

Abstract
H2A.Z mono-ubiquitylation has been linked to transcriptional repression, but the mechanisms associated with this process are not well understood due to the lack of reagents specific for this modified form of H2A.Z. To address this, we developed a biotinylation-based approach to specifically purify ubiquitylated H2A.Z (H2A.Zub) mononucleosomes and characterize their biochemical composition and genomic distribution. We observe that H2A.Zub nucleosomes are enriched for the repressive histone post-translational modification H3K27me3, but depleted of H3K4 methylation and other modifications associated with active transcription. Consistent with these findings, ChIP-Seq analyses reveal that H2A.Zub-nucleosomes are enriched over non-expressed genes. However, the genomics data also suggest that it is the relative ratio of ubiquitylated to non-ubiquitylated H2A.Z, rather than the absolute presence or absence of H2A.Z ubiquitylation, that correlates with gene silencing. Finally, we observe that H2A.Zub-eniched mononucleosomes preferentially co-purify with transcriptional silencing factors as well as proteins involved in higher order chromatin organization such as CTCF and cohesin. Collectively, these results suggest an important role for H2A.Z ubiquitylation in the genome-wide regulation of chromatin and transcription through its recruitment of transcriptional silencing factors and nuclear architectural proteins.
Introduction
Accessibility of the eukaryotic genome is impacted by how it is packaged into chromatin and how chromatin/chromosomes are looped and folded in the 3-dimensional space within the nucleus (1, 2). This organization of the genome regulates many DNA-templated processes including transcription and gene expression. At the simplest level, DNA is wrapped around histone proteins to form nucleosomes, and the ‘nucleosome-on-a-string’ 10 nm fiber can further fold into higher-order structures that not only serve to compact the genome, but also form long-range interactions that bring different parts of the genome together to assemble functional domains. As the repeating unit of chromatin, each nucleosome is made up of ∼ 147 bp of DNA wrapped around a histone octamer containing 2 copies each of the core histones H2A, H2B, H3 and H4 (3). Although most nucleosomes share this same structural architecture, the large repertoire of histone post-translational modifications (PTMs) can confer distinct functional properties to different nucleosomes (4). Common examples of histone PTMs include acetylation, phosphorylation, methylation and mono-ubiquitylation, and many of these PTMs function in a wide variety of nuclear processes such as transcription, DNA replication and repair. These PTMs are deposited or removed by different types of enzymes that either respond to upstream signaling pathways or are constitutively active to maintain the their steady-state levels. The physical and functional effects of histone PTMs vary depending on the type of modification and context, but one frequent outcome is the recruitment of chromatin binding proteins through PTM-binding motifs, and these recruited proteins in turn mediate downstream activities such as chromatin compaction or transcriptional regulation (5, 6). Therefore, histone PTMs can serve as signals to target effector proteins to specific regions of the genome, and relay information from upstream signaling pathways to downstream actions (7, 8). In recent years, it has also been recognized that since nucleosomes contain many different histone PTMs and binding surfaces, the sum of all these binding ligands can together stabilize and regulate interactions between nucleosomes and chromatin binding factors (9, 10, 11). Therefore, determining the combinatorial effects of histone PTMs in the context of the nucleosome is critical for our understanding of the mechanisms underlying chromatin regulated pathways.
In addition to histone PTMs, replacement of core histones with specific histone variants is another cellular mechanism for adding functional diversity to the nucleosome framework. Histone variants are non-allelic isoforms of core histones and are deposited at strategic positions within the genome to perform specialized functions (12). For example, H2A.Z is an H2A-family variant that is often enriched at the nucleosomes flanking transcription start sites (TSSs) of many genes in eukaryotic genomes (13, 14). With the exception of S. cerevisiae, knockout of the H2A.Z gene in other organisms invariably leads to developmental defects (e.g. in mice) or death. The specific functions of H2A.Z that are essential for viability are not known, but H2A.Z is required for transcriptional regulation since its knockdown, or knockdown of the remodeling complexes that deposit H2A.Z to the genome, often leads to dysregulated gene expression (15, 16, 17, 18). H2A.Z ChIP-seq studies in human cells showed that H2A.Z is enriched at active genes (19), suggesting a correlation between H2A.Z and gene activity. Functionally, H2A.Z has been linked to both transcriptional activation and repression, and these distinct activities are likely mediated through its differential PTMs (e.g. acetylation vs. mono-ubiquitylation, 20). For example, H2A.Z is acetylated at multiple lysines at its N-terminus by the NuA4 complex in S. cerevisiae or Tip60 in Drosophila and mammals (21, 22, 23, 24). H2A.Z acetylation directly correlates with transcriptional activation (22) and expression of mutant H2A.Z that cannot be acetylated disrupts expression of the master regulator MyoD and downstream differentiation in myoblast cells (25). A similar mutagenesis approach showed that H2A.Z acetylation is also important for activating androgen-receptor (AR)-regulated genes and for the transcription of eRNAs at active enhancers elements in prostate cancer cells (26). In contrast to acetylation, mono-ubiquitylation of H2A.Z at its C-terminus has been correlated with transcriptional repression. This modification is mediated by the RING1b E3 ligase of Polycomb Repressor Complex 1 (PRC1) and ubiquitylated H2A.Z is enriched on the transcriptionally silent inactive X chromosome in human female cells (27). Ubiquitylated H2A.Z is also found at the promoters of AR-regulated genes prior to hormone induction and transcriptional activation of these genes is accompanied by H2A.Z de-ubiquitylation, suggesting H2A.Zub may antagonize transcription initiation (28). At present, the exact mechanisms of how H2A.Z by itself, or through H2A.Z acetylation and ubiquitylation, participate in transcriptional regulation are not clearly understood. Early studies in S. cerevisiae showed that H2A.Z may affect nuclease sensitivity within the genome and it also functions at chromatin boundaries to antagonize heterochromatin spreading (16, 29). It is also thought that H2A.Z poises inducible promoters for transcription initiation and lowers the stability of the +1 nucleosome at TSSs to facilitate passage of RNA polymerase during transcriptional activation (30). Affinity purification studies found that H2A.Z nucleosomes preferentially bind the bromodomain protein Brd2, and the H2A.Z-dependent recruitment of Brd2 to AR-regulated genes is part of the hormone-activated transcription process in prostate cells (31). The interaction between H2A.Z and Brd2 has also been reported in melanoma and bladder cancer cells (32, 33). In melanoma cells, the H2A.Z.2 isoform recruits both Brd2 and the transcription factor E2F1 to regulate E2F-target genes. Altogether, these studies suggest that H2A.Z-dependent recruitment of interacting proteins, such as Brd2, is a critical mechanistic step in H2A.Z-regulated transcription.
A mechanistic understanding of the role of H2A.Z ubiquitylation has been lacking due to the absence of research tools affording the specific detection and isolation of this modified form of H2A.Z. While multiple antibodies specific for mammalian H2A.Z have been raised, they generally cannot distinguish between non-ubiquitylated and ubiquitylated H2A.Z. Moreover, for some antibodies raised against the C-terminus region where H2A.Z ubiquitylation occurs, their recognition of H2A.Zub may be blocked due to epitope occlusion resulting from the ubiquitin moiety (27). Raising antibodies specific for ubiquitylated proteins is confronted by the additional challenge that they would have to recognize the isopeptide junction between the ubiquitin C-terminus and the sequence flanking the ubiquitylated lysine on the target protein. Lastly, H2A.Z can be mono-ubiquitylated at three possible lysine residues (K120, K121 or K125) (31, 34) all of which would need to be considered when raising specific antibodies.
To advance mechanistic studies of ubiquitylated form of H2A.Z, we have developed a non-antibody-dependent system that affords its specific detection and purification. To this end, we generated and expressed a version of mammalian H2A.Z that auto-biotinylates when it is ubiquitylated, such that streptavidin-coupled reagents can be used for the specific purification of ubiquitylated H2A.Z. This new system is built upon our previous success in developing the Biotinylation-assisted Isolation of CO-modified Nucleosome (BICON) technique for isolating MSK1-phosphorylated H3 (35). Both techniques take advantage of the specific biotinylation of a unique 15 amino acid sequence termed Avi-tag by the E. coli BirA biotin ligase when co-expressed in mammalian cells (36). In this new system, we co-expressed a fusion protein consisting of BirA ligase at the C-terminus of H2A.Z with Avi-tagged ubiquitin, by transfection in human 293T cells. We reasoned that since the ubiquitylation sites on H2A.Z are at its C-terminal end, the BirA placed at the C-terminus of H2A.Z would be in close proximity to the Avi-tag when it is ubiquitylated with Avi-ubiquitin. Moreover, since the biotinylation is an intra-molecular reaction (i.e. both enzyme and substrate are on the same molecule), it is expected to be highly efficient reaction. In this study, we demonstrate the efficacy of this system for the specific purification of Avi-ubiquitylated H2A.Z from transfected mammalian cells. Using this system, we performed biochemical analyses of the purified nucleosomes containing ubiquitylated H2A.Z and identified characteristic combinations of histone PTMs that favour transcriptional repression. We also used the same approach on a genome-wide scale, and identified genomic regions and target genes that are enriched for ubiquitylated H2A.Z, revealing its global association with transcriptional silencing. Finally, we discovered a number of proteins that preferentially co-purify with ubiquitylated H2A.Z-containing nucleosomes, pointing to possible chromatin modifying enzymes that are recruited by these nucleosomes to mediate transcriptional silencing. Importantly, we identified preferential interaction between CTCF and cohesin components with ubiquitylated H2A.Z, raising the intriguing possibility that this form of modified H2A.Z may function in the 3D organization of the mammalian genome and conjunction with its role in transcriptional regulation.
Results
Development and characterization of a ubiquitylation-dependent self-biotinylating system for studying mono-ubiquitylated H2A.Z
We previously developed a method named BICON to specifically purify MSK1-phosphorylated H3 (35). To adapt this method for studying H2A.Z ubiquitylation, we fused the BirA enzyme to the C-terminus of H2A.Z, and placed the Avi-tag sequence on the N-terminus of the ubiquitin molecule (Avi-ub; Fig. 1). We also added a Flag-tag peptide sequence between the H2A.Z and BirA domains to facilitate its detection and purification; the resulting H2A.Z-Flag-BirA fusion is hereafter termed H2A.Z-FB. By co-expressing H2A.Z-FB and Avi-ub in transfected cells, we reasoned that when H2A.Z-FB is mono-ubiquitylated with Avi-ub, the BirA at its C-terminus would preferentially and automatically biotinylate the Avi tag on the conjugated ubiquitin since the enzyme and substrate are now physically on the same molecule (see Fig 1A). Therefore, with this system, we are generating a version of H2A.Z that self-biotinylates when it is ubiquitylated with Avi-ub.

(A) Cartoon diagram illustrating the H2A.Z-Flag-BirA (H2A.Z-FB) fusion and its auto-biotinylation upon ubiquitylation with Avi-tagged ubiquitin (Avi-Ub). (B) 293T cells were co-transfected with wild type (WT) or non-ubiquitylatable (K3R3) H2A.Z-FB with or without Avi-Ub. Whole cell or nuclear extracts were harvested from the transfected cells and total H2A.Z-FB or biotinylated H2A.Z-FB were detected by Western blots using Flag antibody or horse radish peroxidase-couple avidin (Avi-HRP). H3 was used as loading control. (C) Mono-nucleosomes were prepared from 293T cells transfected with the indicated NLS-Flag-BirA, H2A.Z-FB (WT or non-ubiquitylatable KR mutant) and Avi-Ub combinations. The presence of Flag tagged and biotinylated proteins were detected by Western blots using Flag antibody or Avi-HRP.
As a proof of concept, we first co-transfected H2A.Z-FB- and Avi-ub-expression constructs into human 293T cells and examined their expression and biotinylation by Western blot analyses (Fig 1B). As an additional control, we generated a non-ubiquitylatable mutant form of H2A.Z-FB whereby the known sites of potential H2A.Z mono-ubiquitylation (K120, K121, and K125) are mutated to arginines (referred to as the K3R3 mutant) and co-expressed that with Avi-ub in 293T cells. Western blot analysis of whole cell extracts or nuclear extracts of the respective transfected cells using anti-Flag antibody showed strong expression of WT- and K3R3-H2A.Z-FB. The anti-Flag antibody detected two bands (corresponding to the unmodified H2A.Z-FB and the larger mono-ubiquitylated form of H2A.Z-FB) in lysates from cells transfected with WT H2A.Z-FB, but only the lower molecular weight band in cells expressing the K3R3 mutant; thus confirming that the higher molecular weight band is the mono-ubiquitylated H2A.Z-FB. We next examined the presence and detection of biotinylated proteins in the same extracts using horse-radish peroxidase-conjugated avidin (Avi-HRP) and found that the predominant and most abundant protein detected was indeed the Avi-ub-H2A.Z-FB, which co-migrated with the shifted higher molecular weight band detected in the Flag Western blots. The Avi-HRP-detected band in the WT H2A.Z-FB + Avi-ub sample was not present in the H2A.Z-FB K3R3 mutant + Avi-ub sample, further validating that biotinylation specifically occurs on the Avi-ubiquitylated H2A.Z-FB. These results indicate that our experimental system works as predicted and that Avi-ub-H2A.Z-FB is the predominant biotinylated protein in the transfected cells.
We also compared the expression of the transfected H2A.Z-FB + Avi-ub combination in several different mammalian cell lines and found that this system works best in human 293T cells (data not shown). Therefore, all subsequent biochemical and genomic analyses were performed using this cell line. Furthermore, we found that in mono-nucleosome-enriched fractions (harvested from micrococcal-nuclease digested nuclei), in addition to Avi-ub-H2A.Z-FB, Avi-HRP detected a small amount of a lower molecular weight band that is consistent with the size of the endogenous ubiquitylated H2A.Z (Fig 1C). It is likely that this lower band corresponds to Avi-ubiquitylated endogenous H2A.Z or H2A, since H2A.Z-FB potentially can biotinylate any endogenous Avi-ub-H2A.Z/H2A either on the same nucleosome or in close proximity to the H2A.Z-FB. We note that this lower band is also detected by Avi-HRP in cells transfected with Flag-NLS-BirA (BirA fused to Flag and a nuclear localization signal) control, suggesting that the presence of BirA and Avi-ub in cells can result in some background amounts of biotinylation of the Avi-ubiquitylated endogenous H2A.Z or H2A. However, this background amount is minor compared to the biotinylated Avi-ub-H2A.Z-FB in the H2A.Z-FB + Avi-ub transfected cells. Finally, we found that in the K3R3 H2A.Z-FB + Avi-ub transfected cells, only the lower band is detected by Avi-HRP. In this context, there were higher amounts of the biotinylated lower band than found in WT H2A.Z-FB + Avi-ub transfected cells, suggesting that in the absence of a preferred biotinylation site on the Avi-ub-H2A.Z-FB itself, the BirA on this fusion protein can biotinylate less preferred Avi-tags on nearby Avi-ubiquitylated histones. These findings together confirm that wild-type H2A.Z-FB preferentially and predominantly biotinylates the Avi-tagged ubiquitin on itself first, and secondarily biotinylate other nearby Avi-ubiquitylated histones when the main ubiquitylation site on H2A.Z-FB is mutated.
Immunoprecipitation of mono-nucleosomes containing Avi-ubiquitylated H2A.Z-FB or non-ubiquitylated H2.Z-FB
Our previous work, as well as studies by others, has shown that epitope- or GFP-tagging of H2A.Z at the C-terminus does not interfere with its incorporation into nucleosomes (27, 32). The BirA enzyme added to the C-terminus of H2A.Z is similar to the molecular weight of GFP, suggesting that the BirA tag will also not interfere with H2A.Z-FB’s incorporation into chromatin. To confirm this, we harvested micrococcal nuclease-digested chromatin from H2A.Z-FB + Avi-ub-transfected cells and performed mono-nucleosome immunoprecipitation using streptavidin (SA)- or Flag antibody-couple beads (schematically depicted in Fig 2A). As shown in the Coomassie-stained gel (Fig 2B), both the SA- or Flag-immunoprecipitated samples contain stoichiometric amounts of core histones (i.e. roughly equal amounts of H2B, H3 and H4) that co-purify with the immunoprecipitated H2A.Z-FB (lanes 6 and 7 of Fig 2B). Therefore, this indicates that both the non-ubiquitylated and ubiquitylated H2A.Z-FB are assembled into nucleosomes, and that intact nucleosomes are purified by the Flag- or SA-coupled beads. As an additional control, we also co-expressed the H2A.Z-FB with a K10R mutant form of Avi-ub, whereby the lysine within the Avi-tag sequence is mutated to the non-biotinylatable arginine. Mono-nucleosomes harvested from these transfected cells were subjected to immunoprecipitation by SA-beads, and as expected, no detectable amounts of core histones were co-purified. This result confirms that co-purification of the core histones (H2B, H3 and H4) with the Avi-ub-H2A.Z-FB is due to their assembly into nucleosomes, and not non-specific binding of histones to SA-beads.

(A) Cartoon diagram showing the expected types of H2A.Z-FB nucleosomes found in the H2A.Z-FB + Avi-Ub transfected cells and the distinct types of nucleosomes pulled down by SA or Flag antibody immunoprecipitation. (B) Coomassie stained gel of the input mono-nucleosome fraction and SA- or Flag antibody-immunoprecipitated nucleosomes. (C) Western blot analysis of the input and immunoprecipitated nucleosomes using Flag antibody and Avi-HRP. The different types of ubiquitylated and non-ubiquitylated H2A.Z-FB (illustrated on the right hand side and marked by single, double and triple asterisks) were resolved on long SDS polyacrylamide gels and detected by Flag antibody.
We next used Western blot analyses to characterize the SA- or Flag-immunoprecipitated mono-nucleosomes from cells expressing various combinations of H2A.Z-FB and Avi-ub (Fig 2C). By using higher percentage and longer polyacrylamide gels to better resolve the H2A.Z-FB, we detect three distinct bands with the Flag antibody (Fig. 2C); these correspond to H2A.Z-FB that are non-ubiquitylated (the fastest migrating band), ubiquitylated with endogenous ubiquitin (the middle band), and ubiquitylated with Avi-ub (the top band; see nucleosomes depicted by the drawings on the right side of the figure). Most importantly, when we normalized the immunoprecipitated nucleosomes based on the amount of H3 pulled down, we observe that the SA-coupled beads specifically immunoprecipitated the slowest migrating Avi-ub-H2A.Z-FB, whereas Flag-beads mostly immunoprecipitated the non-ubiquitylated H2A.Z-FB and a small amount of H2A.Z-FB mono-ubiquitylated with endogenous (non-Avi-tagged) ubiquitin. From cells expressing the H2A.Z-FB K3R3 mutant and Avi-ub, the Flag-IP only pulled down the non-ubiquitylated H2A.Z-FB as expected. Also, from cells expressing WT H2A.Z-FB and the K10R Avi-ub mutant, the SA-beads did not pull down any H2A.Z-FB, nor endogenous histones, demonstrating specificity of the SA-bead immunoprecipitation. Finally, we note that while Avi-HRP detected both the Avi-ubiqutiylated-H2A.Z-FB and Avi-ubiquitylated-endogenous H2A.Z/H2A in the input mono-nucleosomes, the SA-immunoprecipitation mainly pulled down the Avi-ub-H2A.Z, and only a small amount of the Avi-ub-endogenous H2A.Z/H2A. Together, these results demonstrate that our experimental system and SA-purification scheme highly enriches for ubiquitylated H2A.Z-containing nucleosomes, thus facilitating an in depth study of their interactions and role in chromatin regulation.
Ubiquitylated H2A.Z nucleosomes are enriched for transcriptional repressive histone modifications and depleted of activating histone modifications
Having established that distinct pools of H2A.Z-FB nucleosomes are pulled down by the SA- and Flag-IPs (depicted in Fig. 2A), we next examined the histone modification patterns on these nucleosomes (Fig. 3). We first normalized the samples by H3 levels (H3 Western blot panel of Fig. 3), and used the same amounts of histones/nucleosomes per IP sample for Western blot analyses. As seen before, the Flag antibody preferentially immunoprecipitated mostly non-ubiquitylated H2A.Z-FB (from the WT-H2A.Z-FB-expressing cells) or only non-ubiquitylated H2A.Z-FB (from the K3R3 mutant-expressing cells) whereas the SA-beads immunoprecipitated the Avi-ubiquitylated form of H2A.Z-FB (Flag panel of Fig. 3). Comparison of these two pools of nucleosomes showed similar levels of H3K9me2, H3K9me3 and H4K16ac. However, compared to the nucleosomes that contained mostly non-ubiquitylated H2A.Z-FB, nucleosomes containing Avi-ub-H2A.Z-FB are depleted of H3K4 methylation (most notably for H3K4me2 and H3K4me3), H3K27ac, as well as hyperacetylated H4, all which are general hallmarks for transcriptionally active chromatin. In contrast, the Avi-ub-H2A.Z nucleosomes are enriched for H3K27me3, which is a hallmark for polycomb-silenced facultative heterochromatin. Taken altogether, these characterizations reveal that ubiquitylated H2A.Z-nucleosomes are depleted of histone PTM hallmarks associated with transcriptional activation, but are enriched for H3K27me3 that is associated with the polycomb-silencing pathway.
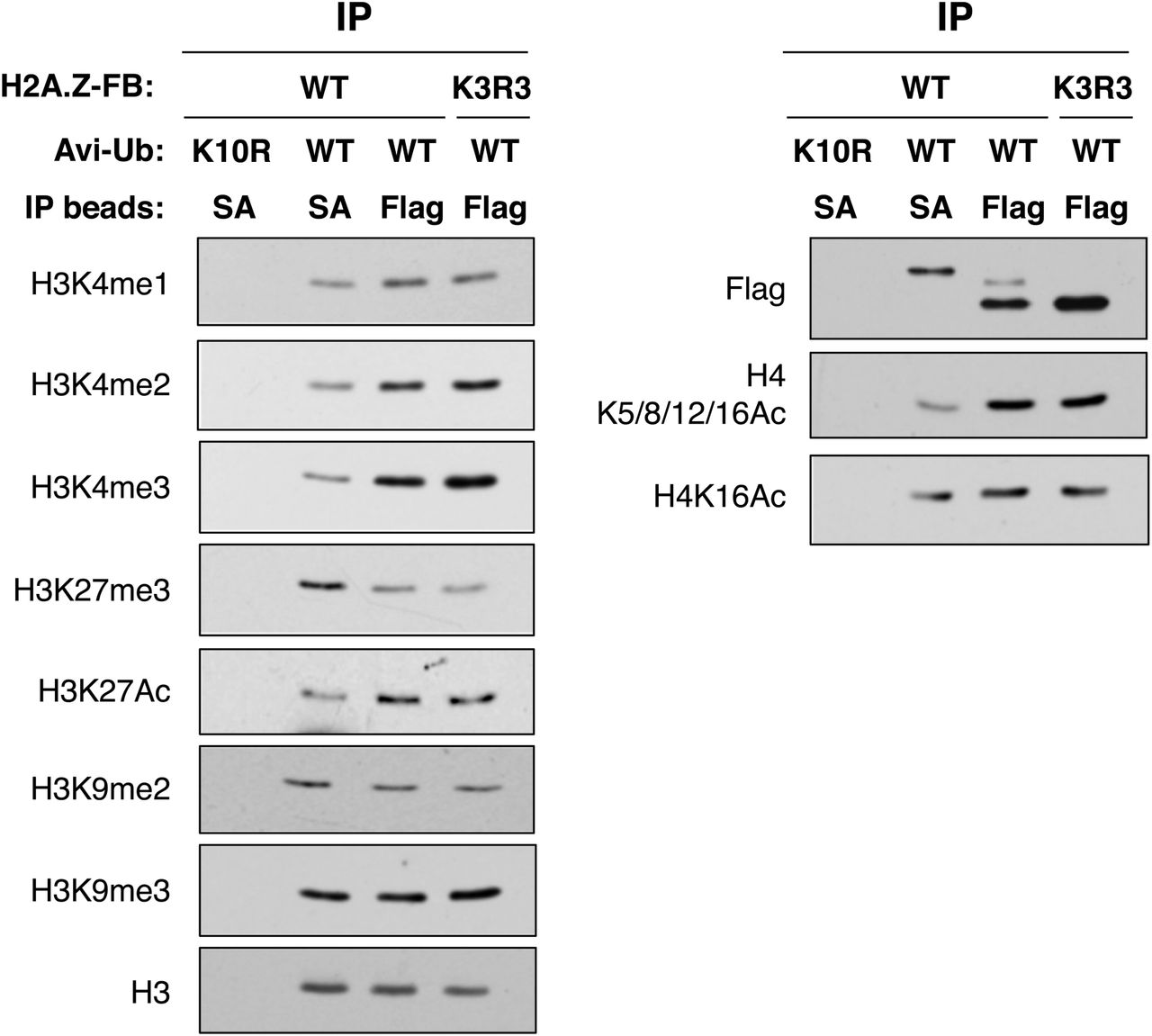
293T cells were transfected with various H2A.Z-FB and Avi-Ub constructs as shown, and the levels of different histone modifications on the SA- or Flag antibody-immunoprecipitated nucleosomes were examined by Western blotting. Equal amounts of the purified nucleosomes were loaded on each lane as indicated by the comparable H3 levels in each lane. The K10R Avi-Ub is the non-biotinylatable Avi-Ub control to show the biotinylation-dependent pulldown of nucleosomes by SA-immunoprecipitation.
Genomic analyses of Avi-ub-H2A.Z-FB-containing chromatin
The nucleosome IP method we used to purify H2A.Z-FB nucleosomes affords the analysis of the distribution of this modified histone variant on a genome-wide scale. Accordingly, using conditions equivalent to native ChIP, we next extracted DNA from the SA- and Flag-immunoprecipitated nucleosomes (from biological replicate preparations) and performed high-throughput sequencing. In parallel, with this ChIP-seq analysis of Avi-ub-H2A.Z-FB- and total H2A.Z-FB-containing chromatin, we performed ChIP-Seq analysis using an antibody against H3 as control for histone density. Overall, we identified ∼ 144,000 peaks in both the SA- and Flag-IP’d samples and found significant (∼ 60%) overlap between them. This is not surprising since the Flag-IP should pull down both non-ubiquitylated and ubiquitylated H2A.Z-FB, whereas the SA-IP specifically enriches the Avi-ub-H2A.Z-FB. H2A.Z has been reported to be enriched at the +1, and to a lesser extent at the −1, nucleosomes flanking transcription start sites (TSSs) of many genes. When the SA- and Flag-IP peaks were mapped to the gene borders (transcription start sites, TSSs, and transcription termination sites, TTSs) we indeed observe a strong peak at +1 nucleosome position and a weaker peak at the −1 nucleosome position for both SA- and Flag-IPs (Fig. 4A), indicating that H2A.Z-FB is correctly targeted to endogenous H2A.Z sites within the genome. Interestingly, when the SA- and Flag-IP peaks were separately mapped to genes with no-, low-, or high-expression (based on analysis of polyA+ RNA-seq data from untransfected 293T cells), there is a comparable degree of enrichment of SA-IP peaks at the TSSs of these three groups of genes, whereas there are significantly higher Flag-IP peaks at low- or high-expression genes compared to non-expressed genes (Fig. 4B). This difference is apparent as a ‘double-dip’ around the TSS when the SA-IP signal is normalized to the Flag-IP signal (3rd panel of Fig. 4B). These findings suggest that while roughly equal amounts of Avi-ub-H2A.Z-FB are found at the +1 nucleosome position of genes irrespective of their expression status, significantly more total H2A.Z-FB (most likely corresponding to non-ubiquitylated H2A.Z-FB) is enriched at the +1 and −1 nucleosome positions of active compared to inactive genes. It is of interest to note that the Flag-IP peaks flanking the TSSs are almost identical for both low- and high-expressing genes, suggesting that while active promoters in general have higher amounts of H2A.Z compared to non-active promoters, the amount of H2A.Z at the +1/-1 nucleosomes does not correlate with gene expression levels (low vs. high expression). Lastly, we also note that higher amounts of both SA- and Flag-IP’d H2A.Z-FB were found at the 3’ end of non-expressing genes compared to low- or high-expression genes, which suggests that there are generally higher amounts of total H2A.Z at gene bodies of non-expressing genes compared to active genes (compare the grey line to the red and yellow lines in Fig 4B), which is consistent with previous H2A.Z ChIP-microarray studies in yeast.

Total genomic DNA extracted from the SA- or Flag antibody-immunoprecipitated nucleosomes were sent for Illumina sequencing and analyzed by bioinformatics tools. (A) Total SA- or Flag-antibody IP peaks were compiled and mapped relative to transcription start sites (TSSs) and transcription termination sites (TTSs) of all genes. (B) SA- and Flag antibody-IP peaks were mapped relative to the TSSs and TTSs of no-expression (grey line), low-expression (red line) or high-expression (yellow line) genes. (C) SA- and Flag antibody-IP peaks were compared relative to one another and statistically determined as either Avi-enriched, Avi-depleted, or no enrichment. These different categories of peaks were mapped to promoters and gene bodies and then stratified based on the RNA-seq determined expression levels of the genes. (D) Expression levels of Avi-enriched (orange), Avi-depleted (red) or no enrichment promoters were analyzed across 55 different tissue or cell types. The fraction of tissues/cells that show greater than the average expression of each gene were plotted and colour coded based on Avi-enrichment, -depletion or no enrichment. (E) Avi-enriched or Avi-depleted peaks were categorized according to ENCODE-defined promoters, enhancers, exons, introns, and intergenic regions. The real estate in each category occupied by Avi-enriched or Avi-depleted peaks were expressed as percent coverage (the percent of total real estate occupied by the respective peak types).
To further analyze the nucleosome IP-seq data, we first merged all peaks detected in the SA- or Flag-IPs, resulting in ∼ 198,000 peaks, and determined the relative enrichment or depletion of the SA-IP signal at each peak by comparing the read density of the SA-IP to the Flag-IP using statistical tests. Peaks where the SA-IP signal is significantly enriched are identified as “Avi-enriched” (12,403 peaks), whereas those with significantly depleted SA-IP signal are identified as “Avi-depleted” (18,881 peaks). Lastly, peaks that do not reach statistical significance for either enrichment or depletion are classified as “no-enrichment”. These three categories roughly correspond to IP-seq peaks enriched for Avi-ubiquitylated H2A.Z-FB, non-ubiquitylated H2A.Z-FB, or roughly equivalent mix of both. By examining the Avi-enriched vs. Avi-depleted peaks at identified gene promoters and stratifying them based on expression levels, we found that > 70% of the Avi-enriched promoters are transcriptionally silent (defined by < 5 RPKM based on RNA-seq data) whereas < 30% of Avi-depleted promoters are transcriptionally silent (Fig. 4C; odds ratio = 7.2, p-value < 2.2e-16, Fisher’s exact test). The distribution of no enrichment peaks across promoters of different expression levels is in between that seen for Avi-enriched and Avi-depleted peaks, which likely reflects the mixture of both ubiquitylated and non-ubiquitylated H2A.Z at these promoters. Lastly, comparison of the Avi-enriched vs. Avi-depleted peaks at gene bodies also showed a higher fraction of Avi-enriched genes at inactive genes; however, the difference is not as striking as seen at promoters (odds ratio = 3.2, p-value < 2.2e-16, Fisher’s exact test). Altogether, these analyses show that ubiquitylated and non-ubiquitylated H2A.Z are respectively enriched on transcriptionally inactive and transcriptionally active promoters, which mirrors the distinct histone PTMs associated with transcriptional repression and activation found on the respective Avi-ub- and non-ubiquitylated-H2A.Z-FB nucleosomes (Fig. 3).
To further characterize the Avi-enriched vs. Avi-depleted promoters, we examined their expression profiles across 55 different tissues/cell types, and expressed the data as the fraction of Avi-enriched or Avi-depleted promoters/genes that is ‘over-expressed’ (i.e. expression > average expression) in different tissues or cell types. As shown in Fig. 4D, we found that Avi-enriched or Avi-depleted promoters (i.e. those enriched for ubiquitylated or non-ubiquitylated H2A.Z respectively at their promoters) have contrasting expression trends. For example, Avi-enriched promoters are not frequently over-expressed in the tissues/cell types tested whereas Avi-depleted promoters (i.e. enriched for non-ubiquitylated H2A.Z) are over-expressed in higher percentages of tissues/cell types tested. These profiles suggest that Avi-enriched genes have a more restricted expression pattern in different cell types, which is consistent with a negative regulatory role of H2A.Z ubiquitylation on gene expression.
Lastly, we compared the distribution of Avi-enriched vs. Avi-depleted peaks across different genomic regions such as promoters, enhancers, exons, introns and intergenic regions. More specifically, we examined the percentage occupancy (i.e. coverage) of these genomic elements by the Avi-enriched and Avi-depleted peaks to determine the relative densities of these peaks within each of the elements (Fig. 4E). Interestingly, Avi-depleted peaks have the highest percentage coverage at promoters and next at enhancers, whereas Avi-enriched peaks have the highest percentage coverage in exons, then at promoters, but least of all at enhancers. The higher occupancy of H2A.Zub (compared to non-ubiquitylated H2A.Z) at exons is intriguing and suggests that it may also have a non-transcription initiation function. The data also found significant occupancy of non-ubiquitylated H2A.Z at predicted active enhancers, but a depletion of H2A.Zub at these elements. Insofar as active enhancers are defined by high levels of H3K4me1 and H3K27ac, this observation is consistent with and possibly explained by our biochemical analyses showing H2A.Zub nucleosomes have low levels of both of these modifications whereas non-ubiquitylated H2A.Z nucleosomes have high levels of these modifications (Fig 3).
Validation and characterization of Avi-enriched and Avi-depleted promoters by gene-specific ChIP
Having identified distinct sets of Avi-enriched and Avi-depleted genes, we chose a number of representative promoters (12 promoters from each group) for validation by gene-specific ChIP. We first performed ChIPs using SA- or Flag-coupled beads, and used qPCR with primers corresponding to regions flanking the TSSs of the 24 tested genes for quantitative analyses. We normalized the SA-IP signals over the Flag-IP signals and plotted the individual gene data as shown in Fig 5A, and we also combined the ChIP data of the two groups of genes in a box plot comparison with statistical analyses (Fig. 5B). In addition, we also examined the H3K4me3, H3K27me3, and total H2A.Z levels at the same promoters by ChIP, and normalized the data to the total H3 ChIP signal (normalizing for nucleosome density) (Figs. 5A and B). By combining the ChIP data from the individual genes in the Avi-enriched or Avi-depleted promoters, we were able to see statistically relevant trends more clearly (Fig. 5B). First, as expected, the Avi-signal (SA-IP normalized to Flag-IP signal) is significantly higher for the Avi-enriched group compared to the Avi-depleted group, indicating an enrichment of Avi-ub-H2A.Z-FB at those promoters (Fig. 5B). In contrast, there was no evidence for differential occupancy of total H2A.Z at the promoters of both groups of genes, and in fact, the H2A.Z ChIP showed an opposite tendency, indicating that the differential Avi-signal seen between the two groups is not simply due to different amounts of H2A.Z at those promoters. Also, consistent with the histone PTM trends observed by our nucleosome-IP-Western blots, the Avi-enriched promoters are hyper-methylated for H3K27 but hypo-methylated for H3K4, whereas the reverse trend is seen at the Avi-depleted promoters. From these data, the most striking feature of the Avi-enriched promoters is the consistent hypo-methylation of H3K4. This observation is not only consistent with our previous biochemical analysis (Fig. 3) and the connections between H2A.Z ubiquitylation and transcriptional silencing, but further suggests a possible incompatibility or mechanistic antagonism between H2A.Z ubiquitylation and H3K4 methylation.
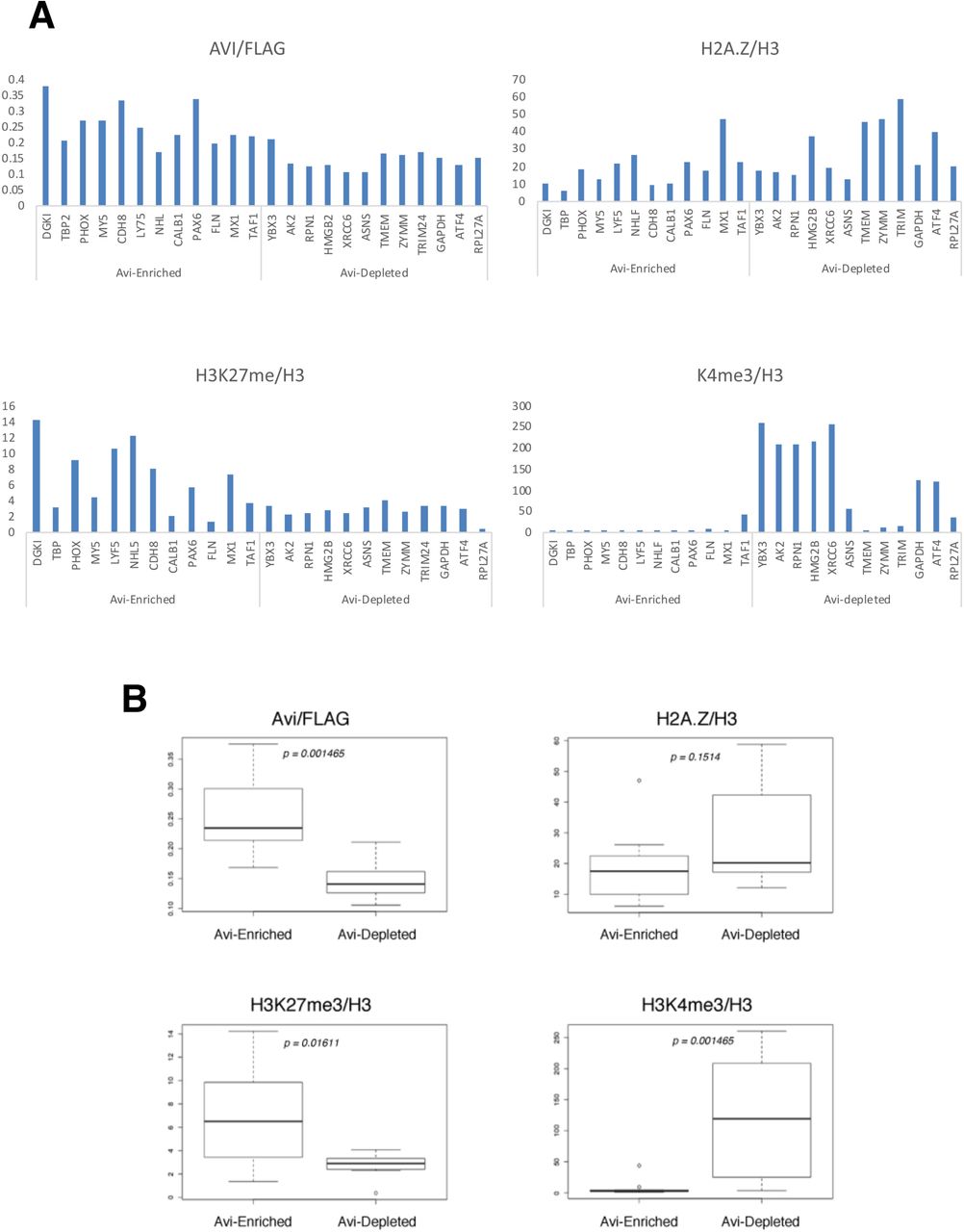
(A) 12 representative gene promoters from the ChIP-seq identified list of Avi-enriched or Avi-depleted promoters were analyzed by ChIP-qPCR assays. The SA-ChIP signal for each gene was normalized to the Flag-ChIP signal, whereas the H2A.Z, H3K27me3 and H3K4me3 ChIP signals were normalized to the total H3 ChIP signal to normalize for the nucleosome densities at the tested promoters. (B) The ChIP data from the individual groups of 12 genes were combined and presented as box plots. The median of each box plot was calculated and marked for statistical comparisons.
Identification of H2A.Zub-nucleosome co-purifying proteins
We next asked whether there are also chromatin binding proteins that differentially associate with ubiquitylated- and non-ubiquitylated H2A.Z-nucleosomes. Accordingly, we co-transfected various combinations of WT or mutant H2A.Z-FB and Avi-ub, harvested mono-nucleosomes from those cells, and performed nucleosome immunoprecipitation using SA-vs. Flag-coupled beads as done in 31. We then normalized the amount of nucleosomes immunoprecipitated and loaded per lane by H3 density and blotted for various chromatin interacting proteins to examine their binding patterns (Fig. 6A). We previously identified Brd2 as an H2A.Z-nucleosome binding protein and found that it is recruited to androgen receptor-regulated genes upon hormone induction in an H2A.Z-dependent manner. Consistent with Brd2 having a role in transcriptional activation, we found that it only co-purifies with the Flag-immunoprecipitated (i.e. H2A.Z-FB-containing) nucleosomes, but not with the SA-immunoprecipitated (i.e. Avi-ub-H2A.Z-FB-containing) nucleosomes. In contrast, menin, a component of the MLL (H3K4 methyltransferase) complex, is found at similar levels in both SA- and Flag-IPs, with a slight increase in level in the SA-IP sample. Given the apparent anti-correlation between H3K4 methylation and H2A.Z ubiquitylation, we next tested whether LSD1, an H3K4me2 de-methylase, specifically co-purifies with the Avi-ub-H2A.Z-FB nucleosomes. Strikingly, both LSD1 as well as DNMT3L, an interacting partner of the de novo DNA methyltransferases DNMT3a/b, both preferentially co-purified with Avi-ub-H2A.Z-FB nucleosomes. The preferential enrichment and recruitment of these proteins may facilitate H3K4 de-methylation and DNA methylation of H2A.Zub-nucleosomes and contribute to transcriptional silencing of H2A.Zub-targeted genes.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Mono-nucleosomes from 293T cells expressing different types of H2A.Z-FB and Avi-Ub were harvested and immunoprecipitated with SA-of Flag antibody-coupled beads. The relative amounts of different chromatin binding proteins co-purifying with normalized amounts of the SA- or Flag antibody-purified nucleosomes were determined by Western blot analyses. (B) Mono-nucleosomes from 293T cells expressing either WT or non-ubiquitylatable (K3R3) H2A.Z-FB and Avi-Ub were immunoprecipitated with either SA- or Flag antibody beads and the relative amounts of SMC1 and CTCF co-purifying with the different immunoprecipitated nucleosomes were determined by Western blotting.
Lastly, given our genomic analyses suggested that H2A.Zub is depleted at enhancer elements, we asked whether enhancer-associated proteins differentially associate with ubiquitylated vs. non-ubiquitylated H2A.Z. To this end, recent studies have found that the cohesion complex and CTCF may have roles in mediating enhancer-promoter interactions and; therefore, we tested whether the cohesion components Rad21 and SMC1 or CTCF differentially associate with the ubiquitylated/non-ubiquitylated H2AZ-FB-containing nucleosomes. Given that cohesion and CTCF are reported to facilitate enhancer-promoter interactions, we initially hypothesized that these factors may preferentially associate with nucleosomes containing the non-ubiquitylated form of H2A.Z-FB. However, to our surprise, we found that Rad21, SMC1, and CTCF were all preferentially recovered in the SA-immunoprecipitates, but minimally in the Flag-immunoprecipitates. This suggests that these chromatin binding proteins preferentially associate with nucleosomes containing the ubiquitylated form of H2A.Z (Fig. 6A). To confirm that the co-purification was dependent on ubiquitylated H2A.Z, we performed SA-immunoprecipitation using nucleosomes harvested from cells transfected with the non-ubiquitylatable K3R3 H2A.Z-FB and Avi-ub. As shown in Fig. 6B, significantly less SMC1 and CTCF were recovered in the SA-immunoprecipitates when the H2A.Z ubiquitylation sites were mutated on the H2A.Z-FB. The residual amounts of SMC1 and CTCF immunoprecipitated with the K3R3 H2A.Z-FB nucleosomes are likely due to their interaction with the biotinylated endogenous H2A.Zub (the lower band detected in the Avi-HRP). This result confirms that cohesion components and CTCF preferentially interact with Avi-ub-H2A.Z-FB nucleosomes. Although the functional significance of these interactions are not yet fully understood, these findings raise intriguing functional and mechanistic possibilities related to H2A.Z ubiquitylation.
Discussion
In this study, we developed an experimental system to specifically purify ubiquitylated H2A.Z-containing chromatin for biochemical and genomic analyses. This method involves fusion of the BirA biotin ligase to the C-terminus of H2A.Z and pairing its expression with Avi-tagged ubiquitin. Consistent with other studies utilizing epitope-tagged histones (e.g. 37), we found that fusion of BirA to H2A.Z does not affect its assembly into nucleosomes (Fig. 1C). Moreover, ChIP-seq analyses showed enrichment of H2A.Z-FB at the +1 nucleosome (and to a lesser extent at the −1 nucleosome) flanking TSSs (Fig. 4A and B), which is same as the known pattern for endogenous H2A.Z, and thus indicates that BirA tagging of H2A.Z does not affect its targeting or deposition into the mammalian genome. With this experimental system, we discovered that ubiquitylated and non-ubiquitylated H2A.Z nucleosomes have differential patterns of histone PTMs. Specifically, the former are enriched for transcriptional repression-associated PTMs such as H3K27me3, as well as hypo-acetylated H2A.Z and H4, whereas the latter are enriched for transcriptional activation marks such as H3K4me1, me2, me3, H3K27Ac, and hyper-acetylated H4 (Fig. 3). These findings thus provide evidence that H2A.Z ubiquitylation co-exists with and possibly co-operates with other histone PTMs to mediate transcriptional silencing on a global scale.
Our results provide new insight into the inter-connections and potential cross-talk among histone modifications within H2A.Z-containing nucleosomes. In previous work, we determined that H2A.Z ubiquitylation is mediated by the RING1b E3 ligase of the PRC1 complex (27), and as such, the coupling of H2A.Z ubiquitylation and H3K27 methylation is consistent with the interplay between PRC2-meditated H3K27 methylation and PRC1 recruitment (38). Previous work also showed an antagonistic switch between H3K27 methylation and acetylation such that loss of H3K27 methylation can lead to increases in the H3K27ac levels (39). The reciprocal patterns of H3K27 methylation and acetylation on ubiquitylated and non-ubiquitylated H2A.Z nucleosomes are consistent with that finding, and further suggest that there may also be coupling of H2A.Z de-ubiquitylation and H3K27 acetylation on the same nucleosome. Lastly, while we have previously shown that H2A.Z-containing nucleosomes have higher levels of H3K4 methylation than H2A-containing nucleosomes (27), we now find that ubiquitylated H2A.Z nucleosomes have lower H3K4me1/2/3 levels compared to those with non-ubiquitylated H2A.Z, suggesting that the connection between H2A.Z and H3K4 methylation may be disrupted or reversed by its ubiquitylation.
ChIP-seq analyses of the SA- and Flag-purified nucleosomes show that the general distributions of ubiquitylated and total H2A.Z across the genome are largely similar. As mentioned above, ubiquitylated and non-ubiquitylated H2A.Z are enriched at the +1 and +1 nucleosomes flanking TSSs (Fig. 4A and B), and our data also show that both types of H2A.Z are more enriched at the gene bodies or 3’ ends of transcriptionally inactive genes compared to active genes (Fig. 4B). This latter observation is consistent with previous ChIP-microarray studies showing accumulation of H2A.Z at the gene bodies of non-expressing genes in yeast, and with a recent report showing increased H2A.Z levels at gene bodies upon transcription inhibition by α-amanitin (40, 41). Our finding is further consistent with the hypothesis that H2A.Z is actively removed from gene bodies by RNAPII during transcriptional elongation and, therefore, the accumulation of H2A.Z nucleosomes at gene bodies of transcriptionally inactive genes is likely due to the lack of transcription-dependent eviction (40). The ChIP-seq analyses in the present study also yielded unexpected but highly informative findings. Insofar as only a small fraction of total H2A.Z in 293T cells is mono-ubiquitylated, it is as yet unknown whether this subpopulation of H2A.Z is restricted to a smaller subset of genes (presumably transcriptionally inactive genes) compared to all genes associated with H2A.Z. Our data indicate that both SA- and Flag-IP peaks co-occupy many genomic loci, suggesting extensive intermingling of both ubiquitylated and non-ubiquitylated H2A.Z throughout the genome. Moreover, when we mapped SA- and Flag-IP peaks across all genes relative to gene features such as TSSs (Fig 4B)., we observe that ubiquitylated H2A.Z (SA-IP peaks) are in similar abundance at the TSSs of both transcriptionally active or inactive genes, whereas total H2A.Z (Flag-IP peaks) is significantly more abundant at the promoters of transcriptionally active genes (Fig 4B). This suggests that, on average, active and inactive genes do not differ in the amount of H2A.Zub flanking their TSSs, but active gene promoters have higher levels of total (presumably mostly non-ubiquitylated) H2A.Z. The enrichment of non-ubiquitylated H2A.Z at active genes fits well with the known association between H2A.Z and transcriptional activity, and we also note that the similar levels of H2A.Z found at the TSSs of both low- and high-expressing genes is consistent with the model that H2A.Z functions to poise genes for transcriptional activation, but may not directly affect the rate of transcription. Altogether, our ChIP-seq data suggest that the transcriptional activity of genes does not correlate with the absolute presence or absence of either non-ubiquitylated or ubiquitylated H2A.Z, but is instead determined by the relative ratios of these different modified forms of H2A.Z at different promoters.
In support of this idea, our statistical identification of Avi-enriched vs. Avi-depleted ChIP-Seq peaks reveal clear correlations between Avi-enrichment (i.e. enriched for H2A.Zub) with gene-silencing, and Avi-depletion (i.e. enriched for non-ubiquitylated H2A.Z) with gene activation (Figs. 4C and D). These correlations are most striking at the promoter regions of genes where > 70% of Avi-enriched promoters are transcriptionally inactive whereas ∼ 70% of Avi-enriched promoters have some transcriptional activities (ranging from low to high expression; Fig. 4C). Gene-specific ChIPs of the promoter regions of representative Avi-enriched- and Avi-depleted-genes also support the correlations between H2A.Zub enrichment/depletion and transcriptional activities. Most strikingly, Avi-enriched promoters are hypo-methylated at H3K4me3 and hypermethylated at H3K27me3 whereas the reverse trend is seen for the Avi-depleted promoters (Fig. 5B). These trends exactly mirror the biochemical analyses showing high levels of H3K27me3 and low levels of H3K4me1/2/3 on H2A.Zub-nucleosomes and the converse low levels of H3K27me3 and high levels of H3K4me1/2/3 on nucleosomes enriched for non-ubiquitylated H2A.Z. Therefore, our study provides corroborating biochemical and genomics data that show reciprocal correlations between H2A.Z ubiquitylation and H3K4-/H3K27-methylation levels, and raise the possibility of functional crosstalks whereby potential antagonism or co-operation of these histone PTMs collaboratively lead to transcriptional silencing.
Further comparison of the genomic distribution of Avi-enriched and Avi-depleted peaks at different functional domains (such as promoters and enhancers etc.) shows interesting differences in their occupancies within promoters, enhancers and exons (Fig. 4E). For example, Avi-depleted peaks cover about 3 times the amount of territories within promoters than Avi-enriched peaks, whereas Avi-enriched peaks occupy about twice the amount of exon territories than Avi-depleted peaks. The larger coverage of promoter sequences by non-ubiquitylated H2A.Z (Avi-depleted peaks) compared to ubiquitylated H2A.Z (Avi-enriched peaks) is consistent with the larger number of Avi-depleted promoters identified (Fig. 4D), and suggests that more genes/promoters are regulated by non-ubiquitylated H2A.Z than those silenced by ubiquitylated H2A.Z. The larger occupancy of H2A.Zub-enriched regions in exons is intriguing and suggests H2A.Z ubiquitylation may have additional non-transcription initiation-related functions.
Lastly, the most striking difference in the genomic distribution of Avi-enriched and Avi-depleted peaks is at enhancers. In this analysis, active enhancers are defined by co-enrichment of H3K4me1 and H3K27ac, and we see about 10 times more coverage of active enhancer regions by Avi-depleted peaks than by Avi-enriched peaks. Given our biochemical analyses showed that H2A.Zub-nucleosomes are depleted of H3K4me1/2/3 and H3K27ac, this could explain the lack of H2A.Zub enrichment at enhancers. H2A.Z has recently been shown to be important for transcription of enhancer RNAs (eRNAs) and for enhancer functions (26), so our data suggest that it is the non-ubiquitylated form of H2A.Z that facilitates eRNA transcription and enhancer functions. More interestingly, given the connections between H2A.Z ubiquitylation and transcriptional silencing, our data also raise the possibility that H2A.Zub may be actively excluded or is functionally antagonistic to eRNA transcription and enhancer activities.
We and others have previously found that H2A.Z-containing nucleosomes bind a number of chromatin binding proteins that mediate downstream functions (31, 33, 32). For example, we found that the transcriptional regulator Brd2 binds H2A.Z nucleosomes through combinatorial interactions with H2A.Z and acetylated H4 (31). More recently, Hake and colleagues also found that PWWP2A binds H2A.Z nucleosomes through multivalent interactions (42). With the system we developed, we examined the binding of various chromatin factors to ubiquitylated and non-ubiquitylated H2A.Z. Consistent with Brd2’s role in transcriptional activation, we found that Brd2 co-purifies with non-ubiquitylated but not with ubiquitylated H2A.Z nucleosomes (Fig. 6A). This result also complements a previous report that suggested H2A.Z ubiquitylation antagonizes Brd2 binding in ES cells (43). In contrast to Brd2, we found several transcription silencing factors, such as LSD1 and DNMT3L, that preferentially interact with ubiquitylated H2A.Z nucleosomes. LSD1, also known as KDM1A, is the first histone de-methylase identified that targets H3K4me2/me1, and functions as a transcriptional co-repressor in specific contexts (44). Therefore, physical recruitment of LSD1 or other H3K4 de-methylases to ubiquitylated H2A.Z nucleosomes may be a direct mechanism leading to the striking anti-correlation between H3K4 methylation and H2A.Z ubiquitylation seen in our biochemical and ChIP analyses (Figs. 3 and 5B). LSD1 is part of two complexes, CoREST and NuRD (45, 46), that both contain histone deacetylases, so the low levels of both H3K4 methylation and H4 acetylation on H2A.Zub nucleosomes could also reflect recruitment of multiple histone modifying activities through these complexes. Finally, LSD1 has also been shown to interact with DNMT1 and DNMT3B, and is important for maintaining global DNA methylation (47, 48). The co-purification of LSD1 and DNMT3L with H2A.Zub nucleosomes suggests that these nucleosomes may also have high levels of DNA methylation. Our original characterization of H2A.Zub found that it was enriched on the inactive X chromosomes in female mammalian cells (27), which are stably silenced by multiple epigenetic mechanisms including H3K27 methylation, loss of H3K4 methylation, H3/H4 de-acetylation, H2A ubiquitylation and DNA methylation (49). Therefore, the chromatin proteins we found co-purifying with H2A.Zub are consistent with the convergence of multiple histone/chromatin-modifying mechanisms on H2A.Z-containing chromatin to mediate stable transcriptional silencing.
Our nucleosome-binding assay also found that CTCF and cohesin components preferentially associate with ubiquitylated H2A.Z nucleosomes. Both CTCF and the cohesin complex are chromosomal architectural proteins that mediate long-range chromatin/chromosome interactions, and they are also important for establishing the boundaries of Topologically Associating Domains (TADs) that define the functional domains within the genome where permissive and high frequencies of chromatin interactions occur (50). Therefore, the co-purification with these proteins suggests that ubiquitylated H2A.Z’s function may also involve higher order chromatin folding/topology. Earlier ChIP-seq studies have found enrichment of H2A.Z at CTCF and Rad21 (a cohesin subunit) binding sites within the human genome (19, 41), and thus our nucleosome-IP results provide direct physical interaction data to confirm and support these previous findings. At present, the functional significance of these interactions are not known. CTCF and cohesins perform multiple cellular functions, including enhancer-blocking/ insulator functions for CTCF, sister cohesion for cohesins, and they are both involved in transcriptional regulation as well (for review, see 51). Given their interactions are specific to ubiquitylated H2A.Z, it is possible that these proteins act together in the context of transcriptional silencing, either directly at the gene level, or through the assembly or maintenance of repressive domains within the genome. CTCF was first identified as a DNA binding protein that binds and represses transcription of the chicken c-myc gene (52). CTCF has also been found to recruit HDACs to silence transcription in transfection-based reporter assays (53), so it is possible that CTCF engage multiple histone modifying mechanisms to silence target genes. Cohesin is often associated with active enhancers and promoters, but there are also connections with gene silencing (54). Early work in S. cerevisae showed that cohesin is targeted to transcriptionally silenced chromatin (55). In multi-cellular eukaryotes, cohesin directly interacts with the PRC1 complex and has been proposed to target Polycomb-group proteins to active genes (14, 56). Therefore, cohesin may physically associate with ubiquitylated H2A.Z through its direct interaction with PRC1. The transcriptional regulatory functions of CTCF/cohesin are complex, involving multiple levels of direct and indirect mechanisms. Therefore, our discovery of their physical association with H2A.Z ubiquitylation raises new questions about their functional links to chromatin and nucleosomes.
Methods
Cell culture, transfection, plasmids, antibodies
HEK293T cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum (DMEM; Wisent). All transfections were carried out using polyethylenimine (PEI; Polysciences). All expression constructs used were based on the pcDNA 3.1 (+) (Invitrogen) backbone with the Flag-BirA cloned in-frame to the C-terminus of H2A.Z-1(hereafter termed H2A.Z-FB) or H2A.Z-K3R3 where all known sites of ubiquitylation on H2A.Z is mutated to arginines. In addition, the AviTag was cloned in-frame to the N-terminus of ubiquitin. To generate H2A.Z-FB-K3R3, K120/121/125 were mutated to R120/121/125. To generate a non-biotinylatable AviTag, K10 of the AviTag sequence (GLNDIFEAQKIEWHE) was converted to R10. In co-transfection experiments, the ratio of H2A.Z-Flag-BirA to AviTag-Ub plasmids was 3:1. Antibodies used in this study are listed in Table 1.
Mononucleosome affinity purification
Generation of mononucleosomes was performed as described previously (31). In brief, HEK293T cells were grown in 15 cm-diameter plates and were transfected with various constructs according to the experiments. Cells were trypsinized, counted, and washed in 1X PBS, 48 hrs following transfection. Cellular pellets were resuspended in buffer A (20mM HEPES, pH 7.5, 10mM KCl, 1.5mM MgCl2, 0.34M sucrose, 10% glycerol, 1mM dithiothreitol, 5mM sodium butyrate, 10mM NEM, and protease inhibitors), pelleted and then resuspended in buffer A containing 0.2% Triton X-100 and incubated on ice for 5 min. The nuclear suspension was centrifuged at 600 × g; nuclei were then washed once in buffer A, then resuspended in cutting buffer (15 mM NaCl, 60 mM KCl, 10 mM Tris pH 7.5, 5mM sodium butyrate, 10mM NEM, and protease inhibitors) plus 2mM CaCl2. Microccocal nuclease (MNase; Worthington) was added at a concentration of 10 units/1.0 × 107 cells then incubated at 37°C for 30 min. The reaction was stopped by the addition of 20mM EGTA (one twenty-fifth of the reaction volume) and immediate gentle mixing by inversion. The MNase-digested nuclei were centrifuged at 1300 × g. The resulting supernatant (S1) was saved and kept on ice. The digested nuclear pellet was subjected to hypotonic lysis by resuspension in TE buffer (10mM Tris-HCL, pH 8.0, 1mM EDTA). Samples were incubated on ice for 1 hr, with occasional mixing by pipette. The suspension was then centrifuged at 16 000 × g and the supernatant (S2) was transferred to a new tube. Salt was adjusted in S1 to 150mM NaCl by adding 2X buffer D (30 mM Tris pH 7.5, 225 mM NaCl, 3 mM MgCl2, 20% glycerol, 0.4% Triton-X 100, 5mM sodium butyrate, 10mM NEM, and protease inhibitors) drop-wise, with constant mixing on a vortex set to low speed. S2 was also titrated to 150mM NaCl by the drop-wise addition of 3X buffer E (60 mM HEPES pH 7.5, 450 mM NaCl, 4.5 mM MgCl2, 0.6 mM EGTA, 0.6 % Triton-X 100, 30% glycerol, 5mM sodium butyrate, 10mM NEM, and protease inhibitors). Insoluble material was pelleted via centrifugation. The clarified supernatants were combined and then used for affinity purification. Streptavidin-agarose (Sigma) or Flag M2-agarose beads (Sigma) were added and incubated overnight at 4°C on an end-over-end rotator. Beads were washed 4 times in 1X Buffer D, followed by 3 washes in 1X Buffer D containing 0.5% Triton X-100. Proteins were eluted from the beads by resuspension in 2X SDS sample buffer and boiled for 10min. For Western blot analysis, samples were run on SDS-polyacrylamide electrophoresis gels according to standard practices.
ChIP-Sequencing
Mononucleosome affinity purification was performed in duplicate as described above using streptavidin-agarose, Flag M2-agarose, or H3 antibody (pulled-down using protein G-coupled Dynabeads; Invitrogen) and eluted in buffer D containing 1% SDS by end-over-end rotation at room temperature for 2 × 10 min. DNA was treated with RNase A and proteinase K, purified by phenol-chloroform extraction, and then re-precipitated with ethanol and resuspended in water. DNA was converted to libraries using Illumina TruSeq ChIP-Seq and sequenced on an Illumina NextSeq500 in single end mode. Reads were converted to FASTQ format and mapped to the human hg19 genome using Bowtie. Duplicate reads were removed. Peaks were called using MACS2 using H3 as a control. Normalized peaks from duplicate experiments were then averaged and matched H3 samples were subtracted to yield fragments per million reads (FPM). The IDR procedure employed by ENCODE was then used to generate a merged peak set for both streptavidin and Flag with all raw peaks as input. At FDR < 0.05, 143k peaks (from 453k and 552k in streptavidin or Flag samples, respectively) were recovered in both. Enhancers were identified as co-localization of H3K4me1, H3K27ac, and DNaseI hypersensitivity sites (HSS) in either HeLa or several other cells and tissues.
ChIP-qPCR
Affinity-purified mononucleosomes were eluted in buffer D-1% SDS as described above. DNA was treated with RNase A and proteinase K, phenol-chloroform extracted from mononucleosomes, re-precipitated with ethanol, and then resuspended in water. Quantitative polymerase chain reactions (qPCR) were assembled in triplicate using PerfeCta SYBR Green SuperMix (Quanta Biosciences) and gene-specific primers. Reactions were run on an Optocon 2 thermocyler (Biorad). Primers used are listed in Table 2.
Author contributions
PC conceived the method and experimental designs, and MKN performed all the experimental work. UB performed all the ChIP-sequencing data analyses. PC wrote the manuscript with assistance and contributions from all co-authors.
Acknowledgements
We thank Keyur Advaryu and Kashif Aziz Khan for scientific discussions and general support. MKN was funded by a Canadian NSERC Postgraduate Scholarship, and this work was funded by in part by operating grants from the Canadian Cancer Society Research Institute and the Canadian Institutes of Health Research awarded to PC, as well as a CIHR Foundation grant awarded to BJB.
References




