

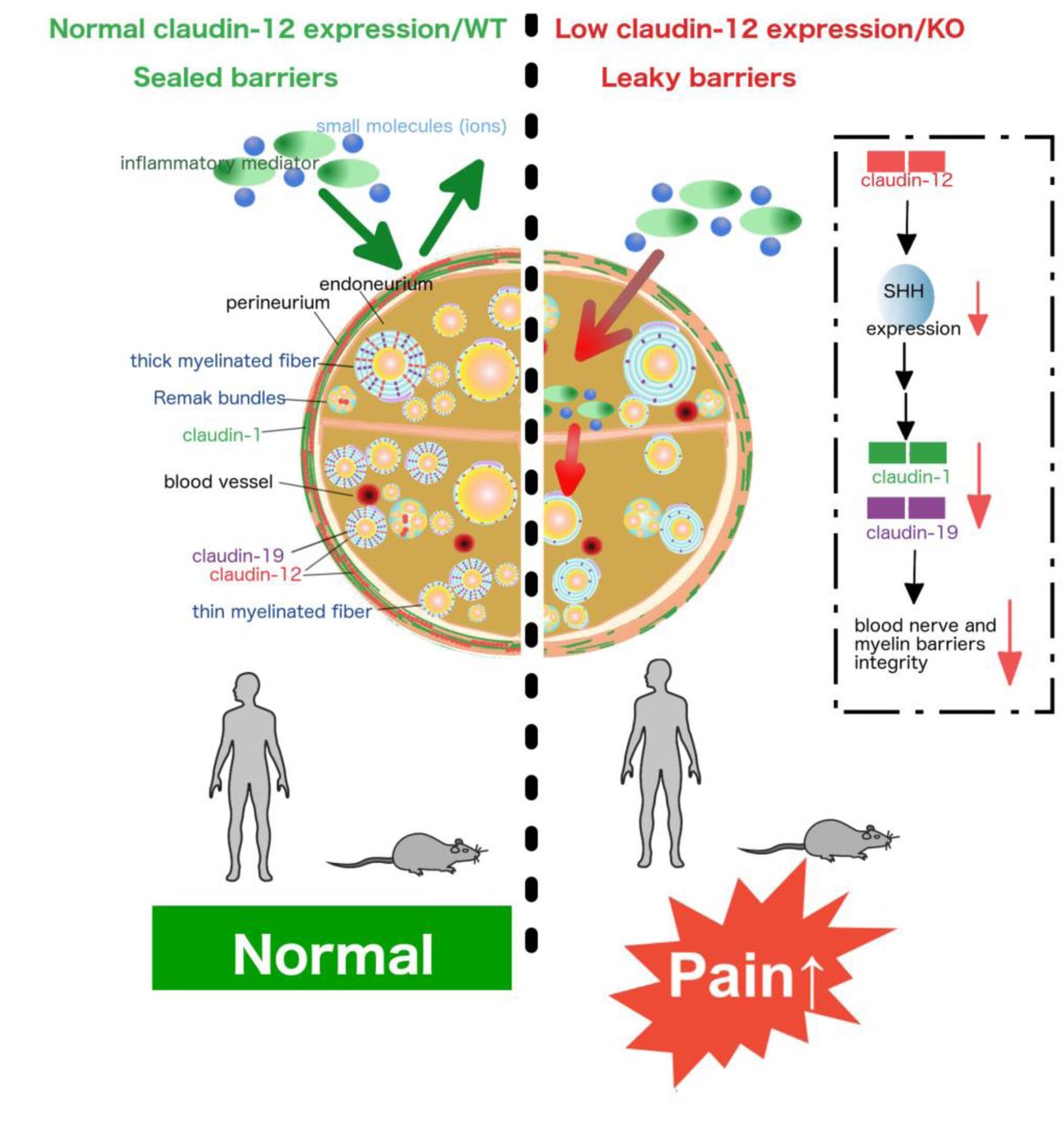
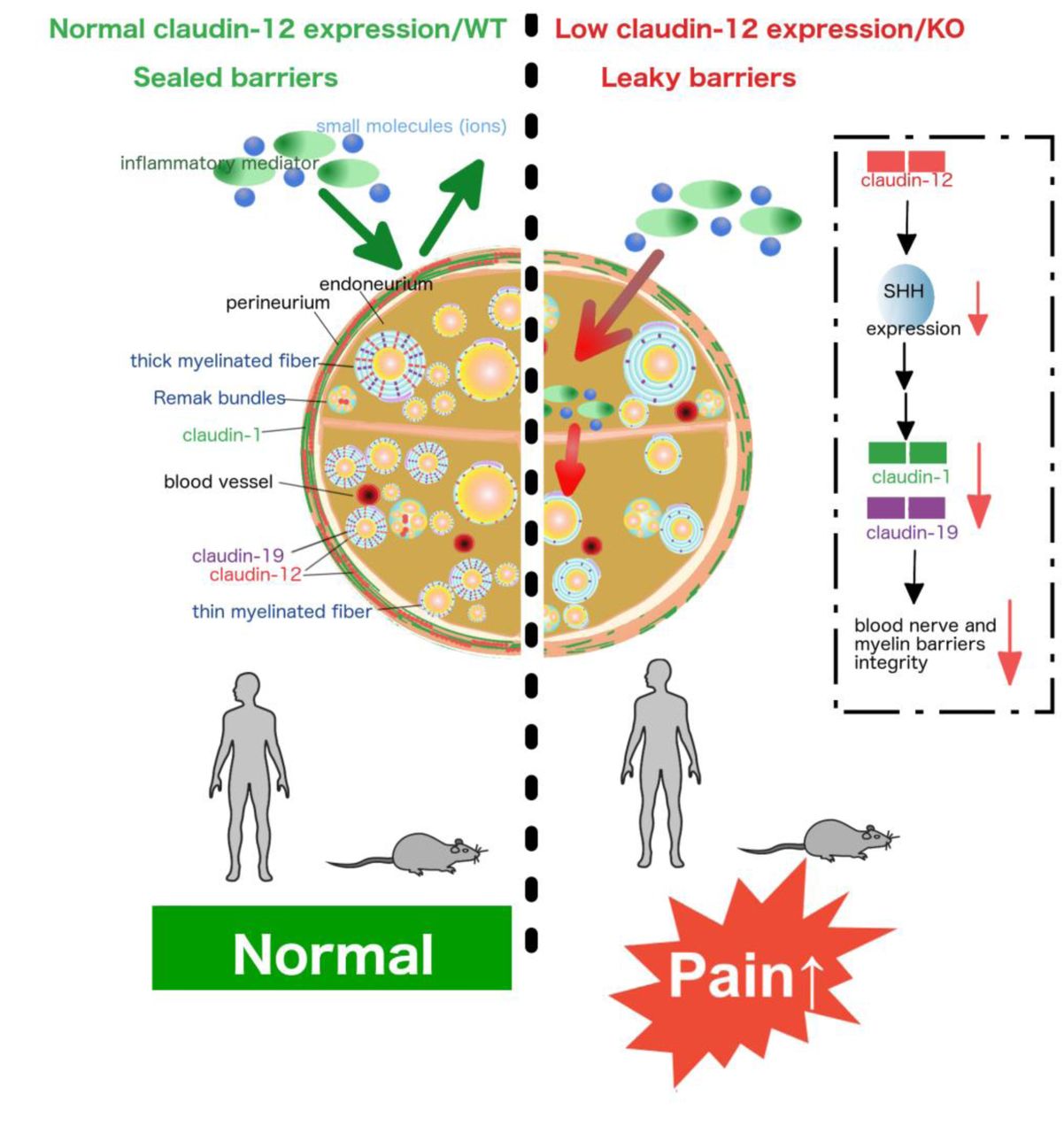
Abstract
Peripheral nerves and their axons are shielded by the blood-nerve and the myelin barrier. Understanding of how these barriers impact nociception is limited, particularly in neuropathy. Here, we identified a regulatory axis of the tight junction protein claudin-12 sex-dependently coordinating perineurial and myelin barrier integrity. Claudin-12 regulated selected tight junction proteins via sonic hedgehog (SHH) and thereby controlled pain sensation and mechanical nociception. In nerve biopsies, only patients with painful polyneuropathy lost claudin-12 in Schwann cells independently of the presence of inflammation or the extent of fiber loss. Global KO of Cldn12 selectively increased perineurial/myelin barrier leakage, damaged tight junction protein expression and morphology and amplified mechanical hypersensitivity in naïve and neuropathic male mice. Other barriers and neurological function remained intact. In vitro transfection studies documented claudin-12 plasma membrane localisation without interaction with other tight junction proteins or intrinsic sealing properties. Rather, claudin-12 had a regulatory tight junction protein function via the morphogen SHH in vivo as observed in Cldn12-KO and after local siRNA application. Female mice were protected against mechanical hypersensitivity, barrier opening, tight junction protein loss and SHH downregulation. Collectively, these studies reveal the critical role of claudin-12 maintaining the myelin barrier and highlight restoration of the claudin-12/SHH pathway as a potential target for painful neuropathy.
Brief summary Claudin-12 regulates perineurial and myelin barrier tightness via sonic hedgehog. Loss of claudin-12 sex-dependently results in mechanical hypersensitivity and pain in inflammatory and noninflammatoy polyneuropathy.
Introduction
Barriers are critical to shield delicate structures in the body. In the PNS, the blood nerve barrier (BNB) consists of the perineurium and endoneurial vessels. It is an important physiological fence which serves to maintain stable environments for axons, Schwann cells, and other endoneurial cells (1, 2). A second barrier in the PNS is the myelin barrier found at the paranode and the mesaxon of Schwann cells. BNB and myelin barrier are formed by several main protein families: tight junction proteins, adherence junction proteins, and cytoplasmic accessory proteins (3). Tight junction proteins are located in cell to cell contacts and tightly connect neighboring cells. They control the passage of molecules and maintain cell polarity. Among the tight junction protein family, claudins are major structural components which form tight junction backbones. Certain key claudins [e.g. claudin-1, claudin-5, claudin-19, occludin and zona occludens (ZO-1, TPJ1)] have been identified as relevant for nerve barriers (4, 5). Interference with them enhances drug delivery of e.g. hydrophilic analgesics to the nerve and thereby induces antinociception (6, 7).
Some neurodegenerative diseases as well as CNS and PNS injury are accompanied by downregulation of tight junction proteins and increased barrier permeability (8, 9). Tight junction protein expression influences nervous system function by controlling the entry of molecules (10). Claudin-1 protects the perineurial barrier (6, 11) and is downregulated after nerve injury (12). Claudin-5, the major tight junction protein in the blood brain barrier (BBB), serves as a neuroprotector in multiple sclerosis brain, but also in the PNS after injury (13). Expression of occludin as well as of ZO-1 is also low in the sciatic nerve (SN) after nerve injury (12–15). Much less examined is the myelin barrier in (painful) peripheral neuropathy. Myelin tight junction proteins such as claudin-19 are responsible for sensory and motor transmissions. Claudin19 is expressed in the perineurium (6) and the paranode (16) of peripheral nerves. A proportion of Cldn19-KO mice have an abnormal conduction velocity in the SN and motor function deficiency (17). Peripheral myelin protein (PMP22) also belongs to the group of tight junction proteins, it is necessary for Schwann cell myelination and dysregulated in inheritable neuropathies. Pmp22 deficiency in mice leads to impaired action potential propagation and myelin barrier breakdown with demyelination, associated with downregulation of Cldn19 and ZO-1/TPJ1 (16).
Claudin-12 is an atypical member of the claudin family because it is one of the few claudins that does not possess a PDZ binding motif. According to phylogenetic analyses, it appears to be only distantly related to all other claudins. It is expressed in the intestine, urinary bladder, BBB and BNB in the nervous system (2, 18–20). Not very much is known about its function, except that claudin-12 is upregulated in some types of cancer (21) and affects barrier integrity and paracellular Ca2+ absorption in intestinal epithelial cells (22). However, its role in the BNB, myelin barrier and nociception are completely unknown.
Polyneuropathy can occur e.g. due to metabolic, genetic, toxic or inflammatory causes. Chronic inflammatory demyelinating polyneuropathy (CIDP) is a heterogenous (auto)immune-mediated inflammatory disease. Patients suffer from weakness, numbness and tingling and, in a subgroup, from pain (23). Cause and pathogenesis of CIDP are only partly understood. In a subgroup of CIDP patients, autoantibodies against paranodal proteins are detected (24, 25). In nerve biopsies, destruction of myelin sheaths and higher expression of inflammatory mediators such as TNF-α and IL-10 are found. Thus, intravenous immunoglobulin therapy is often used (26). Inflammatory mediators can alter barrier function and integrity thereby facilitating their own paracellular passage across barriers further aggravating inflammation as part of a vicious cycle (27, 28). Previous small studies have observed decreased claudin-5 and ZO-1 expression in sural nerve biopsies of CIDP patients (29) and increased endoneurial edema at disease onset (30). This raises the question to the extent of which other tight junction proteins and specifically claudin-1, claudin-5, claudin-12, claudin-19, ZO-1 or occludin are expressed in the BNB and the myelin barrier in CIDP patients and, if so, whether their expression is associated with painfulness.
Different signaling elements contribute to barrier stabilization including the wnt pathway (6, 31), myosin light chain kinase (32) and neuronal guidance molecules (33). The morphogen sonic hedgehog (SHH) on the one hand functions to regulate cell division, vertebra development and differentiation of neurons in the development of the nervous system. On the other hand, it stabilizes endo-and epithelial barriers in the BBB (34, 35) and the BNB (13, 14).
Here, we first set out to study which tight junction proteins are involved in the generation and maintenance of pain in neuropathy. Screening for several tight junction proteins, we specifically detected a downregulation of claudin-12 in patients with painful CIDP or non-inflammatory PNP. Using global Cldn12-KO mice, we uncovered that Cldn12 deficiency resulted in perineurial and myelin barrier breakdown, downregulation of certain other tight junction proteins and exaggerated mechanical allodynia in naïve and neuropathic mice. Moreover, a significant reduction of the morphogen and barrier stabilizer SHH in Cldn12-KO mice could explain the tight junction protein disruption. Together, this study clarifies the role of claudin-12 as a regulatory tight junction protein specifically in the perineurial/myelin but not BBB sealing.
Results
Deficiency of claudin-12 in sural nerves of male or female postmenopausal patients with painful CIDP or non-inflammatory PNP
To describe barrier proteins in detail, we investigated a panel of tight junction protein typical for each barrier in the sural nerve in a cohort of male or female postmenopausal CIDP patients and patients with non-inflammatory idiopathic PNP (Supplementary Table 1). Tight junction proteins included claudin-1, claudin-5, claudin-12, claudin-19, ZO-1 and occludin. Claudin-1-immunoreactivity (IR) was mainly detected in the perineurium in several layers. A significant loss of claudin-1-IR was observed in patients with high fiber loss compared to moderate/low fiber loss independently of the inflammatory status or the presence or absence of pain (Supplemental Figure 1A). Both ZO-1-IR and occludin-IR were found in all compartments of the sural nerve. This included the perineurium and the SN incorporating Schwann cells and endoneurial vessels (Supplemental Figure 1B, D). Similar to claudin-1, ZO-1-IR was downregulated in the sural nerve with high fiber loss independently of pain or inflammation. Claudin-5-IR was detected in the endothelium of endoneurial blood vessels in the sural nerve (Supplemental Figure 1C). Occludin and claudin-5 were not significantly different in the subgroups.
Claudin-19-IR was observed in the perineurium and around nerve fibers co-localizing with the Schwann cell marker S100b (Figure 1A-D) in normal peripheral nerves. In patients with pain, claudin-19-IR was comparable to patients without pain. But, claudin-19-IR was significantly reduced in patients with severe fiber loss independently of inflammation. No differences were found in the clinical characteristics of these groups (Supplemental Table 2). However, significantly more patients with CIDP were in the severe fiber loss group.

(A-D) Sections from dorsal roots of healthy human donors (n = 3) (A) and sural nerves of 23 patients with either non-inflammatory PNP or CIDP were immunolabeled. Representative immunostainings for claudin-19 (red) co-stained with the Schwann cell marker S100b (green) and DAPI for nuclei staining (blue) from one patient with low/moderate fiber loss (B) and one patient with severe fiber loss (C). Higher magnification merged views (white square) are shown in the lower row (Scale bar upper row = 50 µm, scale bar lower low = 25µm). (D) The total claudin-19-IR within nerve was quantified (endoneurium). Patients were grouped according to pain symptoms (red) and extend of fiber loss (blue) and presence/absence of inflammation (grey) (patients characteristics’: Supplementary Table 2 and 3). (E-H) Immunostaining labelled for claudin-12 (red) and S100b (green) as well as DAPI (blue) in the dorsal root of a healthy donor (E), one representative patient with non-inflammatory PNP/CIPD with moderate fiber loss without pain (F) and one with pain symptoms (G). (H) Claudin-12-IR within S100b+ was quantified and compared according to patients’ groups with pain symptoms, extend of fiber loss and inflammation. PNP controls included idiopathic, vasculitic, hereditary and alcoholic PNP. The number of patients in each group are found below the dot plots. *p < 0.05, **p < 0.01, two-sided Student’s t-test. Data are shown as mean ± SEM.
Claudin-12-IR was predominantly located around nerve fibers of healthy humans, CIDP and non-inflammatory PNP patients and was co-expressed with S100b (Figure 1E-H). Some claudin-12-IR was observed in the perineurium. In particular, patients with CIDP and non-inflammatory PNP with pain showed a significant reduction of claudin-12-IR in sural nerve samples compared to patients without pain unrelated to fiber loss and inflammation. No differences were found in the clinical characteristics of these groups except for sex distribution (Supplemental Table 3). In summary, severe PNP with high fiber loss is associated with claudin-1 and ZO-1 downregulation in the surrounding perineurium as well as claudin-19 in Schwann cells themselves. Claudin-12 is unique because its loss parallels pain symptoms independently of fiber loss, inflammation, demyelinating/axonal neuropathy, functional impairment and disease duration.
Claudin-12 and claudin-19 loss after nerve injury
Next, we chose to further examine the well-standardized mononeuropathy CCI in mice, characterized also by an inflammatory reaction due to the ligatures around the SN. Previous data from our and other labs revealed that CCI caused claudin-1, claudin-5, occludin and ZO-1 downregulation in the SN (4, 5, 13). Since claudin-12 and claudin-19 were affected in patients with painful or severe fiber loss CIDP/non-inflammatory PNP, these tight junction proteins were now specifically examined. Cldn19-KO mice have a loss of TJs in Schwann cells and, in a subpopulation, delayed compound action potentials in the SN and reduced motor function (17). However, it is unclear how claudin-12 and claudin-19 are involved in nociception.
In mice, claudin-12-IR was found in myelinating, S100b positive Schwann cells, Remak bundles and the perineurium. Claudin-12-IR was decreased by 78.2% in the SN of male mice with CCI after 7 d, i.e. at the time point of mechanical hypersensitivity (4) (Figure 2A, B). Since claudin-19 is mainly expressed in Schwann cells, we then investigated clnd19-IR in teased nerve fibers. We observed a significant decrease in claudin-19-IR in the paranode of injured mice by 50% (Figure 2C, D). Similarly, Cldn12 mRNA levels in the epiperineurium (EPN), desheathed sciatic nerve (dSN) and dorsal root ganglion (DRG) were significantly lower after CCI by 60%, 80% and 30 %, respectively (Figure 2E). In summary, tight junction protein expression in Schwann cells is diminished in neuropathy.

WT mice underwent chronic constriction injury (CCI). Samples were analyzed before and 7 d after ligation. (A) Double-label immunostaining was performed showing expression of claudin-12 (red) and S100b (green) in the sciatic nerve (SN) of mice [Scale bar image overview: 50 µm, scale bar magnified image (white square): 10 µm]. (B) Claudin-12-IR colocalized with S100b was quantified in the sciatic nerve of untreated naïve mice and CCI mice (**p < 0.01, one-tailed Student’s t-test; n = 4-5). (C, D) Claudin-19-IR was displayed and quantified in the paranodal region of teased nerve fibers of naïve and CCI mice. Nodal regions were identified with an anti-pan-sodium channel Ab (pan-Nav). (***p < 0.001, one-tailed Student’s t-test; n = 40-50 fibers from six mice per group, scale bar: 20 µm) (E) Cldn12 mRNA expression in the desheathed sciatic nerve (dSN), epiperineurium (EPN) and dorsal root ganglion (DRG) was quantified in naïve and CCI mice. (##p < 0.01, EPN, DRG vs. dSN WT; **p < 0.01, ***p < 0.001, naïve versus CCI WT, two-way ANOVA followed by Bonferroni’s post hoc test; n = 6) (F, G) Cldn12-KO mice were generated as described in the methods. Representative examples of Cldn12 mRNA and protein expression in dSN, EPN and DRG tissues in Cldn12-KO and WT are displayed. Uncropped blots are provided in Supplementary. (H) Characteristic images of RNAscope® fluorescent assay for Cldn12 mRNA (red) are presented which were co-stained with immunostaining for claudin-1 (green) and DAPI (blue) in the SN of WT and Cldn12-KO mice (scale bar: 50 µm). P: perineurium, E: endoneurium. (I) Claudin-12-IR was analyzed in the SN of WT and Cldn12-KO mice (scale bar: 50 µm). Data are shown as mean ± SEM.
Mechanical hypersensitivity in male Cldn12-KO mice
Because of the downregulated claudin-12-IR in male or female postmenopausal patients with painful CIDP/PNP and mice with nerve injury, we generated Cldn12-KO mice to investigate the functional role of claudin-12 in the BNB and myelin barrier. We confirmed the deletion of the Cldn12 gene and protein in the PNS (Figure 2F, G) as well as several other tissues of KO mice (data not shown). To localize Cldn12 mRNA expression in the PNS, we labelled SN sections with the Cldn12 RNAscope probe and the anti-claudin-12 Ab (Figure 2H, I). Cldn12 mRNA was lost in KO mice. The claudin-12-IR signal also was absent in the SN of Cldn12-KO mice, while some slight presumably unspecific binding persisted in the perineurium. The summarized data confirmed the successful global KO of Cldn12.
Male Cldn12-KO mice exhibited normal body weight (WT: 23.44 ± 0.71 g vs. Cldn12-KO: 22.13 ± 1.26 g) and normal physical appearance (Supplemental Figure 2). Gross neurological examination did not show any obvious sign of altered sensory or vestibular function in mutants. Specific evaluation of the auditory system using auditory brainstem responses, anxiety-related behavior (open field test, locomotor activity and rears), acoustic startle and pre-pulse inhibition, baseline level of immobility during habituation to the conditioning cages, contextual and cued freezing performance were also comparable between both genotypes. Only, the retinas of Cldn12-KO mice were slightly but significantly thinner than those of their WT littermates.
To determine whether deletion of Cldn12 is involved in motor and sensory function, we performed a battery of behavioral tests in Cldn12-KO and WT mice. Motor strength test for 2-paw and 4-paw handgrip as well as rotarod test showed no significant difference between KO and WT mice (Figure 3A, B). Cldn12-KO mice had significantly higher thresholds for electrical stimulation-elicited vocalization than WT mice, while thresholds for flinch and jump responses were comparable (Figure 3C). This indicates abnormal sensory processing after deletion of Cldn12. In the hot plate test, investigating supraspinal integrated thermal responses, no significant differences were observed (Figure 3C). However, male Cldn12-KO mice were more sensitive to mechanical stimuli (manual von Frey filaments test) at baseline levels and after CCI, while thermal sensation was unaffected (Figure 3E, F). Interestingly, no change in sensation of mechanical stimuli was observed in female Cldn12-KO, while both genotypes of female mice still developed mechanical hypersensitivity after CCI (Supplemental Figure 3A). Together, these results demonstrate that Cldn12 deficiency selectively affects mechanical sensation in male mice.

(A-D) Behavioral tests were performed with naïve male Cldn12-KO mice in comparison with naïve male WT mice. (A) Slip thresholds of two and four paws were analyzed by the grip strength test. (B) The latency to fall of the Rota rod was tested in both genotypes. (C) Vocalization, flinch and jump thresholds to foot shock stimuli were obtained and averaged. (D) Nociceptive response latencies to a hot-plate surface were measured (all above *p < 0.05, unpaired two-sided Student’s t-test, n = 9). (E, F) Mice underwent CCI surgery. Nociceptive thresholds were obtained before and every day after CCI: Thermal hypersensitivity was detected in the Hargreaves test (E) and static mechanical hypersensitivity by Frey filaments test (F) (n = 10, IL: ipsilateral, CL: contralateral, two-way RM-ANOVA, Bonferroni’s post hoc test. ***p < 0.001 IL Cldn12-KO versus IL WT in the von Frey test only. §§§ p < 0.001 IL versus CL Cldn12-KO, ### p < 0.001 IL versus CL WT). Data are shown as mean ± SEM.
Leakiness of the myelin barrier and the perineurium of the BNB in male Cldn12-KO mice
We next explored the morphological organization of the SN in Cldn12-KO (Figure 4A). The total number of axons was significantly lower in male Cldn12-KO, while relative myelin thickness (G-ratio) and the number of undulated fibers were normal. This could point towards a minor loss of small-diameter axons during embryonal development in Cldn12-KO.

(A) Representative images of transverse semithin sections of the sciatic nerve from WT and Cldn12-KO mice including higher magnification (white square) are displayed. The ratio of inner axonal diameter to total outer diameter of nerve fibers (g-ratio), the number of undulated fibers and the number of myelinated axons in the SN were quantified (*p < 0.05, one-tailed Student’s t-test, n = 4, scale bar: 50 µm). (B) Uptake of Na-fluorescein (0.5 mol/kg, i.v.) and Evans blue dye associated to serum or plasma albumin (EBA, 0.26 mol/kg, i.v.; each 10 min after injection) were measured in brain, liver and kidney from WT and Cldn12-KO mice (two-way ANOVA, followed by Bonferroni post hoc test, n = 3). (C-E) Permeabilities of the blood nerve barrier (BNB) and myelin barrier in the SN were evaluated. (C) Examples of EBA penetration in into SN (left) and quantification (right) of fluorescence signals as measurement of the perineurial barrier of the BNB are shown (scale bar: 50 µm). (D) Diffusion of FD (left) and FD fluorescence was quantified in endoneurial microvessels 2 min after i.v. FD injection (right) as part of the BNB (**p < 0.01, two-tailed Student’s t-test, n (EBA) = 4, n (FD) = 5, scale bar: 50 µm). (E) Myelin barrier permeability was assessed by FD penetration into teased sciatic nerve fibers (left). The number of FD positive and negative fibers in the SN (right) was investigated (***p < 0.001, Pearson’s Chi-squared test with Yates’ continuity correction, n = 105-110 fibers from 5 mice/group, scale bar: 20 µm). (F) Freeze-fracture electron microscopy of the tight junction strand network was performed in the SN from WT and KO mice. Tight junction particles associated to the strands of the protoplasmic face (PF) and exoplasmic face (EF) and mesh characteristics were quantified (Supplementary Table 7, scale bars: 250 nm and 100 nm as indicated). n = 3 images from 4 mice/group.
To test barrier function in different organs, we used Evans blue dye associating to plasma albumin (EBA, 68 kDa), FITC dextran (FD, 70 kDa) and Na-fluorescein (368 Da). Cldn12 deficiency did not cause significant changes in barrier permeability of the brain, liver and kidney (Figure 4B). EBA immersion of the SN resulted in WT in bright red fluorescence only in the EPN of WTs. Cldn12 deficiency significantly increased the red fluorescence signal intensity within the nerve (Figure 4C) similar to perineurial leakage after CCI (4). No barrier breakdown was observed in endoneurial vessels after i.v. FD (Figure 4D). Using the FD immersion method for myelin barrier permeability (16), we found that the fluorescence signal significantly accumulated inside of teased nerve fibers from male Cldn12-KO mice demonstrating a leaky myelin barrier (Figure 4E). In contrast, no changes in BNB and myelin barrier permeability were noticed in fertile female Cldn12-KO mice (Supplemental Figure 3B).
In the freeze-fractured electron microscopic analysis, a significant increase in membrane-associated particle density was observed in the protoplasm-face (PF) in the SN of Cldn12-KO compared to WT mice (Figure 4F). In contrast, Cldn12 deficiency resulted in a significant reduction in the exoplasm-face (EF) associated particle density (Supplemental Table 7). Quantification of tight junction structure also revealed a significant increase in mesh length and decrease in mesh diameter in the PF and EF of male Cldn12-KO mice. In summary, deletion of Cldn12 causes an abnormal myelin barrier and perineurium permeability associated with alterations in tight junction morphology in the PNS with no effect on other barriers like the BBB.
No major tight junction protein interactions nor barrier sealing properties of claudin-12
Since tight junction proteins form strands by inter-claudin interactions (36), we further studied whether claudin-12 interacts with other tight junction proteins in an in vitro system. Tight junction-free human embryonic kidney cells (HEK)-293 cells were transfected with yellow fluorescence protein (YFP)- or turquoise (TRQ)-tight junction protein constructs (Figure 5A-D). Cldn5- or Ocln-transfected cells shaped homophilic trans-interactions with a high enrichment factor in tight junctions (positive control) (Figure 5A, C, D). In contrast, while claudin-12 integrated into the plasma membrane no homophilic trans-interactions were made (Figure 5B, D). To investigate heterophilic trans-interactions of claudin-12, we transfected TRQ-Cldn12/YFP-Cldn5 and TRQ-Cldn12/YFP-Ocln (Figure 5C). Contact enrichment of TRQ-Cldn12 and Ocln at cell-cell contacts was slightly but significantly increased compared to TRQ-claudin-12 expressions alone, showing claudin-12 exhibits low grade heterophilic trans-interactions with occludin (Figure 5D). In contrast to trans-interactions, heterophilic cis-associations were neither obtained between claudin-12 and PMP22 nor between claudin-19 and pmp22 (Figure 5E). Cells co-transfected with the two Cldn19 isoforms Cldn19A and Cldn19B served as a positive control and showed strong Förster resonance energy transfer (FRET) signals indicative of substantial cis-interaction.

Tight junction-free HEK-293 cells transfected with yellow fluorescence protein (YFP)- or turquoise fluorescence protein (TRQ)-tagged claudin and occludin are shown, respectively. (A) Yellow arrows indicate claudin-5 enriched in tight junction between transfected cells (red color: trypane blue as membrane marker; live-cell imaging, scale bar: 25 µm). (B) TRQ-Cldn12 distribution was analyzed in cell contacts. Blue arrowheads indicate partial enrichment in tight junctions. (C) Representative images with double transfection are displayed. Green arrows depict contacts between claudin-12 and occludin in the tight junction. Open blue arrows show membrane location of claudin-12 in contacts with non-transfected cells. Yellow arrows indicate contacts between occludin expressing cells. (D) The enrichment factor (EF) between TRQ-Cldn12 in the tight junctions (EF~1), positive control YFP-Cldn5 (EF>1, homophilic trans-interaction between neighboring membranes; n = 5) and TRQ-Cldn12/YFP-Ocln/YFP-Cldn5 heterophilic trans-interactions were quantified (*p < 0.05, compared to claudin-12, Kruskal-Wallis-test and Dunn’s multiple comparison post hoc test, n > 15). (E) Förster resonance energy transfer (FRET) analysis efficiencies measured cis-interaction between claudin-12 and PMP22 (n = 25), claudin-19b and PMP22 (n = 24), claudin-12 and claudin-19a (n = 17) or claudin-19b (n = 15) (single sample student’s t-test with post hoc Bonferroni-Holm analysis). FRET analysis efficiency between claudin-19a and claudin-19b is displayed as positive control (n = 6, single sample student’s t-test with post hoc Bonferroni-Holm analysis; *, p < 0.05). (F) Measurement of TER in MDCK-II cells transfected with YFP-Cldn5 (positive control), YFP (vector control), and YFP-Cldn12 (*p < 0.05, one-tailed Student’s t-test, n = 3). Data are shown as mean ± SEM.
To examine the functional role of claudin-12 in TJs, Madin-Darby canine kidney (MDCK)-II cells were transfected with YFP-Cldn fusion protein constructs. No increase in transepithelial resistance (TER) indicating barrier sealing was observed in YFP-Cldn12 transfected cells compared to the positive control, YFP-Cldn5 transfected cells (Figure 5F). In summary, we confirmed that claudin-12 alone has low barrier sealing function (37) and largely does not interact in homo- or hetero-interactions with other claudins except for some interaction with occludin.
Regulatory function of claudin-12 in male mice
Since claudin-12 seemed not directly involved in barrier sealing, we next asked whether nerve injury further affects mRNA expression of known sealing tight junction proteins in the dSN or EPN of WT and Cldn12-KO mice. In naïve mice, Cldn1 mRNA levels in the EPN as well as Cldn19 mRNA levels in the dSN were significantly decreased in male Cldn12-KO mice (Figure 6A-E). Cldn5 and ZO-1 were unaffected. In contrast, we observed a 3.5-fold upregulation of occludin. Nerve injury caused barrier breakdown and a reduction of Cldn1, Cldn5, Cldn19 and ZO-1 mRNA levels in KO and WT mice by 40-80% mice. In contrast, in isolated brain capillaries no significant differences in tight junction protein mRNA expression, e.g. Cldn1, Cldn3, Cldn5 and Ocln mRNA, were detected between male naïve WT and Cldn12-KO mice (Figure 6F). In line with barrier function and nociceptive threshold determination, no tight junction protein mRNA changes were observed in female Cldn12-KO mice compared to WT mice (Supplemental Figure 3C, D). Specifically, we did not observe a significant downregulation of Cldn19.

The mRNA expression of Cldn1 in the epiperineurium (EPN) (A), Cldn5 (B) Cldn19 (C), Ocln (D) and ZO-1 (TJP1) (E) in the desheathed sciatic nerve (dSN) was quantified in WT or Cldn12-KO mice before and 7 d after CCI (#p < 0.05, ###p < 0.001, naïve Cldn12-KO versus naive WT. *p < 0.05, ***p < 0.001, CCI versus corresponding cohorts from naive WT, n = 5-6). (F) Cldn1, Cldn3, Cldn5, Cldn12 and Ocln mRNA expression was analyzed in isolated brain capillaries of Cldn12-KO and WT mice (n = 4-5, non-detectable, n.d.). (G) Examples of immunostainings for claudin-1 (red) and claudin-5 (green) in the SN are displayed and quantified in naïve Cldn12-KO and WT (bottom, n = 4, scale bar: 50 µm). (H) Representative images of claudin-19-IR and quantification (bottom) in paranode of teased nerve fibers are shown. Nodal regions were identified with a anti pan-sodium channel Ab (pan-Nav). All above two-tailed Student’s t-test. **p < 0.01, naïve KO nerve fibers versus nerve fibers from naive WT, n = 101-104 nerve fibers from four mice per group. Data are presented as mean ± SEM. Scale bar: 20 µm.
Using immunostaining, we examined claudin-1-, claudin-5- and claudin-19-IR in the SN of male KO and WT mice. Cldn1-IR, which was mainly detected in the EPN, was significantly decreased in male Cldn12-KO mice (Figure 6G). No significant changes in claudin-5-IR were detected in endothelial cells of endoneurial vessels of KO mice (Figure 6G). Furthermore, Cldn12 deficiency resulted in a significant downregulation of claudin-19-IR in the myelin barrier compared to WT mice (Figure 6H). As observed with mRNA levels, tight junction protein-IR were comparable in female Cldn12-KO and WT mice (Supplemental Figure 3C, D). In summary, Cldn12 deficiency in male mice attenuates mRNA/protein expression of other specific tight junction proteins sealing the perineurium and the myelin barrier, but not microvessels in the SN and the brain pointing towards a regulatory function in the PNS.
Mechanical allodynia and myelin barrier breakdown after local suppression of Cldn12 transcription in the SN
We next hypothesized that the mechanical allodynia from global Cldn deficiency was a consequence of claudin-12 loss specifically in the SN. To test this hypothesis, we short-term silenced Cldn12 expression in the SN by siRNA. Local Cldn12-siRNA treatment downregulated Cldn12 mRNA and was sufficient to induce myelin barrier breakdown and mechanical allodynia (Figure 7A, B). Similarly, local Cldn19-siRNA application resulted in mechanical hypersensitivity and myelin barrier leakiness with reduced Cldn19 mRNA in the dSN (Figure 7A, C). As a negative control, we applied Clnd5-siRNA, because claudin-5 was unaltered in Cldn12-KO mice. The treatment of SN with Cldn5-siRNA reduced Cldn5 mRNA but did not cause myelin barrier collapse or mechanical allodynia (Figure 7A, D). These results demonstrate that loss of Cldn12 or Cldn19 in the SN was obligatory to the development of mechanical allodynia and myelin barrier leakiness.

(A-D) Mice were treated with a single perisciatic injection of scrambled (scr), Cldn12-, Cldn19- and Cldn5-siRNA. (A) Desheathed sciatic nerves (dSN) were incubated with FICT-dextran (FD) ex vivo after respective in vivo siRNA treatment. Representative images of teased fibers are displayed (scale bar: 50 µm). Ratios of FD positive fibers from Cldn12-(B), Cldn19-(C), Cldn5-(D) or scrambled-siRNA treated sciatic nerve (left panel) were quantified. Mechanical nociceptive thresholds (middle panel, baseline, BL, and after siRNA application) and mRNA expression of Cldn12, Cldn19 and Cldn5 in the SN (right panels) were analyzed following the siRNA injection. (*p < 0.05, **p < 0.01, ***p < 0.001, Cldn-siRNA versus scrambled-siRNA, two-tailed Student’s t-test, n = 5-6; ###p < 0.001, Pearson’s Chi-squared test with Yates’ continuity correction, n = 78-118 fibers from 4-5 mice per groups). Data are presented as mean ± SEM.
Impaired expression of SHH, a master barrier stabilizer, in male Cldn12-KO mice
Because Schwann cells forming the myelin barrier were affected in Cldn12-KO, we further explored myelin associated proteins and transcription factors important for Schwann cell differentiation. The expression of Myelin Basic Protein (Mbp) mRNA, a structural protein in Schwann cells, was unaltered in the dSN of Cldn12-KO mice although these mice had an altered number of axons (Figure 8A). In contrast, Pmp22 mRNA was reduced in Cldn12-KO (Figure 8B). PMP22 is a transmembrane protein related to tight junction proteins, necessary for compact myelin formation and altered in certain forms of hereditary neuropathy (16) The Schwann cell transcription factors HMG-box 10 (Sox10) and growth response protein 2 (Krox20) play a role in nerve injury and are necessary for nerve regeneration after nerve injury (38). No differences of Sox10 and Krox20 mRNA were observed between the genotypes indicating no general defect in Schwann cell differentiation (Figure 8c, d).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) The mRNA expression of myelin basic protein (Mbp), (B) peripheral myelin protein 22 (Pmp22) and the two transcription factors (C) Sox10 and (D) Krox20 were measured in the dSN from WT and Cldn12-KO mice. The mRNA expression was always expressed as fold change to naïve WT mice. (E) mRNA expression of Shh was analyzed in the dSN 7 d after CCI or naïve mice. (F) The Shh mRNA was quantified in the brain cortex, kidney and liver from WT and KO mice. (G) Representative images of Western blot analysis of SHH protein expression in the dSN from WT and KO mice. Bands were analyzed by densitometry (intensity density value, IDV). Uncut blots are found in Supplementary. *p < 0.05, ** p < 0.01, naïve Cldn12-KO versus naïve WT, all two-tailed Student’s t-test, PCR n = 6, Western blot n = 8). Data are presented as mean ± SEM.
Neuronal barriers are stabilized by the morphogen SHH (34). In the dSN from Cldn12-KO mice, Shh mRNA and SHH protein were significantly downregulated in naïve animals and after nerve injury (Figure 8 E, G, H). In contrast, Shh mRNA expression was normal in the cortex, liver and spleen of KO mice in accordance with unaltered tight junction protein mRNA expression in these tissues (Figure 8F). In the same line, Shh mRNA in female mice was unaffected (Supplemental Figure 3E, F). Collectively, these results demonstrate that Cldn12 deficiency is associated with a reduction of SHH causing tight junction protein loss in the SN and myelin barrier only in the PNS, which lowers mechanical nociceptive thresholds in male animals.
Discussion
In this study, we investigated the newly-discovered tight junction protein claudin-12 in peripheral nerves of patients, animals and in transfected cells. In the human sural nerve, claudin-12 was significantly lost only in patients with painful CIDP or non-inflammatory PNP compared to the non-painful group. Injury-naïve Cldn12-KO mice were hypersensitive to mechanical nociceptive stimuli and suffered from a breakdown of the perineurial barrier and myelin barrier in the PNS. Cldn12 deficiency lowered mRNA and protein expression of other tight junction proteins namely claudin-1 and claudin-19, and the number of myelinated axons. Loss of claudin-12 in the SN also reduced the morphogen and barrier stabilizer SHH explaining the effect on other tight junction proteins. The regulatory and barrier stabilizing role of claudin-12 was confirmed by local siRNA treatment. Cldn12 transfected cells were incapable of intense heterophilic cis- and trans-interactions nor did they have direct sealing properties. Together, our results suggest, for the first time, that Cldn12 expression in the PNS regulates barrier permeability and mechanical nociception via other tight junction proteins such as claudin-19, claudin-1 and SHH in male but not female mice.
Increasing evidence suggests that barrier integrity and tight junction protein expression affect the progression of several neurological diseases such as Alzheimer’s or multiple sclerosis, as well as disease of the kidney, e.g. familial hypomagnesaemia, or gut diseases, e.g. inflammatory bowel disease (39, 40). Cldn12 is expressed in low amounts in microvascular cells in the brain (41, 42), lung (43), intestine (43), SN (20) and cancer cells (21). Besides controlling Ca2+ absorption in the gastrointestinal system, the function of claudin-12 in the nervous system is unknown. It was previously postulated that claudin-12 is important for BBB formation. Since all neurological tests except for mechanical nociceptive threshold were normal in naïve Cldn12-KO mice in our study, Cldn12 seems at least not to be crucial for CNS function.
In our study, claudin-12 was found in myelinating and non-myelinating Schwann cells and the perineurium. Cldn12 deficiency suppressed Cldn1 in the EPN and Pmp22 and Cldn19 in the dSN in male mice. In line with this, perineurium and myelin barrier were more permeable to medium-sized dyes. Cldn5 in endoneurial vessels and its barrier function were unaltered. Ocln mRNA was upregulated, but this seems to have no functional consequence. tight junction proteins are differentially expressed throughout peripheral nerves (2, 44). For example, claudin-1 is the major protein in the perineurium (6, 11). Cldn5 is mainly expressed in the endothelial layer of blood vessels in the SN regulating this part of the BNB (12, 13). Cldn19 is found in the paranode of nerve fibers (16, 17). Thus, the Cldn12 deficiency-induced leaky barrier in the SN may derive from the downregulation of claudin-1 in the perineurium and claudin-19/PMP22 in the dSN, suggesting that claudin-12 could be involved in the integrity of perineurial barrier of the BNB and myelin barrier. The presented data also imply that Cldn12 has a more prominent role in the PNS, since we observed no difference in mRNA expression of Cldn1, Cldn5 and Cldn19, or barrier function in the brain, liver and kidney of male Cldn12-KO mice. It has been observed before that tight junction proteins influence the expression of other tight junction proteins. For example, knockdown of CLDN4 suppresses the expression of CLDN1, CLDN9, CLDN12 in opossum kidney cells (45). However, in the latter study the exact mechanism behind how knockdown of Cldn4 affected tight junction protein mRNA expression was not evaluated.
In addition, tight junction ultrastructure in the perineurium of claudin-12-free peripheral nerves was impaired. The number of particles of the tight junction strands, formed by tight junction proteins, oriented to the extracellular space of membrane in the cell-cell contact, was drastically reduced in the absence of Cldn12. That is, there were less particles between neighboring cells in Cldn12-KO mice. This finding would explain increased tracer uptake into nerve fibers, and that the deficiency of Cldn12 causes loss of tightening and particle forming tight junction proteins, as for instance of claudin-1 (36) or claudin-19 (17). Reversely expressed, claudin-12 possesses a tight junction-forming and tight junction-maintaining function, respectively, by supporting tightening tight junction proteins. Moreover, claudin-12 is able to modulate cell barrier stabilizers, such as SHH, or other regulators of tight junction proteins like occludin (46). Changes in the latter is also associated with Cldn12 deficiency. Therefore, a general regulatory role of claudin-12 is assumed in peripheral nerves.
The morphogen SHH is known to regulate barrier tightness in the BBB and the BNB. SHH increases the expression of Cldn5 and Ocln in brain microvascular cells (34, 47). In the BNB, Shh is upregulated after nerve injury as a compensatory mechanism (13). Repressing the hedgehog/smoothened (smo) pathway via the smo receptor antagonist cyclopamine suppresses Cldn5 and Ocln mRNA expression in the SN and elicits mechanical hypersensitivity (12–14). We did not see downregulation of Cldn5 and Ocln mRNA in male Cldn12-KO mice but rather Cldn19, Cldn1 and Pmp22 mRNA. At least Pmp22 is also induced by SHH in vitro (48). It is therefore conceivable that SHH regulates other tight junction proteins in peripheral nerves as well. In our study, Cldn12 deletion reduced Shh expression exclusively in the SN in male but not in female mice. Also, Shh expression in the brain, liver and kidney of Cldn12-KO mice were unaffected. Interestingly, Shh expression can be stimulated by estrogen in estrogen receptors positive gastric cancer cells (49). After nerve injury, estradiol improves nerve recovery, at least in part, by increasing SHH signaling and SHH-induced angiogenesis after crush injury. This is not a direct effect but rather due to a reduced expression of the SHH inhibitor hedgehog-interacting protein (HIP) in endothelial and Schwann cells (50). Thus, our data point at a role for claudin-12 in setting neuronal barrier function and imply that in its absence and with low estrogen SHH is downregulated selectively in the PNS in mice. This ultimately reduces the expression of selected tight junction proteins in the peripheral nerve forming the BNB and myelin barrier.
Deletion of Cldn12 gene via KO or siRNA in the SN led to mechanical allodynia. Although nerve injury drives loss of tight junction protein expression and barrier opening in the PNS and spinal cord (4, 5, 13, 51), it is unknown whether this is sufficient to cause pain. Indeed, barrier opening by tissue plasminogen activator (4), by siRNA or peptides against Cldn1 opens the perineurial barrier without causing hypersensitivity (6, 11). Using the fluorescence tracer EBA and FD in permeability assays, we showed a higher permeability in the perineurial barrier and myelin barrier of male KO mice after Cldn12 and Cldn19-siRNA but not Cldn5-siRNA. Therefore, hypersensitivity after siRNA application is unique to certain tight junction proteins presumably sealing the myelin barrier. Barrier breakdown could result in the dysregulated entrance of ions as well as pro-nociceptive and inflammatory mediators (e.g. IL-1β and TNF-α) (52). These could, for example, activate transient receptor potential vanilloid 1 (TRPV1) or transient receptor potential ankyrin 1 (TRPA1) expressed along the axonal membrane and thereby increase excitability causing hypersensitivity (53, 54).
A few tight junction protein KO models collectively implicate critical roles of tight junction proteins in nociception in the PNS. For example, loss of junctional adhesion molecule-C (JAM-C, Jam3) specifically in Schwann cells results in mechanical hypersensitivity (55). Because Jam3 regulates tight junction protein assembly and sealing in epithelia (56), it is highly likely that mechanical allodynia is actually a result of abnormal tight junction proteins in the SN. Indirect evidence comes from the Schwann cell-specific low density lipoprotein receptor-related protein 1 (Lrp1) KO mice (57). These mice display abnormalities in axon myelination and ensheathment of axons by non-myelinating Schwann cells in Remak bundles. They also exhibit mechanical allodynia, even in the absence of nerve injury. We have previously shown that tight junction protein expression in the SN is dependent on LRP1 (11). Knockdown of Cldn11 and Cldn19, also expressed in Schwann cells, resulted in delayed nerve conduction velocity, abnormal behavioral responses and motor function deficiencies but nociception was not examined (17, 58, 59). Here, we now provide genetic evidence via full KO or siRNA that Cldn12 as well as Cldn19 are critical for the myelin barrier and normal mechanonociception.
Cldn12-KO mice had a reduced number of myelinated axons, lower Pmp22 mRNA expression as well as diminished exoplasm-face-associated particle density in the PNS. This reduction could also be mimicked by perineurial Cldn12- and Cldn19-but not Cldn5-siRNA application. Pmp22, a membrane protein, is mainly expressed in Schwann cells (60). Its expression reflects de/re-myelination in peripheral nerve fibers after injury (61). Indeed, loss of Pmp22 causes myelin junctions to collapse, axons to degenerate and nerve fibers to swell (62). Pmp22-KO mice exhibit dysregulated myelination, downregulated Cldn19 and ZO-1 (Tjp1), and importantly, a leaky myelin barrier (16). So, reduced expression of Pmp22 can explain some parts of the Cldn12-KO phenotype. Myelinated Aβ and Aδ-fibers react to mechanical stimulation and carry touch sensation to the central terminal. In contrast, non-myelinated C-fibers mainly carry the signals for noxious heat stimuli and pain sensation (63). Given that Cldn12 specifically regulates sensory transmission in myelinated sensory fibers, the normal thermal sensitivity observed in Cldn12-KO mice could be explained by the fact that Cldn12 deletion does not influence nociceptive C-fibers organized in Remak bundles.
Three tight junction proteins were lowered in patients with CIDP or non-inflammatory PNP with high fiber loss, both in males and in postmenopausal females. These include claudin-1 in the perineurium and claudin-19 and ZO-1 in the nerve. Cldn5 and occludin were not different between groups. This pattern of tight junction protein loss could simply be an associated sign of severe neuropathy and nerve destruction. Alternatively, early barrier breakdown might fuel the autoimmune attack in CIDP or the diffusion of other exogenous toxic mediators e.g. autoantibodies resulting in accelerated nerve destruction. However, the two groups did not differ in functional impairment measured by overall disability sum score (ODSS) arguing against a significant impact of clinical severity, at least. The clinical phenotype still seems to be important for other tight junction proteins, because sera from CIDP patients can in vitro open the barrier and reduce claudin-5 expression in human peripheral microvascular cells dependent on disease severity (64). Earlier, Kanda et al. described downregulated claudin-5 and ZO-1 in the biopsies of ten CIDP patients independent of the clinical phenotype (29). However, in the present larger study, tight junction protein alterations were independent of the presence of inflammation (CIDP vs. non-inflammatory PNP). No difference in claudin-5-IR was observed. Most importantly, claudin-12 expression discriminated painful from non-painful PNP. Specifically, claudin-12 expressed in Schwann cells was critical. This indicates a central role of the myelin barrier in painful CIDP or non-inflammatory PNP. Our data support that downregulation of Schwann cell-derived claudin-12 may contribute to abnormal sensory (e.g. pain) processing in myelinated Aδ- or Aβ-fibers but maybe not the case for C-fibers in Remak bundles in CIDP and non-inflammatory PNP patients. The presented study has limitations. The first one is the small number of patients included in this study. So, a selection bias is likely. The second one is that the gender influence is clearer in mice than in patients. So, although it will be a challenge due to the disease epidemiology, pre- and postmenopausal female PNP patients should be examined. The third weakness is that we used patients of non-inflammatory PNP of different etiology, because we decided to match for other factors like age. Fourthly, barrier function using our functional assays would have been helpful to determine the significance of our findings. These assays, however, will be needed to be established in human material. Perineurial permeability might have to be measured differently because of more layers of perineurial cells in humans. Last but not least, we did not include nerve samples from healthy donors, because of ethical reasons for sural nerve biopsy in healthy individuals. Nevertheless, in the future, our results should be confirmed on a larger more defined cohort in comparison the samples healthy donors or donors with unrelated dieses.
In summary, loss of Cldn12 sex-dependently causes mechanical allodynia, operating via myelin dysfunction/degradation and SHH suppression-induced loss of selected tight junction proteins in the PNS in mice. Further, claudin-12 is decreased in CIDP or non-inflammatory PNP patients with pain. Future studies will be aimed at the exact cellular function of claudin-12 in Schwann cells to understand its contribution to pain, sensory processing and barrier function. In this line, results of this study might explain the frequent side effect of pain in commonly used hedgehog/smo inhibitors for cancer treatment like sonidegib or vismodegib. Finally, pushing myelin barrier sealing via the hedgehog pathway including possible small molecules that could be used therapeutically [e.g. activation of smo with smoothened agonist (SAG) (16), purmorphamine (65) or oxysterols] could open new avenues in developing drug treatments for neuropathy.
Methods
Patients and tissue donors
Patients with a diagnosis of CIDP fulfilling the Inflammatory Neuropathy Cause and Treatment (INCAT) criteria (66) who attended the Dept of Neurology of the University Hospital Würzburg were included in the study. Sural nerve biopsies were obtained as part of the routine workup. The resulting cohort comprised of 10/12 patients with CIDP and 10/12 control patients with non-inflammatory PNP, respectively. The diagnosis of neuropathy was based on patient history and neurological examination and included pain assessment, quantitative sensory testing and electrophysiological tests. In addition, the overall disability sum score (ODSS) was obtained. It is composed of an arm and leg disability scale with a total score ranging from 0 (“no signs of disability”) to 12 (“most severe disability score”) (67).
Demographics and clinical characteristics of the different cohorts are given in the Supplemental Table 1-3. Female patients were only included if postmenopausal. Sural nerve biopsies were routinely analyzed for fiber loss (low, medium, high) and type of damage (axonal, demyelinating, mixed).
Because sural nerve biopsies form healthy donors are difficult to obtain in sufficient numbers, we used dorsal root nerves for qualitative comparison. Dorsal root nerves were obtained from the Netherland tissue bank (NBB) from tissue donors without concomitant disease affecting the PNS. No ethical approval was necessary for this analysis (Ethical committee of the University of Wuerzburg, ref. 20180430 01).
Animals
For Cldn12-KO and WT mice, chimera with Cldn12 gene ablating deletion were first generated by transfer of the murine embryonic stem cell clone 13208A (VelociGene; http://www.velocigene.com/komp/detail/13208) into blastocysts from albino-C57BL/6J-mice (B6(Cg)-Tyrc-2J/J) as provided by the Transgenic Core Facility, Max Planck Institute of Molecular Cell Biology and Genetics, Dresden/Germany. Then, the Cldn12-KO line (C57BL/6-Cldn12tm1(KOMP)Vlcg/Ph) was back-crossed ten times with C57BL/6 mice. From mating of the resulting heterozygous mice, KO and WT mice were investigated and revealed normal body-, behavior- and reproduction properties. The strain (EM:11196) is now available at EMMA strain search - Infrafrontier GmbH, Neuherberg/Germany (https://www.infrafrontier.eu/search). The animals were kept in small groups using standard cages, on 12:12 h dark–light cycle and standard diet according to the German animal welfare law approved by the animal ethics committee of Berlin (G0030/13, LaGeSo).
Chronic constriction injury (CCI) and nociceptive tests
We housed mice in a light and temperature-controlled room under specific pathogen free conditions. Mice had free access to food and water. Male and female Cldn12-KO and WT-C57BL/6 mice (8-10 weeks) were used. Male and female mice were randomized to the treatment vs. control. Under deep anesthesia (2% isoflurane), we placed four loose silk ligatures (6/0) (with 1 mm spacing) around the SN at the level of the right mid-thigh in Cldn12-KO and WT mice (4). Ligatures were tied until they elicited a brief twitch in the respective hind limb. The muscle layer and the incision in the shaved skin layer were closed with suturing material and metal clips separately after the surgery. Mice were monitored and maintained in a controlled temperature (37 °C) until fully recovered from anesthesia. The experimenter was blinded to the genotype.
Thermal nociceptive behavior responses were assessed by the Hargreaves method. Mice were placed on the glass plate of Hargreaves’s apparatus set to 29 °C (IITC, USA). The radiant heat source was applied to the plantar part of the ipsilateral and contralateral hind limb paw. The latency before the first reaction (licking, lifting, and moving the paws) was recorded with a cutoff time of 20 s. The latency time was taken three times from both hind paws with at least a 5-min interval between each test.
Static mechanical hypersensitivity was measured using von Frey filaments (Ugo Basile SRL, Italy). Filaments were applied in ascending order from 0.008 g to 1.4 g. In general, the filaments were applied to the plantar surface of the ipsilateral and contralateral hind paw and were held for 1–3 s until the filaments were bent to an angle of 45°. We determined the withdrawal threshold of the hind-limbs to a mechanical stimulus by using 50% paw withdrawal threshold (PWT) method (4, 51).
All mice were sacrificed by cervical dislocation after the behavioral test; ipsilateral and contralateral SNs as well as DRGs were harvested for the biochemical and molecular experiments by removing connective tissue and ligatures. All experiments were performed by an experimenter blinded to the genotype.
Freeze-fracture electron microscopy
Approximately 4-6 mm of the SN (proximal to the sciatic trifurcation) from naïve WT and Cldn12-KO was gently freed from the surrounding connective tissue. Then, SN were fixed with 2.5% glutaraldehyde in PBS with Ca2+/Mg2+ (PBS+/+) for 2 h at RT. Afterwards, nerves were washed twice with PBS+/+ to be processed for freeze-fracturing as described earlier (68). Quantification of TJs was conducted directly from the original images taken from 3 representative nerve preparations deriving from 4 individual animals in each group.
RNAScope method
Cryosections of 10 µm thickness were cut at −20°C in a cryostat (Leica Biosystems CM3050 S Research Cryostat, Leica Biosystems Nussloch GmbH, Nussloch, Germany). Until further use, slides were stored at −80°C. Tissue sections were put in precooled 4% PFA in DEPC-treated distilled water. All steps were performed with DEPC-treated reagents. After 15 min of incubation at 4°C, samples were dehydrated in ethanol (50%, 70%, 100% x 2; 5 min each) at RT. Hydrophobic barriers measuring approximately the same area, were drawn around the tissue sections and allowed to dry completely. Afterwards, each section was incubated with two drops of RNAscope Protease IV reagent (15 min, RT). The RNAscope assay (Advanced Cell Diagnostics, Bio-Techne, Minneapolis, USA; 320850) was performed according to manufacturer’s instructions. All probes are found in the Supplemental Table 4. After blockage with 10% donkey serum in PBS, sections were immunostained with anti-claudin-1 Ab and secondary Ab to mark the perineurium. Finally, sections were mounted using VECTASHIELD Hardset Antifade Mounting Medium (H-1400, Vector Laboratories, Burlingame, USA). After 15 min of drying and hardening at RT, the slides were stored at 4°C until imaging.
All stainings (RNAscope, nerve permeability) were viewed using a KEYENCE BZ-9000 immunofluorescence microscope (Osaka, Japan) fitted with BZ-II analyzer software. The acquired images were analyzed using a free software package (ImageJ/Fiji). To analyze the fluorescence intensity of the immunostaining, the background was first measured in every picture. Using this corrected total cell fluorescence (CTCF) method, we calculated the integrated density, area and mean gray values of the fluorescence signal from red, green and blue channels separately in each on region of interest (ROI).
Nerve permeability
Permeability of the BNB (perineurium)
To analyze the permeability of the perineurium, we used Evans blue albumin (EBA) (4, 11). EBA was prepared in 5% BSA with 1% EB dye (Sigma-Aldrich, St. Louis, USA) in sterile distilled PBS and filtered through a 0.20 µm filter (Sartorius stedim Biotech GmbH, Germany). Dissected SNs (1 cm length, proximal to the sciatic trifurcation) from Cldn12-KO and WT were ex vivo immersed in 2 ml of EBA for 1 h and then fixed with PFA (4% paraformaldehyde) overnight. Afterwards, tissues were embedded in Tissue–Tek, cut into 10 µm thick sections and mounted on microscope cover glasses with permaFluor Mountant (Thermo Scientific, Fremont, USA). The permeability of the perineurium was determined by the diffusion of EBA into the endoneurium of the SN.
Permeability of the myelin barrier
The whole SN was dissected into epiperineurium (EPN) and the desheathed SN (dSN). The dSNs were sealed with vaseline in both ends and incubated in artificial cerebrospinal fluid (as following (mM): 10 HEPES, 17.8 NaCl, 2 NaHCO3, 4 MgS04, 3.9 KCl, 3 KH2PO4, 1.2 CaCl2, 10 Dextrose; pH 7.4), containing fluorescein isothiocyanate-dextran (FD; 5 mg/ml; MW: 70 kDa; Sigma) for 1 h at 37 °C. Afterwards, nerves were fixed in 4% PFA for 5 min at RT, then placed on glass slides and teased into individual fibers under a dissecting microscope. We then mounted these teased fibers by VECTASHIELD Antifade Mounting Medium and covered them with microscope coverslips. The green fluorescence signal was determined.
Permeability of the BNB (endoneurial vessels)
Anaesthetized mice were laid down in a supine position on a pad. The 5th intercostal space was opened and enlarged by a retractor to open the thorax. The pericardium was stripped exposing the heart anterior wall and 50 µl of FD solution was injected into the left ventricle using an insulin syringe. The dye-injected mice were sacrificed by decapitation after 2 min. SNs were dissected and embedded in Tissue-Tek. Frozen samples were cut into 12 µm-thick sections on a cryostat at −20°C. Without any fixation, microscope glass slides containing tissue sections were mounted and were imaged by fluorescence microscopy.
Brain microvessel isolation and qRT-PCR
Brain capillaries were isolated from cortices of 8-20 weeks-old Cldn12-KO and WTmice by homogenization in Dulbecco’s modified Eagle’s medium (DMEM, 4.5 g/l glucose; Life Technologies, Darmstadt, Germany) with a dounce tissue grinder (Wheaton, Millville, USA) on ice. Myelin was removed by adding dextran (60-70 kDa, 16% (w/v) final concentration; Sigma-Aldrich) and centrifugation (15 min, 4,500×g, 4 °C). The resuspended pellet was filtered (40 µm nylon mesh, Merck Millipore) and the remaining capillaries were rinsed off with DMEM and 1% (v/v) fetal calf serum (Life Technologies) (69).
RNA was extracted from isolated murine brain capillaries by GeneMATRIX Universal RNA Purification Kit (EURx, Gdansk, Poland). The cDNA was synthesized with the Maxima First Strand cDNA Syntheses Kit (ThermoFisher). qRT-PCR was performed with StepOne Real-Time PCR system, 48/96 well (Life Technologies, Darmstadt, Germany) using the Luminaris Color HiGreen high ROX qPCR Master Mix (ThermoFisher) as described in the manual. Primer sequences with a melting temperature of around 60 °C (BioTeZ, Berlin, Germany) designed with Primer3Plus.com (Supplemental Table 5). The mRNA expression was normalized to β-actin and displayed as fold change expression calculated by the 2−ΔΔCT method as described above (70).
Cell culture
Cell lines from human embryonic kidney (HEK-293; (37)) and Madin-Darby canine kidney (MDCK-II) were grown in 6-well plates until 80% confluency and transfected using polyethylenimine (PEI; Polysciences Inc, Warrington, USA) (36). Constructs used were N-terminally fused with yellow fluorescent protein (YFP) or pmTurquoise2 (TRQ): YFP-Cldn5 (71), YFP-Cldn12, TRQ-Cldn12 and YFP-Ocln (72). For each well, 10 µl of PEI (1 mg/ml) was added to 250 µl Opti-MEM (ThermoFisher); separately, 2 µg plasmid DNA was mixed with another 250 µl Opti-MEM. 5 min after incubation at RT, both solutions were combined and carefully mixed. Following 25 min incubation at RT, the mixture was added drop wise to each well. Transfection medium was removed after incubation overnight and cells were transferred into a 25 cm² flask. Cells were cultured in DMEM (1 g/ml glucose) containing 10% fetal calf serum and 1% penicillin/streptomycin (Thermo Fischer) at 37 °C and 10% CO2. For transfected cells, G418 was added to the medium (70).
Live cell imaging, trans-interaction, and transepithelial electrical resistance (TER)
Cells were seeded onto glass coverslips coated with poly-L-lysine (Sigma-Aldrich), washed with HBSS+/+ (with Ca2+ and Mg2+; ThermoFisher) and imaged in HBSS+/+ using a confocal microscope (Zeiss NLO with a Plan-Neofluar 100x 1.3 Oil objective, Zeiss, Oberkochen, Germany) (36). Plasma membranes were visualized by trypan blue (0.05%; Sigma-Aldrich) in HBSS+/+. For trans-interactions, the enrichment factor EF was detected. Half the fluorescence intensity (I) of a YFP- or TRQ-tagged protein at cell contacts between two transfected cells was divided by YFP or TRQ fluorescence at contacts between expressing and non-expressing cells (EF=Icontact/2Ino contact) (73).
To measure the TER with a Cellzscope (nanoAnalytics, Muenster, Germany), cells were seeded onto hanging PET cell culture inserts (Merck Millipore) at a density of 80,000 cells/insert. After TER values had reached a stable plateau, the cells were transferred into a 24-well plate and washed twice with HBSS+/+ warmed to 37 °C (74).
FRET measurements
HEK 293 cells (CRL-1573, A.T.C.C., Manassas, VA, USA; grown in Minimum Essential Medium Eagle (MEM; Sigma M0446) supplemented with 10% (v/v) FBS (Gibco 10270106) and 1% penicillin/streptomycin (Corning 30-002-CI) were transiently transfected with pcDNA 3.1 plasmids containing cDNA for N-terminally enhanced yellow or cyan fluorescent protein (EYFP or ECFP)-tagged murine Cldn12, human CLDN19a (NP_683763), human CLDN19b (NP_001116867) or human PMP22 (NP_000295), respectively. To this end, HEK 293 cells were seeded onto poly-lysine (Sigma P48323) coated 32 mm diameter cover slips (Thermo Scientific/Menzel) in six well plates (Falcon 353046). On the following day, cells were co-transfected with two plasmids encoding a YFP- and a CFP-fusion protein, respectively. The total sum of plasmid DNA amounted to 4 µg/well but YFP/CFP ratios varied. DNA was mixed with 10 µl PEI (polyethylenimine, 1 mg/ml, Polysciences Europe GmbH, Eppelheim, Germany) in 500 µl MEM without supplements. After 30 min incubation, the mixture was added to the cells. They were used for live cell imaging in a Zeiss LSM 780 after 24 to 48 h. CFP and YFP fluorescence signals were excited at 458 nm (detection 463 − 507 nm) and 514 nm (detection 518 - 588 nm), respectively. FRET was determined as percent increase in CFP signal intensity before and after acceptor bleaching with 20 pulses of 514 nm laser radiation at 100% laser power (75).
siRNA treatment via perineurial injection
Male mice were deeply anaesthetized with 2% isoflurane. We use commercial siRNA to suppress local mRNA expression. Cldn5-targeting siRNA (Silencer® Pre-designed siRNA, siRNA ID: s64050, Ambion), Cldn12-targeting siRNA (Silencer® Pre-designed siRNA, siRNA ID: s82332, Ambion), Cldn19-targeting siRNA (Silencer® Pre-designed siRNA, siRNA ID: s202784, Ambion) or scrambled-siRNA-Cy3 (Mission®siRNA universal negative controls, ProducNo. SIC003, Sigma) alone were applied on the surface of the SN after surgical preparation as outlined above. For siRNA delivery, 2 µg of the target siRNA or scrambled-siRNA was mixed with i-Fect™ transfection reagents (Neuromics, Edina, MN) in a ratio of 1:5 (W:V) to a final concentration of 400 mg/L.
Fluorescence quantification
ImageJ/Fiji was used to analyze our targeting fluorescence signals. To measure FD signals in teased fibers, we scanned and imaged nerve fibers from Cldn12-KO and WT mice by fluorescence microscopy at 40X magnification. Microscope and camera settings (e.g., light level, exposure, gain, etc.) were identical for all images. The number of individual fibers with positive FD penetration and the total number of individual fibers were counted and calculated. The total number of fibers was approximately 100 in 5 mice from WT and KO groups. After background subtraction in every picture, bright field images were used to guide manual selection of individual nerve fiber outlines. 20 µm2 ROI were saved onto ROI manager. ROIs were applied on the green channel, and the selected FD signals were analyzed using the built-in plugin of ImageJ/Fiji. The mean gray values, area and integrated density of ROIs were obtained and analyzed. When the mean gray value of the ROI was bigger than 10, the related fiber was considered to be a positive FD penetration fiber. Finally, a ratio was calculated by dividing the number of positive FD penetration fibers by the total number of individual fibers.
To analyze claudin-12-immunoreactivity (IR) signals in the SN of human and mice, co-expression regions of s100 and claudin-12 staining were selected and taken from coronal sections of the SN. S100-IR signals were used to determine location of Schwann cells, which were saved to the ROI manager. These ROIs were applied in the red channel and claudin-12-IR signals in these ROIs were analyzed using the Measure built-in plugin of ImageJ/Fiji. The integrated densities of ROIs were counted and exported to an Excel sheet for statistical analysis.
Statistical analyses
Statistical analysis was done using SigmaPlot or Excel software. When comparisons were more than two groups, one- or two-way ANOVA followed by the Bonferroni post-hoc test was used. For pain behavior tests, two-way RM ANOVA with Bonferroni post-hoc test was employed. The two-tailed Student’s t-test was applied to examine differences between two groups. For transfected cell culture dataset, comparisons were performed using the Kruskal-Wallis-test and Dunns multiple comparison post hoc test. Differences between groups were considered significant if p ˂ 0.05. All data are expressed as mean ± SEM.
Human and animal study approval
This study was approved by the Ethical committee of the University of Würzburg (238/17). The study is registered at the German clinical trial register (DRKS00017731) All patients gave informed, written consent to participate. Dorsal root nerves were obtained from the Netherland tissue bank (NBB) from tissue donors without concomitant disease affecting the PNS. No ethical approval was necessary for this analysis (Ethical committee of the University of Wuerzburg, ref. 20180430 01). Mouse experiments were performed in accordance with ARRIVE guidelines and approved by the Institutional Animal Care and Utilization Committee of the Government of Unterfranken, Germany (REG 2-264).
Author Contribution
JTC and HLR were responsible for the study concept and design. JTC, XH and IUO conducted the PNS barrier analyses and pain behavior experiments. IEB, RB, OBK generated and analyzed the claudin-12-KO. DG, LW and SD performed the in vitro experiments. PFB accomplished the tight junction morphology experiments. CS and KD conducted the clinical study. JS, AKR and XH analyzed the patients’ samples. CS, AB and IEB provided critical revision and important intellectual content. HLR, AKR and XH obtained funding for the study. JTC, MKH and HLR drafted and edited the article.
Acknowledgement
This study was supported by the Else–Kröner–Fresenius Foundation and the German Research Foundation (Ri817/13-1) (HLR). XH received founding from the China Scholarship Council. AKR was supported by the Interdisciplinary Center of Clinical Research (IZKF). We are grateful to our patients who consented to be a part of this study. We also acknowledge the invaluable assistance in animal husbandry of the ZEMM.
We are grateful to Jianghui Hou, Washington University, St. Louis, USA, and Nina Himmerkus, Institute of Physiology, Christian-Albrechts-University of Kiel, Germany, for providing the claudin-19 Ab. We thank Roland Naumann, TCF, MPI-CBG Dresden for stem cell microinjection/transfer and Mohammed Selloum, Mouse Clinical Institute, Illkirch/France for providing us with the phenotyping data of general sensory-motor function functions of Cldn12-KO and WT littermates.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
References




