

Abstract
Surface attachment, an early step in the colonization of multiple host environments, activates the virulence of the human pathogen P. aeruginosa. However, the signaling pathways and downstream toxins specifically induced by surface association to stimulate P. aeruginosa virulence are not fully understood. Here, we demonstrate that alkyl-quinolone (AQ) secondary metabolites are rapidly induced upon surface association and represent a major class of surface-dependent cytotoxins. AQ cytotoxicity is direct and independent of other AQ functions like quorum sensing or PQS-specific activities like iron sequestration. Furthermore, the regulation of AQ production can explain the surface-dependent virulence regulation of the quorum sensing receptor, LasR, and the pilin-associated candidate mechanosensor, PilY1. PilY1 regulates surface-induced AQ production by repressing the AlgR-AlgZ two-component system. AQs also contribute to the known cytotoxicity of secreted outer membrane vesicles. These findings collectively explain previously mysterious aspects of virulence regulation and provide new avenues for the development of anti-infectives.
Introduction
The opportunistic human pathogen P. aeruginosa infects a wide range of hosts such as mammals, plants, insects, and fungi (Rahme et al., 1995), and is a major contributor to the morbidity of cystic fibrosis patients (Nixon et al., 2001) and hospital-acquired infections (Richards et al., 1999). P. aeruginosa uses a large set of secreted proteins and secondary metabolites to carry out the multiple requirements necessary for a successful infection, including host colonization, immune evasion, nutrient acquisition, and host cell killing (cytotoxicity) (Valentini et al., 2018). Given the multiple activities involved in pathogenesis, we recently developed a quantitative imaging-based host cell killing assay to specifically study the factors acutely required for killing host cells during short timescales (Siryaporn et al., 2014). This assay revealed that cytotoxicity is activated by attachment of P. aeruginosa to a solid surface (Siryaporn et al., 2014). This surface-induced cytotoxicity does not require the Type-IV Pilus (TFP), TFP-associated signaling complexes (PilA-Chp-Vfr/cAMP), or Type III Secretion Systems (T3SS), but does require two regulatory proteins, LasR and PilY1 (Siryaporn et al., 2014). Since well-characterized cytotoxins such as T3SS and Vfr targets are not necessary for surface-induced host-cell killing in this assay, we sought to address the outstanding questions of which specific toxins mediate host cell killing in response to surface attachment and how these toxins are regulated by LasR and PilY1.
LasR is an important component of the complex network of P. aeruginosa quorum sensing (Lee and Zhang, 2015). Quorum sensing (QS) is the process by which bacteria synthesize and secrete autoinducer signaling molecules that accumulate and activate their receptors as a function of bacterial cell density. There are at least three QS systems that have been previously implicated in regulating P. aeruginosa virulence: the las, rhl, and pqs QS systems (Lee and Zhang, 2015). These systems form a complex and interconnected network with extensive regulatory cross-talk (Maura et al., 2016). For example, LasR transcriptionally upregulates the autoinducer synthase enzymes of the rhl and pqs systems (Xiao et al., 2006b; Farrow et al., 2008), which in turn activate numerous downstream factors (Farrow et al., 2008). As a result, identifying the specific contribution of LasR dependent QS to the phenomenon of surface-induced virulence has been challenging.
Besides LasR, the other factor known to be required for surface-induced virulence is PilY1 (Siryaporn et al., 2014), a minor pilin-associated protein with a putative mechanosensory domain (Bohn et al., 2009). PilY1 promotes several surface-dependent behaviors including virulence induction (Siryaporn et al., 2014), twitching motility (Bohn et al., 2009), and biofilm formation (Kuchma et al., 2015; Luo et al., 2015). Like QS, there are multiple signaling pathways that have been proposed to function downstream of PilY1 (Luo et al., 2015). However, disrupting the key effectors of the two best-characterized pathways downstream of PilY1, c-di-GMP or cAMP production, does not influence surface-induced virulence (Siryaporn et al., 2014).
Understanding the signaling pathways that LasR and PilY1 use to trigger surface-induced virulence has been particularly challenging because the relevant output of these pathways, such as the cytotoxin(s) that P. aeruginosa secretes to kill host cells when surface-associated, has remained unknown. P. aeruginosa possesses numerous candidate toxins that could mediate surface-induced virulence, including the type III secretion system (T3SS) and numerous other secreted proteins and secondary metabolites (Valentini et al., 2018). Many of these candidates were previously found not to be required for surface-induced virulence (Siryaporn et al., 2014), which could reflect functional redundancy or the existence of a previously-overlooked cytotoxin.
Here we identify the pathways that activate surface-induced virulence by first showing that a single family of cytotoxins, the alkyl-quinolones (AQs), are both necessary and sufficient to explain the surface-induced killing of Dictyostelium discoideum by P. aeruginosa. We demonstrate that surface association triggers increased AQ secretion, which requires both LasR and PilY1. We show that these findings are also relevant to mammalian host cells and demonstrate that AQs are a major cytotoxic component of secreted outer membrane vesicles. Together our data support the conclusion that surface-induced virulence results from induction of AQs, which act as toxins that directly kill host cells.
Results
Alkyl-quinolones are necessary and sufficient for surface-induced virulence of P. aeruginosa towards D. discoideum
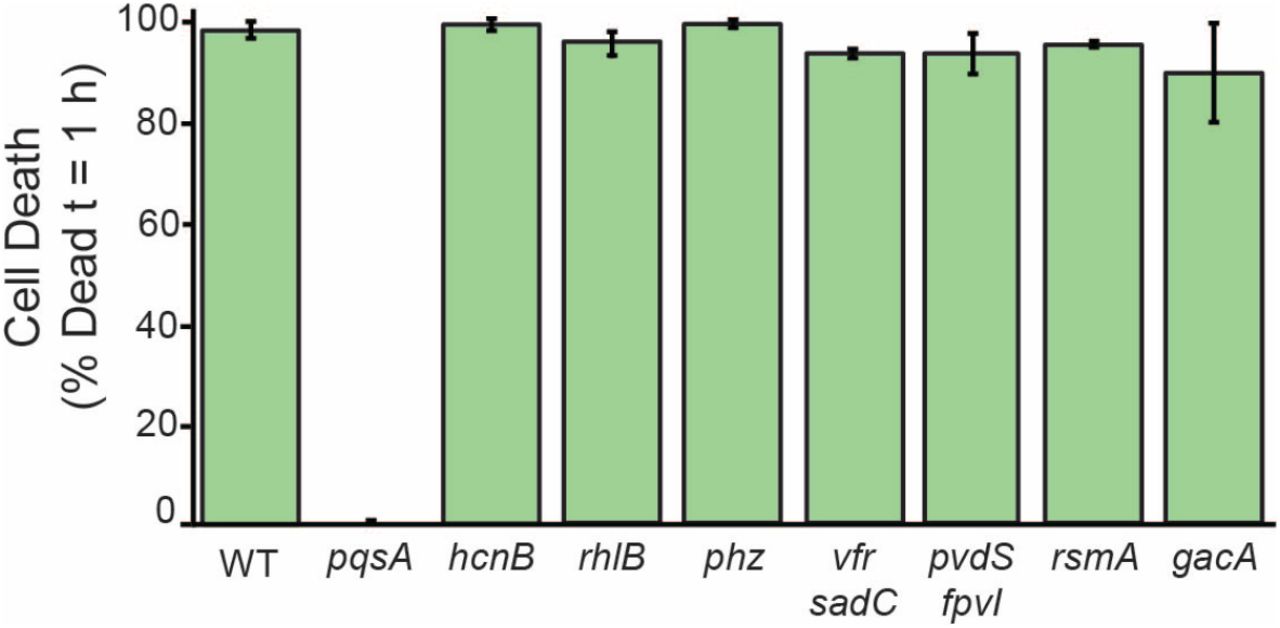
Surface attachment strongly stimulates the ability of P. aeruginosa PA14 to kill D. discoideum amoebae (Siryaporn et al., 2014). To identify the factors required for virulence of surface-associated P. aeruginosa we used our surface-induced virulence assay to screen a number of mutants in secreted effectors known to promote pathogenesis (Figure 1-S1). specifically, we grew each mutant to the same density, allowed it to associate with a glass surface for 1 hour, added D. discoideum host cells, and monitored host cell death by fluorescence microscopy using the live-cell-impermeant dye, calcein-AM. Loss of many candidate P. aeruginosa cytotoxins did not reduce surface-induced killing of D. discoideum, including phenazines, rhamnolipids, and hydrogen cyanide (Figure S1). In contrast, pqsA was absolutely required for surface-induced virulence (Figure 1A). PqsA is an enzyme required for the biosynthesis of AQs such as PQS, HHQ, and HQNO (Coleman et al., 2008), suggesting that AQs play a key role in surface-induced virulence.

Quantification of D. discoideum killing by surface-attached P. aeruginosa mutants after 1 hour of co-culture. Cell death was indicated by positive staining by the fluorescent dye calcein-AM. Values are averages of three independent experiments and error bars represent standard error. Approximately 150-300 cells were analyzed for each measurement.

Alkyl-quinolone production is necessary and sufficient for surface-induced killing of D. discoideum by P. aeruginosa. (A) D. discoideum feeding on surface-attached wild type and ΔpqsA P. aeruginosa after 1 h co-culture (scale bars = 30 μm). Fluorescent calcein-AM staining indicates cell death. (B) Schematic of the PQS pathway depicting the functions of relevant genes. Solid and dotted arrows represent biosynthetic reactions and gene regulation, respectively. (C) Quantification of D. discoideum killing by PQS pathway mutants. Expression of pqsABCDE is driven by the endogenous pqsA promoter or a strong constitutive promoter inserted upstream of the pqsA gene (Pconst − pqsABCDE) (D) Cytotoxicity of purified HHQ to D. discoideum under conditions of the cell death assay in (C). (E) Axenic growth of D. discoideum in the presence of purified HHQ or PQS. Values are mean ± SD (n = 5). Values in (C) and (D) are mean ± SE (n = 3). For statistical analysis in (C), mutants were compared to wild type (Student’s t-test, two-tailed, * = p<0.05, ***p<0.001). Approximately 200-300 cells were analyzed for each measurement in (C) and (D).
Figure 1–Figure supplement 1. Virulence of various P. aeruginosa mutants towards D. discoideum.
Figure 1–Figure supplement 2. Virulence of pqsA and pqsH mutants supplemented with HHQ.
Figure 1–Figure supplement 3. Quantification of AQ production in Pconst − pqsABCDE strains by LC/MS
The alkyl-quinolone (AQ) family of small molecules in P.aeruginosa performs a diverse set of virulence-related functions including quorum-sensing signaling (Rampioni et al., 2016), iron acquisition (Diggle et al., 2007), immune suppression (Kim et al., 2010), and anti-bacterial activities (Hazan et al., 2016). One AQ species, PQS, has been suggested to possess cytotoxic activity towards some mammalian cell types (Abdalla et al., 2017) and HQNO can disrupt electron transport in vitro (Reil et al., 1997), but it has remained unclear if AQs are important for surface-induced virulence and if so, which AQ species and activities mediate this virulence. To determine the parts of the AQ pathway responsible for surface-induced virulence of P. aeruginosa towards D. discoideum, we assayed mutants deficient in the four keys steps of the AQ pathway (Figure 1B): 1) converting the anthranilate precursor to HHQ (mediated by PqsABCD), 2) converting HHQ into PQS and HQNO (by PqsH and PqsL, respectively), 3) feedback regulation onto pqsABCDE expression (by PQS and HHQ activating the transcriptional regulator PqsR, also known as MvfR), and 4) stimulating RhlR-dependent QS (by PqsE). Neither the production of HQNO (ΔpqsL) nor the activation of rhlR-dependent targets (Δ pqsE) was required for the killing of D. discoideum by P. aeruginosa (Figure 1C). However, pqsA, pqsH and pqsR mutants showed reduced ability to kill D. discoideum (Figure 1C).
The reduced virulence of Δ pqsH suggested that PQS contributes to surface-induced virulence, which could be due to its role in iron acquisition, its QS ability to activate PqsR, or its activity as a cytotoxin. To address the potential role of iron binding, we relied on the fact PQS binds iron while its HHQ precursor does not (Diggle et al., 2007). Deleting pqsH, the enzyme that converts HHQ to PQS, results in reduced levels of all AQs due to the absence of PqsR-mediated feedback induction. We thus transiently supplemented pqsH and pqsA mutants with HHQ for 1 hour at concentrations sufficient to induce PqsR (50 μM). We then washed away the HHQ before exposing these bacteria to D. dictyostelium, such that during the virulence assay the pqsA mutants would have no HHQ or PQS while pqsH mutants would have HHQ but no PQS. We found that transient HHQ addition was sufficient to restore WT-levels of virulence to pqsH mutants but not to pqsA mutants (Figure 1-S2), indicating that HHQ is required for surface-induced virulence but PQS is not.

Quantification of D. discoideum killing by surface-attached P. aeruginosa pqsH and pqsA mutants supplemented with exogenous HHQ or PQS. HHQ, PQS, or DMSO was added to cultures after the initial 1:100 dilution, and then cultures were grown following the procedures for the D. discoideum cell death assay, as described in Materials and Methods. Surface attached and planktonic bacteria were washed twice in PSDB to remove any exogenous HHQ or PQS prior to incubating the bacteria with D. discoideum. Values are averages of three independent experiments and error bars represent standard error. Approximately 300-500 cells were analyzed for each measurement
Having eliminated PQS-specific roles of AQs such as iron sequestration, we next tested if surface-induced virulence requires PqsR activation to achieve high levels of the AQs themselves or if additional PqsR targets (Maura et al., 2016) might be required. Consequently, we replaced the endogenous pqsA promoter with a strong constitutive promoter (POXB20), which we refer to as (Pconst). This construct was sufficient to fully restore the virulence of pqsR and pqsH mutants to WT levels (Figure 1C). These results suggest that the requirement of PQS production for full virulence is due to the ability of PQS to promote high levels of pqsABCDE by activating the PqsR-dependent positive feedback loop. To further eliminate the possibility of PqsE-mediated QS, we simultaneously deleted pqsE, pqsH, and pqsR in the (Pconst − pqsABCDE) background and showed that these bacteria retained PqsA-dependent virulence (Figure 1C). To confirm that each of the strains analyzed retained the expected ability to produce HHQ and/or PQS, we used LC/MS to directly quantify these molecules in the bacterial supernatants and found the expected results in all cases (Figure 1-S3).

Quantification of alkyl-quinolone concentrations in wild type and mutant P. aeruginosa cultures using LC/MS. Stationary phase cultures were extracted with ethyl acetate, dried, and resuspended in methanol. Samples were analyzed by LC/MS, and AQ concentrations were calculated using a standard curve constructed from commercial AQ standards. Values are averages of three independent samples and error bars represent standard error (n.d.=not detected).
The ability of high expression of pqsABCDE to induce virulence in the absence of pqsH suggested that surface-induced virulence is mediated either by HHQ itself, or by another previously uncharacterized product of pqsABCDE. To determine if HHQ is not just required, but also sufficient for killing D. discoideum we obtained commercially-purified HHQ (Sigma Aldrich, St. Louis, MO) and determined its lethal dose under the conditions used in the surface-induced virulence assay (Figure 1D). Purified HHQ induced rapid cell death in D. discoideum at concentrations above 30 μM (Figure 1D), and inhibited growth of D. discoideum at concentrations as low as 1 μM when added to axenic D. discoideum cultures (Figure 1E). While PQS was not necessary for virulence, PQS also acted as a direct cytotoxin as purified PQS killed D. discoideum (Figure 1E). Together, our results demonstrate that HHQ and PQS are cytotoxic towards D. discoideum, that AQ production is both necessary and sufficient for surface-induced virulence of P. aeruginosa, and that AQs other than PQS can mediate this toxicity.
AQ regulation can explain the effects of known surface-induced virulence regulators
We previously showed that LasR and PilY1 are required for surface-induced virulence (Siryaporn et al., 2014). To understand whether the virulence defects of these mutants are due to loss of AQ production, we determined if they can be rescued by replacing the endogenous pqsA promoter with the POXB20 strong constitutive promoter (Pconst). This pqsABCDE overexpression restored full virulence to surface-associated lasR and pilY1 deletion mutants (Figures 2A and 2B). Furthermore, constitutive pqsABCDE expression was sufficient to induce detectable virulence in planktonic bacteria (Figures 2A and 2B). While the virulence achieved by expression of pqsABCDE in planktonic cells did not reach the same levels as the surface-attached bacteria, the conversion of avirulent cells to a virulence state with only the expression of a single operon is notable.

PilY1 and LasR promote surface-induced virulence through pqsABCDE expression.(A) Representative images of D. discoideum feeding on surface-attached and planktonic P. aeruginosa expressing pqsABCDE genes under control of the endogenous pqsA promoter (PpqsA − pqsABCDE) or a strong constitutive promoter (Pconst − pqsABCDE) after 1 hour of co-culture (scales bars = 30 μm). Fluorescent calcein-AM staining indicates cell death. (B) Quantification D. discoideum killing by surface-attached and planktonic wild type, ΔpilY1 and ΔlasR P. aeruginosa after 1 h co-culture. (C) Mean fluorescence intensity per cell (>500 cells) of surface-attached P. aeruginosa expressing a (PpqsA − mCherry) promoter fusion. Values are mean ± SE (n = 3). (D) LC/MS-based quantification of PQS in extracts of wild type, ΔpilY1, and ΔlasR in stationary-phase liquid cultures. Values are mean ± SE (n = 3), and concentrations were calculated using a standard curve constructed from purified PQS standards (n.d.= not detected). (E) Quantification D. discoideum killing by mutants in the AlgR-PilY1 pathway after 1 h co-culture. Values in (B) and (E) are mean ± SE (n = 3). Statistical analysis mutants in (C) were performed against wild type (Student’s t-test, two-tailed, * = p<0.05, ** p <0.01, ***p<0.001). Approximately 150-300 cells were analyzed for each measurement in (B) and (E).
We next sought to determine if pqsABCDE expression is altered in lasR and pilY1 mutants using a fluorescent reporter fusion to the pqsABCDE promoter. We fused a 500-bp fragment upstream of the pqsA gene to a promoterless mCherry gene, and integrated this construct at a neutral chromosomal locus. Wild type, ΔlasR, and ΔpilY1 bacteria expressing this reporter were grown to mid-exponential phase (OD600nm = 0.6), and OD-matched cultures were allowed to attach to a surface for 1 hour. We measured the mean fluorescence intensity of individual surface-attached bacteria and normalized them to a constitutively expressed fluorescent reporter. The pqsABCDE promoter activity of both ΔlasR and ΔpilY1 was significantly lower than that of wild type (Figure 2C). To determine if the decreased promoter activity impacts PQS production, we measured PQS levels in ethyl acetate extracts of ΔlasR and ΔpilY1 cultures using LC/MS. This revealed that PQS production is decreased in both ΔlasR and ΔpilY1 compared to wild type (Figure 2D). Together these results suggest that the effects of known virulence regulators can be explained by their effects on pqsABCDE operon expression.
LasR has previously been show to induce the PqsR pqsABCDE regulator (Xiao et al., 2006b), but the connection between PilY1 and pqsABCDE expression has not been previously reported. Since the well-characterized c-di-GMP and cAMP pathways do not appear responsible for PilY1-mediated virulence induction (Siryaporn et al., 2014), we compared the previously-reported surface-dependent transcriptional changes in WT and pilY1 mutants (Siryaporn et al., 2014). We found that both the AlgR-AlgZ TCS and its associated regulon are repressed in a PilY1-dependent-manner (Siryaporn et al., 2014; Belete et al., 2008). To test if AlgR can regulate virulence we generated a phosphomimetic mutation in algR (algRD54E) and overexpressed it from a multi-copy plasmid. AlgRD54E overexpression strongly inhibited virulence (Figure 2E). To determine if AlgR functions downstream of PilY1 we turned to epistasis analysis and generated a pilY1 algR double mutant, which restored virulence to the avirulent pilY1 single mutant (Figure 2E). Thus, algR is epistatic to pilY1 and likely functions downstream of it. To determine if PilY1 acts on the levels or phosphorylation state of AlgR, we replaced the chromosomal copy of algR with a phosphomimetic algRD54E allele. Unlike in the case of overexpressing algRD54E from a plasmid, chromosomal expression of algRD54E did not affect virulence (Figure 2E), which suggests that PilY1 functions by transcriptionally repressing the levels of algR.
Virulent surface-attached cells secrete more AQs than avirulent planktonic cells
AQs have not been previously described to be surface-regulated, but our findings above suggested that surface attachment might stimulate AQ production. AQ quantification under the conditions of our surface-induced virulence assay is challenging using traditional MS-based techniques. Consequently, we developed a fluorescence-based AQ biosensor that can be used to measure PQS and HHQ levels under the exact conditions of the surface-induced D. discoideum cell death assay. Specifically, we engineered a reporter strain with three features: 1) it is unable to synthesize AQ’s itself (due to deletion of pqsA), 2) it linearly responds to PqsR activation without quorum-sensing feedback (due to replacement of the pqsR promoter with a constitutive Ptac promoter), and 3) it has a plasmid containing both a fluorescent YFP reporter for PqsR activation by AQs (PpqsA − YFP) and a constitutive mKate reporter (PrpoD−mKate) to normalize for plasmid copy number.
We validated our AQ biosensor by analyzing its response to purified AQ standards (Sigma-Aldrich, St. Louis, MO) in a 96-well format and by examining it in mutants with known effects on AQ production. The AQ biosensor responded linearly to PQS and HHQ across the range of AQ levels previously reported (Xiao et al., 2006a) for P. aeruginosa cultures (Figure 3A). Consistent with the known binding affinities of PqsR (Xiao et al., 2006a) the sensor responded more strongly to PQS than HHQ and did not respond to HQNO (Figure 3A and 3B). To use the biosensor to monitor AQ production by P. aeruginosa we doped the AQ reporter 1:100 into surface-attached wild type, ΔpilY1, ΔalgR, and ΔpilY1 ΔalgR. We note that the AQ biosensor is itself avirulent such that doping it at low levels (1:100) enabled us to quantify AQ production without disrupting the assay. Consistent with the epistasis results obtained with respect to virulence (Figure 2E), the AQ reporter showed reduced AQ levels upon deletion of pilY1, and this decrease was suppressed in a pilY1 algR double mutant (Figure 3-S1).

Biosensor-based quantification of AQ levels in surface-attached P. aeruginosa populations. AQ biosensor was doped into P. aeruginosa samples as described in Figure 3. Mean YFP/mKate fluorescence intensity per cell was calculated for approximately 500 cells, and values were normalized by the mean YFP/mKate fluorescence of biosensor doped into the surface-attached pqsA mutant. Values are averages of three independent experiments and error bars represent standard error. (B) Representative images of samples described in (A) (scale bars = 10 μm).
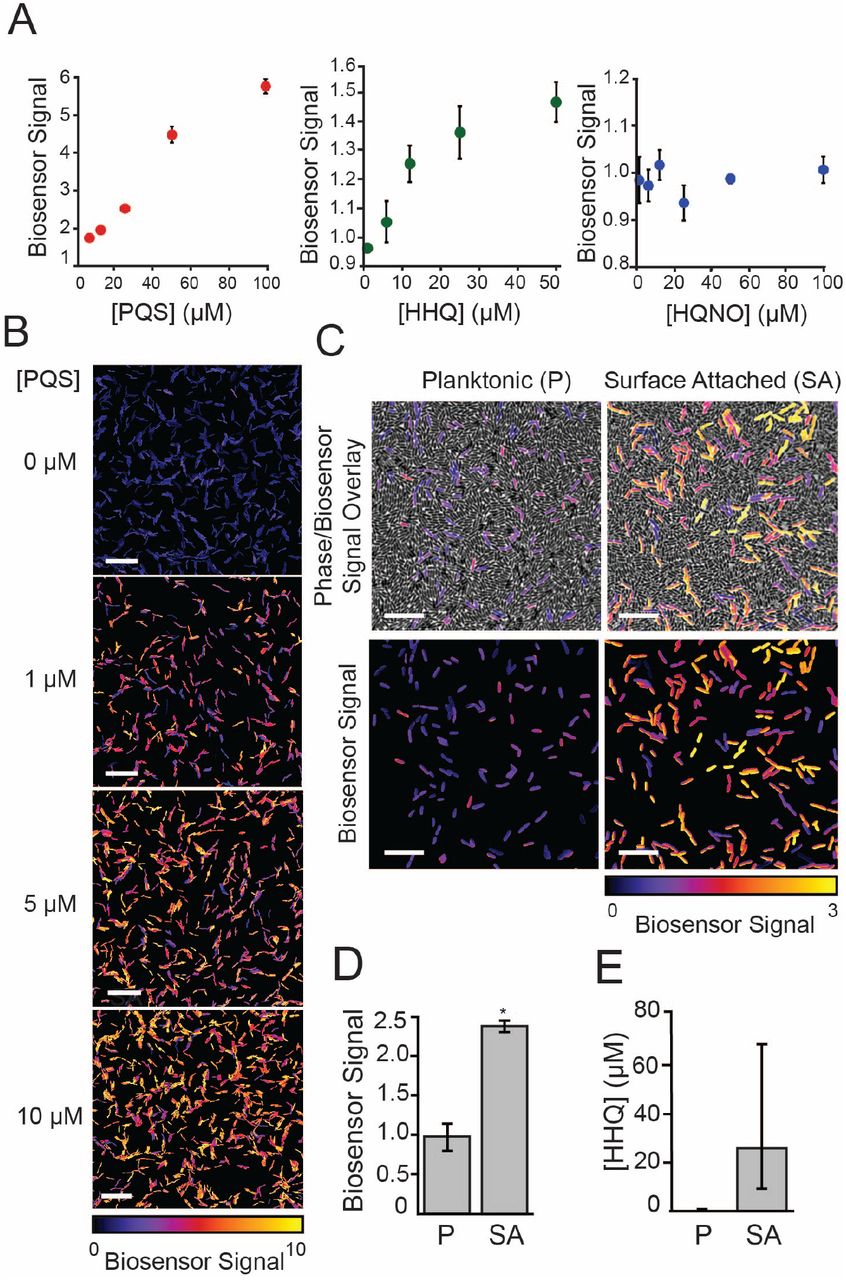
Biosensor-based detection of alkyl-quinolones in planktonic and surface-attached P. aeruginosa populations. (A) Response of AQ biosensor to increasing concentrations of purified PQS, HHQ, and HQNO in a 96-well microplate-based assay. Biosensor signal was calculated by normalizing YFP/mKate fluorescence by a DMSO control. Values are mean ± SD (n = 6). (B) Representative images of biosensor signal of single cells in response to addition of purified PQS added to 1 % agar pads used for imaging (scale bars = 10 μm). (C) Representative images of biosensor doped 1:100 into planktonic or surface-attached ΔpqsH P. aeruginosa grown under conditions of the D. discoideum cell death assay (scale bars = 5 μm). Top images show an overlay of phase images and biosensor signal and bottom images show only the biosensor signal. (D) Quantification of biosensor signal in (C). Biosensor signal is the mean YFP/mKate fluorescence per cell (>500 cells) subtracted by the value from biosensor doped into surface-attached pqsA mutant, as describe in the material and methods. (E) HHQ concentration calculated from biosensor signal in (D) using an HHQ standard (Figure 3-S1). Values in (D) and (E) are mean ± SE (n = 4-6). Statistical analysis in (D) is Student’s t-test (two-tailed, ** p <0.01).
Figure 3–Figure supplement 1. Biosensor-based quantification of AQ production in algR mutants
Figure 3–Figure supplement 2. HHQ standard curve for biosensor-based AQ quantification of surface-attached P. aeruginosa
Having validated our AQ biosensor, we used it to compare AQ levels between planktonic and surface-attached P. aeruginosa populations. Because the AQ biosensor responds to both HHQ and PQS, we focused on quantifying AQs from ΔpqsH, which makes HHQ but not PQS. We note that this strain is less virulent than wild type but retains 40% of its virulence and its virulence is still specifically induced by surface-association (Figure 1C). We doped the AQ biosensor (1:100) into cultures of DpqsH at the time of D. discoideum addition and observed a signi1cant increase in biosensor signal as compared to planktonic ΔpqsH populations (Figures 3C and 3D). Conversion of biosensor signal to HHQ concentration using an HHQ standard curve (Figure S5) indicated that surface-attached pqsH bacteria secrete at least 20-fold more HHQ than planktonic cells (Figure 3D).
AQ cytotoxicity promotes surface-induced virulence towards mammalian cells and is important for outer membrane vesicle toxicity
To determine if our findings using D. discoideum host cells can be applied to mammalian hosts, we assayed the toxicity of purified HHQ, PQS, and HQNO towards adherent TIB-67 mouse monocytes and A549 human lung epithelial cells. Specifically, we added various concentrations of each purified AQ to the mammalian cells under standard culture conditions in a 96-well format and assessed viability after 48 hours using a water-soluble tetrazolium assay (Figure 4A). Both HHQ and PQS inhibited growth of TIB-67 in range of the concentrations observed in P. aeruginosa cultures by mass spectrometry, while HQNO did not (comparing Figure 4A to Figure 1-S3). PQS was significantly more toxic to TIB-67 than HHQ (Figure 4A). Both PQS and HHQ were also cytotoxic towards the A549 lung epithelial cell line, but this activity was lower than that against TIB-67 (Figure 4A).

Cytotoxicity of alkyl-quinolones to TIB-67 mouse monocytes and human A549 epithelial cells. (A) MTT cell viability assay of A549 human bronchial epithelial cells or TIB-67 mouse monocytes after 48 h of treatment with various concentrations of the alkyl-quinolones HHQ, PQS, or HQNO in a 96-well format. Percent survival is relative to an untreated control. Values are mean ± SE (n = 3). (B) Percent survival of TIB-67 monocytes after 18 h co-cultured with surface-attached P. aeruginosa. Values are mean ± SE (n = 5). (C) Representative images of TIB-67 monocytes co-cultured with surface-attached P. aeruginosa or treated with exogenous HHQ under conditions of the microscopy-based virulence assay. Propidium iodide (PI) staining of DNA of non-viable cells indicates cell death (scale bars = 50 μm). (D) Representative images of TIB-67 monocytes following treatment with outer membrane vesicles (OMV) purified from wild type or ΔpqsA P. aeruginosa culture supernatants. (E) Quantification of OMV toxicity towards TIB-67 monocytes. Percent death is the increase in PI-stained nuclei divided by the total PI-negative cells at 1 h. Values are mean ± SE. Approximately 2500-3000 cells were analyzed for each measurement in (B) and (E).
Figure 4–Figure supplement 1. Schematic of the monocyte cell death assay.
We also developed an imaging-based virulence assay adapted from previous studies (Siryaporn et al., 2014) (Figure 4-S1) to determine if AQs are necessary for surface-induced P. aeruginosa virulence towards mammalian cells. Comparing the wild type and ΔpqsA P. aeruginosa revealed significantly more surface-induced monocyte killing by the wild type bacteria (Figure 4B). To more precisely distinguish the effect of AQ cytotoxicity from other activities mediated by PQS, we assayed the virulence of pqsH pqsE pqsR Pconst − pqsABCDE and an isogenic, pqsA null, derivative (Figures 4B and 4C). In the absence of PqsH, PqsE, and PqsR, any difference in virulence between these strains should be solely dependent on the ability of the bacteria products of PqsABCD such as HHQ. The strain producing HHQ caused significantly more killing than the pqsA mutant (Figures 4B), indicating that PQS-independent AQ activity increases the cytotoxicity of P. aeruginosa towards TIB-67 mouse monocytes. Thus, AQs represent important cytotoxins that promotes the surface-induced ability of P. aeruginosa to kill both amoebae and monocytes and this activity does not require PQS. The fact that loss of AQ production completely eliminates surface-dependent virulence towards amoebae (Figure 1C) but not towards monocytes (Figure 4B) indicates that there are also additional factors that can partially compensate for the loss of AQs in the context of monocyte infection.

(A) Representative images of AQ biosensor doped into samples of surface-attached ΔpqsA P. aeruginosa treated with 10 μM HHQ or DMSO added to 1% agar pads used for imaging (scale bars = 5 μm). (B) Mean biosensor signal per cell calculated from approximately 500 individual cells for each HHQ concentration. Standard curves were constructed for each independent experiment and used to calculate HHQ concentrations in Figure 3. Error bars represent standard error. Curve fit was computed using KaleidaGraph software (Synergy Software, Reading, PA).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Schematic of the monocyte cell death assay described in Materials and Methods. (B) Representative images of TIB-67 monocytes treated with surface-attached, wild type P. aeruginosa at a multiplicity of infection of approximately 1:50 at 1 and 15 hours of incubation at 30°C. Cell death is indicated by propidium iodide staining.
An intriguing and poorly understood feature of P. aeruginosa virulence is the ability of these bacteria to secrete cytotoxic outer membrane vesicles (OMVs), packed with multiple virulence factors (Bomberger et al., 2009). PQS stimulates OMV production and is known to be abundant in OMVs (Mashburn-Warren et al., 2009). Given our identification of AQs as potent cytotoxins, we hypothesized that they could play a role in mediating both OMV production and cytotoxicity. We therefore treated monocytes with equal amounts of OMVs purified from wild type or ΔpqsA P. aerguinosa, and monitored monocyte death (Figure 4D). OMVs containing AQs were significantly more cytotoxic than those that did not (Figure 4E), indicating that AQs are a major contributor to the cytotoxicity of OMVs, not just their production.
Discussion
Multiple lines of evidence support our conclusion that host-cell killing in response to P. aeruginosa surface association is mediated by induction of AQ cytotoxins. Loss of PqsABCDE, which inhibits AQ production, leads to loss of surface-induced virulence towards D. discoideum and significantly reduces the surface-induced virulence towards monocytes. Thus, AQ production is important for surface-induced virulence towards multiple host types. Restoring PqsABCD in the absence of PqsE, PqsH, or PqsR rescues surface-induced virulence, indicating that a PQS-independent AQ, such as HHQ, is sufficient to induce virulence even in the absence of genes responsible for AQ-mediated QS or iron-dependent signaling. Finally, purified HHQ and PQS are sufficient to directly kill host cells in the absence of bacteria, indicating that these factors are themselves cytotoxins as opposed to regulators of additional factors. These studies reinforce recent suggestions that AQs serve multiple important functions in virulence (Lin et al., 2018) and add surface-induced host cytotoxins to this growing list.
Our findings also implicate AQ production as a powerful reporter for dissecting the signal transduction pathways responsible for surface-induced virulence. Our analysis of the two known regulators of surface-induced virulence in the Dictyostelium cytotoxicity assay, PilY1 and LasR, showed that both regulators control virulence by activating pqsABCDE expression. Specifically, deletion of pilY1 and lasR reduced pqsA promoter activity, PQS production, and surface-induced virulence. Meanwhile, constitutive expression of pqsABCDE was sufficient to restore the surface virulence of pilY1 and lasR mutants and to increase the virulence of planktonic cells. Epistasis studies on both virulence and AQ production revealed that the repression of AlgR by PilY1 promotes high levels of pqsABCDE expression in surface-attached cells. A similar epistatic relationship between PilY1 and the AlgR-AlgZ system was recently implicated in the virulence of P. aeruginosa towards C. elegans hosts (Marko et al., 2018), suggesting that this pathway is also important in other virulence contexts. In the future it will be important to determine how AlgR/Z directly or indirectly regulate pqsABCDE expression. Additional outstanding questions include how LasR- and PilY1-dependent signaling is actually modulated by surface association and how these pathways intersect with other surface-dependent signaling pathways such as those mediated by TFP/cAMP or c-di-GMP (Bohn et al., 2009; Luo et al., 2015; Persat et al., 2015; Laventie et al., 2019).
More than 50 distinct AQs have been identified in P. aeruginosa cultures (?). Historically, the bulk of the work on AQs has focused on PQS and its roles in quorum sensing and the iron starvation response (Lin et al., 2018). More recently, a greater appreciation has emerged that AQs can carry out diverse functions that range from signaling (Lee and Zhang, 2015; Rampioni et al., 2016), iron scavenging (Diggle et al., 2007), antibacterial tolerance (Hazan et al., 2016), OMV induction (Mashburn-Warren et al., 2009), immune suppression (Kim et al., 2010), and cytotoxicity towards host cells or other bacteria (Abdalla et al., 2017; Wu and Seyedsayamdost, 2017). Our work supports this expanded view of the functional repertoire of AQs, representing evidence that AQ production is important for surface-induced virulence and demonstrating that HHQ can function as a cytotoxin that directly kills host cells independently of PQS.
The fact that AQs are sufficient to explain surface-induced D. discoideum killing was surprising because P. aeruginosa is generally thought to kill hosts using a large and redundant set of cytotoxins (Valentini et al., 2018; Streeter and Katouli, 2016). The large number of toxins present in P. aeruginosa could be due to the preferential ability of different toxins to kill different host cell types. For example, A549 epithelial cells appeared less sensitive to AQ cytotoxicity in our assays, such that other cytotoxins like T3SS could take precedence in targeting these cells. While future studies will be needed to dissect the specific contributions of each virulence factor in different contexts such as with different hosts and in the presence of an intact immune system, our study highlights the value of quantitative assays that can define the specific capacities of different factors.
D. discoideum cells are highly phagocytic, suggesting that host delivery might be a limiting step given the poor solubility of AQs. Our data suggest that HHQ and PQS are more active when delivered to hosts by bacteria, as the AQ biosensor indicated that lower levels of HHQ are required to kill D. discoideum when the HHQ is produced by bacteria than when supplemented exogenously. The increased cytotoxicity of AQs when produced by bacteria could be related to OMVs. PQS is a strong stimulator of OMV production (Mashburn-Warren et al., 2009), and our work shows that AQs are necessary for OMV toxicity. OMVs could thus represent a built-in cytotoxin delivery system that increases the ability of P. aerguniosa to use AQs to both promote virulence and mediate intercellular signaling.
Given that multiple AQs can act as cytotoxins, which AQ species is likely to mediate host cell killing in vivo? HHQ and other AQs are present in the serum, urine, and sputum of CF patients with P. aeruginosa infections (Collier et al., 2002; Barr et al., 2015), and have been shown to correlate with clinical progression (Barr et al., 2015). The conversion of HHQ to downstream AQs requires oxygen (Schertzer et al., 2010), and many infection sites are microaerophilic (Kamath et al., 2017; Hassett et al., 2009). Indeed, HHQ levels were found to be considerably higher than PQS or HQNO in a mouse burn infection model (Xiao et al., 2006a). Since bacterial biofilms on surfaces such as the CF lung are also oxygen-limited (Kamath et al., 2017; Hassett et al., 2009), HHQ may warrant particular attention as a candidate AQ cytotoxin in these conditions. In addition to being oxygen-insensitive, HHQ is a poor agonist of PqsR, with 100-fold weaker activation than PQS (Xiao et al., 2006a) (also see Figure 3A). Consequently, blocking the conversion of HHQ to PQS could represent a mechanism for P. aeruginosa to specifically induce a cytotoxic AQ without inducing competing PQS-dependent targets such as pyocyanin. Given that PQS is the most potent PqsR agonist among AQs (Xiao et al., 2006a) and that a recent study found that HQNO is the most potent antibiotic (Thierbach et al., 2017), we suggest that AQs could be functionally specialized with PQS serving primarily as a QS signaling molecule, HQNO primarily for inter-bacterial competition, and HHQ for host cell cytotoxicity under oxygen-limited conditions. Finally, we note that the different activities of AQs are not mutually exclusive such that induction of AQs upon surface association could represent a powerful strategy to simultaneously initiate cytotoxicity to ward off engulfment by phagocytic cells, suppress immune function (Kim et al., 2010), and signal additional downstream factors to promote factors associated with later stages of infection (Rampioni et al., 2016).
Methods and Materials
Bacterial Strains, Plasmids, and Growth Conditions
The strains and plasmids used in this study are described in Tables 1 and 2. Bacterial cultures were routinely grown in lysogeny broth (LB) broth at 37°C with aeration or on LB solidified with 1.5% agar (BD Biosciences, San Jose, CA). When stated, bacteria were grown in PS:DB media, which consists of development buffer (DB) (5 mM KH2PO4, 2 mM MgCl2, pH 6.5) and 10% (v/v) PS medium (10 g L−1 Special Peptone (Oxoid, Hampshire, United Kingdom), 7 g L−1 Yeast Extract (Oxoid, Hampshire, United Kindom), 10 mM KH2PO4, 0.45 M Na2HPO4, 15 g L−1 glucose, 20 nM vitamin B12, 180 nM Folic Acid, pH 6.5). Antibiotics were added at the following concentrations when appropriate: carbenicillin 300 μg mL−1, gentamycin 30 μg mL−1, and tetracycline 200 μg mL−1 for P. aeruginosa; 100 μg mL−1, gentamycin 30 μg mL−1, and tetracycline 15 μg mL−1 for E. coli. Expression of Ptac - or Plac -controlled genes was induced with 1 mM IPTG. When indicated, cultures were supplemented with HHQ, PQS, or HQNO (Cayman Chemicals, Ann Arbor, MI). Unless otherwise stated, chemicals and reagents were purchased from Sigma Aldrich (St. Louis, MO).
Bacterial strains and cell lines used in this study.
Plasmids used in this study.
Strain Construction
Primers used in this study are described in Table 3. All gene deletions described here are unmarked, in-frame deletions generated by two-step allelic exchange, as described previously (Hmelo et al., 2015). Briefly, upstream and downstream homology arms flanking the relevant gene were amplified with primer pairs (-KO1,-KO2 and -KO3,-KO4; Table 3), fused through overlap extension PCR (OE-PCR), and cloned into restriction sites of plasmid pEXG2. The pEXG2 plasmid was integrated into P. aeruginosa through conjugation using the donor strain E. coli S17. Exconjugants were selected on gentamycin and then the mutants of interest were counterselected on 5% sucrose. Transposon insertions obtained from the PA14 Transposon Mutant Database (Liberati et al., 2006) were transferred between strains using the λ-Red recombination system (Lesic and Rahme, 2008).
Primers used in this study.
To generate the pqsABCDE overexpression strain, the POXB20 promoter was ampli1ed from the plasmid pSF-OXB20 (Oxford Genetics, Cambridge, MA) using primer pair (OXB20-5 and OXB20-3) and spliced between two 400 bp fragments that flank the pqsA promoter, which were amplified from gDNA using primers (pqsUP-5, pqsUp-3) and (pqsDOWN-5, pqsDOWN-3), respectively. The resulting construct was cloned into pEXG2 and integrated onto the chromosome using allelic exchange. To generate the inducible pqsR expression construct, the pqsR gene was amplified from gDNA with primer pair (pqsR-5, pqsR-3) and cloned into pUC18-mini-Tn7T-LAC. Proper gene orientation was confirmed by restriction mapping, and the resulting construct was integrated onto the chromosome by co-electroporation with pTNS2 using methods described previously (Choi and Schweizer, 2006).
To generate the algRD54E mutation and overexpression construct, the algR gene was amplified from gDNA using primers (algR-pUC-5, algR-pUC-3) and subcloned into pUC19 (New England Biolabs, Ipswich, MA). Site-directed mutagenesis was performed by amplification of pUC19::algR with primers (algR-D54E-Fw, algR-D54E-Rv), and the mutant allele was either cloned into pEXG2 and integrated into the P. aeruginosa chromosome by allelic exchange, or cloned into pBBRMC3 to obtain the overexpression construct. The PpqsA − mCherry promoter fusion construct was generated by amplifying an approximately 500 bp fragment upstream of the pqsA gene with primer pair (P1pqsA-1, P1pqsA-2) and fusing it by OE-PCR to a promoterless mcherry gene, amplified from mini-CTX-2::PA1/04/03-mCherry with primer pair (P1pqsA-3, P1pqsA-4). The resulting fragment was cloned into the mini-CTX-2 plasmid and integrated into the chromosome at the CTX-2 phage attachment site (attB).
To generate the fluorescent AQ biosensor, a fragment containing the pqsA promoter and PqsR binding site (−19 bp to −219 bp) was amplified from gDNA using primer pair (pqsA-PaQa-5, pqsA-PaQa-3), and cloned into the BamHI/XhoI sites of pUCP18::PpqsA − YFP PrpoD − mKate. This plasmid was then transformed into a PAO1 strain containing a deletions in pqsA, and replacement of the pqsR promoter with a constitutive Ptac promoter. We note that while our virulence assays were all performed with the PA14 strain of P. aeruginosa, we used the PAO1 strain for the biosensor since the reporter is avirulent and does not interfere with PA14 virulence while PAO1 maintains plasmids at higher levels than PA14.
D. discoideum and mammalian cell culture
D. discoideum AX3 was maintained axenically as described previously (Siryaporn et al., 2014; Fey et al., 2007). Briefly, frozen stocks were inoculated into overnight cultures of E. coli B/r, and plated on GYP plates. After incubation for 4-6 days at 22°C, individual spores were inoculated into PS media supplemented with Antibiotic-Antimycotic (AA) solution, and incubated at 22°C 100 rpm. When cultures reached approximately 1 × 106 cells mL−1, cells were back-diluted 1:100 in fresh PS media. Axenic cultures were maintained for up to 1 month.
J774A.1 mouse monocytes (ATCC® TIB-67) and A549 (ATCC® CCL-18) (verified mycoplasma free) were grown at 37°C with 5% CO2 in Dulbecco’s Modified Eagle’s Medium (Gibco, Dublin, Ireland) supplemented with 10 % fetal bovine serum and Penicillin-Streptomycin solution (Invitrogen, Grand Island, NY). Cells were passaged according to the ATCC protocols.
D. discoideum cell death assay
Cell death assays were performed as described previously with minor modifications (Siryaporn et al., 2014). Overnight cultures of P. aeruginosa were diluted 1:100 in PS:DB media and grown to OD600nm = 0.6-0.8 at 37 °C with aeration. Cultures were transferred to glass-bottom dishes (Mattek, Ashland, MA) and incubated for an additional 1 hour at 100 rpm on a rotary shaker. For the planktonic condition, aliquots of culture media were washed with PS:DB, concentrated 20-fold, and plated onto fresh glass-bottom dishes. For the surface-attached condition, culture media was aspirated and surface-attached cells were washed with PS:DB. Aliquots of D. discoideum culture (between 2-5 × 105 cells mL−1) were washed with PS:DB, and added to planktonic and surface-attached bacteria to achieve the appropriate multiplicity of infection (MOI), which ranged between roughly 500:1 to 1000:1 (P. aeruginosa to D. discoideum). See Siryaporn et al. (2014) for details regarding assay validation and MOI quantification. The combined samples were covered with a 1% agar pad, prepared by pouring molten 1% agar in PS:DB containing 1 μM calcein-AM (Invitrogen, Grand Island, NY) on a glass surface, and cutting the solidified pad into 1 cm × 1 cm sections. Samples were analyzed by imaging cells with phase contrast and FITC channels after 1 hour of incubation at 25 °C using fluorescence microscopy. Cell death was quantified by counting the total number of calcein-AM-positive and -negative cells. All reported values of percent cell death are averages of at least three independent experiments. Each set of experiments included wild type and pqsA mutant controls, and each measurement was of 150-500 cells.
For quantifying cytotoxicity of purified AQs under conditions of the microscopy-based cell death assay (Figure 1D), AQs were diluted 1:200 into molten 1% agar in PS:DB and pads were prepared as described above. Samples of D. discodium were transferred to glass-bottom dishes and covered with a 1 cm × 1 cm agar pad and incubated at 25°C for 1 hour. For quantifying cytotoxicity of purified AQs in axenic D. discoideum cultures (Figure 1E), D. discoideum was subcultured to 10,000 cells mL−1 in fresh PS media with antibiotics and varying concentrations of AQs. Cultures were incubated at 22°C with shaking at 450 rpm, and cell density was measured by counting with a hemocytometer. Experiments were performed with five biological replicates.
Quantification of fluorescent reporter expression
Reporter strains were grown according to the procedures of the D. discoideum cell death assay. Bacterial cultures were transferred to glass-bottom dishes when OD600nm reached 0.6, and incubated at 37°C with shaking (100 rpm) for 1 h. Cells were washed and isolated as described above, and single cells were imaged immediately after addition of the 1% agar pad using fluorescence microscopy. Mean fluorescence intensity per cell was computed for 500-1000 cells, depending on the experiment. When comparing pqsA promoter activity between planktonic and surface-attached bacteria, mean fluorescence intensity per cell was normalized by the expression of a constitutive Ptac −mCherry reporter, which was measured in the same manner as the PpqsA) reporters. All reported values are averages from at least three independent experiments.
LC/MS-based quantification of AQ production
Overnight cultures of P. aeruginosa were subcultured 1:100 into 20 mL PS:DB and grown for 18 h 200 rpm. Cultures were extracted with equal volumes of ethyl acetate, dried and resuspended in methanol. Samples were analyzed by HPLC-MS on a 1260 Infinity Series HPLC system (Agilent, Santa Clara, CA) equipped with an automated liquid sampler, a diode array detector, and a 6120 Series ESI mass spectrometer using an analytical Luna C18 column (5 m, 4.6 × 100 mm, Phenomenex, Torrance, CA) operating at 0.6 mL min−1 with a gradient of 25% MeCN in H2O to 100% MeCN over 18 minutes. A standard curve constructed from commercial AQ standards was used to calculated AQ concentrations in cultures.
Plate-based AQ biosensor assay
Overnight cultures of the AQ biosensor was diluted 1:100 in LB with 1 mM IPTG and grown to late exponential phase (OD600nm = 1.0-2.0). Bacteria were resuspended in fresh LB to OD600nm = 1.0, and added to equal volumes of LB supplemented with various concentrations of AQs (purchased from Sigma-Aldrich) in a 96-well plate. Plates were sealed with a breathable membrane (DivBio, Dedham, MA) and incubated at 37°C 450 rpm while monitoring fluorescence in the YFP (500 nm excitation/ 540 nm emission) and mKate (590 nm excitation/ 645 nm emission) channels. Biosensor signal was calculated by normalizing the YFP signal by the mKate signal and subtracting the baseline expression from a DMSO control.
Biosensor-based AQ quantification of surface-attached bacteria
Overnight cultures of pqsH mutant P. aeruginosa were grown following the procedures of the D. discoideum cell death assay. The AQ biosensor was prepared according to the procedures of the plate-based assay, but resuspended to OD600nm = 0.2 in PS:DB. For planktonic samples, biosensor was added to equal volumes of planktonic cell suspensions. For surface-attached samples, biosensor was added to washed, surface-attached cells. Cells were covered with a 1% agar pad in PS:DB (1 mM IPTG), and incubated at 25 °C for 1-2 hours. Single-cell YFP and mKate fluorescence was measured by microscopy, and biosensor signal was calculated by normalizing the YFP signal by the mKate signal, and subtracting by the baseline expression of biosensor doped into a lawn of surface-attached ΔpqsA P. aeruginosa. For clarity, biosensor signal in representative microscopy images are normalized by the mean biosensor signal of biosensor doped into a lawn of surface-attached ΔpqsA. All reporter biosensor signal measurements are averages from at least three independent experiments, and approximately 500 individual cells were analyzed for each condition. An HHQ standard curve was generated and used to convert biosensor signal to HHQ concentration. Biosensor was added to surface attached ΔpqsA P. aeruginosa and covered with 1% agar pads made with PS:DB, supplemented with various concentrations of HHQ. Biosensor signal in response to HHQ standards was calculated as described above. A new standard curve was constructed for each independent experiment.
Mammalian cytotoxicity assay of purified AQs
TIB-67 monocytes or A549 epithelial cells were seeded at a density of 150,000 cells or 75,000 cells cm−2 in a 96-well plate and incubated at 37°C 5% CO2 for 24 hours. Cells were treated with purified AQ compounds dissolved in DMSO such that the final concentration of DMSO was less than 0.5%. After 48 hours, media was aspirated and the WST-8 reagent EZQuant (Alstem Bio, Richmond, CA) was used to assess cell viability. Values reported are averages of three biological replicates. At least three independent experiments were performed, and the trends observed in Figure 4A was observed across all experiments.
Microscopy-based TIB-67 cell death assay
For the microscopy-based assay, TIB-67 monocytes were seeded at a density of 75,000 cells cm−2 and incubated at 37°C 5% CO2 for 24 hours. TIB-67 monolayers were washed twice with phosphate buffered saline (PBS) (Gibco, Dublin, Ireland) and combined with P. aeruginosa samples. To prepared P. aeruginosa samples, cultures were grown following the procedures of the D. discoideum cell death assay. When cultures reached OD600nm = 0.5-0.6, cultures were transferred to petri dishes coated with a thin layer of 1% agar in PS:PBS (10% (v/v) PS media in PBS with 1 mM MgSO4 and 0.1 mM CaCl2, pH = 7.2) and grown for an additional 1 hour at 37°C on a rotary shaker (100 rpm) to allow surface attachment. Surface-attached bacteria were washed twice with PS:PBS. The density of surface-attached bacteria could be adjusted based on force applied during washing steps using an automated pipette, and density was optimized to achieve a final multiplicity of infection (MOI) of 1:50 to 1:150 (P. aeruginosa to TIB-67). Propidium iodide (PI;1 μM final) and sub-MIC dose of tetracycline (5 μg mL−1 final) was added to prepared monocyte samples. Agar pads were excised and inverted onto prepared monolayers of TIB-67 monocytes. Concentrations of PI and tetracycline were calculated based on the volume of the agar pad. Samples were incubated at 30°C in an incubated chamber, and cells were tracked over the course of 24 hours using fluorescence microscopy. Cell death was monitored by PI staining of the DNA of non-viable cells. Bacterial co-culture with monocytes resulted in substantial monocyte lysis (see Figures 4C) and subsequent diffusion of the nuclear PI stain. Therefore, P. aeruginosa virulence was most precisely quantified by counting viable monocytes (PI-negative) as opposed to dead monocytes (PI-positive). PI-negative cells were counted at various timepoints and divided by the number of PI-negative cells at 1 h in the same field of view. Treatment of monocytes with AQs or purified in OMVs did not result in lysis (see Figures 4C and 4D), and therefore virulence was measured by counting dead, PI-positive cells (Figure 4E).
OMV isolation and quantification
Outer membrane vesicles (OMV) were isolated as described previously (Bauman and Kuehn, 2006). Because OMVs are not significantly produced in the absence of PQS, we used the approach of Bauman and Kuehn (2006) to stimulate OMV production in the pqsA mutant, and kept conditions the same for wild type cultures to minimize differences between samples. Briefly, overnight cultures of P. aeruginosa were subcultured 1:200 in LB and grown at 37°C 250 rpm to O600nmD = 0.3-0.4. Polymyxin B was added to a final concentration of 4 μg mL−1, and cultures were grown at 37°C 250 rpm until they reached an OD600nm between 0.8-1.0. Culture supernatants were filtered through a 0.45 μm filter (Millpore, Burlington, MA), then centrifuged at 40,000 x g for 1 hour. Pellets were resuspended in PBS, filtered through a 0.45 μm filter, and centrifuged for an additional 1 hour at 100,000 x g. Samples were resuspended in PBS and concentrated with a 10,000 MWCO centrifugal filter device (Millipore, Burlington, MA). Filtrate was used as a vehicle control. OMV concentrations were measured by addition of the lipophilic dye FM-464 (Fisher Scientific, Hampton, NH) and measuring fluorescence (506 nm excitation/ 750 nm emission). The OMV concentration of the wild type sample was adjusted to match the concentration of the sample. Final OMV stocks were approximately 5000-fold concentrated compared to culture supernatant, and diluted 1:200 (based on the volume of the agar pad) into each sample of TIB-67 monocytes.
Competing Interests
The authors declare no competing interests.
Acknowledgments
We wish to thank members of the Gitai and Shaevitz labs for helpful discussions and comments on the manuscript, the Bassler and O’Toole labs for strains and reagents, and Ben Bratton for technical assistance and data analysis. ZG is supported by an NIH Pioneer Award DP1-AI124669 and an award from the Princeton Catalysis Initiative.
References




