenic wild-type cell labelled with Dextran-OG and anti-GlCHC antibodies to illuminate PV lumina and the PV-PM interface, respectively. Scale bar: 1 µm. (C) Schematic reconstruction of a surface view (left panel) of the PV system associated to clathrin assemblies (blue) and GlPXD3 coats (red), based on data presented in this report and in [13]. PV membranes and lumina are represented in dark and light green, respectively. Cross-sections at (1) and (2) yield views in the right panel, highlighting foci of clathrin assemblies beneath the PM, above GlPXD3’s coat-like deposition pattern surrounding PVs. (D) An overview of the G. lamblia PIP-binding interactome associated to PVs. All represented PIP-binding proteins were found to contact clathrin assemblies (GlCHC) in either reciprocal (double-headed arrows) or one-way (single-headed arrows) modes of interaction, following filtering of co-IP data either at high (black solid lines) or low (grey dashed lines) stringency. (E-G) Nanoenvironments defined by specific sets of interaction partners including clathrin assemblies, PIP-binding proteins, SNARES and proteins of currently unknown function.
The PV-associated PIP-binding protein interactome appears as a tightly knit molecular network with GlCHC at its center (Fig 10D and S5). Despite the high level of interconnectivity of distinct PIP-binder interactomes (Fig S5), specific molecular circuits such as the ones defined by the SNARE quartet (Fig 10E), pseudokinase Gl15411 and novel DLP Gl9605 (Fig 10F), as well as StAR-related lipid-transfer protein Gl16717 (Fig 10G), can be recognized. Notably, GlPXD1 and 2 are the only PIP-binders whose extended interactomes include the G. lamblia clathrin light chain (Fig 10E and S5), arguably GlCHC’s closest binding partner. The GlPXD1 interactome further stands out for enrichment of proteasome-associated components (Table S1), invoking scenarios concerning clathrin assembly turnover in G. lamblia. Although previous data show that clathrin assemblies are long-lived stable complexes [19], they would still require remodeling, degradation, and substitution with new components. In the absence of classical components as well as C-terminal motifs on GlCHC for ordered disassembly of clathrin coats, GlPXD1’s proteasome-enriched interactome points to proteasome-mediated degradation of GlCHC assemblies as an alternative process to achieve turnover albeit without recycling of coat components.
In the context of clathrin assembly dynamics, GlNECAP1 once again comes to the forefront. NECAP1 is characterized as an AP2 interactor and an important component of CCVs in the assembly phase [53]. Given that CCVs have not been detected in Giardia, this begs the question of the functional role of a NECAP1 cardiolipin-binding orthologue in G. lamblia which was found to interact with G. lamblia AP2 subunits and GlFYVE. Recent developments in gene knock-out [63] and CRISPR-Cas9-based knock-down [64, 65] methodologies tailored to G. lamblia will be instrumental towards a full functional characterization of GlNECAP1’s function(s)
Perturbation of PIP binding homeostasis affects fluid-phase uptake
We initially hypothesized that perturbation of either PIP saturation or PIP-binding activity would elicit fluid-phase uptake phenotypes by impacting PV functionality. The hypothesis tested positive for the saturation of PI3P, PI(3,4,5)P3, and PI(4,5)P2. A significant effect on cell width was also detected when PI(3)P binding sites were saturated by overexpressing 2xFYVE::GFP (Figs 3B and F), linking PIP residues to both endocytic homeostasis and overall maintenance of cell size, possibly in connection to membrane turnover. Complementing these data, ectopic expression of both GlFYVE and GlNECAP1 significantly impacted fluid-phase uptake. Furthermore, ectopic expression of GlNECAP1 induced an enlarged PV phenotype similar to that induced by expression of a predicted GTP-locked GlDRP mutant [66]. Ectopic expression of a truncated GlFYVE deprived of its ARM repeats induced the formation of membrane-bounded compartments of undefined origins. ARM folds are superhelical structures mostly involved in protein-protein interactions [67], suggesting that a loss of these domains may impact GlFYVE function and protein complex formation. In line with this hypothesis, the NT-GlFYVE epitope-tagged reporter loses association to PVs. In contrast to the GlFYVE-induced uptake phenotype and despite a severe PV clustering phenotype, HA::GlPXD2-expressing cells still appear to perform fluid-phase uptake comparably to wild-type cells. This suggests that PV morphology can be decoupled from effective PV-mediated uptake. Taken together, these data link PIPs to clathrin assemblies and fluid-phase PV-mediated uptake, providing new insights on clathrin’s hitherto unclear role in Giardia endocytosis.
Beyond clathrin assemblies
Investigation of the molecular milieu within which clathrin-associated PIP-binding proteins operate in G. lamblia revealed two protein sets of special interest. Four predicted SNARE proteins were detected in both the GlPXD2 and GlPXD3 interactomes. Further investigations will be necessary to determine whether the function of this SNARE quartet is indeed fusing PM and PV membranes at contact sites, thereby allowing entry of fluid-phase material into PV organelles.
Another finding of special interest concerns Gl9605, a hitherto unrecognized DLP found in the interactome of GlPXD3 with similarity to bacterial DLPs (BDLPs; Fig S6). Similar to their eukaryotic counterparts, BDLPs are capable of helical self-assembly and tubulation of lipid bilayers, and were shown to be most closely related to the mitofusins FZO and OPA (Fig S6) [24, 25], but only distantly related to classical dynamins [26]. BDLPs were also found in the Archaea class Methanomicrobia [68], making the family ubiquitously distributed across all kingdoms. These data show how the DLP/DRP family in G. lamblia has now expanded to include the previously unidentified endocytosis-associated Gl9605 BDLP homologue. GlDRP plays a role in the regulation of PV and encystation-specific vesicle (ESV) size [66]. Although its role in fluid-phase uptake has not been determined, expression of a GTP-locked GlDRP mutant inhibited endocytosis of biotinylated surface proteins [66]. On the other hand, a similar mutational analysis of Gl9605 shows that this DLP variant can elicit a dominant-negative fluid-phase uptake phenotype. Although we did not test Gl9605 involvement in surface protein uptake, the data so far suggest that two distinct DLPs play independent albeit complementary roles in the regulation of PV-mediated fluid-phase uptake and organelle homeostasis.
In this work, we report on the detailed functional characterization of PIP-binding proteins in G. lamblia that associate to clathrin assemblies. Our data reveals a previously unappreciated level of complex interplay between lipid residues and their protein binders in marking and shaping endocytic compartments in this parasite. However, several identified PIP-binding modules appear to associate to PVs independently of clathrin. Their extended interactomes and their involvement in fluid-phase uptake have yet to be investigated but current data point towards a complex network of PIP binders of varying binding preference and affinity, all working in the same subcellular environment, yet, in some cases (GlFERM, GlBAR1 and 2, GlPROP1 and 2, Gl16801), not directly linked to clathrin assemblies. The only known exception is Glepsin whose localization remains controversial due to conflicting reports [21, 69]. We systematically did not detect Glepsin in any of the interactomes for clathrin-associated PIP binders, in line with its localization at the ventral disk [21]. Altogether, the variety of PIP residues and PIP-binding modules in the G. lamblia cortical area containing endocytic PVs underscores their necessity for correct functioning of membrane traffic even in a protist so clearly marked by reduction in endomembrane complexity.
Materials and Methods
Giardia lamblia cell culture, induction of encystation and transfection
G. lamblia WBC6 (ATCC catalog number 50803) trophozoites were cultured and harvested applying standardized protocols [52]. Encystation was induced by the two-step method as previously described [70, 71]. Transgenic cell lines were generated using established protocols by electroporation of linearized or circular pPacV-Integ-based plasmid vectors prepared from E.coli as described in [72]. Transgenic lines were then selected for puromycin resistance (final conc. 50 µg ml −1). After selection, transgenic trophozoites carrying integrated or episomal reporter constructs were further cultured with or without puromycin, respectively.
Construction of expression vectors
Oligonucleotide sequences used for cloning in this work are listed in Table S8. pPacV-Integ-based [34] expression of epitope tagged reporter constructs was driven using either putative endogenous (pE) or encystation-dependent (pCWP1) promoters. Constructs 2xFYVE::GFP and GFP::P4C [39] were kindly provided by Dr. H. Hilbi (University of Zurich).
PV labelling using fluid-phase markers
Fluid-phase uptake assay in G. lamblia was performed as described previously [26] using dextran coupled to either Oregon Green 488 (Dextran-OG) (Cat. Nr. D-7171, Thermo Fisher Scientific) or Texas-Red (Dextran-R) (Cat. Nr. D-1863, Thermo Fisher Scientific) fluorophores, both at 1mg/ml final concentration.
Co-immunoprecipitation with limited cross-linking
Co-immunoprecipitation GlPXD1-6, GlNECAP1, and GlFYVE was done as previously reported [19, 42]. Protein input was standardized to 0.8mg/ml total protein.
Protein analysis and sample preparation for mass spectrometry (MS)-based protein identification
Protein analysis was performed on 4%/10% polyacrylamide gels under reducing conditions (molecular weight marker Cat. Nr. 26616, Thermo Scientific, Lithuania). Immunoblotting was done as described in [73]. Gels for mass spectrometry (MS) analysis were stained using Instant Blue (Expedeon, Prod. # iSB1L) and destained with ultra-pure water.
Mass Spectrometry, protein identification and data storage
MS-based protein identification was performed as described in [19]. Free access to raw MS data is provided through the ProteomeXchange Consortium on the PRIDE platform [74]. Accession numbers for datasets derived from bait-specific and corresponding control co-IP MS analyses are the following: PXD013890 for GlPXD1, 3 and 6, PXD013897 for GlFYVE, PXD013896 for GlNECAP and PXD013899 for GlPXD2 and 4.
In silico co-immunoprecipitation dataset analysis
Analysis of primary structure and domain architecture of putative components of giardial PIP--binding proteins was performed using the following online tools and databases: SMART for prediction of patterns and functional domains (http://smart.embl-heidelberg.de/), pBLAST for protein homology detection (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins), HHPRED for protein homology detection based on Hidden Markov Model (HMM-HMM) comparison (https://toolkit.tuebingen.mpg.de/#/tools/hhpred), PSORTII for sub-cellular localisation prediction (https://psort.hgc.jp/form2.html), TMHMM for transmembrane helix prediction (http://www.cbs.dtu.dk/services/TMHMM/), RCSB for 3D structure of homologues (https://www.rcsb.org/), and the Giardia Genome Database to extract organism-specific information such as protein expression levels, predicted molecular sizes and nucleotide/protein sequences (www.giardiaDB.org). The generated co-IP datasets were filtered using a dedicated control-co-IP dataset generated using non-transgenic wild-type parasites. Filtration of the bait-specific co-IP and control-co-IP datasets was done using Scaffold4 (http://www.proteomesoftware.com/products/) with high stringency parameters (95_2_95, FDR 0%) and low stringency parameters (95_2_50, FDR 0%). Furthermore, exclusive hits for bait-specific datasets were manually curated using the following criteria for inclusion into the interactome model: i) exclusive detection with > 3 spectral counts in bait-specific datasets or ii) an enrichment of peptide counts >3 with respect to the ctrl. co-IP dataset. Data presented in Tables S1-7 show exclusive and non-exclusive protein hits filtered using both stringency levels.
Immunofluorescence analysis (IFA) and light-microscopy
Samples for immunofluorescence and analysis of subcellular distribution of reporter proteins by wide-field and laser scanning confocal microscopy (LSCM) were prepared as described previously [33, 35]. Nuclear DNA was labelled with 4’, 6-diamidino-2-phenylindole (DAPI). The HA epitope tag was detected with either the anti-HA antibody (1:50 or 1:100; Anti HA high affinity 3F10, Cat. Nr. 11867423001, Roche), anti-V5 (1:50 or 1:100; V5 Tag Monoclonal Antibody, Cat. Nr. R960-25, Thermo Fisher Scientific) or self-made antibodies raised against GlCHC (dilution 1:1000) followed by an anti-rat antibody coupled to fluorochrome in case of wide-field or confocal microscopy (1:200; Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488, Cat. Nr. #A11006, Invitrogen) and for STED microscopy (Goat anti-Rat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 594, Cat. Nr. A11007, Invitrogen). Specific PIP residues were detected using anti-PI(3)P (1:100; Purified anti-PI(3)P IgG, Z-P003 Echelon Biosciencies), antiPI(4,5)P2 (1:100; Purified anti-PI(4,5)P2 IgM, Z-P003 Echelon Biosciencies) and anti-PI(3,4,5)P3 (1:100; Purified anti-PI(3,4,5)P3 IgM, Z-P045 Echelon Biosciencies) followed by an anti-mouse antibody coupled to fluorochrome in all three cases (Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa flour 594, Cat. Nr. A-11005, Thermo Fischer Scientific or Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa flour 488, Cat. Nr. A-11017, Thermo Fischer Scientific). Cells were generally imaged at maximum width, with nuclei and the bare-zone at maximum diameter. Deconvolution was performed with Huygens Professional (Scientific Volume Imaging). Three-dimensional reconstructions and signal overlap quantification (Mander’s coefficient) in volume images of reconstructed stacks were performed using IMARIS x64 version 7.7.2 software suite (Bitplane AG) or FIJI [75], respectively.
Super resolution (gSTED) microscopy
Sample preparation was done as described for wide field microscopy and LSCM. For imaging, samples were mounted in ProLong Gold antifade reagent (Cat. Nr. P36934, Thermo Fisher Scientific). Super resolution microscopy was performed on a LSCM SP8 gSTED 3x Leica (Leica Microsystems) at the Center for Microscopy and Image Analysis, University of Zurich, Switzerland. Nuclear labelling was omitted due to possible interference with the STED laser. Further data processing and three dimensional reconstructions of image stacks were done as described for LSCM.
Sample preparation for transmission electron microscopy
Transgenic trophozoites expressing GlPXD2 (GL50803_16595) and non-transgenic line were harvested and analysed by transmission electron microscopy (tEM) as described previously [66].
DAB staining in APEX2 expressing cells
Transgenic trophozoites expressing GlNECAP1::APEX2-2HA, GlCHC::APEX2-2HAand GlCLC::APEX2-2HA were harvested and washed with PBS followed by fixation in 2.5% EM grade glutaraldehyde in cacodylate buffer (100 mM cacodylate (Cat. Nr. 20838), 2mM CaCl2 (Cat. Nr. 21097, Fluka) in PBS) for 1h at RT. Samples were washed twice before and after quenching for 5 min in 20mM glycine/cacodylate buffer. For staining, cells were resuspended in 500ul substrate solution containing 1.4mM DAB tetrahydrodhloride (Cat. Nr. D5637, Sigma) with 0.3mM H2O2 (Cat. Nr. H1009, Sigma) in cacodylate buffer and incubated for 15 min. The reaction was terminated by washing thrice in cacodylate buffer and prepared as described for tEM.
Chemical fixation of DAB-stained cells
DAB stained cell suspicions were post-fixed with 1% aqueous OsO4 for 1 hour on ice, subsequently rinsed three rimes with pure water and dehydrated in a sequence of ethanol solutions (70% up to 100%), followed by incubation in 100% propylene oxide and embedding in Epon/Araldite (Sigma-Aldrich, Buchs, Switzerland). Samples were polymerised at 60°C for 24h. Thin sections were imaged pre- and post-staining with aqueous uranyl acetate (2%) and Reynolds lead citrate.
Expression and Purification of Bacterial Fusion Proteins
For each candidate PIP-binding protein, corresponding nucleotide stretches coding for selected amino acid residues (Table S9) were modified by including an HA-coding sequence either at the 5’ end or the 3’ end and then subcloned into the pMal-2Cx E. coli expression vector (New England Biolabs). The resulting recombinant variants were expressed as maltose-binding protein (MBP) fusions in E.coli (strain Bl21) and grown in LB medium either at 37°C (MBP-GlPXD1, MBP-GlPXD2, MBP-GlPXD3, MBP-GlPXD6, MBP-GlNECAP1 and MBP-GlFYVE) or 30°C (MBP-GlPXD4 and MBP-GlPXD5) to an OD600=0.4. Induction of expression was performed by adding0.2 mM IPTG (Isopropyl β-D-1-thiogalactopyranoside, Cat. Nr. 15529019, Thermo Fischer Scientific) to the cultures and incubating for a further 4 hours. Cells were harvested at 4°C (4,000 x g) and bacterial pellets were resuspended in 5 ml of cold column buffer with 1x PIC (Protease inhibitor cocktail set I; Cat. Nr. 539131-10VL, Merck) and 200 mM PMSF (Cat. Nr. 329-98-6, Sigma Aldrich). Cells were lysed by sonication and centrifuged (20 min, 9,000 × g, 4°C). Cleared supernatant was incubated with amylose resin slurry (Amylose Resin High Flow, Cat. Nr. E8022L, BioLabs) for 4 hours at 4°C on a turning wheel, washed with column buffer and then transferred to an empty column (BioRad 15 ml). Unbound protein was washed using until background OD280 reached ~0.06. Protein fractions were eluted using 10mM maltose solution and pooled for overnight dialysis in a dialysis cassette (Slide-a-Lyzer, Cat. Nr. 66380, Thermo Fischer Scientific) against 25mM NH4Ac at 4°C and later lyophilized. Protein fractions were stored at −80°C.
Protein lipid overlay (PLO) assay
E. coli-derived lyophilized proteins were reconstituted in 1x PBS and protein concentration was measured using the Bradford assay. PIP strips (PIP strips, Cat. Nr. P-6001 and P-6002, Echelon) or PIP arrays (PIP strips, Cat. Nr. P-6100, Echelon) were first floated on ultrapure water for 5 min before incubation in blocking buffer (1xPBS containing 0.1%v/v Tween-20 and 3% fatty-acid free BSA (Sigma A7030)) at RT for 1h. Thereafter 0.5 μg/ml of protein in PBS containing 3% fatty acid free BSA were incubated for 1h at RT with gentle agitation. After washing with 1xPBS containing 0.1% v/v Tween-20, PIP–strips were incubated (1h, RT, agitated) with a monoclonal anti-HA antibody (clone 3F10, monoclonal antibody from Roche) at a dilution of 1:500 in blocking buffer. Subsequently strips were washed and incubated (1h, RT, agitated) with a goat-derived polyclonal anti-rat antibody conjugated to HRP at a dilution of 1:2000 in blocking buffer (Cat. Nr. 3050-05, Southern Biotech). After further washing, strips were developed using a chemiluminescent substrate (WesternBright ECL HRP Substrate, Cat. Nr. K-12045-D50).
Densitometric analysis of lipid strips and arrays
Relative quantification of immunoblotting signal intensity on PIP strips and arrays overlaid with PIP-binding proteins was performed using FIJI [75]. For each strip or array, the spot with the highest pixel number was set as a reference for 100% binding; signals coming from all other spots were normalized against it. The data were visualized as bar charts of relative signal intensity as a measure of lipid-binding preference for each PIP-module.
Identification of giardial orthologues of known PIP-binding domains
PIP-binding domain representatives were used as bait for in silico searches within the Giardia genome database (GDB) (http://giardiadb.org/) using the online tool HHpred (https://toolkit.tuebingen.mpg.de/) to detect remote giardial homologues using hidden Markov models (HMMs; Table 1) [25]. Outputs were firstly evaluated based on the calculated probability and the corresponding E-value for the prediction, with cut-offs for probability and e-value set to 90 and 1e-10, respectively. Sequence identity and similarity were also considered. To validate the prediction, candidate giardial PIP-binding proteins were then utilized as baits to search PDB databases using HHpred to retrieve orthologous PIP-binding proteins/modules. For additional validation, I-TASSER [29–31]was also used to predict hypothetical structures of putative giardial PIP-binding domains next step validation.
Multiple sequence alignment analysis
Multiple sequence alignment using two or more sequences was performed with the Clustal Omega sequence alignment algorithm [76, 77]. The sequences used to compile the alignments shown in supplementary figure 1 were chosen based on representative members for each PIP-binding domain type [1, 10, 78]. Alignments for figures 6 and 8 were based on previously characterized G1-G4 GTP binding motifs [50] and NECAP1 proteins [53], respectively.
De novo structural modeling and analysis
Ab-initio prediction of hypothetical 3D models presented in supplementary figure 1 was done using I-TASSER [29–31]. The best model was chosen based on the C-score predicted by the algorithm. A C-score is a measure of confidence for a model based on the significance of threading template alignments and the convergence parameters of the structure assembly simulations. It ranges from −5-to 2, with higher C-scores indicating higher confidence. The final 3D structures were displayed using PyMOL (The PyMOL Molecular Graphics System, Version 2.0 Schrödinger, LLC.). The superimposition of Giardia PIP-binding proteins with their closest structural orthologue are based on I-TASSER predictions, with structural similarities expressed by TM-score and RMSDa values. The TM-score is computed based on the C-score and protein length. It ranges from 0 to 1, where 1 indicates a perfect match between two structures. RMSDa is the root mean square deviation between residues that are structurally aligned by TM-align [79]. Specifically for GlBAR1 and 2, the structural overlap analysis was performed by selecting positively-charged residues from previously characterized BAR domains shown to play a role in lipid binding [80]. These were manually superimposed on corresponding residues in the predicted GlBAR1 and 2 structures.
Phylogenetic analysis
Subjected sequences of GTPase domains were aligned using Clustal Omega tool. The tree construction was submitted to a PHYLogeny Inference Package (PHYLIP) program [81, 82] using random number generator seed set to 111 and number of bootstrap trials set to 10000. The tree was visualised using the on-line tool iTOL and includes branch lengths as a measure of evolutionary distance [83].
Supplemental Tables 1-7: Proteins identified in the interactomes of GlPXD1-4 and 6, GlFYVE and GlNECAP1
Supplemental Table 8: List of oligonucleotide names and sequences for construct synthesis
Supplemental Table 9: Amino acid sequences of lipid-binding modules used in vitro for protein lipid-overlay assay
Supplemental Figure 1: Multiple sequence alignment and structural prediction analysis of G. lamblia PIP-binding domains.
Supplemental Figure 2: Lipid-binding properties of Giardia-lipid binding domains.
Supplemental Figure 3: Subcellular distribution of PI(3)P, PI(4,5)P2 and PI(3,4,5)P3 in G. lamblia trophozoites
Supplemental Figure 4: APEX-mediated electron microscopy analysis of GlNECAP1 subcellular deposition.
Supplemental Figure 5: Overview of core protein interactomes determined from co-IP analyses
Supplemental Figure 6: Phylogenetic analysis and tree reconstruction for the predicted GTPase domain of the novel dynamin-like protein Gl9605.
Acknowledgements
ABH was supported by Swiss National Science Foundation grants 140803 and 125389. Florian Schmidt and Ritta Rabbat are acknowledged for their technical assistance. Dr. Hubert Hilbi (University of Zurich) is gratefully acknowledged for sharing Legionella-derived constructs 2xFYVE::GFP and GFP::P4C.
Footnotes
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.↵
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.
- 15.↵
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.↵




