

Summary
Ischaemic cardiac injury results in a complex coordinated cellular response that enables effective damage control and recovery. Intercellular communication is integral to the coordination of these repair responses in complex tissue contexts. Extracellular vesicles (EVs) facilitate molecular transport across extracellular space, allowing local and systemic signaling during homeostasis and in disease. Extensive in vitro studies have described the biogenesis, cargos and functional roles of EV populations but the characterisation of endogenously produced EVs in vivo is still in its infancy. Here we use larval and adult transgenic zebrafish to characterize endogenous cardiovascular EVs. Using a membrane-tethered fluorophore reporter system, cell-type specific EVs can be tracked in vivo by high spatiotemporal resolution light sheet imaging and modified flow cytometry methods allows these EVs to be extracted and assessed ex vivo. Importantly, we show that ischaemic cardiac injury models dynamically alter EV production during repair.
Introduction
Extracellular vesicles (EVs) collectively describes all plasma membrane-bound vesicles produced and released by most cell types. An assortment of lipids, proteins and nucleic acids can be loaded into EVs either as luminal cargo or as a component of the EVs membrane providing signaling capacity for intercellular communication. EVs can be broadly split into three main classes: exosomes, formed from an endocytic pathway, microvesicles that are directly shed from the plasma membrane, and apoptotic bodies, which are shed from cells undergoing apoptosis (Caruso and Poon, 2018; van Niel et al., 2018). EVs can be trafficked both locally and systemically and have been isolated from a wide range of biological fluids, including blood (Crawford, 1971; Emanueli et al., 2016) and pericardial fluid (Beltrami et al., 2017; Kuosmanen et al., 2015). Various surface glycans, lipids and proteins work in combination to target EVs to regions of extracellular matrix (ECM) (Rilla et al., 2019) and recipient cells, and these interactions are widely recognised to play integral roles in various communicatory pathways (Colombo et al., 2014). EVs are most often considered within pathophysiological contexts, having been implicated in the progression of many diseases, including cancer and cardiovascular disease (Boulanger et al., 2017; Tkach and Thery, 2016), however EVs also play roles in homeostasis (Ridger et al., 2017; Yanez-Mo et al., 2015).
Cardiovascular disease remains the single biggest killer in the western world Myocardial infarction (MI), often as a result of coronary heart disease, drives ischaemia and ultimately cardiomyocyte cell death in affected regions of the heart, with the ensuing cellular events, including the inflammatory response and revascularisation, serving to repair the injured region and restore tissue function, but ultimately resulting in the formation of irreversible scarring and reduced function of the injured region (Li and Izpisua Belmonte, 2016). These repair processes require the concerted effort of multiple different cell types within the heart, implying the need for a complex communication system. In humans the dead myocardium is replaced primarily by fibrotic scar tissue limiting the functional capacity of the heart and leaving the patient vulnerable to future complications (Cahill et al., 2017; Li and Izpisua Belmonte, 2016; Talman and Ruskoaho, 2016). In regenerative species such as the zebrafish, however, the heart follows a similar initial repair process, but the deposited scar tissue is quickly resolved and the functional myocardium is fully replaced by cardiomyocyte dedifferentiation and re-entry into the mitotic cycle (Chablais et al., 2011; Gonzalez-Rosa et al., 2011; Poss et al., 2002; Schnabel et al., 2011). Zebrafish have emerged as a powerful model of human disease and have many advantages as a vertebrate model including fecundity, external development, genetic tractability and unrivalled cellular level in vivo imaging due to their transparency during embryonic and larval stages (Lieschke and Currie, 2007). Currently, little is known about the role of endogenous EVs in regenerative contexts despite the potential to identify pro-regenerative signals being exchanged between cell types.
The potential of EVs as biomarkers of disease and as novel therapeutic delivery vehicles has generated significant interest in recent years, including applications in tissue (predominantly bone) regeneration (Wiklander et al., 2019). However, the ability to reliably define the heterogeneous spectrum of EV subtypes, and further, to ascribe functional significance to that specific subtype in vivo, is still in its infancy (Beer and Wehman, 2017; Hyenne et al., 2017). The majority of EV characterization to date, by necessity, has been performed in vitro (van Niel et al., 2018). Strategies have been developed to obtain EV populations from culture medium, providing pure samples of EVs derived from known cell types, or from biological fluid samples, producing EVs from specific sites but from heterogenous origins, allowing the molecular components of these EVs to be determined and compared (Gyuris et al., 2019; Kalra et al., 2012; Thery et al., 2006; van Niel et al., 2018). Recent studies have developed novel ways to label exogenous EVs to investigate the biodistribution of these EV populations and are starting to dissect the functional changes elicited in recipient cells resulting from their addition to cell culture or in vivo systems (Beltrami et al., 2017; Hyenne et al., 2019; Lai et al., 2014; Wiklander et al., 2015). Whereas these studies using exogenous EVs are beginning to identify important roles in multiple different tissues and disease states, they fail to address the full complexity of endogenous in vivo EV populations. A recent report has started to bridge this gap by investigating endogenously produced EVs in a novel zebrafish model (Verweij et al., 2019b). However, in vivo studies of endogenous cardiovascular EV function during disease (and potentially tissue regeneration) remain limited.
Here, we describe techniques to stably label endogenous cell-type specific EVs with fluorescent reporters in vivo in larval and adult zebrafish, allowing these vesicles to be tracked, extracted and validated. We demonstrate that global zebrafish EVs (either labelled through a ubiquitous promoter or by secreted Annexin-V binding) as well as endothelial- and cardiomyocyte-derived EVs can be observed in the peripheral circulation and the pericardial space, respectively. Using adapted flow cytometry techniques (including fluorescence activated vesicle sorting – FAVS (Cao et al., 2008)) and imaging flow cytometry (Lannigan and Erdbruegger, 2017)) we can analyse and purify these cell-type specific EVs from tissue samples and evaluate different EV populations ex vivo. Most importantly, we demonstrate that models of ischaemic injury, in larvae and adults, result in dynamic changes to the EV profile both systemically and specifically in the heart.
Results
Stable labelling and imaging of endogenous EVs in vivo
EVs have been shown to be involved in multiple different developmental, homeostatic and disease situations including cancer and cardiovascular disease (Boulanger et al., 2017; Tkach and Thery, 2016). Despite recent advances in labelling methods, the distribution and function of cell type specific EVs in vivo remains incompletely understood largely due to the technical challenges of studying these small vesicles. Using EV labelling methods similar to those previously described for exogenous vesicles (Lai et al., 2015), we have made use of stable transgenic zebrafish to label endogenous EVs. Using different promoters to drive the expression of prenylated fluorophores that are incorporated into the plasma membrane ubiquitously or in a cell type specific manner, we show that this label is incorporated into the
EVs produced by the labelled cells. Initially, we used a transgenic line where prenylated GFP is driven by a near-ubiquitous promoter, tethering the fluorophore to the inner leaflet of the plasma membrane and, consequently, incorporating GFP in the EVs from the majority of cells (Tg(Ola.Actb:Hsa.HRAS-EGFP) (referred to as Tg(actb2:HRAS-EGFP)); Figure 1A,B; (Cooper et al., 2005)). EVs labelled in this way were observed in the peripheral circulation and in the pericardial space of larval zebrafish at 3 days post fertilisation (dpf) ((Figure 1C,G) and supplementary videos 1 & 2). The pericardial space is a relatively large extracellular space in larval zebrafish (Figure 1D-F) and GFP+ EVs were observed moving within the pericardial fluid, influenced by the movement of the heart (supplementary video 2).

(A) Schematic representation of the cell/EV labelling strategy. The fluorophore is tethered to the inner leaflet of the plasma membrane via a CAAX or HRAS motif. (B) Overview image of a Tg(actb2:HRAS-EGFP) larval zebrafish at 3 dpf. The pericardial wall and the DA are outlined in magenta and blue, respectively. The boxed areas define the regions shown in the image sequences in C and G, as indicated. (C) Image sequence of GFP labelled EVs (arrowed) moving through the DA. Blue dashed lines demark the endothelium lining the vessel. Insets show higher magnification views of the arrowed EV. (D) Schematic showing the position of the larval heart and pericardial space in a ventral view of a 3 dpf fish. (E) Image of the heart in ventral view with fluorescently labelled cardiomyocytes of a Tg(myl7:GFP) fish (green) and injected Dextran to demonstrate the extent of the pericardial space (red) at 3 dpf. (F) Overview image of the heart of a Tg(actb2:HRAS-EGFP) larval zebrafish at 3 dpf. (G) Image sequence of the boxed region in F showing a GFP labelled EV (arrowed and inset) moving through the pericardial space. The magenta dashed line in D-G demarks the outer pericardial wall. The orange dashed line in G demarks the bulbus arteriosus. The white dashed line in G demarks the ventricle and atrium. Anterior is to the left in B,C. V = ventricle, A = Atrium, PS = Pericardial space. Scale bars: B = 200 µm; C = 5 µm; insets in C,G = 2 µm; E,F = 50 µm; G = 20 µm.
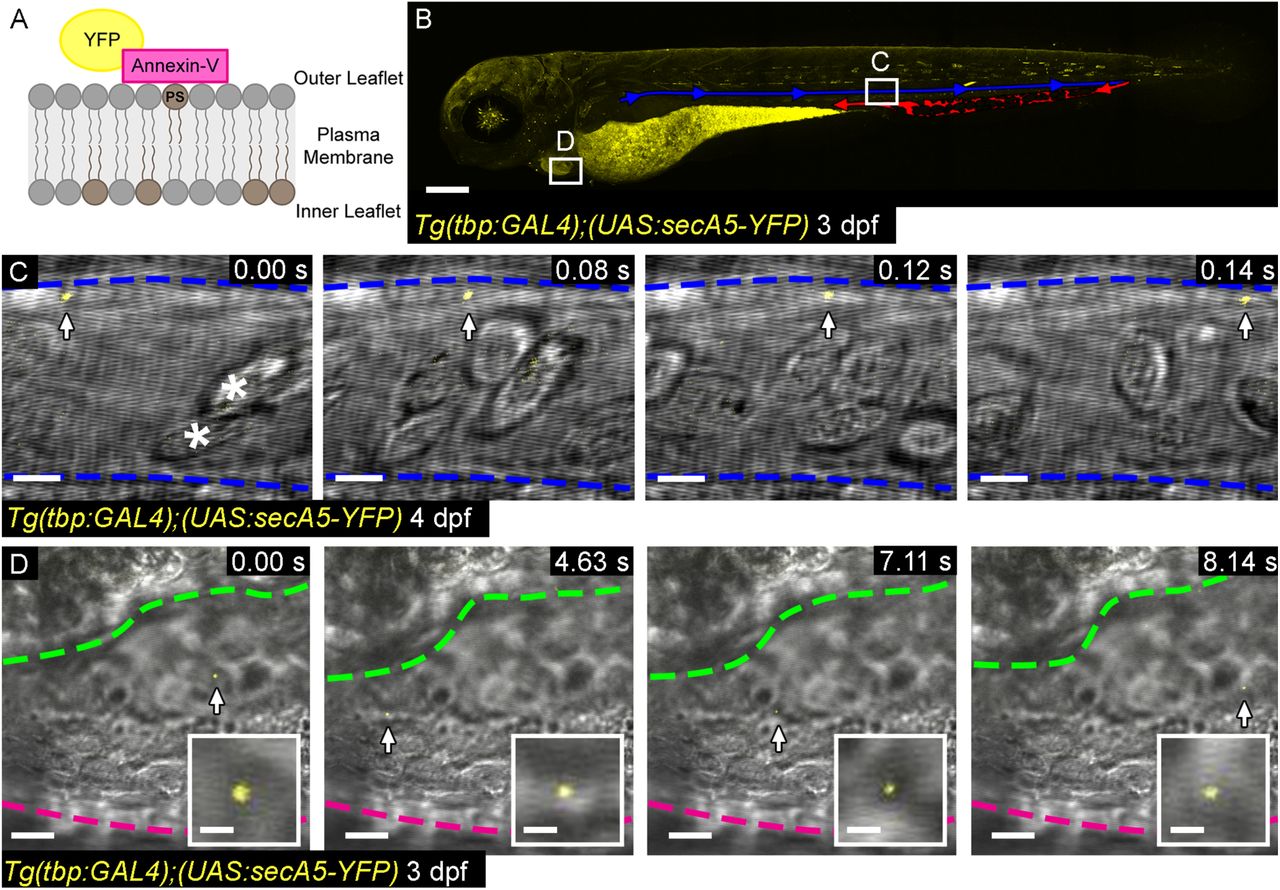
Additionally, we made use of a ubiquitously expressed secreted Annexin-V line which binds with high affinity to the phosphatidylserine expressed on the outer surface of apoptotic cells and populations of EVs (referred to as Tg(tbp:GAL4);(UAS:secA5-YFP) (van Ham et al., 2010); Figure S1), including those relevant in cardiovascular disease (Jansen et al., 2017; Jansen et al., 2012). As with the actb2 line we observed Annexin-V labelled EVs in both the peripheral circulation and pericardial space of larval zebrafish (Figure S1C,D).
Although these systems allowed us to successfully identify endogenous EVs in vivo, they do not allow us to determine the cellular origin of the EVs. To do this we made use of two more transgenic lines that express a prenylated fluorophore driven by cell specific promoters, whereby the fluorophore is incorporated into the EVs in a cell-type specific manner (Figure 2). Analysis of larval zebrafish expressing endothelial (Tg(kdrl:mCherry-CAAX); EC-EVs) or cardiomyocyte (Tg(myl7:HRAS-mCherry); CM-EVs) specific promoters driving membrane tethered mCherry (Fujita et al., 2011; Yoruk et al., 2012) revealed the presence of cell-derived EVs in the peripheral circulation and pericardial space, respectively (Figure 2C,G and supplementary videos 3,4). EC-EVs moved rapidly in the peripheral circulation with the blood flow and were often observed moving more slowly close to the endothelial wall of the dorsal aorta (DA), suggesting EV-cell interactions (Figure 2C and supplementary video 3). CM-EVs were observed moving within the pericardial fluid, as observed for Tg(actb2:HRAS-EGFP) labelled EVs (supplementary video 4). Static CM-EVs were also observed interacting with the pericardial wall, potentially with regions of ECM (Figure 2H-J and supplementary video 5).

(A) Overview image of a Tg(kdrl:mCherry-CAAX) fish at 3 dpf. All endothelial cells are labelled with mCherry. The boxed area defines the region shown in the image sequence in B,C. (B) Brightfield image of the DA. Red dashed lines demark the vessel walls and blue dashed lines outline blood cells. (C) Image sequence of mCherry+ EC-EVs moving through the DA (arrows and inset). (D) Ventral view of 3 dpf Tg(myl7:HRAS-mCherry) zebrafish, boxed region highlights position of E,F. (E,F) Overview images of the entire hearts of Tg(myl7:HRAS-mCherry) fish in ventral view. E shows a maximum projection of a fixed fish; F shows a single plane of live light sheet imaging. (G) Image sequences of higher magnification views of the colour coded boxed areas in F. mCherry+ CM-EVs are observed moving through the pericardial space (arrowed and inset). (H-J) A maximum intensity projection of deconvolved images of the ventricle and internal surface of the pericardial wall (H) reveals static CM-EVs as shown with digital zoom of boxed region (I). The orthogonal view (YZ) of this region suggests the EVs are interacting with a layer of unmarked ECM rather than direct contact with underlying cells (J). Arrows indicate static EVs. Anterior is to the left. Scale bars: A = 200 µm; B,C,H,J = 5 µm; insets in C,G = 2 µm; D = 100 µm; E,F = 50 µm; G = 20 µm; I = 2 µm.
Validation of endogenous EVs
To further characterise these endogenous EVs we sought to validate the identified populations further (Figure 3). We used a tissue dissociation protocol followed by differential centrifugation and filtration to separate a crude “large” EV fraction (centrifugation at 20,000 g) and a crude “small” EV fraction (centrifugation at 100,000 g) from entire zebrafish larvae (Figure 3A). Analysis of these different fractions using Dynamic Light Scattering (DLS) supported the isolation of different size groups (Figure 3B). To allow us to obtain and verify our endogenous fluorescently labelled EVs, we made use of a modified flow cytometer to analyse ubiquitous (actb2(GFP)+) EVs extracted from whole zebrafish larvae at 3 dpf (Figure 3C-H). Similar modified flow cytometers have previously been used to assess populations of small vesicles (van der Vlist et al., 2012). Isolated EVs were labelled with calcein violet 450 AM, which labels intact EVs (Gray et al., 2015), prior to analysis by flow cytometry. Total populations of calcein+ and actb2(GFP)+ EVs were assessed from EVs extracted from pools of Tg(actb2:HRAS-EGFP) larvae (Figure 3C-H). Control experiments with samples extracted from non-transgenic zebrafish and singly labelled EVs (GFP or calcein) from transgenic and non-transgenic zebrafish allowed double positive (i.e. cell type specific (fluorophore+) and intact (calcein+)) EV specific gates to be assigned, avoiding any background signal (Figure 3C-F). Analysis of EV fractions following detergent treatment confirmed their lipidaceous nature (Figure 3G,H). Flow cytometry analyses suggest that 29 % ± 8.6 (SD) of total events are labelled with calcein, representing intact EVs (Figure 3H). Similarly, flow cytometry analysis suggests that 30 % ± 6.9 (SD) of calcein+ vesicles are also actb2(GFP)+ (Figure 3F). We also analysed calcein+ EVs extracted from Tg(actb2:HRAS-EGFP) larvae using an imaging flow cytometry ImageStream®xMk II system, confirming the presence of individual single or double labelled vesicles (Figure S2B,C). Imaging flow cytometry suggests 65 % of total events are calcein(violet)+ (concentration as per routine 300 µl resuspension volume = 2.14 x 108 EVs/ml) demonstrating the enhanced sensitivity and value of the ImageStream system (Erdbrugger et al., 2014). This analysis suggests 34 % of calcein+ vesicles are also actb2(GFP)+, supporting our previous flow cytometry assessments.

(A) Schematic describing the centrifugation steps taken to isolate EV fractions following cell dissociation of whole zebrafish larvae. (B) Histogram of DLS analysis of different EV fractions from whole 5 dpf larvae indicating that smaller EVs are pelleted at 100,000 g than at 20,000 g. (C-G) Typical flow cytometry scatter plots showing the gates used to sort EVs and the controls used to define these gates: An extraction buffer only control plus calcein AM reveals background noise (C), EVs extracted from non-transgenic (wildtype) fish and labelled with calcein AM allow us to assign a calcein+ gate (D), EVs extracted from Tg(actb2:HRAS-EGFP) fish without calcein AM indicates GFP+ EVs and analysis of EVs extracted from Tg(actb2:HRAS-EGFP) fish with calcein AM labelling identifies a gate of GFP+ calcein+ EVs (F). Treatment of transgenic EVs labelled with calcein AM plus detergent destroys the majority of EVs, confirming their lipidaceous structure (G). Similar gating strategies can be used to analyse mCherry+ EVs from Tg(kdrl:mCherry-CAAX) and Tg(myl7:HRAS-mCherry) fish. (H) Plot of the number of calcein+ EVs of total events from untreated and detergent treated samples. (I) Total Internal Reflection (TIRF) microscopy allows high resolution imaging of extracted EVs from Tg(actb2:HRAS-EGFP) fish (green) alongside 60 nm synthetic fluorescent beads (magenta). (J) TIRF imaging of a single EV derived from a Tg(actb2:HRAS-EGFP); Tg(kdrl:mCherry-CAAX) double transgenic fish stained with calcein AM. (K) Western analysis of protein extracted from cell and EV fractions confirms expression of the EV component Alix. Scale bars: I,J = 5 µm.
Live imaging analysis cannot determine the true size of the EVs observed in the DA and pericardial space as these are likely to be below the resolution limit of light sheet microscopy (≥250 nm). To investigate the size of the extracted EVs further, we first analysed isolated fluorescently labelled EVs from Tg(actb2:HRAS-EGFP); (Tg(kdrl:mCherry-CAAX) double transgenic larvae by high resolution Total Internal Reflection (TIRF – approx. max resolution <100 nm) microscopy together with 60 nm synthetic beads revealing actb2(GFP)+ EVs of a similar size to the synthetic beads (Figure 3I,J). Despite the fact these beads (and likely most EVs) are below the resolution limit, they can be visualised by their fluorescence, although their true size still cannot be accurately determined. TIRF imaging of a single sorted (FAVS) vesicle confirms triple labelling for calcein (intact vesicle), GFP (near-ubiquitous marker) and mCherry (endothelial specific) (Figure 3J). Finally, western blot analysis with an antibody against a known EV component demonstrated expression in isolated small and large EV fractions (Figure 3K).
Ischaemic injury models result in changes to the EV profile of zebrafish larvae
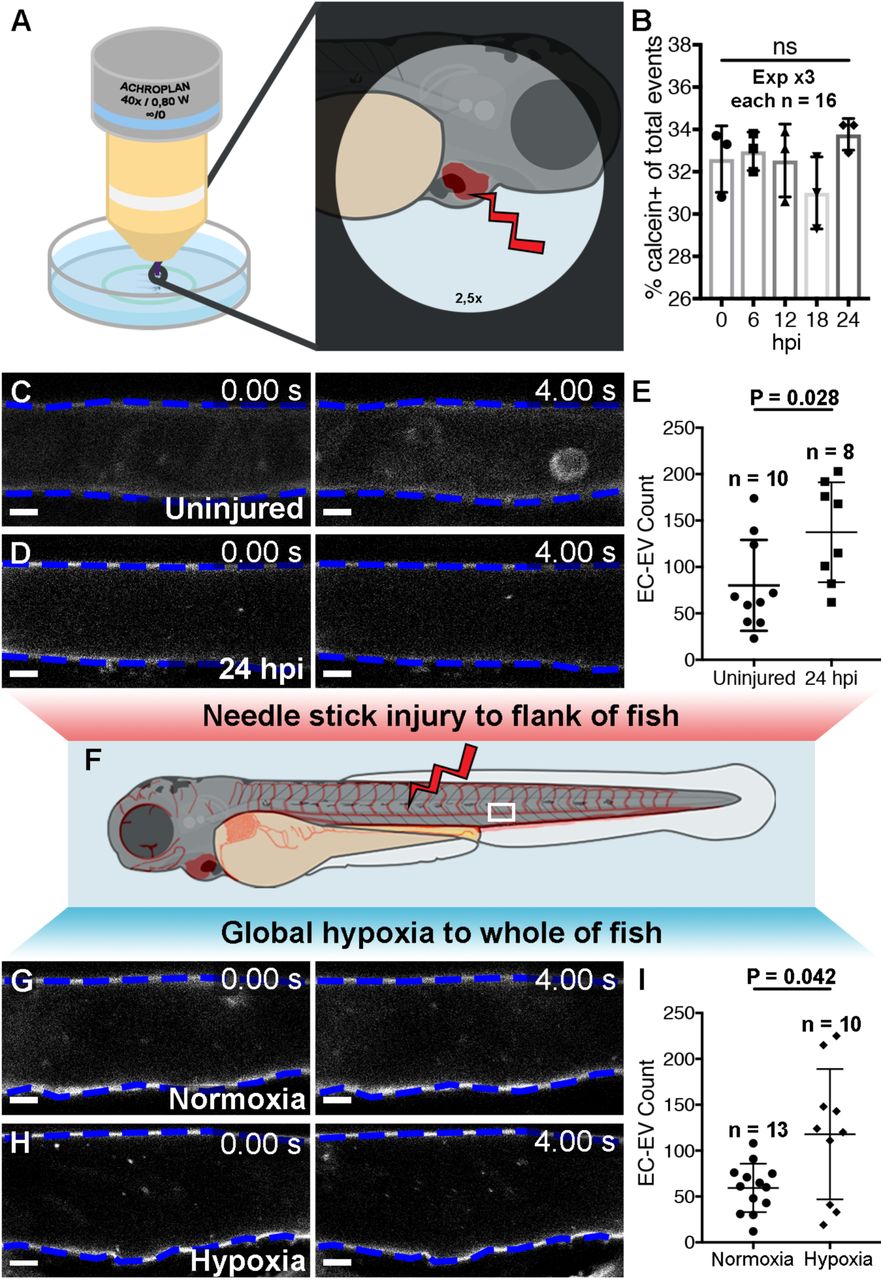
We can observe and isolate EVs derived from specific cardiovascular cell types using our zebrafish model, but we wanted to determine if and how these vesicles are affected by models of cardiovascular disease and cardiac damage. Initially, we used a MicroPoint ablation laser to induce damage specifically to the surface of the ventricle in 3 dpf zebrafish larvae and assessed the number of EVs produced by flow cytometry (Figure 4A,B). We did not see changes to the overall number of calcein+ EVs at 6, 12, 18 or 24 hours post heart injury (hphi), perhaps due to the minor nature of the induced injury and/or limits to the sensitivity of flow cytometry techniques to detect subtle changes to EV populations. Next, we used an established larval injury model (Gurevich et al., 2016; Gurevich et al., 2018) to induce more significant damage to the flank of Tg(kdrl:mCherry-CAAX) fish at 3 dpf using a 30-gauge needle and performed manual counts of kdrl(mCherry)+ EC-EVs in the peripheral circulation at 24 hpi (Figure 4C-F). This flank injury resulted in significantly more EC-EVs in the DA compared to uninjured control fish (Figure 4C-E). To produce a strong, global injury response in the whole fish we mimicked aspects of ischaemic injury by inducing hypoxia in whole larval zebrafish. Incubating 3 dpf Tg(kdrl:mCherry-CAAX) larvae in 5 % oxygen for 18 hours significantly increased the number of EC-EVs observed in the peripheral circulation when compared to control larvae maintained in normoxic conditions (Figure 4F-H; Supplementary videos 6,7). This suggests that both a substantial flank injury and a global ischaemic/hypoxic environment can induce increased EC-EV release into the peripheral circulation. Collectively, these data suggest dynamic EV responses following models of tissue injury in larval zebrafish, however, we also wanted to assess endogenous cardiovascular EVs from fully differentiated, adult tissues and to determine if a model of MI in adult zebrafish hearts resulted in changes to cardiovascular EVs.

(A) Schematic depicting the laser cardiac injury method. (B) Quantification of the number of calcein+ EVs in uninjured (0 hpi) and injured fish at 6,12,18 and 24 hpi as determined by flow cytometry. (C,D) Image sequences of the DA of a uninjured (C) and an injured Tg(kdrl:mCherry-CAAX) fish at 24 hpi (D) (E) Quantification of the number of EC-EVs in the DA in uninjured larvae and at 24 hpi. (F) Schematic depicting additional injury methods. (G,H) Image sequences of the DA of a 4 dpf Tg(kdrl:mCherry-CAAX) fish under normoxic (G) or hypoxic (5 % oxygen) conditions (H). (I) Quantification of the number of EC-EVs in the DA under normoxic or hypoxic conditions. Statistical analysis in B,E,I: Two-tailed Mann-Whitney tests. Scale bars: C,D,G,H = 5 µm.
Characterisation of EVs from adult cardiac tissue reveals dynamic changes following a model of MI
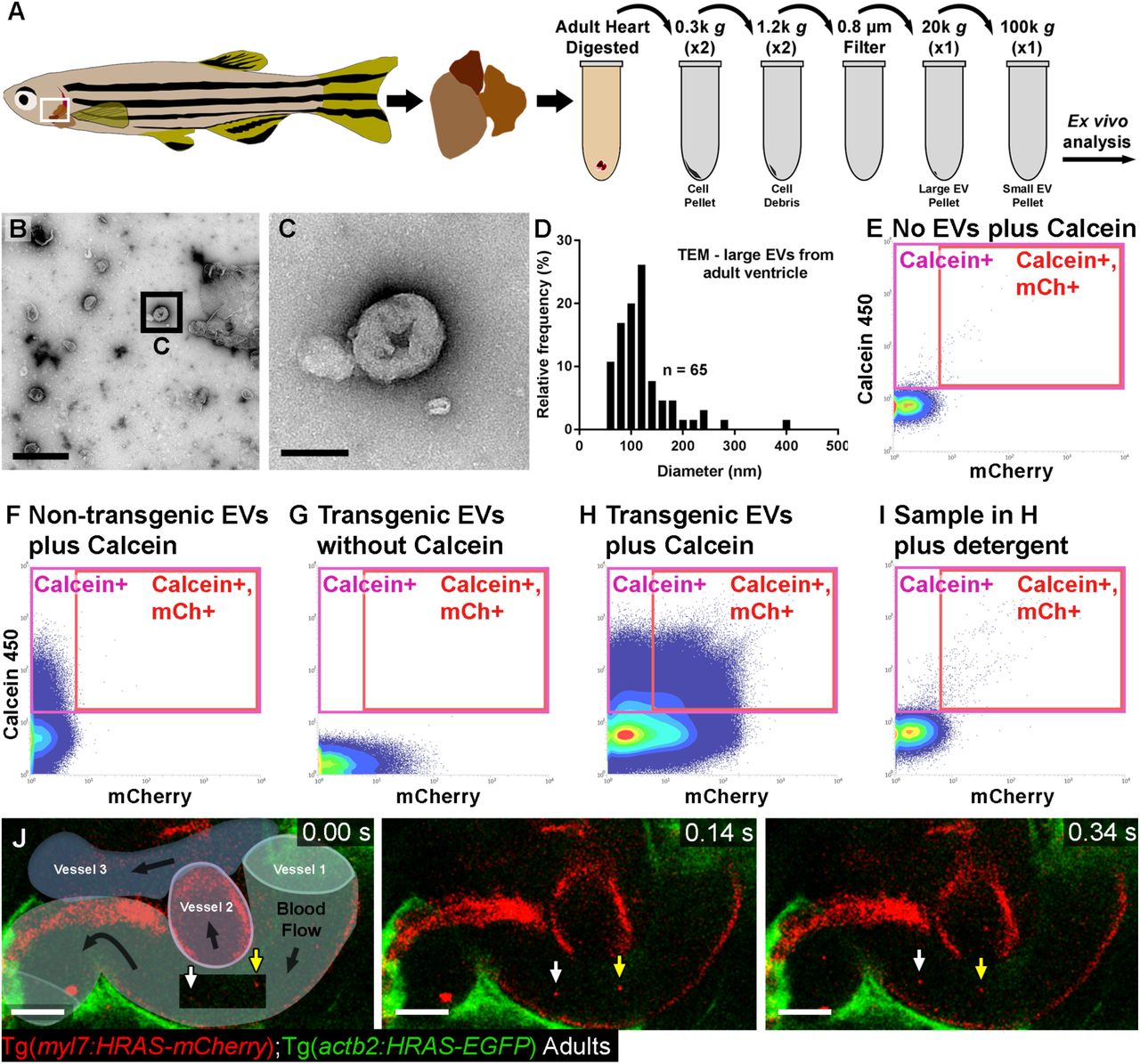
To validate EVs from adult cardiac tissue firstly we extracted EVs from isolated ventricles of unwounded adult fish and analysed these ex vivo using similar techniques described for larval samples (Figure 5A). To further validate these adult cardiac EVs, we isolated total EVs from ventricles and visualised them by transmission electron microscopy (TEM; Figure 5B,C). This analysis revealed EVs with a characteristic cup-like structure in sizes ranging from ∼50-300 nm (Figure 5C,D), supporting our DLS data on size distribution of EVs in larvae. Flow cytometry analysis revealed populations of intact, cell-type specific EVs derived from adult ventricles that could be obtained in much higher numbers than from entire larval fish (Figure 5E-I). To observe cell type specific EVs in vivo, we performed live imaging of superficial vessels of adult Tg(kdrl:mCherry-CAAX) fish revealing EC-EVs present in the peripheral circulation (Figure 5J and supplementary video 8).

(A) Schematic of an adult zebrafish, adult heart and centrifugation process to obtain adult cardiac EV fractions. Boxed region demarks the approximate position of the live imaging shown in K. (B,C) TEM negative stain micrograph of an isolated large EV fraction from a pool of adult ventricles (n = 3). C shows a higher magnification view of the boxed region in B. (D) Histogram of the size distribution of EVs from a large EV pellet from adult zebrafish ventricles analysed by TEM. (E-I) Typical flow cytometry scatter plots showing the gates used to sort adult cardiac EVs and the controls used to define these gates. (J) Schematic overlay describing the position of the three vessels visible in the integrated time series of live imaging of EC-EVs in the peripheral circulation of an adult Tg(actb2:HRAS-EGFP); Tg(kdrl:mCherry-CAAX) double transgenic fish. White and yellow arrows indicate two EC-EVs moving with the blood flow. Scale bars: B = 500 nm; C = 100 nm; J = 10 µm.
To assess the EV response to a model of MI, we performed cryoinjury on the hearts of adult zebrafish and extracted EVs from isolated ventricles of unwounded and 24 hpi fish and analysed them via flow cytometry and DLS (Figure 6). Flow cytometry did not reveal significant changes to the overall number of calcein+ or intact CM-EVs in the heart following cardiac injury (Figure 6A,B), however there were significantly fewer EC-EVs as a proportion of calcein+ EVs (Figure 6C) suggesting an increase in EVs derived from a cell type not assessed here e.g. interstitial fibroblasts or inflammatory cells. Interestingly, DLS revealed a significant shift in the size distribution of total EVs at 24 hpi when compared to unwounded hearts (Figure 6D) suggesting a dynamic EV response following cardiac injury.

(A-C) Quantification of the number of calcein+ (A), myl7(mCherry)+ (B) and kdrl(mCherry)+ EVs in uninjured and injured hearts at 24 hpi. (D) Histogram of DLS analysis reveals a significant shift in the size of overall EVs at 24 hpi compared to uninjured hearts. Statistical analysis in A-C: Two-tailed Mann-Whitney tests. Statistical analysis in D: a custom permutation test using total variation distance was used to test the null hypothesis that control and 24 hpi distributions were the same.
Discussion
Injury to cardiac tissue as a result of cardiovascular disease and MI results in extensive repair mechanisms that involve multiple different cell types and require complex cell-cell communication to coordinate this response. Cell-cell communication roles for EVs have been defined in different homeostatic and pathophysiological contexts but there is still relatively little known about the detailed in vivo function of these small vesicles. Here we have described methods to fluorescently label cell-type specific endogenous EVs and visualised these in vivo in a vertebrate zebrafish model system. Our findings complement recent studies that describe the labelling, biodistribution and potential functional roles of exogenous tumour derived and endogenous CD63+ EVs in larval zebrafish (Hyenne et al., 2019; Verweij et al., 2019b). The described techniques and advancements in the use of zebrafish for in vivo EV research pave the way for investigations into multiple homeostatic and pathophysiological states (Verweij et al., 2019a) and the involvement of endogenous vesicles in cellular processes such as tissue regeneration.
Here we describe techniques to stably label endogenous cardiovascular EVs. Endothelial cell derived EVs were observed in the peripheral circulation of larval and adult zebrafish, moving rapidly with the blood flow, but also exhibiting reduced speed when observed close to the vessel wall, as has been previously described (Hyenne et al., 2019; Verweij et al., 2019b). Cardiomyocyte-derived EVs were observed in the pericardial fluid of zebrafish larvae, where they were influenced by the movement of the heart. Static CM-EVs were also observed, potentially interacting with cells or ECM of the pericardial wall. We further analysed these endogenous EV populations ex vivo and show that large numbers of endogenous EVs can be isolated from zebrafish tissues using a modified FACS procedure. We have also demonstrated that models of ischaemic injury result in dynamic changes to different EV populations.
The labelling strategy described here allows the identification of the cellular origin of EVs and to visualise these in vivo but the proportion of EVs that incorporate fluorophore labelling during biogenesis is incompletely understood, potentially limiting the number of EVs produced by each cell type that we can successfully identify (Shen et al., 2011). Indeed, flow cytometry assessments suggest that approximately 30-65 % of events are labelled with calcein (e.g. intact vesicles that contain esterases) and that approximately 30 % of these are labelled with actb2(GFP)+ (thought to be a near-ubiquitous promoter), suggesting relatively restricted fluorophore and esterase incorporation and/or limited preservation of intact EVs during isolation and/or sorting. Nevertheless, shared fluorescence between the selected cell types and EVs observed within extracellular space confirm the cellular origin of these vesicles and sufficient numbers of intact vesicles can be obtained for further studies, even when selecting only EVs that are both calcein+ and membrane-fluorophore labelled.
Larval in vivo imaging studies revealed static CM-EVs on the pericardial wall. Analysis of the pericardium of Tg(actb2:HRAS-EGFP); Tg(myl7:HRAS-mCherry) larvae failed to show any direct interaction between CM-EVs and the cells lining the pericardial wall, potentially suggesting these EVs are interacting with an ECM component of the pericardium (Rilla et al., 2019). The larval zebrafish model described here is well placed to allow the study of these EV-ECM and EV-recipient cell interactions further. Additionally, future studies in larvae and/or adults would allow the functional significance of this interaction to be studied during repair and regeneration. Interestingly, we did not observe
CM-EVs in the peripheral circulation or EC-EVs in the pericardial space of larvae, even following injury to the heart, suggesting localised control of EV release or rapid local uptake.
We have also demonstrated that we can visualise and extract EVs from adult zebrafish. EC-EVs could be observed moving rapidly with the blood flow in superficial vessels of adult fish and TEM reveals a characteristic cup-shaped appearance of extracted vesicles from isolated adult hearts. TEM measurements and DLS analysis reveal similar size distributions for total EVs extracted from adult hearts and from whole larvae, and are in line with previous reports of EVs from zebrafish, mice and humans (Akbar et al., 2017; Emanueli et al., 2016; Hyenne et al., 2019; Lawrie et al., 2009; Sahoo et al., 2011).
Most importantly, we have demonstrated that models of cardiovascular injury can induce changes to EV number and size, suggesting a dynamic intercellular communication response that involves EVs, supporting previous in vitro models, clinical findings and studies in non-regenerative rodent models of MI (Akbar et al., 2017; Beltrami et al., 2017; Boulanger et al., 2017; Emanueli et al., 2016; Gupta and Knowlton, 2007; Loyer et al., 2018; Malik et al., 2013). Similar numbers of overall EVs and CM-EVs following cardiac injury but a reduction in EC-EVs, as a proportion of total (calcein+) EVs, suggest an increased proportion of EVs deriving from an, as yet underdetermined, cell-type. Indeed, CM-EVs and EC-EVs represent approximately 30 % of total calcein+ EVs in the adult heart, suggesting a significant contribution from other cell types. Interstitial cardiac fibroblasts, and/or inflammatory cells are potential candidates and future studies will determine the contribution of these other cell types to the EV composition of the repairing and regenerating heart.
Interestingly, we observed an increase in EC-EVs following larval injury models and a reduction in EC-EVs following an adult MI model, suggesting dynamic changes to EVs that may be highly tissue- and time-point dependent. Further studies will be required to determine if the reduction in EC-EVs results from reduced numbers of ECs in the injured heart at 24 hpi or from dynamic changes to EC-EV production following injury. Our data further suggest that there is a significant shift to smaller EVs at 24 hpi potentially indicating increased exosome release. This is similar to what has been described for clinical plasma samples in patients undergoing cardiac surgery (Emanueli et al., 2016) and may represent an active shift in EV biogenesis in response to MI. Additionally, further studies to define the cargo and composition of these post-ischaemic EVs could reveal their roles in tissue repair and regeneration and identify potential therapeutic targets.
In summary, we have shown that endogenously produced cell-type specific EVs can be stably labelled with fluorophores, revealing their cellular origin and allowing them to be observed and tracked in vivo. Large numbers of cell-type specific EVs can be obtained from adult hearts allowing future evaluations of cargo during homeostasis and in models of disease. Finally, models of ischaemic injury produce dynamic changes to EV size and numbers produced by different cell types demonstrating the role of these small vesicles in these processes.
Author Contributions
AS planned and performed experiments, analysed data and wrote the manuscript. LSB and AH planned and performed flow cytometry/FAVS with AS. MB and DP planned, advised and performed EM evaluations of EVs with AS. AP and JL planned, advised and performed ImageStream assessments of EVs with AS. CE and RR jointly conceptualised the project. RR supervised experiments and wrote the manuscript. All authors contributed to drafting the manuscript.
Declaration of Interests
The authors declare no competing interests
Materials and Methods
Zebrafish lines and procedures
The Tg(actb2:HRAS-EGFP) (Cooper et al., 2005), Tg(tbp:GAL4);(UAS:secA5-YFP) (van Ham et al., 2010), Tg(kdrl:mCherry-CAAX) (Fujita et al., 2011), Tg(myl7:GFP) (Lepilina et al., 2006) and Tg(myl7:HRAS-mCherry) (Yoruk et al., 2012) transgenic zebrafish lines have been described previously. For larval cardiac injury a MicroPoint pulsed nitrogen pumped tuneable dye laser (Andor/Oxford Instruments) connected to a Zeiss Axioplan II microscope (Zeiss Microimaging) was used as previously described (Otten and Abdelilah-Seyfried, 2013). Briefly, a 40X water immersion objective and a double laser pulse at a wavelength of 435 nm was used to damage the surface of the apex of the ventricle. Larval flank needle-stick injuries were carried out as described previously (Gurevich et al., 2016). Briefly, 3 dpf larval zebrafish were anesthetised in 0.13% MS-222 (Sigma; A5040), placed on 24.5 x 76.2 mm glass slides in a drop of Danieau’s medium and positioned laterally, anterior to the left. A single needle-stick injury into the dorsal epaxial musculature anterior to the cloaca was performed at 75°, using a 30-gauge needle. For hypoxia experiments, 3 dpf larval zebrafish in Danieau’s medium were incubated at 28°C in a hypoxia workstation (InvivO2 300, Ruskinn), deoxygenised by positive infusion of 5% CO2/95 % N2 gas mixture to maintain 5% oxygen (Santhakumar et al., 2012) for 18 hours. Cardiac injuries on adult zebrafish were carried out as described previously (Gonzalez-Rosa and Mercader, 2012). Briefly, fish were anaesthetised in 0.13% MS-222 (Sigma; A5040) and placed ventral side up in a pre-cut sponge soaked in aquarium water containing anaesthetic. A 4 mm incision was made through the skin and the pericardial sac directly above the heart to expose the ventricle. The ventricle was dried using a sterile cotton swab and a liquid nitrogen cooled probe applied to the exposed ventricle for 30 seconds. Adult fish used were aged 4 - 18 months and fish were randomly assigned to control or test experiments using a random number generator. All lines are maintained according to standard procedures and all animal work is carried out in accordance with UK Home Office and local University of Bristol regulations.
Imaging
3/4 dpf and adult zebrafish were anaesthetised in 0.01 mg.ml-1 MS-222 and mounted in 1% low-gelling agarose (Sigma). Live imaging was performed on a Zeiss Z.1 light sheet system with a 40x W Plan Apochromat objective. Additionally, some imaging was performed on a Leica TCS SP8 AOBS confocal laser scanning microscope with a 25x/0,95 W HC FLUOTAR objective. The resonant scanner option was used for high speed imaging (frame intervals of 0.02-0.04 seconds) of EVs in the peripheral circulation and the pericardial space. The conventional scanner was used for imaging of fixed samples. Images were processed using Fiji (Schindelin et al., 2012) and Imaris. Deconvolution was carried out using Huygens Professional version 16.10 (Scientific Volume Imaging, The Netherlands) using the CMLE algorithm. To achieve optimal results images were acquired at a sampling density that satisfied Nyquist-Shannon sampling theorem. Deconvolved images are noted in the figure legends. For manual counts of EVs, all analysis was blinded and positive events were counted from 1-minute videos of the DA above the cloaca (region highlighted in Figure 2C).
Cell Dissociation and EV Isolation
Adult or 4-5 dpf larval zebrafish were treated with an anaesthetic overdose before adult hearts were dissected (1-3 hearts pooled per experiment) or whole larvae (16-20 pooled per experiment) were placed into a digestion buffer consisting of perfusion buffer (PBS plus 10 mM HEPES, 30 mM Taurine and 5.5 mM Glucose) plus 0.25% Trypsin, 12.5 µM CaCl2 and 5 mg/ml Collagenase II (Worthington Biochemical Corp; LS004176). A stopping buffer consisting of perfusion buffer plus 10% (vol/vol) FBS and 12.5 µM CaCl2 was added to stop the digestion. EVs were isolated from the single cell suspension by differential centrifugation at 300 g (2 x 10 minutes), 1200 g (2 x 10 minutes), before being passed through a 0.8 µm sterile filter and a final centrifugation step of either 20,000 g (30 minutes) or 100,000 g (70 minutes) was used to crudely pellet large EVs and/or small EVs respectively, which were then resuspended in 300 µl sterile filtered PBS. Samples were either used immediately or snap frozen in liquid nitrogen, stored at −80 °C and later thawed at 37 °C for 2 minutes before use.
Dynamic Light Scatter (DLS)
Hydrodynamic particle size of crude EV samples in PBS was measured by DLS. All measurements were obtained in triplicate using a Zetasizer Nano-ZS equipped with an internal Peltier temperature controller (Malvern Instruments, Malvern Hills, UK).
Flow Cytometry
From crude EV samples, intact EVs were labelled with calcein violet 450 AM (eBioscience; 65-0854-39) as previously described (Gray et al., 2015). Detergent treated negative controls were mixed with 0.05% Triton X-100 (Sigma: T8787). Flow cytometry analysis and FAVS was carried out using a BD Influx system, with optimisations made to allow for the reliable detection of fluorescently labelled EVs (van der Vlist et al., 2012). Briefly, a high power 200 mW 488 nm laser was used for optimal excitation of small particles and GFP, a 50 mW 405 nm laser was used to excite calcein 450, a 100 mW 552 nm laser was used to excite mCherry and the following bandpass filters were used to detect the respective emissions: 530/40 nm, 460/50 nm and 610/20 nm. A small-particle detector allowed for high sensitivity in detecting forward scatter (FCS) and a 0.45 threshold on a logarithmic scale was used. A 4 mm obscuration bar was used to optimally detect submicron particles. For flow cytometry of EVs, a 100 µm nozzle and 21 PSI was used. To increase sorting speed and throughput for FAVS, a 70 µm nozzle and 42.9 PSI was used following confirmation that the signal was not compromised by the reduced time travelling through the laser. EVs were sorted into 100 µl PBS. All flow cytometry experiments were performed at 4 °C.
Imaging flow cytometry analysis was carried out as previously described (Lannigan and Erdbruegger, 2017). Briefly, analysis was performed using a fully calibrated (ASSIST tool) ImageStream®xMk II (Amnis-Luminex, Seattle, USA). 405 nm and 488 nm excitation lasers, a 785 nm side scatter (SSC) laser, brightfield illumination and a six channel charge-coupled device camera with time delay integration were used. For maximum resolution and high sensitivity, fluidics were set at low speed and magnification was set at 60x (0.3 mm2/ pixel). Speed beads (1.5 µm diameter, carboxylated polystyrene microspheres) run continuously during data acquisition to maintain focus and image synchronicity. They form a discrete population based on their SSC and fluorescence properties and can therefore be easily identified (Figure S2A) and eliminated from the sample post-acquisition (Figure S2B,C). The ImageStream Data Exploration and Analysis Software (IDEAS®6.2 EMD Millipore, Seattle) was used to perform data analyses. A compensation matrix was applied to adjust for spectral overlap between channels.
Transmission Electron Microscopy (TEM)
Samples were prepared for TEM in alignment with established methods (Rikkert et al., 2019). Five microlitres of isolated EV sample was placed onto Formvar-carbon coated, glow-discharged, 300 mesh copper grids (prepared in house by Wolfson Bioimaging Facility staff) and left for 30 seconds to allow the contents to adhere to the carbon support, the buffer was then removed using blotting paper. 1% uranyl acetate was then placed onto the grid and left to negatively stain the sample for 30 seconds, before being removed using blotting paper. The grids were left to air-dry before being visualised on a FEI Tecnai 12 120kV BioTwin Spirit TEM and images acquired using a FEI Eagle 4k x4k CCD camera.
Total Internal Reflection (TIRF) Microscopy
EV prep for TIRF microscopy was performed as previously described (Ter-Ovanesyan et al., 2017). Briefly, 50 µl sorted EV samples plus 0.06 µm far-red labelled carboxylate-modified microspheres (Sigma; T8870) (1:10000 final concentration) were placed onto 35 mm glass bottom dishes (Mattek; P35G-1.5-14-C) and left for 2 minutes to allow the contents to settle. Images were then acquired using a Leica AM TIRF multi-colour system attached to a Leica DMI 6000 inverted epifluorescence microscope.
Western Blot
Western blot analysis was carried out using standard methods. Briefly, cells, large EV and small EV pellets were lysed on ice for 10 min with lysis buffer consisting of basic buffer (125 mM NaCl, 20 mM TRIS pH 7.4, 1% Nonidet P40 (Sigma Aldrich; 11754599001) and 10% Glycerol) plus 0.1 mg.ml-1 Phenylmethylsulfonyl fluoride, protease cocktail inhibitor (Sigma Aldrich; 04693159001), 50 mM NaF and 10 mM Na3O4V. Equal protein concentrations were resolved via 8 % SDS–PAGE, transferred to an Immobilon-P polyvinylidene difluoride membrane (Sigma Aldrich; IPVH00010) and stained with an anti-ALIX antibody (Sigma Aldrich; SAB4200476).
Statistics
In all cases n numbers refer to biological replicates. All experiments were repeated at least twice. Raw data recording and analysis was conducted using GraphPad Prism6/7. Statistical significance was determined via nonparametric Mann-Whitney or Kruskal-Wallis/Dunn’s multiple comparison tests or a randomised permutation test using total variation distance between two groups (details provided in figure legends). All statistical analysis was blinded. In all cases error bars represent SD. For all data sets a Grubb’s outlier test was performed and any significant outliers (alpha = 0.05) were removed. Precise p values are included in all graphs.

(A) Schematic representation of the Annexin-V labelling strategy. (B) Overview image of a larval Tg(tbp:GAL4);(UAS:secA5-YFP) zebrafish at 3 dpf. Blue indicates the DA and red the caudal haematopoietic (venous) tissue (CHT). Boxes depict the approximate position of the image sequences in C and D. (C) Image sequence of an Annexin-V labelled EV moving through the DA. Blood cells can be clearly seen in brightfield (asterisks). (D) Image sequence of an Annexin-V labelled EV (also inset) moving through the pericardial space. Anterior is to the left. The magenta line demarks the outer wall of the pericardial space, green outlines the ventricle. Scale bars: B = 200 µm; C = 5 µm; D = 10 µm; insets in D = 2 µm.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A-D) ImageStream analysis and quantification of EVs from a pool of 30 whole Tg(actb2:HRAS-EGFP) larvae. Control experiments using PBS, PBS plus calcein AM, actb2(GFP+) EVs with and without calcein AM and speed beads (1.5 µm diameter, carboxylated polystyrene microspheres, see methods for details) were used to set correct gates for double positive (fluorescence and calcein) EVs (A). Analysis of EVs from Tg(actb2:HRAS-EGFP) fish demonstrates different populations of EVs which can be individually visualised (lower panels) (B). Detergent treatment destroys EV integrity further confirming their lipid nature (C,D).
Video 1. Live imaging of actb2+ EVs in the peripheral circulation of a larval zebrafish. Image sequence (751 frames = 15 seconds) of actb2+ EVs passing through the DA of a 3 dpf Tg(actb2:HRAS-EGFP) larval zebrafish. Scale bar = 5 µm.
Video 2. Live imaging of actb2+ EVs in the pericardial space of a larval zebrafish. Image sequence (191 frames = 3.28 seconds) of actb2+ EVs travelling through the pericardial fluid of a 3 dpf Tg(actb2:HRAS-EGFP) larval zebrafish. Scale bar = 50 µm, 5 µm.
Video 3. Live imaging of EC-EVs in the peripheral circulation of a larval zebrafish. Image sequence (751 frames = 15 seconds) of mCherry+ EC-EVs passing through the DA of a 3 dpf Tg(kdrl:mCherry-CAAX) larval zebrafish. Scale bar = 5 µm.
Video 4. Live imaging of CM-EVs in the pericardial space of a larval zebrafish. Image sequence (669 frames = 12 seconds) of mCherry+ CM-EVs travelling through the pericardial fluid of a 3 dpf Tg(myl7:HRAS-mCherry) larval zebrafish. Scale bar = 50 µm, 5 µm.
Video 5. 3D reconstruction of the pericardial space of a Tg(actb2:HRAS-EGFP); Tg(myl7:HRAS-mCherry) larval zebrafish. 3D view of CM-EVs in the pericardial space of a Tg(actb2:HRAS-EGFP); Tg(myl7:HRAS-mCherry) at 3 dpf. Images were deconvolved using the Huygens CMLE algorithm and a 3D reconstruction was created with Imaris software. Scale bar = variable with zoom, see video.
Video 6. Live imaging sequence of EC-EVs in the peripheral circulation of larval zebrafish injury models. Image sequences (522 frames = 10.44 seconds) of EC-EVs passing through the DA of 4 dpf Tg(kdrl:mCherry-CAAX) larval zebrafish. Injured larvae at 24 hpi and uninjured controls and normoxia and hypoxia treated fish (18 hours at 5% Oxygen) are shown. Scale bar = 5 µm.
Video 7. Live imaging sequence of EC-EVs in the peripheral circulation of adult zebrafish. Image sequences (837 frames = 16.62 seconds) of mCherry+ EC-EVs passing through a superficial blood vessel near the gills of a 12-month Tg(actb2:HRAS-EGFP); Tg(kdrl:mCherry-CAAX) adult zebrafish. Scale bar = 5 µm.
Acknowledgements
This work was supported by a BHF Intermediate Fellowship (FS/15/2/31225) to RR, a BHF studentship for AS awarded jointly to RR and CE (FS/18/34/33666), the BHF Oxbridge Centre of Regenerative Medicine (RM/13/03/30159) (jointly to RR and CE), and a BHF Chair award grant (CH/15/31199) and Leducq Transatlantic Network MIRVAD (both to CE), Wellcome Trust funding of a Zeiss light sheet Z.1 system and MRC funding of a pre-clinical In-vivo functional imaging platform for translational regenerative medicine. The authors wish to thank the Wolfson Bioimaging Facility for imaging expertise and Professor Jonathan Rougier (University of Bristol) for statistical advice.
References




