

Abstract
The endoplasmic reticulum (ER) is a key organelle of membrane biogenesis and crucial for the folding of both membrane and secretory proteins. Stress sensors of the unfolded protein response (UPR) monitor the unfolded protein load in the ER and convey effector functions for the maintenance of ER homeostasis. More recently, it became clear that aberrant compositions of the ER membrane, referred to as lipid bilayer stress, are equally potent activators of the UPR with important implications in obesity and diabetes. How the distinct signals from lipid bilayer stress and proteotoxic stress are processed by the highly conserved UPR transducer Ire1 remains unknown. Here, we have generated a functional, cysteine-less variant of Ire1 and performed systematic cysteine crosslinking experiments to establish the transmembrane architecture of signaling-active clusters in native membranes. We show that the transmembrane helices of two neighboring Ire1 molecules adopt an X-shaped configuration and that this configuration is independent of the primary cause for ER stress. Based on these findings, we propose that different forms of stress converge in a single, signaling-active conformation of Ire1.
Introduction
The endoplasmic reticulum (ER) marks the entry-point to the secretory pathway for soluble and membrane proteins. Under adverse conditions, accumulation of unfolded proteins causes ER stress and initiates the unfolded protein response (UPR) mediated by the Inositol-requiring enzyme 1 (Ire1) in budding yeast, and by the troika of IRE1α, the PKR-like Endoplasmic Reticulum Kinase (PERK), and the activating transcription factor 6 (ATF6) in vertebrates (1). Once activated, the UPR attenuates the production of most proteins and initiates a wide transcriptional program to upregulate ER chaperones, ER-associated degradation (ERAD), and lipid biosynthesis (2). Through these mechanisms, the UPR is centrally involved in cell fate decisions between life, death, and differentiation (3). Insulin-producing β-cells, for example, rely on UPR signals for their differentiation into professional secretory cells, while chronic ER stress caused by an excess of saturated fatty acids kills them (4). The transcriptional program of the UPR controls to expression of more than five percent of all genes with tremendous impact on virtually all aspects of cell physiology (2). Consistent with these broad effector functions, the UPR is associated with numerous diseases including diabetes, cancer, and neurodegeneration (5).
Ire1 is highly conserved among eukaryotes and represents the only transducer of ER stress in baker’s yeast (6, 7). It is a type I transmembrane protein equipped with an ER-luminal sensor domain and two cytosolic effector domains: a kinase and an endoribonuclease (RNase) (8–10). How exactly unfolded proteins activate the UPR via direct and indirect mechanisms is a matter of active debate (11–15). ER stress caused by the accumulation of unfolded proteins leads to the oligomerization of Ire1 (16), which activates the cytosolic effector kinase and RNase domains (17). The unconventional splicing of the HAC1 precursor mRNA initiated by the RNase domain facilitates the production of an active transcription factor that controls a broad spectrum of UPR-target genes via unfolded protein response elements (UPRE) in the promotor region (2, 18).
Lipid bilayer stress due to aberrant compositions of the ER membrane is equally potent in activating the UPR (19–21). This membrane-based mechanism is conserved throughout evolution (20, 22, 23) and has been implicated in the lipotoxicity associated with obesity and type II diabetes (4, 24). We have shown that Ire1 from baker’s yeast inserts an amphipathic helix (AH) into the luminal leaflet of the ER-membrane, thereby forcing the adjacent TMH in a tilted configuration that locally squeezes the bilayer (25). Aberrant stiffening of the ER membrane during lipid bilayer stress would increase the energetic penalty for membrane deformation and thus stabilizes an oligomeric state of Ire1 via a membrane-based mechanism (25, 26).
The accumulation of unfolded proteins in the ER can be triggered with dithiothreitol (DTT) and Tunicamycin (TM), which interfere with protein folding in the ER by inhibiting disulfide bridge formation and N-linked glycosylation, respectively. Lipid bilayer stress is caused, for example, by an accumulation of saturated lipids or by inositol-depletion (19, 21). While both proteotoxic and membrane-based stress lead to the formation of Ire1 clusters (16, 25, 27), it remains unexplored if there is only one signaling-active configuration of Ire1 is or if distinct conformations trigger custom-tailored responses.
Here, we report on a systematic dissection of Ire1’s TMH region domain in signaling-active clusters. We have engineered a cysteine-less variant for genomic integration at the endogenous locus, generated a series of constructs featuring single cysteines in the TMH region, and developed a new crosslinking approach to study the transmembrane configuration of Ire1 in the natural environment of ER-derived membrane vesicles featuring the native complexity of lipids and proteins. This approach uncovers the transmembrane architecture of Ire1 in signaling-active clusters and suggests – irrespective of the primary cause of ER-stress (i.e. unfolded proteins or lipid bilayer stress) an X-shaped configuration of the TMHs from neighboring Ire1 molecules. Our findings underscore the crucial importance of Ire1’s highly bent configuration in the TMH region for stabilizing an oligomeric state via a membrane-mediated mechanism.
Results
We employed systematic cysteine-crosslinking in the TMH region of Ire1 to gain insight into the structural organization of its signaling-active clusters during ER-stress. Recognizing that Ire1 is activated by aberrant physicochemical membrane properties (25, 26), which are exceedingly hard to mimic using bottom-up approaches, we performed these crosslinking experiments in microsomes exhibiting the natural complexity of proteins and lipids of the ER membrane.
Cysteine-less Ire1 is functional
We have generated a cysteine-less version of Ire1 that would allow us to introduce single cysteine residues in the TMH region for a subsequent crosslinking using copper sulfate (CuSO4). The cysteine-less construct is based on a previously established knock-in construct of IRE1 that provides a homogeneous, near-endogenous expression level (25) and encodes for a fully-functional, epitope-tagged variant of Ire1, with an 3xHA tag and monomeric, yeast-enhanced GFP (yeGFP) inserted in a flexible loop at the position H875 (25, 28) (Figure 1A). To generate a cysteine-less version, we have substituted each of the twelve cysteines in the luminal, transmembrane and cytosolic regions of Ire1 with serine. Two cysteines in the signal sequence, which are co-translationally removed, remained in the final construct to ensure correct ER-targeting and membrane insertion (Figure 1A). Cysteine 48 of yeGFP (C48yeGFP) was mutated to serine, while C70yeGFP is present in the cysteine-less construct to ensure correct folding of the fluorescent protein (29). Notably, C70yeGFP is buried inside the fluorescent protein (30) and thus inaccessible for crosslinking agents under non-denaturing conditions.

(A) Schematic representations of the knock-in construct IRE13xHA-GFP indicating the position of cysteine residues and topology. All twelve cysteines of Ire1 and C48 yeGFP of yeGFP were substituted to serine to generate a cysteine-less variant. Cysteine 70 of yeGFP (C70 yeGFP) is functionally relevant (29) and remained in the final construct. The two cysteines in the signal sequence of Ire1 have not been replaced, as the signal sequence is removed upon ER-translocation. (B) Resistance of the indicated yeast strains to chronic ER-stress. Stationary overnight cultures of the indicated yeast strains were used to inoculate a fresh culture in full or minimal media to an OD600 of 0.2. After cultivation for 5 to 7 h at 30°C the cells were diluted with pre-warmed full or minimal media to an OD600 of 0.1. Cells were cultivated for 18 h at 30°C in the indicated media and stressed with DTT. The density of the resulting culture was determined using the OD620 or OD600. (C) Relative levels of spliced HAC1 mRNA in acutely stressed cells normalized to the degree of HAC1 slicing in stressed cells expressing IRE13xHA-GFP wildtype. Exponentially growing cells of the indicated strains were used to inoculated fresh YPD media to an OD600 of 0.2, cultivated in YPD and acutely stressed with either 4 mM DTT (left panel) or 1.0 µg/ml Tunicamycin (right panel) for 1 h. The relative level of spliced HAC1 in these cells was analyzed by RT-qPCR and quantitated using the comparative ΔΔCT method using normalization to ACT1 levels. The data were normalized to the splicing of HAC1 in stressed cells carrying the IRE13xHA-GFP wildtype construct. All error bars in this figure represent the mean ± SEM of at least three independent experiments. Significance was tested by an unpaired, two-tailed Student’s t test. ***p<0.001, **p<0.01, *p<0.05.
The steady-state level of wildtype and cysteine-less Ire1 are comparable (Figure S1A) and both proteins are properly integrated into the membrane as shown by subcellular fractionation (Figure S1B) and extraction assays (Figure S1C). The functionality of cysteine-less Ire1 was analyzed using a sensitive assay scoring the growth of cells exposed to chronic ER stress (25). Liquid cultures in minimal (synthetic complete dextrose; SCD) and full (yeast peptone dextrose; YPD) media were exposed to different concentrations of the reducing agent DTT and cultivated for 18 h prior to determining of the optical density (OD) of the culture. Cells producing either the wildtype or cysteine-less Ire1 are phenotypically indistinguishable and substantially more resistant to DTT than cells lacking IRE1 (Figure 1B). This suggests that the cysteine-less variant of Ire1 is functional and capable to mount an adaptive UPR.

(A) Protein levels of cells expressing either IRE13xHA-GFP WT or the cysteine-less (cysteine-less) variant. The isogenic wildtype strain BY4741 that does not express a HA-tagged variant of IRE1 was used as a specificity control (NC). Stationary overnight cultures were used to inoculate a fresh culture in SCD complete to an OD600 of 0.2 and cultivated until an OD600 of 1 was reached. 0.1 OD equivalents of cell lysates were immunoblotted using anti-HA and anti-Pgk1 antibodies. (B) Subcellular fractionation of exponentially growing cells expressing cysteine-less IRE13xHA-GFP by differential centrifugation at 5,000 x g and 100,000 x g. Stationary overnight cultures were used to inoculate a fresh culture in SCD complete to an OD600 of 0.2 and cultivated until an OD600 of 1 was reached. 80 OD600 equivalents were harvested and used for microsomal membrane preparation. The individual supernatant and pellet fractions were analyzed by immunoblotting using anti-HA, anti-Pgk1 and anti-Dpm1 antibodies by loading 0.4 OD equivalents. (C) Extraction assay of microsomes. Carbonate and urea extraction validate proper membrane integration of cysteine-less IRE13xHA-GFP (cysteine-less). Samples of each step corresponding to 0.2 OD equivalents were analyzed by immunoblotting using anti-HA, anti-Pgk1 and anti-Dpm1 antibodies. (D) PDI levels in acutely stressed cells normalized to the fold change of unstressed cells expressing IRE13xHA-GFP wildtype. Exponentially growing cells of the indicated strains were used to inoculated fresh YPD media to an OD600 of 0.2, cultivated in YPD and acutely stressed with either 4 mM DTT (left panel) or 1.0 µg/ml Tunicamycin (right panel) for 1 h. The relative level of PDI in these cells was analyzed by RT-qPCR and quantitated using the comparative ΔΔCT method using normalization to ACT1 levels. The data were normalized to the PDI level in unstressed cells carrying the IRE13xHA-GFP wildtype construct. All error bars in this figure represent the mean ± SEM of at least three independent experiments. Significance was tested by an unpaired, two-tailed Student’s t test. **p<0.01, *p<0.05. (E) Analysis of fluorescent clusters by microscopy in the indicated cells expressing either IRE13xHA-GFP WT or the cysteine-less construct. The number of clusters per cell was determined for the indicated conditions and plotted in the pie chart. The total number of analyzed cells is given for each condition.
The functionality of the cysteine-less variant during ER-stress was further validated by an alternative assay based on the degree of HAC1 splicing (Figure 1C) and the mRNA level of the UPR-target gene PDI1 (Figure S1D). Exponentially growing cells were acutely stressed for 1 h with either DTT or Tunicamycin, characterized by RT-qPCR, and compared to unstressed cells. Expectedly, the spliced HAC1 mRNA was upregulated several-fold in stressed cells compared to unstressed cells and this upregulation was observed in both wildtype and cysteine-less Ire1-producing cells. (Figure 1C). Consequently, we also observed an upregulation of the mRNA of PDI1 in response to ER-stress albeit to slightly lower degree for the cysteine-less version compared to the wildtype construct (Figure S1D). The number of signaling-active clusters per cell during ER-stress was almost identical for cells expressing either wildtype and cysteine-less Ire1 (Figure S1E). We conclude that the cysteine-less variant of Ire1 is functional during chronic and acute ER stress.
Crosslinking of Ire1’s TMH in ER-derived microsomes
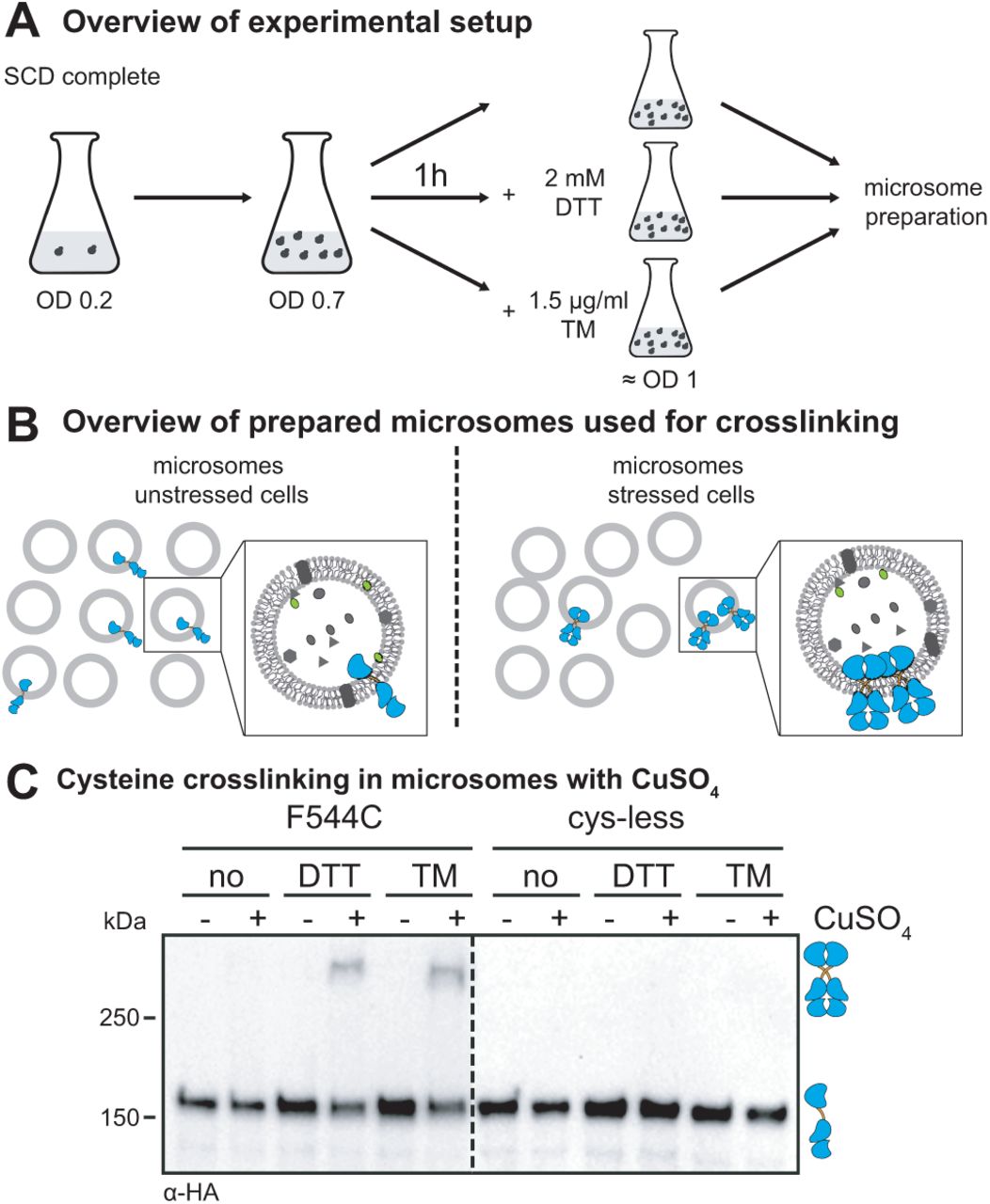
We established a strategy to crosslink a series of single-cysteine variants of clustered Ire1 via copper sulfate (CuSO4) in microsomes derived from the ER of stressed cells (Figure 2A-C). This approach has several advantages over previous attempts and provides unprecedented insights into structure-function relationships of this UPR-transducer. Ire1 is studied 1) in the context of the full-length protein, 2) in ER-derived microsomes featuring the native lipid environment, and 3) at a physiological protein-to-lipid ratio with the full range of transmembrane proteins being present in the same membrane. Using this system, we have studied the configuration of Ire1’s TMH in UPR-signaling clusters. Notably, these long-lived clusters are stable for several minutes (16, 31). Thus, CuSO4-mediated crosslinking that occurs on the same time-scale can provide useful structural information even though it leads to the irreversible formation of disulfide bonds under our experimental conditions.

(A) Overview of the cultivation of yeast cells for cysteine crosslinking. Stationary cells were used to inoculate a fresh culture in SCD complete media to an OD600 of 0.2. After cultivation at 30°C to an OD600 of 0.7, Ire1-clustering was induced either by DTT (1h, 2 mM, SCD) or TM (1h, 1.5 µg/ml, SCD). After harvesting the unstressed and stressed cells, cells were lysed and used for microsomal membrane preparation by differential centrifugation. (B) Overview of cysteine crosslinking with CuSO4. Microsomes from unstressed cells and microsomes from stressed cells containing Ire1 clusters were incubated with CuSO4 to induce cysteine crosslinking of Ire1. (C) Crosslinking of single cysteine variants of Ire1 in microsomes. The indicated strains were cultivated in the presence and absence of ER-stressors. Cells were cultivated and treated as described in (A). 80 OD equivalents of cells were harvested and disrupted prior to the preparation of crude microsomes by differential centrifugation. The crosslinking of juxta-posed cysteines was induced by 10 mM CuSO4 on ice and an incubation for 5 min. 8 µl microsomes with a typical protein concentration of 1 mg/ml were incubated with 2 µl 50 mM CuSO4. The reaction was stopped by the addition of 2 µl 1 M NEM, 2 µl 0.5 M EDTA and 4 µl membrane sample buffer. The resulting samples were subjected to SDS-PAGE corresponding to 0.25 OD equivalents and analyzed by immunoblotting using a monoclonal anti-HA antibody.
Cells expressing either a cysteine-less variant of Ire1 or a variant containing a single-cysteine in the TMH region (F544C) were cultivated to the mid-exponential phase in minimal medium. These cells were either left untreated or stressed for 1 h with either DTT (2 mM) or TM (1.5 µg/ml) to cause ER-stress, which leads to the formation of Ire1-clusters (16, 25). We then isolated crude microsomes from these cells and incubated them on ice for 5 min either in the presence or absence of 10 mM CuSO4 to catalyze the formation of disulfide bonds by oxidizing nearby sulfhydryl groups (32). Given the low copy number of ∼260 for Ire1 (33) and the fragmentation of the ER during microsome preparation, we expect to detect crosslinking of single-cysteine variants of Ire1 only when it was pre-clustered prior to the preparation.
Immunoblotting of the resulting samples revealed a prominent, HA-positive signal corresponding to monomeric Ire1 and for some samples, a less-pronounced HA-positive signal from a band with lower electrophoretic mobility that was only observed when 1) Ire1 contained a single-cysteine in the TMH region (F544C), 2) the microsomes were prepared from stressed cells (either DTT or TM), and 3) when crosslinking was facilitated by CuSO4 (Figure 2C). This suggests a remarkably specific formation of covalent, disulfide bonds between two Ire1 molecules in the TMH region, despite the presence of numerous other, potentially competing membrane proteins with exposed cysteines in the ER. A Co-IP analysis using Flag-tagged and HA-tagged Ire1 variants co-produced in a single cell and crosslinked in microsomes via the native cysteine in the TMH region (C552) verified that the additional band with low electrophoretic mobility represented presumably disulfide-linked, SDS-resistant dimers of Ire1 (Figure S2). We conclude that Ire1 variants with single cysteines in the TMH region can be specifically cross-linked via CuSO4, when present as pre-formed clusters in microsomes isolated from stressed cells.

A crosslinking experiment using CuSO4 was performed with microsomes prepared from cells expressing a HA-tagged variant of Ire1 from endogenous locus (IRE13xHA-GFP) and a Flag-tagged variant (IRE13xFlag-GFP) from a CEN-based plasmid. A yeast culture in selective SCD-LEU was inoculated to an OD600 of 0.2 from a stationary overnight culture and cultivated at 30°C until an OD600 of 0.7 was reached. The cells were either stressed with 2 mM DTT or left untreated and were further cultivated for 1 h. 80 OD600 equivalents from these cultures were harvested by centrifugation. Microsomal membranes were isolated by differential centrifugation. Microsomes prepared from cells expressing only one of the two tagged variants of Ire1 served as controls. Both constructs contained a single cysteine in the TMH region at the position 552 (C552). After incubation of the microsomes with 10 mM CuSO4 on ice for 5 min, the crosslinking reaction was stopped by the addition of NEM in a final concentration of 111 mM and EDTA in a final concentration of 50 mM. The microsomes were then solubilized using 2% Triton X-100 and subjected to an IP using anti-Flag beads. Both the input and IP samples were analyzed by immunoblotting using anti-Flag and anti-HA antibodies.
A crosslinking screen identifies an X-shaped configuration of Ire1’s TMH
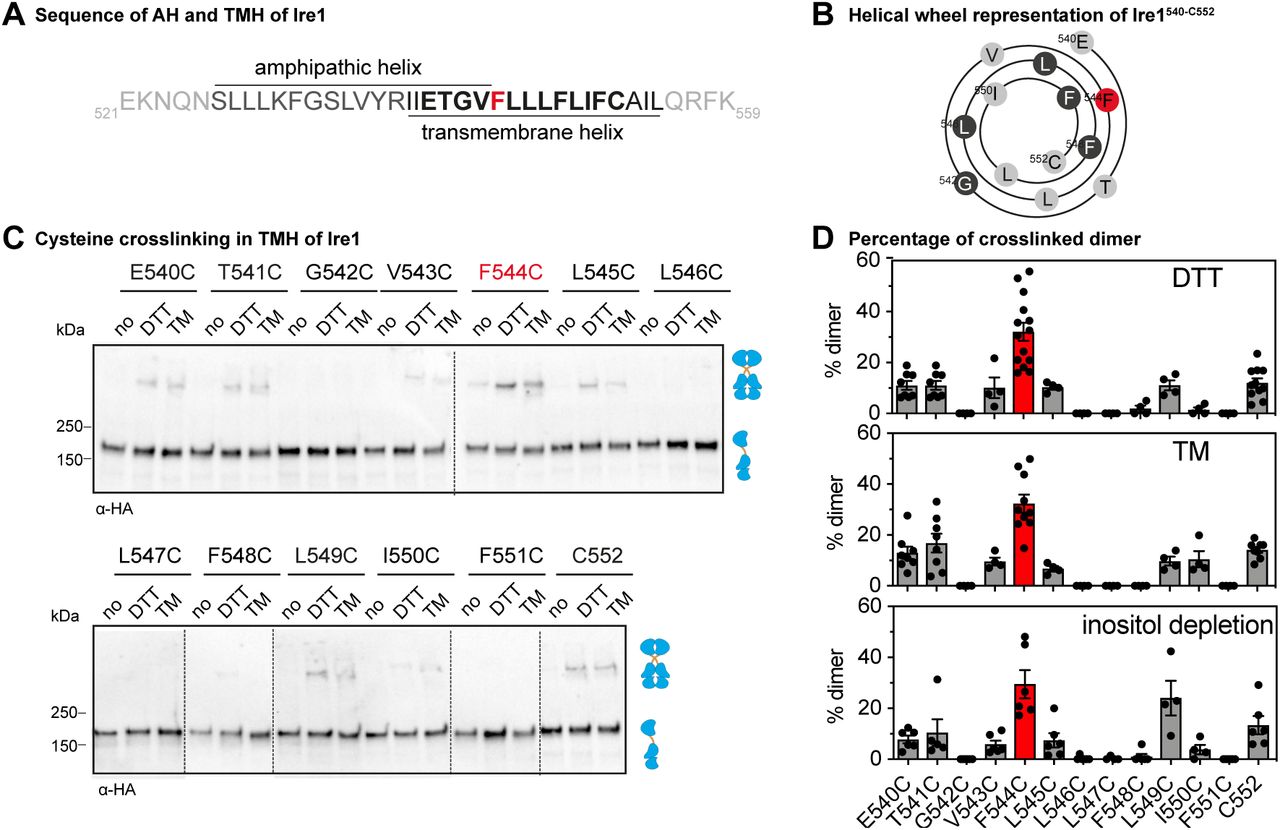
Next, we systematically introduced single cysteine-residues in the TMH-region of Ire1 in order to characterize the structural configuration of Ire1’s TMH in signaling-active clusters. We generated a total of thirteen variants each containing a single cysteine in the TMH region starting with E540C at the transition between the AH and the TMH (Figure 3A) and ending at the residue C552, a native TMH residue that is substituted to serine in the cysteine-less Ire1. Consequently, this scanning approach covered more than three helical turns and almost the entire short TMH of Ire1 (Figure 3A, B). All variants with engineered cysteine residues (E540C to F551C) were subjected to a sensitive, cell-based assay to ascertain the functionality of the UPR under condition of prolonged ER stress (Figure S3A). Consistent with the functional role of the AH in the ER-luminal membrane leaflet (25), we found that the substitution of an AH-residue to cysteine (E540C, T541C, or G542C) impaired the stress response of Ire1 as evident from the increased sensitivity of the respective cells (Figure S3A). Notably, this increased sensitivity to DTT is indeed due to a functional defect because the mutations do not alter the steady-state level of the Ire1 variants (Figure S2B). In contrast, substitution of TMH residues (V543C-F551C) did not result in an impaired resistance to DTT in the respective cells (Figure S3A). This suggests that these TMH residues have no specific relevance for Ire1 activation (Figure S3A). We then subjected this set of single cysteine variants to the established cysteine-crosslinking procedure (Figure 3C). While some variants (e.g. G542C or L546C) showed no detectable crosslinking, a significant portion of other variants (e.g. T541C or L549C) was crosslinked under the given experimental conditions (Figure 3C,D). The F544C variant consistently exhibited the highest crosslinking efficiency (Figure 3C,D). The overall pattern of crosslinking residues in the TMH residues was independent of the condition of ER stress (Figure 3D). Proteotoxic stress induced by either DTT, TM or lipid bilayer stress caused by inositol-depletion (19, 34) showed essentially the same crosslinking pattern (Figure S3C, 3D). These data suggest that the structural organization of Ire1 in signaling-active clusters is comparable for different types of stress, at least in the TMH region.

(A) The resistance to ER-stress was investigated for the indicated yeast strains. Stationary overnight cultures of the indicated yeast strains were used to inoculate a fresh culture in full or minimal media to an OD600 of 0.2. After cultivation for 5 to 7 h at 30°C the cells were diluted with fresh minimal media to an OD600 of 0.1. Cells were cultivated for 18 h at 30°C and stressed with DTT. The density of the resulting culture was determined using the OD620 or OD600. The error bars represent the mean ± SEM of at least two independent clones. (B) Protein levels of cells expressing different IRE13xHA-GFP variants. The lysates of exponentially growing cells were immunoblotted using anti-HA and anti-Pgk1 antibodies. (C) Crosslinking of single cysteine variants of Ire1 in microsomes derived from cells grown in lipid bilayer stress conditions. Exponentially growing cells in SCD complete media were washed and used to inoculate a fresh media w/o inositol to an OD600 of 0.5. To induce lipid bilayer stress, the cells were grown in inositol depleted SCD complete media for 3 h. 80 OD equivalents were harvested and used for microsomal membrane preparation. CuSO4 induced crosslink was performed by incubating 8 µl of microsomes with 2 µl of 50 mM CuSO4 for 5 min on ice. After stopping the reaction with NEM and EDTA, samples were subjected to SDS-PAGE with a subsequent immunoblotting with anti- HA antibody.

(A) Primary structure of TMH region of Ire1 including the adjacent ER-luminal amphipathic helix (25). Almost all residues of the short TMH (shown in bold) were systematically mutated to cysteines for CuSO4 crosslinking and are illustrated as helical wheel representation in (B). (B) Helical wheel representation of the TMH residues (Ire1540-552) subjected to the cysteine scanning approach. (C) Crosslinking of single cysteine variants of Ire1 in microsomes. Stationary cells were used to inoculate fresh SCD complete media to an OD600 of 0.2. After cultivation to an OD600 of 0.7 at 30°C, cells were treated either with 2 mM DTT or 1.5 µg/ml TM in SCD medium for 1 h. Microsomes from unstressed cells (no) and stressed cells were prepared by differential centrifugation and subjected to a CuSO4-mediated crosslinking procedure. 8 µl microsomes were incubated with 2 µl 50 mM CuSO4 (final concentration 10 mM) for 5 min on ice. After the reaction was stopped with NEM, EDTA and membrane sample buffer, the samples (0.25 OD equivalents) were subjected to SDS-PAGE followed by immunoblotting using anti-HA antibodies. (D) Quantification of tested UPR stress conditions. Cells were cultivated and treated as described in (C). For inositol depletion, exponentially growing cells were inoculated to an OD600 of 0.5 and cultivated for 3h in inositol depleted media. Cells were harvested and treated as described in (C). Immunoblots of crosslinked microsomes from cells depleted from inositol are shown in Fig. S3. The percentage of crosslinked dimer from at least three independent experiments was determined using the densiometric signals of the bands illustrated in (C) (ER stressors: DTT and TM) and Fig S3. (ER stressors: inositol depletion) corresponding to the monomeric and covalently dimeric protein, which were determined using ImageJ (n > 4, mean ± SEM).
Cysteine crosslinking can be used to infer structural models. The observed pattern of crosslinking residues in the TMH of Ire1 is very distinct to those observed in the TMH region of the growth hormone receptor (35) and the thrombopoietin receptor (36), which form parallel dimers leading to a helical periodicity of crosslinking. Our data also disfavor a model of the two TMHs - attached to the ER-luminal and cytosolic portions of Ire1 via flexible loops – forming dynamic complexes in an entirely random orientation. Instead, our crosslinking data suggest an X-shaped configuration of the TMHs with the best-crosslinking residue F554 positioned at the crossing point. Intriguingly, such an arrangement would be consistent with the previously reported, highly tilted orientation of the monomeric TMH of Ire1, which is enforced by the adjacent, ER-luminal AH (25).
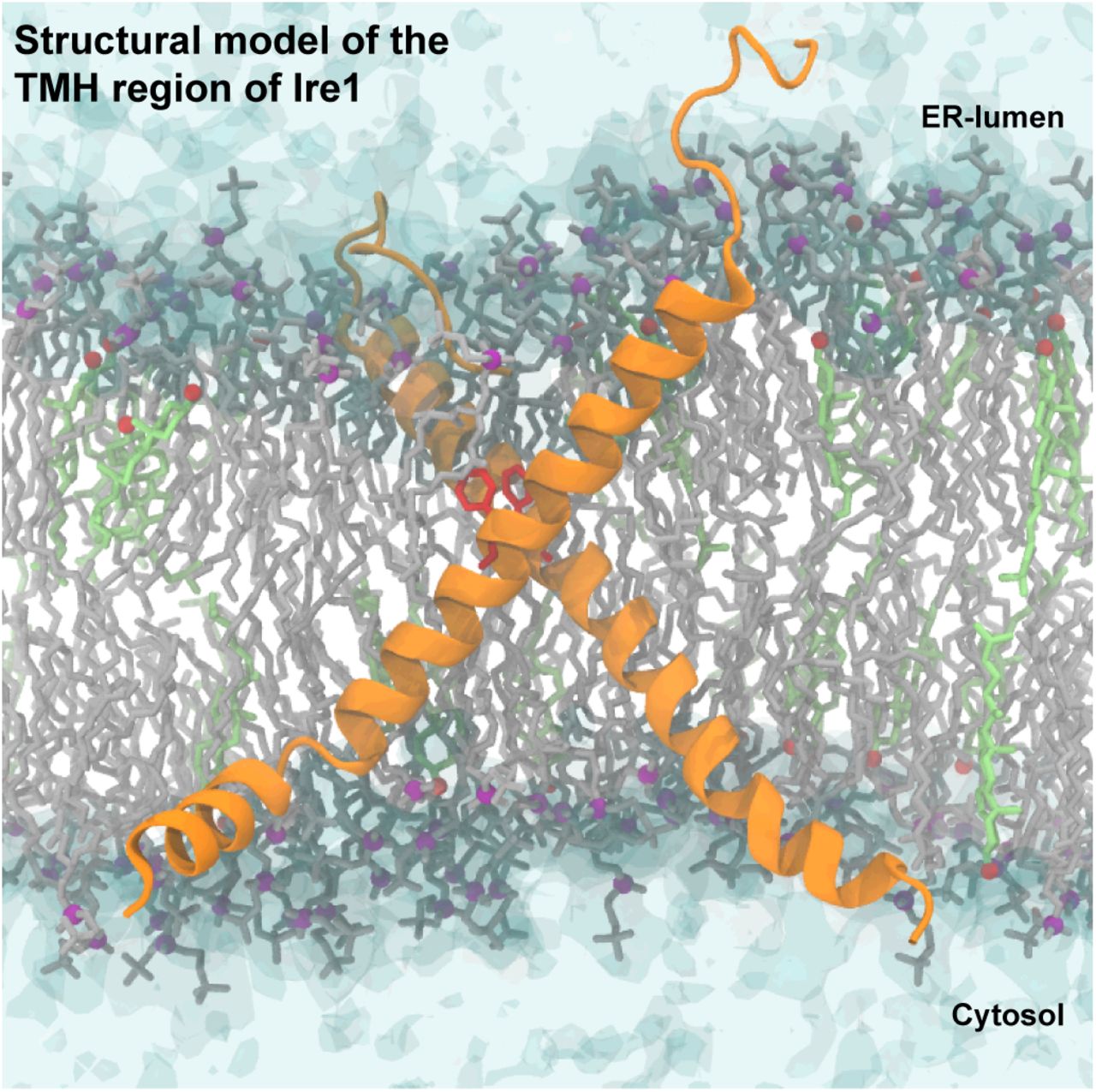
In order to obtain a structural representation of these crosslinking data, we modeled a dimer of the TMH region of Ire1 based on extensive molecular dynamics (MD) simulations in lipid membranes that integrated the crosslinking data, and in particular the contact between the two F544 (Figure 4, Supplementary Movie) and restrained the two F544 residues to face each other. The resulting model of the TMH region highlighted the overall, highly bent architecture of the TMH region of each protomer leading to an X-shaped configuration of the dimer (Figure 4, Supplementary Movie). Substantial membrane thinning and water penetration, which were previously observed around the monomeric TMH region of Ire1 (25), become clearly apparent and are most significant in the region of the AHs (Figure S4, Supplementary Movie).

Configuration of a model TMH dimer obtained from atomistic molecular dynamics simulations. Protomers are shown as an orange ribbon, with the residues F544 highlighted in red. The phosphate moieties of POPC are shown as purple beads. Rhe hydroxyl groups of cholesterol molecules are shown as red beads. Water is shown in a transparent surface representation.

Configuration of a model TMH dimer obtained from atomistic molecular dynamics simulations. Protomers are shown as an orange ribbon, with the residues F544 highlighted in red. POPC lipids and their phosphate moiety are shown in gray and purple, respectively. Cholesterol molecules and their hydroxyl groups are shown in light green and red, respectively. Water is shown in a transparent surface representation.
Validating the structural model of the TMH region of Ire1
Our crosslinking approach has identified the TMH residue F544 as a candidate residue that might stabilize the transmembrane configuration of Ire1 in signaling-active clusters. Interestingly, an aromatic residue in the TMH region of the mammalian IRE1α (W547) was reported to stabilize the oligomeric state of IRE1α (37). Hence, we embarked on testing a similar role for F544 in Ire1 from baker’s yeast. We generated a F544A mutation, which contained the native C552 in the TMH. A cell-based assay revealed that the F544A mutant was phenotypically indistinguishable from cysteine-less Ire1 (Figure 5A) just as the F544C mutant (Figure S1A), thereby suggesting that F544 is not functionally relevant. This finding was corroborated by Cu2+-mediated crosslinking of C552 in microsomes isolated from cells stressed either with DTT or TM. The intensity of the band corresponding to crosslinked Ire1 was unaffected by the F544A mutation (Figure 5B). Thus, F544 does not contribute to the stability of Ire1-oligomers even though it is placed in close proximity to equivalent residues on the TMH of neighboring Ire1 molecules.

(A) The ER-stress resistance of cells expressing the F544A variant of Ire13xHA-GFP containing the native cysteine 552 was scored using a yeast growth assay. Stationary overnight cultures of the indicated yeast strains were used to inoculate a fresh culture to an OD600 of 0.2. After cultivation for 5 to 7 h at 30°C, the cells were diluted with fresh media to an OD600 of 0.1. Cells were cultivated for 18 h at 30°C in the presence of the indicated concentrations of DTT. The density of the resulting culture was determined using the OD600. (B) The impact of the F544A mutation in the TMH of Ire1 on the degree of crosslinking via cysteine 552 was determined using the microsome-based crosslinking assay. Stationary cultures were used to inoculate fresh SCD complete media to an OD600 of 0.2 and cultivated to an OD600 of 0.7. Cells were stressed by the addition of 2 mM DTT or 1.5 µg/ml TM for 1 h. Microsomes of 80 OD units of the indicated yeast strains were prepared by differential centrifugation and used for cysteine crosslinks. 8 µl microsomal membranes were incubated with 2 µl CuSO4 (final concentration 10 mM) and incubated for 5 min on ice. The reaction was stopped by adding 8 µl stopping buffer. Samples corresponding to 0.25 OD equivalents were subjected to SDS PAGE and a subsequent immunoblotting with anti-HA antibodies. (C) The ER-stress resistance of cells expressing the AH-disrupting F531R variant of Ire13xHA-GFP containing the native cysteine 552 was scored using a yeast growth assay. The indicated cells were cultivated and treated as in (A). (D) The impact of the AH-disrupting F531R mutation of Ire1 on the degree of crosslinking via cysteine 552 was determined using the microsome-based crosslinking assay. Cells were cultivated and further treated as described in (B). The data are represented as the mean ± SEM and are derived from at least three independent experiments. Significance was tested by an unpaired, two-tailed Student’s t test. **p<0.01.
Our structural model of the monomeric TMH (25) and the dimeric TMH (Figure 4, Supplementary Movie) suggested an unusual, tilted architecture of the TMH region, which is stabilized by the ER-luminal AH. We proposed that this configuration of the TMH region facilitates Ire1 to sense aberrant membrane properties by contributing to the stability of oligomeric Ire1 during lipid bilayer stress (25, 38). We put this model to the test, by combining the single-cysteine F544C for crosslinking with the AH-disruption F531R mutation. The resulting F531R/F544C double mutant exhibited only a very mild functional defect as implied by the cellular resistance to prolonged stress caused by DTT (Figure 5C). Strikingly, the disruption of the AH by the F531R mutation greatly impaired the crosslinking propensity of the F544C thereby suggesting an important contribution of the AH to the transmembrane architecture of Ire1 (Figure 5D). An analogous set of experiments was performed with a construct that contained an AH-disrupting F531R mutation (25) and, as a single cysteine the native TMH residue C552. This F531R/C552 construct was functionally impaired (Figure S5A) and showed a significantly reduced crosslinking propensity compared to the equivalent construct with an intact AH (Figure S5B), thereby highlighting structural and functional importance of the ER-luminal AH.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) The ER-stress resistance of cells expressing the F544A variant of Ire13xHA-GFP containing the native cysteine 552 was scored using the yeast growth assay as described in Figure 5. (B) The impact of the F544A mutation in the TMH of Ire1 on the degree of crosslinking via cysteine 552 was determined using the microsome-based crosslinking assay. The data are represented as the mean ± SEM and are derived from at least three independent experiments. Significance was tested by an unpaired, two-tailed Student’s t test. *p<0.05.
Discussion
Here, we establish a structural model for the TMH region of Ire1 in signaling-active clusters (Figure 4). Employing a systematic cysteine crosslinking approach in native membranes and aided by molecular dynamics simulations, we show that neighboring TMHs of clustered Ire1 are organized in an X-shaped configuration during ER-stress. Based on the remarkable similarity of the crosslinking patterns observed in the context of lipid bilayer stress caused by inositol-depletion and proteotoxic stress caused by DTT or TM (Figure 3, Figure S3C), we propose that different forms of ER-stress converge in a single, signaling-active configuration in the TMH region. Our structural and functional analyses reveal a crucial role of the transmembrane architecture in controlling the oligomeric state of Ire1. The finding that the F544A mutation of the best-crosslinking residue at the intersection of neighboring TMHs causes no functional defect (Figure 5A,B) reinforces our interpretation that Ire1 senses lipid bilayer stress via its unusual architecture rather than by stereospecific, structural elements in the TMH.
Our findings are consistent with a crucial role of the ER-luminal AH in forcing the adjacent TMH in a highly tilted and bent configuration and deforming the membrane (25) (Figure 4, Figure S4, Supplementary Video). We show that disrupting the functionally important AH by incorporation of a charged residue into the hydrophobic face substantially reduces the crosslinking efficiency, thereby implying a significant structural change (Figure 5C,D and Figure S5A,B).
Modeling the dimeric transmembrane organization aided by the crosslinking data provides intriguing insights into membrane-deforming potential of Ire1 (Figure, 4, Figure S4, Supplementary Video). The ER-luminal AH and a cluster of charged residues at the cytosolic end of the TMH locally distort the bilayer, which is best evidenced by a decreased membrane thickness in the proximity of the monomeric (25) and dimeric transmembrane domain of Ire1 (Supplementary Video). Such membrane deformations also cause disordering of lipid acyl chains and come at an energetic cost, which is controlled by the composition and physical properties of the surrounding bilayer (38, 39). The higher this cost e.g. due to increased lipid saturation or inositol-depletion, the higher is the energetic gain upon coalescing these regions and thus the propensity of Ire1 to oligomerize.
This membrane-based activation of the UPR is not restricted to yeast, but has also been reported for UPR transducers in worms (23, 40), fishes (41), and mammals (20, 37, 42). In fact, all three transducers of the UPR from mammals can be activated by aberrant membrane signals albeit via distinct mechanisms (20, 37, 43). The IRE1α- and PERK-dependent branches of the UPR respond to increased lipid saturation even when the ER-luminal domain is removed (20). While some studies suggested that stress-sensing relies on generic features in the TMH of mammalian IRE1α, which are independent of the exact amino acid sequence in the TMH region (20, 44), a more recent study proposed a mechanism of sensing that is based on specific residues in the TMH (37). Cho et al. scored the oligomerization propensity of IRE1α in cells via bimolecular fluorescence complementation in palmitate-treated cells and suggested that the increased lipid saturation might induce a conformational switch in the TMH region, which relies on a tryptophan (W457) as putative sensing residue and a conserved leucine zipper motif (SxxLxxx) involving serine 450 (37, 44). Intriguingly, such a rotation-based mechanism of sensing would be reminiscent of the lipid saturation sensor Mga2 from baker’s yeast controlling the expression of the essential fatty acid desaturase-encoding gene OLE1 (45, 46).
We propose that Ire1 from baker’s yeast uses a mechanism distinct from that proposed for mammalian IRE1α based on the following evidence: 1) Mutagenesis in the entire TMH of Ire1 from baker’s yeast causes no functional defect unless the integrity of the AH is disrupted (25). 2) Mutagenesis of individual TMH residues (V543-F551) including three aromatic residues lining one side of the TMH – one of which being the best-crosslinking residue F544C - causes no relevant functional defects (Figure S3A). These findings are notable because aromatic residues have been implicated in lipid/membrane sensing in other systems (37, 45, 46). 3) Our crosslinking data provide no evidence for a rotational re-organization of the TMHs during lipid bilayer stress (Figure 3C,D, Figure S3C). 4) Our cell-based assays and crosslinking data underscore the central importance of an intact AH adjacent to the TMH for normal UPR activity and crosslinking in the TMH region.
Based on similar physicochemical features of the TMH region including the evolutionary conserved AH, we propose that the mammalian PERK might use a similar mechanism as Ire1 from baker’s yeast, while the mammalian IRE1α -as a variation to a common theme-seems to employ a distinct mechanism (37, 44). Clearly, more studies with an increased temporal and spatial resolution that do not interfere with the dynamic properties of Ire1 will be required to identify the unique specializations of individual UPR transducers and to unequivocally establish their structural and dynamic features that govern their membrane sensitivity.
Our combined results lead to the following model. The activity of Ire1 requires an oligomerization of its cytosolic effector domains, which mediate the splicing of the HAC1 in yeast of XBP1 in mammals for signal propagation (1). This oligomerization involves the entire protein as a response to divergent activating signals, which are sensed by different portions of the protein.
Unfolded proteins are sensed by the ER-luminal domain via a binding groove (12), which is formed by two interacting ER-luminal domains and is further facilitated by a subsequent formation of higher oligomers (47). Under these conditions, the transmembrane domain and the cytosolic effector domains ‘follow’ the oligomerization of the ER-luminal domains. A large diversity of ER-luminal and cytosolic interactors including chaperones can tune and specify the activity of mammalian UPR transducers (14, 48). This may reflect a way to custom-tailor the globally-acting UPR to different cell types with distinct protein folding requirements during steady-state and particularly during differentiation. Lipid bilayer stress activates the UPR in both yeast and mammals via a membrane-based mechanism and does not require the binding of unfolded proteins to the ER-luminal domain and/or associated chaperones (19, 20, 25).
We propose that prolonged and chronic forms of ER-stress, which can be mimicked by sub-lethal doses of DTT or TM, activate the UPR predominantly via this membrane-based mechanism. When Ire1 is rendered insensitive to proteotoxic stress by mutations in the binding groove for unfolded proteins (19, 25) and even when the entire ER-luminal domain of IRE1α was substituted by a zipper domain (49) the resulting proteins are activated under these conditions. We suggest that all portions of Ire1, the ER-luminal domain, the TMD and the cytosolic domains cooperate in determining the stability of oligomeric configurations of Ire1. An intriguing implication of this model is that different forms of ER-stress would be independent of each other, but inter-dependent in a somewhat additive fashion. Lipid bilayer stress would sensitize Ire1 to unfolded proteins and, conversely, an increased load of unfolded proteins in the ER lumen would increase the sensitivity of Ire1 to aberrant lipid compositions (20, 38). The intriguing hypothesis that chemically distinct signals from the lumen and the membrane of the ER cooperate in UPR activation remains to be experimentally tested.
Clearly, more studies are required to better understand the molecular mechanisms of signal integration by UPR transducers particularly in the context of prolonged and chronic ER stress. The clinical relevance is obvious as ER-stress and UPR activation have been implicated the inflammatory response (50), B-cell differentiation into plasma cells (51), insulin resistance, and pancreatic β-cell failure (4). It has been speculated that the sensing of lipid bilayer stress by IRE1α and PERK might account for instances, where relatively modest levels of unfolded protein load lead to substantial UPR signaling and has been termed anticipatory UPR (52). An exciting alternative possibility is that the membrane-based UPR may be caused by an overcrowding of the ER membrane with membrane proteins (38, 53).
We have established a cysteine-less, functional variant of Ire1 allowing for a detailed structure-function analysis of this UPR transducer in its native membrane context. This construct and the established protocol for cysteine-crosslinking can be readily adapted to address other central questions regarding the structural organization of signaling-active clusters of Ire1, which may be inaccessible by other approaches.
Materials and Methods
Reagents and Antibodies
All chemicals and reagents used in this study were purchased from Sigma Aldrich, Carl Roth or Millipore and are of analytical or higher grade. The following antibodies were used: mouse anti-Flag monoclonal (M2) (Santa Cruz), rat anti-HA monoclonal (3F19) (Roche), mouse anti-Dpm1 monoclonal (5C5A7) (Life Technologies), mouse anti-Pgk1 (22C5D8) (Life Technologies), mouse anti-MBP monoclonal (NEB), anti-mouse-HRP (Dianova), anti-rat-HRP (Dianova).
Strains and plasmids
All strains used in this study are listed in Table 1, plasmids are listed in Table 2.
Yeast strains of used in this study
Plasmids used in this study
Genetic manipulation of S. cerevisiae
For genetic manipulation of S. cerevisiae, we used a previously described knock-in strategy (25). This construct comprised the endogenous promoter of IRE1 (−1 to −551 bp), the sequence of IRE1 with a sequence coding for a 3xHA tag and a monomeric version of yeGFP (A206R yeGFP) inserted at the position of H875, and the endogenous 5′ terminator sequence on the plasmid pcDNA3.1-IRE13xHA-GFP (25). A cysteine-less variant of this construct was generated by substituting all 12 cysteines in IRE1 with serine using a strategy based on the QuikChange method (Stratagene). Additionally, the cysteine 48 (C48 yeGFP) of the monomeric yeGFP was substituted to serine, while the inaccessible, functionally relevant cysteine 70 (C70 yeGFP) remained in the final construct (29, 30). Derivatives of this constructs were generated by substituting individual codons of TMH residues to cysteine. All mutants used in this study are listed in Table 2. These plasmids were linearized using HindIII and XhoI restriction enzymes and used for transforming a previously established cloning strain lacking both the gene of IRE1 and its promotor (25). Strains used in this study are listed in Table 1.
Additionally, a Flag-tagged cysteine-less Ire1 version based on the CEN-based Ire1 construct from the pPW1628/pEv200 plasmid (28) was generated. The 3xHA epitope tag in the knock-in construct was replaced by a 3xFlag epitope tag using the Q5 site-directed mutagenesis kit (NEB). The newly generated knock in sequence was amplified in a multi-step PCR reaction adding the terminator sequence from the pEv200 plasmid and BssHI and HindIII restriction site. The transfer of the IRE13xFlag-GFP sequence in the CEN-based vector pPW1628/pEv200 (28) was performed using BssHI/HindIII restriction sites.
Cultivation of S. cerevisiae
The yeast strains were cultivated at 30°C on agar plates containing SCD complete medium or selection medium. Liquid yeast cultures were inoculated with a single colony and typically cultivated at 30°C for a minimum of 18 h to reach the stationary phase. This overnight culture was used to inoculate a fresh culture, which was inoculated to an OD600 = 0.2 and cultivated until the mid-exponential cultures.
For microsomal membrane preparation, stationary cells were used to inoculate a fresh culture in SCD complete medium to an OD600 of 0.2. After cultivation at 30°C to an OD600 of 0.7, the cells were either left untreated or stressed with either 2 mM DTT or 1.5 µg/ml Tunicamycin for 1 h. For inositol depletion, exponentially growing cells were washed with inositol-depleted media and used to inoculate the main culture to an OD600 of 0.5 in SCD complete w/o inositol, which was further cultivated for 3 h.
Live cell confocal microscopy
A fresh culture in SCD medium was inoculated to an OD600 = 0.2 and cultivated for 5.5 h. To induce ER-stress, DTT was added to a final concentration of 2 mM followed by additional cultivation for 1 h. The cells were harvested placed on microscopic slides coated with a thin layer of SCD containing 1.5% agarose for immobilization. Microscopy was performed using a Zeiss LSM 780 confocal laser scanning microscope (Carl Zeiss AG) with spectral detection and a Plan-Apochromat 63x 1.40 NA oil immersion objective. GFP fluorescence was excited at 488 nm and the emission was detected between 493 and 598 nm. Transmission images were simultaneously recorded using differential interference contrast (DIC) optics. Z-stacks (450 nm step-size, 62.1 µm pinhole size) were recorded. Ire1-clusters were identified by automated segmentation using CellProfiler (54). In brief, cellular areas of the field of view were determined based on sum projections of recorded z-stacks. Clusters within these cells were identified by adaptive thresholding of the respective maximum intensity z-projections.
Preparation of cell lysates and immunoblotting
Lysates were prepared from exponentially growing cells, which were harvested by centrifugation (3.000xg, 5 min, 4°C) and then washed once with ddH2O and once with PBS. During washing, the cells were transferred into 1.5 ml reaction tubes allowing for a more rapid centrifugation (8.000xg, 20 sec, 4°C). The tubes with the washed cell pellet were placed in a −80°C freezer and stored until further use.
For preparing a cell lysate, either 5 or 20 OD equivalents were resuspended in 400 µl or 1000 µl lysis buffer (PBS containing 30 µg/ml protease inhibitor cocktail), respectively. After addition of either 100 µl or 500 µl of zirconia beads, respectively, the cells were disrupted by bead beating for 5 min at 4°C. Four volumes units of the resulting lysate were mixed with one volume of 5x reducing sample buffer (8M urea, 0.1 M Tris-HCl pH 6.8, 5 mM EDTA, 3.2% (w/v) SDS, 0.15% (w/v) bromphenol blue, 4% (v/v) glycerol, 4% (v/v) β-mercaptoethanol) and then incubated at 95°C for 10 min for fully unfolding and solubilizing the proteins therein. 0.1 OD equivalents of the resulting sample was subjected to SDS-PAGE and the proteins were separated on 4-15% Mini-PROTEAN-TGX strain-free gels (BioRad). For subsequent immuno-blotting, proteins were transferred from the gel to methanol-activated PVDF membranes using semi-dry Western-Blotting. Specific proteins were detected using antigen-specific primary antibodies, HRP-coupled secondary antibodies, and chemiluminescence.
Assaying the resistance to ER-stress
Stationary overnight cultures were used to inoculate a fresh culture to an OD600 of 0.2. After cultivation for 5 to 7 h at 30°C the cells were diluted with pre-warmed medium to an OD600 of 0.05. 50 µl of these diluted cultures were mixed in a 96-well plate with 180 µl of medium and 20 µl of a DTT dilution series leading to a final concentration of DTT between 0 and 2 mM and 0 and 4 mM, respectively. After incubation at 30°C for 18 h, the cultures were thoroughly mixed and 200 µl of the cell suspension were transferred to a fresh 96-well plate for determining the density of the culture via a photospectrometer using the OD600/OD620.
RNA preparation, cDNA synthesis and quantitative real-time (qPCR) PCR analysis
Exponentially growing cells were used to inoculate a 20 ml culture to an OD600 of 0.2 in YPD. After cultivation for 3 h, the cells were either left untreated or stressed with either 4 mM DTT or 1.0 µg/ml Tunicamycin. After an additional cultivation for 1 h, 5 OD equivalents were harvested by centrifugation (3.000xg, 5 min, RT). The supernatant was discarded while the pellet was snap frozen in liquid N2 and stored at −80°C. The RNA was prepared from these cells using the RNeasy Plus RNA Isolation Kit (Qiagen). 500 ng RNA of the total isolated RNA were used as a template for the synthesis of cDNA using Oligo(dT) primers and the SuperscriptTM II RT protocol (Invitrogen). qPCR was performed using ORA qPCR Green ROX L Mix (HighQu) in a Piko Real PCR system (Thermo Scientific). The following primers were used to amplify DNA sequences of interest:
HAC1s forward primer: 5’ – CTTTGTCGCCCAAGAGTATGCG – 3’
HAC1s reverse primer: 5’ – ACTGCGCTTCTGGATTACGC – 3’
ACT1 forward primer: 5’ – TGTCACCAACTGGGACGATA – 3’
ACT1 reverse primer: 5’ – AACCAGCGTAAATTGGAACG – 3’
All reactions were performed in technical duplicates. Non-template controls (RNA) and non-reaction controls (H2O) were additionally performed. The qPCR program included the following steps: 1) 95°C, 15 min; 2) 95°C, 20 sec; 3) 58°C, 20 sec; 4) 72°C, 30 sec; 5) 72°C, 5 min; steps 2-4 were repeated 40 times. The relative quantification of HAC1 splicing levels was based on the comparative ΔΔCT method using normalization to ACT1 levels (StepOnePlusTM user Manual, Applied Biosystems).
Microsomal membrane preparation
80 OD600 equivalents were harvested from a mid-exponential culture by centrifugation (3.000xg, 5 min, 4°C), washed with PBS, and stored at −80°C. All steps of membrane fractionation were performed on ice or at 4°C with pre-chilled buffers. The cell pellets were resuspended in 1.5 ml lysis buffer (50 mM HEPES pH 7.0, 150 mM NaCl, 1 mM EDTA, 10 μg/ml chymostatin, 10 μg/ml antipain, 10 μg/ml pepstatin). For cysteine crosslinking experiments, a lysis buffer without EDTA was used. Cells were disrupted by using zirconia beads (Roth) and a bead beater (2 x 5 min). Cell debris was removed from the lysate by centrifugation (800x g, 5 min, 4°C) and further cleared from major mitochondrial contaminations (5,000 x g, 10 min, 4°C). The pellet was discarded, and the supernatant was spun again (100.000x g, 45 min, 4°C) to obtain a crude microsomal fraction in the pellet. The microsomes were resuspended in 1.4 ml lysis buffer, sonicated for homogenization (50%, 5×1sec, MS72 tip on a sonifier cell disrupter from Branson Ultrasonic) snap frozen in liquid N2 in small aliquots to avoid freeze-thaw cycles, and stored at −80°C for further use.
Test of membrane association
The cleared supernatant of a 5.000xg step was divided into equal parts, which were then mixed with an equal volume of lysis buffer supplemented with 0.2 M Na2CO3 resulting in a final pH of 11, 5 M urea, 2% Triton X-100 or without additional additives. After incubation for 1 h on a rotator, these samples were centrifuged (100,000x g, 45 min, 4°C) to separate soluble from insoluble material. The supernatant and pellets from these fractions corresponding to 0.2 OD equivalents were further analyzed by SDS-PAGE and immunoblotting.
CuSO4-induced cysteine crosslinking
For cysteine crosslinking with CuSO4, the microsomes were thawed on ice. 8 µl of microsomes with a protein concentration of 1 ± 0.2 mg/ml were then incubated with 2 µl of 50 mM CuSO4 in ddH2O resulting in a final concentration of 10 mM CuSO4 and incubated for 5 min. A sample supplemented with 2 µl ddH2O served as a non-crosslinking control. The reaction was stopped with 8 µl of membrane sample buffer (4 M urea, 50 mM Tris-HCl pH 6.8, 1.6 % (w/v) SDS, 0.01% (w/v) bromophenol blue, 2% (v/v) glycerol) including 125 mM EDTA and 250 mM NEM. Samples were heated for 5 min at 95°C and further analyzed by SDS-PAGE and immunoblotting. The percentage of dimer formation was determined using the densiometric signals of the bands corresponding to the monomeric and covalently dimeric protein, which were determined using ImageJ.
Immunoprecipitation from microsomes after CuSO4-induced cysteine crosslinking
300 µl of microsomes with a typical protein concentration of 1 mg/ml were incubated with 12.5 µl 250 mM CuSO4 (final concentration of 10 mM) for 5 min on ice. The reaction was stopped by adjusting the sample to a final concentration of 50 mM EDTA and 111 mM NEM by adding 30 µl of 0.5 M EDTA stock solution and 44 µl of 1 M NEM stock solution, respectively. The final volume was adjusted to 1.3 ml with lysis buffer with a final concentration of 5 mM EDTA. The CuSO4 concentration was thus reduced to 2.4 mM and the NEM concentration to 33.6 mM, respectively.
After crosslinking, the microsomes were solubilized using 2% Triton X-100 and incubated for 1 h at 4°C under constant agitation. Insoluble material was removed by centrifugation (20.000x g, 10 min, 4°C). The resulting supernatant was incubated with 8 µl Flag beads (Sigma Aldrich), equilibrated with IP wash buffer (lysis buffer + 5 mM EDTA + 0.2 % Triton X-100), for 3 h under constant shaking. Flag beads were washed five times with IP wash buffer by centrifugation (8.000xg, 30 sec, 4°C). For elution, the Flag beads were incubated with 10 µl IP-Wash and 10 µl 5x reducing sample buffer for 5 min at 95°C, which did not disrupt the disulfide bond formed between to protomers of Ire1. These samples were analyzed by SDS-PAGE and immunoblotting.
Modelling of the dimer
We extracted an equilibrated conformation of a monomeric 56 amino-acid long sequence 516-SRELD EKNQNSLLLK FGSLVYRIIE TGVFLLLFLI FCAILQRFKI LPPLYVLLSK I-571 from a previously performed 10 µs long equilibrium MD simulation. We duplicated the configuration in order to create a new system containing two identical protomers. We then rotated and translated one of the two protomers to form a dimer structure, such that the two F544 faced each other with the distance between their Cβ atoms at around 0.7 nm. A short energy minimization in solution resolved all steric clashes between side-chains. The structure of the model dimer was prepared by using gromacs/2019.3 tools (55) and VMD (56). We used Charmm-GUI (57, 58) to reconstitute the dimer in a bilayer containing 248 POPC and 62 cholesterol molecules modelled in the Charmm36m force-field (59–61). We solvated the system with 24813 TIP3P water molecules, 72 chloride and 66 sodium ions, corresponding to a salt concentration of 150 mM.
Equilibrium and restrained simulations of the dimer model
After an initial energy minimization and quick relaxation, we carefully equilibrated the dimer model in the bilayer. We first ran a 50 ns long simulation restraining the position of protein atoms by using harmonic potentials with force-constants (in units of kJ mol-1 nm2) of 500 for backbone atoms and 200 for side-chain atoms. We then ran further 50 ns lowering the force-constants to 200 and 50, respectively. After this equilibration, we relieved all restraints and ran two independent 500 ns long MD simulations. In the first simulation, the system evolved according to its unbiased dynamics; in the second, we restrained the distance between the two Cβ atoms of F544 on each protomer, gradually driving them closer in time. We first centered the restraining harmonic potential around 0.5 nm with a force-constants (in units of kJ mol-1 nm2) of 1000. We then lowered the equilibrium distance of the potential and decreased the force-constants (in units of kJ mol-1 nm2): 0.4 nm and 750 after 10 ns from the beginning of the run; 0.3 nm and 500 after 20 ns; and 0.2 nm and 500 after 30 ns and for the remaining duration of the simulation.
We ran both the restrained equilibration and unbiased production simulation in gromacs/2019.3, and the restrained production simulation in gromacs/2018.7 patched with the open-source, community-developed PLUMED library (62) version 2.5 (63), using a time step of 2 fs. Electrostatic interactions were evaluated with the Particle-Mesh-Ewald method (64). We maintained a constant temperature of 303 K (65), applying separate thermostats on the protein, membrane, and solvent with a characteristic time of 1 ps. We applied the semi-isotropic Berendsden barostat (66) for the restrained equilibration, and the Parrinello-Rahman barostat (67) for the production runs, acting separately on the x-y plane and z direction to maintain a constant pressure of 1 atm, and with a characteristic time of 5 ps. We constrained all hydrogen bonds with the LINCS algorithm (68). Molecular visualizations were obtained with VMD and rendered with Tachyon.
Data analysis, representation, and statistical tests
The amount of crosslinked dimer was determined using the densiometric signals of the bands corresponding to the monomeric and covalently dimeric protein. The percentage was calculated from the ratio of dimer signal to whole Ire1 signal. The software Image J was used for the determination if the densiometric signal.
Throughout the manuscript, the data are represented as the average ± SEM. All experiments were performed at least in triplicates. The significance was tested by unpaired Student’s t tests. Further information regarding statistical analysis can be found in the figure legends. In the following, the exact value of n is provided for every dataset of this paper.
Fig. 1B: left panel: ΔIRE1, WT: n=21 (technical replicates from four individual colonies); cysteine-less: n=12 (technical replicates from four individual colonies)
right panel: ΔIRE1: n=15 (technical replicates from three individual colonies); cysteine-less: n=12 (technical triplicates from four individual colonies); WT: n=9 (technical triplicates from three individual colonies)
Fig. 1C: left panel: WT -DTT: n= 4; WT +DTT: n=6; cysteine-less -DTT: n=6; cysteine-less +DTT: n=5
Right panel: WT -TM: n=4; WT +TM: n=5; cysteine-less -TM: n=6; cysteine-less +TM: n=4
Fig. 3D: DTT: E540C, T541C: n=8 (technical duplicates from four individual colonies); G542C, V543C, L545C, L546C, L547C, F548C, L549C, I550C, F551C: n=4 (technical duplicates from two individual colonies); F544C: n=14 (technical duplicates from seven individual colonies); C552: n=11 (technical duplicates from six individual colonies)
TM: E540C, T541C: n=8 (technical duplicates from four individual colonies); G542C, V543C, L545C, L546C, L547C, F548C, L549C, I550C, F551C: n=4 (technical duplicates from two individual colonies); F544C: n=10 (technical duplicates from five individual colonies); C552: n=8 (technical duplicates from four individual colonies)
Inositol depletion: n=6 (technical duplicates from three individual colonies)
Fig. 5A: ΔIRE1, cysteine-less: n=12 (technical replicates from two individual colonies), F544A C552: n=12 (technical triplicates from four individual colonies)
Fig. 5B: DTT: C552: n=11 (technical duplicates from six individual colonies), F544A C552: n=4 (technical duplicates from two individual colonies)
TM: C552: n=8 (technical duplicates from four individual colonies), F544A C552: n=4 (technical duplicates from two individual colonies)
Fig. 5C: ΔIRE1, cysteine-less: n=12 (technical replicates from two individual colonies), F531R F544C: n=12 (technical triplicates from four individual colonies)
Fig. 5D: DTT: F544C: n=14 (technical duplicates from seven individual colonies), F531R F544C: n=4 (technical duplicates from two individual colonies)
TM: F544C n=10 (technical duplicates from five individual colonies), F531R F544C: n=4 (technical duplicates from two individual colonies)
Fig. S1B: left panel: WT -DTT: n= 5; WT +DTT: n=5; cysteine-less -DTT: n=6; cysteine-less +DTT: n=6
Right panel: WT -TM: n=5; WT +TM: n=5; cysteine-less -TM: n=6; cysteine-less +TM: n=3
Fig. S3A: OD620: ΔIRE1: n=9 (technical triplicates from three individual colonies): cysteine-less: n=12 (technical triplicates from four individual colonies); E540C – L546C: n=6 (technical triplicates from two individual colonies) OD600: ΔIRE1: n=10 (technical replicates from three individual colonies): cysteine-less: n=22 (technical triplicates from four individual colonies); L547C – F551: n=6 (technical triplicates from two individual colonies)
Fig. S4A: ΔIRE1, cysteine-less: n=12 (technical replicates from two individual colonies), F544R C552: n=9 (technical triplicates from three individual colonies)
Fig. S5B: DTT: C552: n=11 (technical duplicates from six individual colonies), F531R C552: n=4 (technical duplicates from two individual colonies)
TM: C552: n=8 (technical duplicates from four individual colonies), F531R C552: n=4 (technical duplicates from two individual colonies
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft (SFB807 ‘Transport and Communication across Biological Membranes’ to R.E. and G.H.; SFB894 ‘Ca2+-Signals: Molecular Mechanisms and Integrative Functions’ to R.E). R.C. and G.H. were supported by the Max Planck Society. We thank Carsten Mattes for excellent technical support and especially Kristina Halbleib for her important contributions during the early phase of the project.
References




