

ABSTRACT
It is difficult to discern the relative contributions of ultraviolet-A (UVA; 320-400nm) and ultraviolet-B (UVB; 280-320nm) radiation to human melanoma development. Here, we compared the tumorigenic consequences of a single UVA or UVB exposure in mouse models predisposed to Braf- or Nras-mutant melanoma. Exposures approximated the amount of UVA or UVB energy contained in ∼40 minutes of summer sunlight. While UVA accelerated melanoma onset in a subset of mice, UVB universally reduced tumor latency and induced gene mutations relevant to the human disease. Genomic analyses uncovered distinct mutational signatures specific to each UV spectrum. The UVB-specific signature was biased for mutations on the untranscribed DNA strand and closely mirrored mutational signatures enriched in human cutaneous melanoma. The UVA-specific signature mimicked SBS51, a mutational signature found in human uveal melanoma. Distinctions in the trinucleotide patterns of the UVA and UVB signatures suggest that cytosine deamination plays a key role in UVB-mediated melanomagenesis.
INTRODUCTION
Genomic and epidemiological data suggest that ultraviolet radiation (UV) is the major etiologic agent in melanoma. Two UV spectra reach Earth’s surface: UVA (320-400 nm) and UVB (280-320 nm) 1. UVA is the most prevalent form of UV in terrestrial sunlight; however, it is poorly absorbed by DNA 1-4. By contrast, UVB can directly damage DNA and is the major form of UV responsible for erythema 4. The mutational effects of UV irradiation are apparent in human melanoma genomes, which exhibit one of the highest somatic mutation rates of any tumor type 5,6. UV has also been linked to melanoma risk through epidemiological studies showing increased incidence in individuals with a history of childhood sunburns, tanning bed use, or intense, intermittent UV exposures 7. While these observations establish correlative relationships between UV irradiation and melanoma formation, there is limited understanding of the genetic events and relative risk conveyed by different UV spectra.
Genetically engineered mouse models (GEMMs) provide a controlled genetic background in which the genomic and phenotypic effects of specific UV exposures can be studied. It is known that neonatal or chronic UV treatment accelerates the formation and progression of melanoma in a variety of GEMMs 8-12. Tumors from several UV-treated GEMMs have been sequenced, including those representative of the most common genetic subtypes of melanoma: NRAS- and BRAF-mutant 8,12,13. However, the only study that has sequenced both UVA- and UVB-induced tumors used a chronic UV dosing scheme and was unable to identify any mutational patterns specific to each spectrum 8.
Here, we used a single-dose UV irradiation scheme to characterize the phenotypic and genomic effects of UVA and UVB exposure in Nras- and Braf-mutant mouse models. We exposed these animals to a single dose of UVA or UVB, approximating the amount of energy from each UV spectrum in 40 minutes of intense sunlight. Then, we monitored the mice for melanoma development. Tumors from these animals were sequenced to gain insight into the genomic mechanisms by which each UV spectrum influences melanoma formation.
RESULTS
UV exposure alters Nras- and Braf-mutant melanomagenesis
We generated TN and TB mice to model the major genetic subtypes of human melanoma 14 (Figure 1A, B). TN mice are homozygous for the LSL-NrasQ61R allele 10,15, whereas TB animals carry a heterozygous, conditional Braf V637E allele (Braf CA15). Oncogene expression is driven by the endogenous gene promoter in both models, and is activated by a melanocyte-specific, tamoxifen-inducible Cre recombinase (Tyr::CreER(T2)17). Therefore, the expression of oncogenic Nras or Braf in these mice mimics the presence of NRAS and BRAF mutations in most benign human nevi 18. Mice carrying only the Braf CA allele rarely develop melanoma 19. Therefore, p16INK4a conditional knockout alleles (p16L, 20) were included in both the TN and TB models. While p16INK4a loss-of-function is an early event observed in >60% of human melanomas, germline mutations affecting p16INK4a are insufficient to drive the disease in mice or humans 10,21,22.

A, TN mice are homozygous for a melanocyte-specific, tamoxifen-inducible CRE transgene (Tyr::CreER(T2)), a conditional p16INK4a knockout allele (p16L), and a conditional NrasQ61R knock-in allele (LSL-NrasQ61R). Open triangles represent Lox P sites. A star indicates the location of the Nras Q61R mutation. B, TB mice carry a single, Braf V637E conditional allele (LSL-Braf CA) and are homozygous for Tyr::CreER(T2) and p16L. Note that BRaf V637E is the murine equivalent of human BRAFV600E. Open triangles represent Lox P sites and the location of the V637E mutation is indicated by a star. C & D, Kaplan-Meier curves depicting the melanoma-free survival of TN (C) and TB (D) mice treated on postnatal day three with a single dose of ambient light (No UV), UVA or UVB. Statistical significance was determined by comparing values from control (0 kJ/m2) and UV-irradiated animals of the same genotype using Gehan-Breslow-Wilcoxon tests. † = p < 0.0001, ** = p < 0.01. E, Total tumor burden of control and UV-irradiated TN and TB mice at euthanasia. Each circle represents a single mouse. Boxes represent the mean and interquartile range for each group. Whiskers span from the minimum to the maximum value. Statistical significance was determined using two-tailed, unpaired t-tests with Welch’s correction. ** = p < 0.01 vs. control (0 kJ/m2) animals of the same genotype. F, Average tumor growth rates for UVB- and mock-exposed TN and TB mice. Each circle represents a single tumor (TN: 0 kJ/m2 n=17; 70 kJ/m2 UVA n=21; 150 kJ/m2 UVA n=21; 4.5 kJ/m2 UVB n=41; TB: 0 kJ/m2 n=27; 70 kJ/m2 UVA n=10; 4.5 kJ/m2, n=41). Data are presented as described in E.
TN and TB mice were treated with 4-hydroxytamoxifen (4OHT) on postnatal days one and two to induce Cre activity and stimulate recombination of the conditional p16INK4a knockout and LSL-Nras61R or Braf CA alleles. On postnatal day three, the mice were exposed to a single dose of ambient light (“No UV”), UVA or UVB irradiation. The amount of UVB delivered approximated that which is contained in 40 minutes of summer sunlight (4.5 kJ/m2; see: Methods), whereas the amount of UVA employed models a short, indoor tanning session (70 or 150 kJ/m2; see: Methods). These dosing schemes approximate sun exposures of a similar duration, as the UVB to UVA ratio in sunlight is ∼1:20, but varies based on season, cloud cover and latitude 23.
The onset of spontaneous melanoma was compared among mice exposed to No UV, UVA or UVB irradiation. Exposure to a single dose of 4.5 kJ/m2 UVB did not cause erythema or blistering, but dramatically accelerated melanoma onset and decreased overall survival in both the TN and TB models (Figures 1C-D and Suppl. Figure S1). Exposure to 70 kJ/m2 UVA also enhanced melanoma formation, but with greatly reduced penetrance (Figure 1C-D). Doubling this dose of UVA in the TN model did not further facilitate melanoma formation, suggesting that 70 kJ/m2 UVA was sufficient to elicit the maximal response achievable with a single treatment (Figure 1C). Together, these results reveal the potent ability of UVB to stimulate melanoma formation in TN and TB mice. Furthermore, our findings suggest that UVA can facilitate melanoma onset in some settings, albeit to a much lesser extent than UVB.

A&B, Kaplan-Meier curves depicting the overall survival rates of TN and TB mice treated on postnatal day three with a single dose of ambient light (No UV), UVA or UVB. Mice were euthanized after meeting pre-determined exclusion criteria due to tumor burden or malaise. Statistical significance was determined by comparing values from control (0 kJ/m2) and UV-treated animals of the same genotype using Gehan-Breslow-Wilcoxon tests. † = p < 0.0001, ** = p < 0.01
We next examined the incidence and growth phenotypes of tumors arising in each of our experimental cohorts. Tumor incidence increased in UVB-exposed TN mice, but was not significantly altered in TN animals treated with UVA or TB mice exposed to any form of UV (Figure 1E). Tumor distribution and burden were also similar between male and female TN and TB mice regardless of exposure, with ∼59% of tumors arising on the trunk, ∼13% on the head and ∼16% on the ears or tail (data not shown). Once established, TN and TB tumors grew at the same rate regardless of exposure (Figure 1F). Therefore, enhanced tumorigenesis, rather than more rapid melanoma growth, is responsible for the reduction in overall survival observed in UVB-exposed TN and TB mice.
We postulated that melanomas arising in UVA- or UVB-exposed mice would exhibit distinct histopathological features. Therefore, we examined hematoxylin and eosin stained tumor sections representative of the rate of onset and body site distribution of melanomas from each cohort. Tumors from both models contained variable percentages of myxoid and spindle cells with comparable degrees of invasion, mitosis and granulocyte infiltration regardless of treatment (Suppl. Figure S2A-C, data not shown). A paucity of pilosebaceous units and hyperplasia of the overlying epidermis was also observed in UVA, UVB and unexposed mice of both genotypes (Suppl. Figure S2A&F). Most tumors from the UVB-treated TN cohort contained neoplastic cells with plasmacytoid features that were not prevalent in TN melanomas from the UVA and No UV cohorts (6 of 7 vs. 2 of 6 and 0 of 5 tumors, respectively; Suppl. Figure S2D). Fibroblastic features were seen in UVA-treated TN melanomas (3 of 6), but were not overtly apparent in tumors from other TN mice (Suppl. Figure S2E). Unlike the TN model, tumor samples from TB mice contained areas of pigmentation, typically characterized by multiple clusters of melanophages distributed at the dermal-hypodermal interface with or without associated neoplastic cells and occasionally within the tumors (Suppl. Figure S2F). These results show that although the histopathological features of cutaneous murine and human melanomas differ, a single UVA or UVB exposure can promote the formation of cutaneous, murine tumors with distinct morphologic features.

A, TN and TB tumors, regardless of UV exposure history, contain variable proportions of spindle (left, cellular) and myxoid (right, spaces) morphologies. Bar = 200 μm. B&C, Higher magnification images of the myxoid (‘B’, bar = 20 μm) and spindle (‘C’, bar = 20 μm) morphologies depicted in ‘A’. Arrows = mitotic figures. D, Representative image of the neoplastic cells with plasmacytoid features seen in the majority of TN tumors evaluated. (circles; bar = 20 μm). E, Half of the tumors in UVA-treated TN mice exhibited a fibroblastic phenotype with abundant collagen (indicated by *; bar = 20 μm). F, Tumor samples from TB mice contained areas of pigmentation, typically appearing as clusters of melanophages at the dermal-hypodermal interface (boxes, bar = 200 μm). All images are of H&E stained tissues.
UVB exposure humanizes the genome of TN and TB melanomas
In contrast to human melanomas, GEMM-derived tumors are frequently characterized by a high burden of genomic copy number alterations (CNAs) and few single nucleotide variants (SNVs) 6,14,24-26. Thus, we used whole exome sequencing (WES) to determine whether a single UVA or UVB exposure would promote the formation of murine melanomas with ‘humanized’ cancer genomes containing a high frequency of SNVs. The burden of CNAs was reduced in all TN tumors exposed to UVB and in 5 of 7 TN tumors exposed to UVA (Figure 2A, B). Conversely, neonatal UV treatment had little effect on the number of CNAs detected in TB-derived melanomas (Figure 2A, B). The most commonly observed CNA in TB tumors was a gain in chromosome 6, the region in which Braf resides (Figure 2A). Consistent with this observation, 5 of 12 TB melanomas showed increased Braf protein expression as compared to normal, murine skin (Figure S3). Recurring copy number gains in chromosomes 1 and 10 were also observed in tumors from the TB-UVA, TN-No UV and TN-UVA groups (Figure 2A).

A, Immunoblot of Braf and β-Actin expression in representative TB tumors. B, Quantification of the immunoblot shown in ‘A’.

A, Heatmap showing areas of genomic gain or loss within each sequenced tumor. Columns correspond to individual tumors and rows correspond to genomic bins. B, Fraction of each sequenced melanoma genome exhibiting a CNA is graphed as a box plot with whiskers set at the 5th and 95th percentiles. Dots represent individual tumors. p-values determined by ANOVA with Dunn’s correction. C-D, SNVs (C) and indels (D) per megabase of captured genome are plotted and analyzed as in ‘B’. E, Relative enrichments of the indicated IPA disease functions in mutations from each tumor group. For A-D, * = p < 0.05, ** p<0.01, *** = p < 0.001.
To determine the SNV burden of tumors from our UVA- and UVB-treated GEMMs, we aligned each melanoma genome to the mm10 reference genome, removed variants observed in pooled normal controls, and excluded polymorphisms observed in dbSNP 27. The average SNV burden of melanomas from both TN and TB mice increased as a result of prior UVB exposure, whereas the SNV burden of UVA-irradiated TN and TB tumors was slightly higher, but not statistically different, than that observed in tumors from the No UV groups (Figure 2C). The frequency of insertion and deletions (indels) did not differ in tumors from irradiated and unirradiated TB mice, but increased slightly in TN UVB melanomas as compared to unirradiated controls (Figure 2D).
WES also revealed UV-dependent changes in the specific gene ontologies mutated in TN and TB melanomas. In both models, more of the SNVs and indels discovered in the UVB groups overlaped with genes frequently altered in human melanoma than SNVs and indels identified in the No UV tumors (Suppl. Figure S4). Overlapping genes included those encoding protein products involved in cell adhesion (cadherins, demoplakin), vesicular trafficking (dyneins, myosins, actin remodeling enzymes), the extracellular matrix (collagens, laminins), epigenetic gene regulation (Brd1, Chd8, Kdm2c, Kmt2c, Kmd2d), and receptor tyrosine kinase signaling (Erbb4, Igfr, Mtor, Pten) (Suppl. Figure S4). In contrast to published sequencing studies of melanomas broad-spectrum or UVB-irradiated GEMMs 8,12,13, Trp53 mutations were extremely rare in our cohort (1 in 36 tumors).

Shown are genes that were mutated in at least one murine tumor sample and altered at a frequency of 15% or more in the TCGA human melanoma provisional dataset.
We next used Ingenuity Pathway Analysis (IPA) to identify mutated gene ontologies enriched in tumors from each of our experimental cohorts. Genes mutated in TN and TB tumors from the No UV and UVA cohorts were rarely linked to melanoma, whereas the top five disease and biological functions altered in melanomas derived from UVB-exposed TN and TB mice included “cutaneous melanoma”, “melanoma”, “skin cancer”, and “skin tumor” (Figure 2E, Suppl. Table 1). Together, these data establish that a single dose of UVB induces a high burden of disease-relevant SNVs in TN and TB tumors.
C>T transitions are the predominant mutation type driven by UVA and UVB
The elevated frequency of cytosine to thymidine (C>T) transitions in human melanoma is often attributed to UVA and UVB irradiation 28. To test whether UV-treatment enriched for C>T transitions in our GEMMs, we quantified the burden of each SNV type (C>A, C>G, C>T, T>A, T>C or T>G) in melanomas from TN and TB mice treated with No UV, UVA or UVB. As anticipated, melanomas from UVA- and UVB-treated TN and TB mice had a greater number of C>T transitions than unirradiated controls (Figure 3A and Suppl. Table 2A; p < 0.043 for each group vs. No UV). Since methylated cytosines form cyclobutane pyrimidine dimers (CPDs) with higher efficiency 29, we separately analyzed C>T alterations occurring within a CpG site. We found that the majority of C>T alterations occurred within non-CpG sites, suggesting that methylated cytosines may not be preferentially mutated in our models (Figure 3A). UVB also increased the total number of T>C (A>G) and T>A mutations in TB tumors (p < 0.032 for both groups vs. No UV); however, these mutation types were not similarly elevated in melanomas from UVB-irradiated TN mice (Suppl. Table 2A).

A-B, Absolute mutation burden (A) and frequency (B) of each mutation type in TN and TB tumors arising after mock (No UV), UVA or UVB exposure. Note that C>T transitions are broken into two classes: those that occur within CpG sites (light red) and those that do not (dark red). See Suppl. Table 2A-B for a complete listing of p-values for all comparisons.
We further evaluated the mutation type data, accounting for variances in tumor mutational load by determining the contribution of individual SNV types to the total mutational burden of each experimental group. Consistent with our analysis of mutational burden, C>T transitions at non-CpG sites were enriched in tumors from both UVA- and UVB-treated models as compared to No UV controls (Figure 3B and Suppl. Table 2B; p < 0.045 for all UV-No UV pairs). This enrichment of C>T mutations was accompanied by global decreases in the frequency of most other SNV types. An exception to this observation was the enrichment of T>A transversions in melanomas from UVA-treated TN mice (p < 0.007 for UVA vs. No UV). T>A transversions were also more frequent in melanomas from UVA-irradiated TB mice, but this enrichment did not reach statistical significance (p < 0.16 for UVA vs. No UV).
We looked for evidence of oncogene-dependent mutational enrichments and found that C>T transitions were both more abundant (p < 0.05) and frequent (p < 8.20×10−5) in TB-UVB tumors than TN-UVB tumors (Figure 3, Suppl. Table 2A-B). Other mutation types did not differ significantly in number or frequency between the two genotypes. Together, our findings highlight the dominance of C>T transitions in melanomas from both UVA- and UVB-treated mice and suggest that an underlying Braf mutation may promote further accumulation of these mutations. Furthermore, we provide evidence that UVA irradiation may drive the acquisition of both C>T transitions and T>A transversions.
UVA and UVB drive C>T mutations in distinct trinucleotide contexts
CPD-associated C>T lesions are considered classical ‘UVB signature mutations’ and occur preferentially at dipyrimidine sites 30. C>T transitions in other, non-cutaneous cancers lack this specificity 31. We used lego plots to visualize the trinucleotide context of C>T mutations in tumors from our TN and TB models (Figure 4A). Most C>T transitions occurred at dipyrimidine sites in UVA- and UVB-induced melanomas from both models (Figure 4B-C, red bars). However, UVB-associated C>T transitions were enriched in TCG and CCA sites, whereas UVA-associated C>T mutations were more evenly distributed amongst the eight potential dipyrimide triplets (Figure 4B-C, back two rows of red bars).
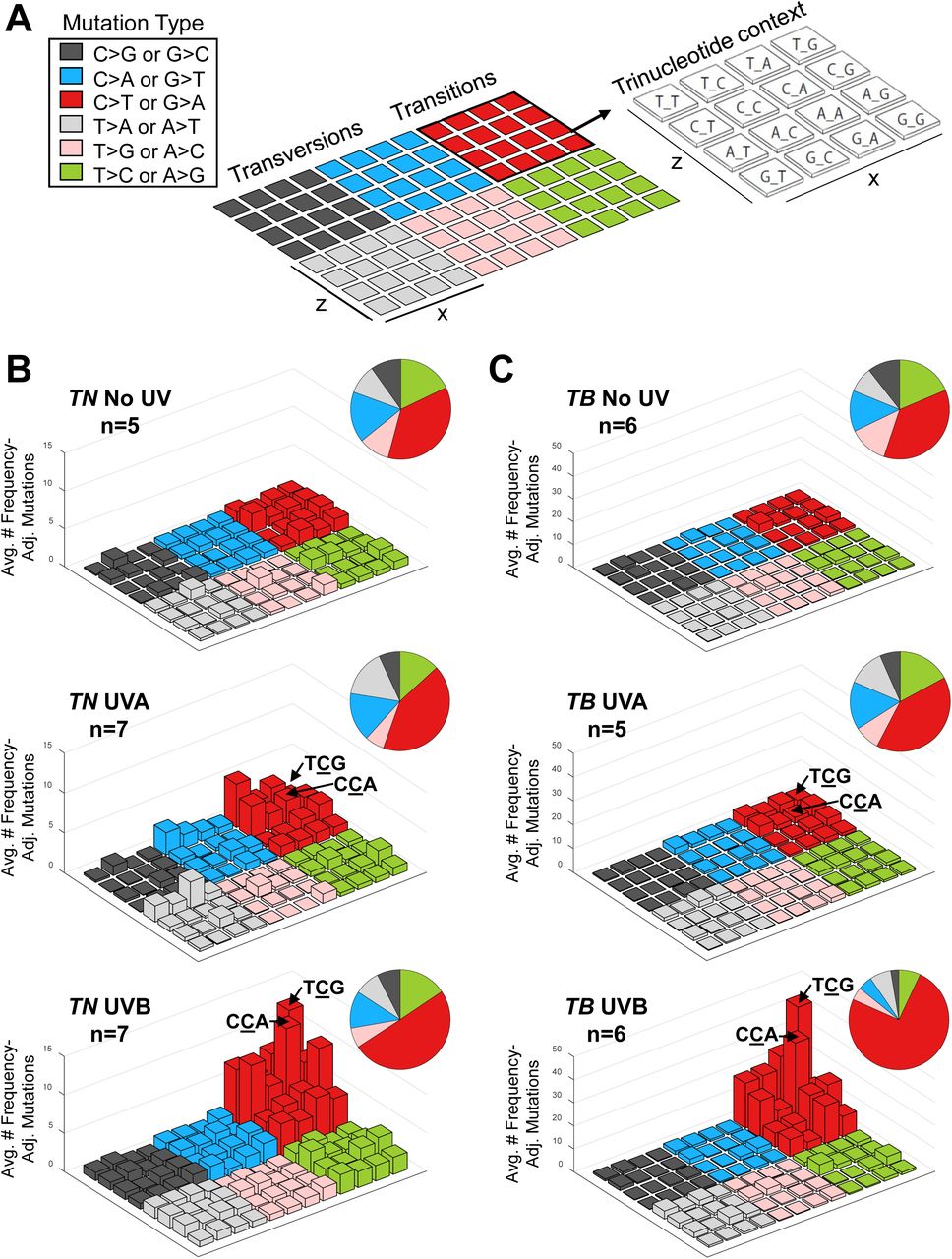
A, Diagram showing the layout of trinucleotide mutations in each lego plot. The mutated base is indicated by the color of the bar. The location of the bar within the 4 × 4 color block grid is indicative of the nucleotide sequence surrounding each mutation. The height of each bar corresponds to the average number of mutations in the indicated experimental group, normalized to the relative frequency of the relevant trinucleotide sequence in the mouse exome. B-C, Lego plots showing the average frequency of mutations within each trinucleotide context for the indicated experimental cohorts. Total frequencies of each mutation type are shown in the associated pie charts.
The enrichment of T>A transversions in our UVA-induced tumors (Figure 3 and Suppl. Table 2A-B) led us to investigate the preferred trinucleotide context of these mutations. The majority of UVA-induced T>A transversions followed a 5’ thymidine residue and, remarkably, the same nucleotide preference was observed for C>A transversions in these tumors (Figure 4B-C). These data highlight the preferential mutation of pyrimidines with a 5’T in melanomas from UVA-treated TN and TB mice. Furthermore, our results show that UVA and UVB promote C>T mutations with distinct trinucleotide contexts.
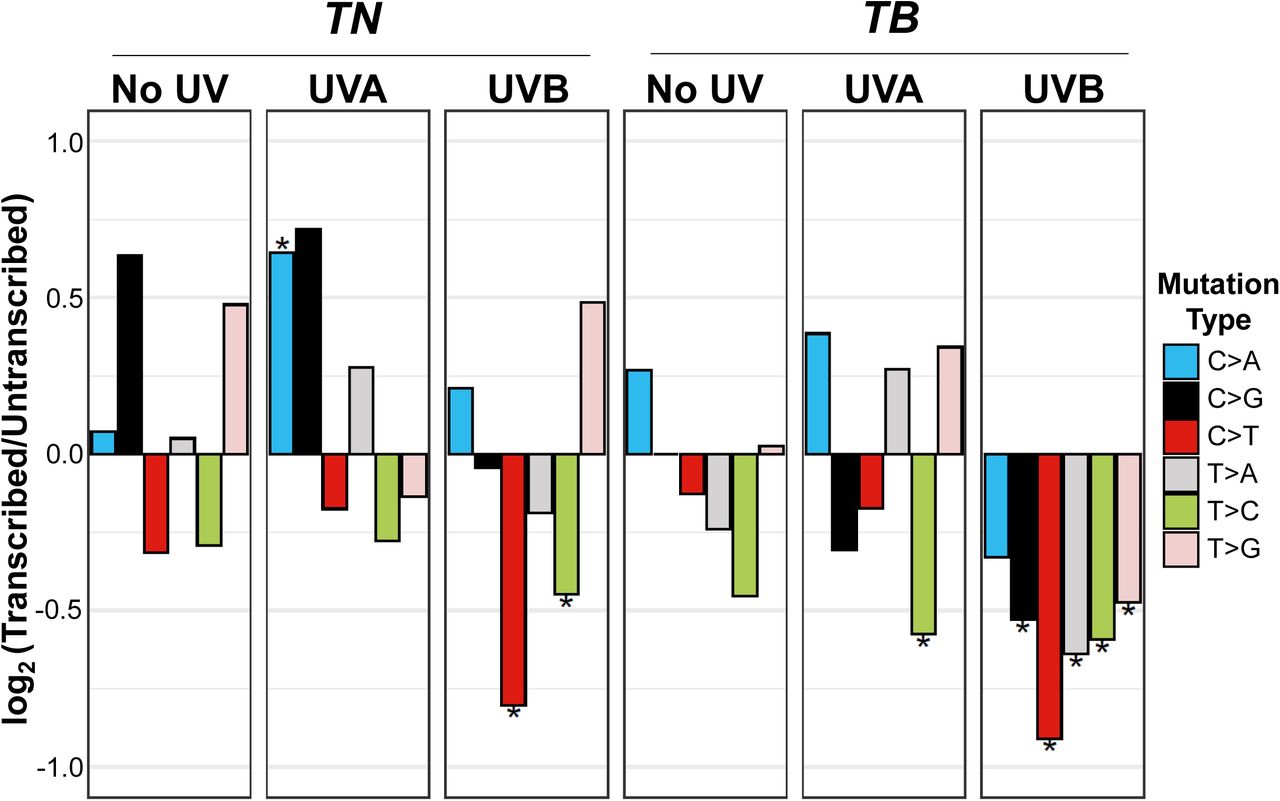
UVB-induced mutations localize to the non-transcribed strand
Studies in cultured fibroblasts and model organisms implicate transcription-coupled repair (TCR) in the rapid resolution of UV-induced DNA lesions 32. Therefore, we investigated whether the SNVs observed in our TN and TB tumors exhibited a strand bias. C>T and T>C mutations were biased towards the untranscribed strand in all tumors regardless of exposure or model (Figure 5, Suppl. Table 2C). By contrast, C>A transitions were enriched on the transcribed strand, and the remaining mutation types (C>G, T>A, T>C) showed variable strand bias, dependent on the experimental group (Figure 5). All mutation types were enriched on the untranscribed strand in tumors from UVB-treated TB mice, and TN UVB tumors showed a similar shift in strand enrichment compared to unirradiated controls (Figure 5). These data support a role for TCR in the resolution of both UV-induced and naturally occurring C>T and T>C mutations. Our results also show that unlike UVA, UVB promotes the accumulation of a variety of SNV types on the untranscribed DNA strand.

The strand location of each mutation type was determined from aggregate data from the indicated mouse models and exposures. Plotted are the log-transformed ratios of transcribed versus untranscribed mutations. Statistical significance of strand bias was assessed using a Poisson test, where an * indicates significant enrichment. Complete p-value listings can be found in Supplemental Table 2C.
Identification of distinct UVA and UVB mutational signatures
Non-negative matrix factorization was used to identify the three most prevalent signatures in our complete dataset: Signatures A, B and C (Figure 6A). Signature A was enriched in UVB tumors from both models as compared to melanomas from mock and UVA irradiated mice of the same genotype (p < 0.01 for all comparisons; Figure 6B, Suppl. Table 2D). Signature B was lower in TN UVA tumors than in TN No UV or UVB melanomas (p < 0.002 for both comparisons), but did not change in TB tumors facilitated by different forms of UV (Figure 6B, Suppl. Table 2D). Interestingly, Signature C was enriched in TN UVA (p < 1.0×10−8) and TB UVA (p < 0.006) tumors as compared to melanomas from No UV and UVB mice of the same genotype (Figure 6B, Suppl. Table 2D). Thus, Signature A appears to define melanomas from UVB-treated TN and TB mice, whereas Signature C is specifically linked to tumors arising after UVA exposure.

A, Non-negative matrix factorization (NMF) was used to identify de novo mutational signatures in sequenced TN and TB melanomas. Three signatures were selected based on explained variance and cophenetic score. The relative contribution of each mutation type and trinucleotide context is shown for each signature. Mutations occurring on the transcribed strand are indicated by open bars, whereas mutations occurring on the untranscribed strand are depicted as closed bars. B, Enrichment of identified signatures in tumors from each model and exposure type. Significant differences between groups were assessed using student’s t-tests with Holm-Bonferroni correction. Complete p-value listings appear in Supplemental Table 2D. C, Strand bias of the indicated mutation types within each de novo signature shown in (A). p-values were generated using a Poisson test as in Figure 5. * = p < 0.05
Mutational signatures enriched in human skin cancer are biased towards the untranscribed DNA strand 30. A similar strand bias was seen in our UVB signature, Signature A, whereas our UVA-associated signature, Signature C, exhibited variable strand bias (Figure 6C). These data are consistent with the idea that transcription coupled repair (TCR) plays a larger role in the resolution of DNA lesions induced by UVB than UVA.
Murine UVA and UVB signatures mimic distinct COSMIC mutational signatures
The COSMIC mutational signature most closely associated with human skin cancers and melanoma prognosis is Signature 7, which has recently been divided into four sub-signatures: SBS7a-d (See: 8,33). Interestingly, Signature A in our dataset had a high degree of similarity with SBS7a and SBS7b (cosine of similarity = 0.83, p < 6.98×10−48; cosine of similarity = 0.86, p < 6.06×10−55, respectively), however, Signatures C best aligned with SBS50, SBS51 and SBS58 (Figure 7A; cosine of similarity = 0.68, p < 8.00×10−19; cosine of similarity = 0.61, p < 1.23×10−14; cosine of similarity = 0.66, p < 1.23×10−17 respectively). The etiology of SBS50, SBS51 and SBS58 is unknown 33.

A, Heatmap depicting the cosine similarity score between de novo signatures discovered in our dataset and COSMIC signatures derived from human tumors. B, Shown is the relevant enrichment of each COSMIC mutational signature in TN and TB tumors from mice exposed to No UV (pink), UVA (green), or UVB (blue). Complete p-value listings appear in Supplemental Table 2E. C, Shown in blue is the proportion of acute myeloid leukemia (AML, n = 121), cutaneous melanoma (CM, n = 466) and uveal melanoma (UVM, n = 79) samples in the TCGA database that contain the indicated COSMIC mutational signature. D, The proportion of AML, CM and UVM samples from (B) that contain the indicated mutational signature, shown in Figure 6A and derived from the TN and TB mouse models.
We next performed the converse analysis, looking to see if any COSMIC signatures were enriched in our murine melanoma datasets. While SBS7a contributed equally to the mutational patterns of No UV, UVA and UVB tumors, SBS7b was significantly enriched in TN UVB and TB UVB melanomas (Figure 7B, Suppl. Table 2E). Other signatures increased in tumors from both UVB-accelerated models included: SBS2 which may be linked to APOBEC deamination, SBS10b, which is associated with hypermutated samples, and SBS19 which is of unknown etiology. SBS2 was also enriched in UVA-induced tumors, however, SBS51 showed the highest specificity for our TN UVA and TB UVA samples.
Because only UVA can pass through the cornea 34, we hypothesized that both SBS51 and our UVA-associated Signature C would be enriched in a subset of uveal melanomas. Compared to acute myeloid leukemia (AML) and cutaneous melanoma (CM), a greater percentage of uveal melanoma samples within the TCGA dataset exhibited the SBS51 signature (Figure 7C; p < 0.041 and p < 3.3×10−5 respectively). Signature C was similarly enriched in the same uveal melanoma samples as compared to CM, but no difference was seen between UVM and AML samples (Figure 7D; UVM vs AML p < 0.45; UVM vs CM p < 6.6×10−11). These data support a potential role for UVA in the formation of a subset of uveal melanomas.
DISCUSSION
Human melanoma has one of the highest mutational burdens of any tumor type 30. Yet, tumors arising in most melanoma GEMMs, including our unirradiated TN and TB mice, are largely characterized by CNAs rather than SNVs 6,14,24-26. Here we find that a single UVB exposure can resolve this conundrum and effectively recapitulate the high burden of SNVs in human melanoma. Moreover, tumors from UVB-irradiated TN and TB mice are enriched for somatic alterations and mutational signatures characteristic of human melanoma (Figs. 2E, 6B). These data are consistent with genomic analyses in other UVB-accelerated GEMMs and support the use of irradiated models in pre-clinical melanoma research 8,12,13. Our results indicate a disparity in the melanomagenic potential of UVA and UVB. Along with prior publications 8,35, these data provide additional evidence that UVA exposures can increase melanoma risk, but not to the same extent as UVB. What distinguishes this study from past investigations is that we have used our GEMMs to identify and characterize distinct mutational signatures arising as a consequence of each UV type. These signatures provide critical insights into the differences between UVA and UVB carcinogenesis.
As might be expected, C>T transitions are the prevailing mutation type in melanomas from TN and TB mice treated with either UVA or UVB (Fig. 3). This observation corroborates previous conclusions that dipyrimidine photoproducts are the major lesions involved in both UVA- and UVB-dependent melanocyte carcinogenesis. HPLC-MS/MS data from human skin and primary keratinocyte cultures estimate the CPD yield from UVB to be 103-105 fold greater per J·m−2 than UVA 36. Thus, we expected our UVB dose of 4.5 kJ/m2 to induce at least 240 times more dipyrimidine photoproducts than our UVA dose of 70 kJ/m2. However, we find that the total number of C>T mutations in tumors from our UVA- and UVB-irradiated TN and TB GEMMs differs by only 2- and 7-fold, respectively (Fig. 3A). There are several possible explanations for this observation which are not mutually exclusive. First, UVA penetrates deeper into the skin than UVB and may reach melanocytes within the hair follicle and dermis with higher efficiency 1. Alternatively, the majority of melanocytes may die as a consequence of UVB, but not UVA irradiation. Several reports support this model showing that melanocytes are extremely resistant to UVA-mediated cell death 37-39. Interestingly, we were unable to detect TUNEL positive melanocytes in our models following 4.5 kJ/m2 of UVB irradiation (data not shown), making this mechanism unlikely. Different rates of photoproduct repair could also explain the lower than expected ratio of UVB:UVA-induced C>T mutations in our GEMMs. UVA has been implicated in the oxidation of a number of nucleotide excision repair (NER) components, including RPA, PCNA, XPA, XPE and XPA 1. A reduction in the activity of one or several of these components may compromise photoproduct repair in UVA-irradiated cells and explain why UVA-induced CPDs take longer to resolve 36. UVB damage also causes keratinocytes to produce paracrine factors like α-Melanocyte Stimulating Hormone (αMSH) and Endothelin-1 (End-1), which activate ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR), resulting in a DNA damage response which could enhance the recognition and repair of UV photolesions 40. Thus, in vivo CPD repair rates may be faster and more efficient in UVB-versus UVA-irradiated melanocytes. Finally, differences in the types of dipyrimidine photoproducts induced by each UV source could explain why UVB is less mutagenic than predicted. For example, TT CPDs, which are reportedly the least mutagenic dipyrimidine dimer, appear to make up 90% of CPDs in UVA-irradiated DNA, but only 54% of CPDs in UVB-irradiated DNA 23. Determining the relative role of each of these processes in UVB carcinogenesis will require further investigations using tissue-specific knockouts and rapid melanocyte isolation protocols.
Our observation that TCG triplet-associated C>T mutations are enriched in tumors from TN and TB mice irradiated with UVB, but not UVA (Fig. 4B-C), provides insight into how UVB drives melanocyte carcinogenesis. Experimental models employing the lacZ transgene as a mutagenic reporter show that TCG-associated C>T mutations are more prevalent when mice are irradiated with longer UV wavelengths 41. Therefore, we expected C>T mutations embedded in a TCG triplet to be more frequent in our UVA-irradiated models. This was not the case (Fig. 4B-C). TCG sites are known hotspots for UV-mutagenesis for several reasons. UVA and UVB induce TC dimers at twice the frequency of CC or CT dimers 23,42. Once dimerized, cytosines within a CPD lesion appear to deaminate a million-times faster than those contained in undamaged dsDNA 43. This process is further enhanced by a catalysis reaction involving the carbonyl group of a 3’ guanine residue 43. Deamination of cytosine to uracil and mC to thymidine results in TU and TT dimers, which cause a C>T transition upon repair by the bypass DNA polymerase, Pol η 44. Because the t1/2 of TCG-associated CPDs seems to be in the range of several hours 45, the longer these lesions go unresolved, the more likely that deamination will occur. These observations have led us to consider the possibility that differences in the frequency of TCG-associated T>C mutations among UVA and UVB tumors reflect the prolonged chemical or physical persistence of CPDs in UVB-irradiated melanocytes from our models. Persistence could be due to a number of UVB-specific challenges, including: 1) a higher initial burden of lesions, 2) altered melanocyte proliferation, and 3) differences in the types of CPDs elicited. It is interesting to note when pol η is deleted from murine cells, C>T transitions decrease in TCG triplets and become enriched within CCA sites 46. This suggests that the CCA bias observed in our UVB-irradiated tumors could result from the use of other translesion synthesis enzymes.
Efforts to parse the mutational signatures of UVA and UVB have been hindered by a lack of data in which melanoma onset can be attributed to a single form of UV irradiation. However, exploiting the controlled genetic background and tunable UV sensitivity of our models, we were able to derive distinct mutational signatures induced by UVA (Signature C) or UVB (Signature A) (Fig. 6A-B). Signature A is enriched in human cutaneous melanomas and exhibits high concordance with COSMIC mutational signatures associated with UV damage (SBS7a, SBS7b; Fig. 7A, D). Signature C, however, aligns closely with human mutational signatures of unknown origin (SBS51, SBS53; Fig. 7A). Increased frequencies of C>A and T>A transversions distinguish our UVA-enriched Signature C from Signature A (Figs. 4B-C, 6A-B). While C>A transversions likely result as a consequence of UVA-induced guanine oxidation, the origin of T>A mutations is unclear. Of note, >90% of BRAFV600E mutations are caused by T>A transversions, suggesting a possible role for UVA in the etiology of these alterations 47.
The potential enrichment of Signature C in human uveal melanomas is interesting, as UVA has superior tissue penetrance and is more likely than UVB to reach melanocytes within the eye 34. Unfortunately, there is no available uveal melanoma dataset large enough to separate tumors of the choroid from those of the iris and ciliary body to which UVA can penetrate 34. The fact that uveal melanomas lack conical UV mutation signatures (e.g. SBS7a/b; Figure 7b) has led to speculation that UV irradiation may play a minor role in this disease 48. However, welders, who are occupationally exposed to full-spectrum UV irradiation, are at increased risk for uveal melanoma 49. Whether this risk could be due to UVA is unclear. Using polysulphone dosimeters it is estimated that shielded welders still experience up to 90-150 kJ/m2 of facial UV irradiation in an 8 hour day 50,51. However, polysulphone dosimeters do not measure UVA, which can penetrate materials like glass. Therefore, UVA exposures are likely to be high and should be further explored as an occupational hazard in welders.
Mutations in Signature A and SBS7a/b are biased to the untranscribed DNA strand. This bias has been attributed to differences in the efficiency of transcription-coupled repair (TCR) as compared to global genome repair (GG-NER). TCR rapidly recruits NER components to UV-induced DNA lesions within actively transcribed genes, whereas GG-NER is responsible for resolving mutations throughout the genome 52. Why our UVA-enriched mutational signatures, SBS51, SBS53 and Signature C, exhibit inconsistent transcriptional strand bias is unclear (Fig. 6C). It is similarly possible that the damage response, burden and types of lesions induced by each form of UV underlie this observation.
Together, our data suggest a model in which C>T transitions are at the crux of both UVA- and UVB-mediated carcinogenesis. The limited frequency of TCG-associated C>T mutations in melanomas from UVA-versus UVB-irradiated mice, supports a model in which melanocyte carcinogenesis and melanoma formation is associated with the persistence of CPDs after irradiation. Since the melanoma onset is more dramatically impacted by UVB than UVA, methods to enhance the rate of CPD resolution should mitigate the ability of sunlight to facilitate skin cancer formation.
MATERIALS AND METHODS
Mouse models
All animal research protocols were reviewed and approved by The Ohio State University Institutional Animal Care and Use Committee (Protocol #2012A00000134). The TN and TB models employ a tamoxifen-inducible transgenic allele (Tyr::CreER(T2)) to drive melanocyte-specific CRE recombinase activity 17. In these models, the endogenous Nras or Braf and Cdkn2a loci are modified such that CRE activation leads to the production of mutant NrasQ61R 15 or BrafV637E (equivalent to human BRAFV600E) 16 and loss of p16INK4a expression 20 (Figure 1A, B). All mice were backcrossed >7 generations to pigmented, C57Bl/6J mice.
Cre induction and UV exposure
Inducible knock-in and knockout alleles were activated with 20 mM 4-hydroxytamoxifen on post-natal (p.n.) days one and two as described 15. Subjects from each litter were randomly assigned to receive either ambient light (No UV), UVA or UVB on p.n. day three. Mice are unfurred at three days of age. Therefore, the use of a depilatory agent was unnecessary. UVB was delivered to the dorsal side of each animal as a single dose of 4.5 kJ/m2 using a fixed position, 16W, 312nm UVB light source (Spectronics #EB-280C). [See 10 for additional source information.] UVA was similarly delivered using a 16W source containing two BLE-8T365 bulbs (Spectronics). Based upon the spectrum of these bulbs, the calculated McKinlay-Diffey erythemal effective energy (EEE) of the original 70 kJ/m2 dose is 14.2 mJ/cm2 53. The average tanning parlor dose is 4.5 Standard Erythema Doses (SEDs; 54). One SED is equivalent to 10 mJ/cm2 EEE-weighted UV light 53. Therefore, an individual receives >3 times more UVA in an average tanning session than a mouse in our 70 kJ/m2 experimental protocol (45 mJ/cm2 / 14.2 mJ/cm2 = 3.17).
Outcome monitoring and histopathology
Mice were randomly numbered following treatment and blindly monitored three times a week for tumor formation. Established melanomas were measured by calipers at least three times per week and tumor size (width × length (mm)) recorded until protocol exclusion criteria were met. Representative tumors were harvested from each cohort, fixed in 10% neutral buffered formalin, routinely processed and embedded in paraffin wax. Sections (4 μm) were stained with hematoxylin and eosin (H&E), and evaluated by a veterinary pathologist, certified by the American College of Veterinary Pathologists (KMDL), using an Olympus BX45 light microscope with attached DP25 digital camera (B&B Microscopes Limited, Pittsburgh, PA). Tumor morphology was assessed as described by Banerjee and Harris 55. In each sample the extent of skin and subcutis tumor invasion, tumor pigmentation and the maximum number of mitotic figures were determined from three different fields of view using a 40x objective and 10x ocular lens with a field number of 22 mm.
Sample preparation and whole exome sequencing
Tumor DNA was isolated from flash frozen tissue using the Quick-DNA Miniprep Plus Kit (Zymo Research). Tissues were placed in 2 mL tubes containing 190 μL of diluted Zymo Solid Tissue Buffer and 3.0 mm zirconium beads (Sigma Aldrich Cat# Z763902). Samples were then subjected to homogenization using the Precellys Evolution Homogenizer (Bertin Instruments) using the preset elastic setting: speed: 6,800 RPM, cycle: 4 × 30 sec, pause: 45 sec. Homogenized samples were then incubated in 20 mg/mL of Proteinase K overnight at 55°C before continuing with the Solid Tissues protocol as described for the Quick-DNA Miniprep Plus Kit. Control DNA was generated from toe clips or splenic tissue derived from ten representative TN and ten representative TB animals. These controls were then combined at an equal ratio and concentrated using the Genomic DNA Clean & Concentrator-10 kit (Zymo Research).
The integrity and concentration of resulting genomic DNA was confirmed on an Aglient TapeStation and Life Technologies Qubit. Indexed libraries were generated from 200 ng of genomic DNA using the Kapa Hyper Prep and Agilent SureSelectXT Mouse All Exon target enrichment systems. Exome hybridization was conducted using 500 ng of each DNA library and the resulting target-enriched fragments PCR-amplified (11 cycles). Indexed libraries were pooled and subjected to paired-end 150 bp sequencing on an Illumina HiSeq4000. Average target coverage was 75X (range 53 -107X). Raw data files were deposited in the NCBI Sequence Read Archive (SRA) under accession #PRJNA574176.
Sequence Alignment, Processing and Variant Calling
Sequences were aligned to mm10 using BWA (version 0.7.15) 56. Duplicates were removed using Picard (version 2.17.11) 57 and the resulting sequences re-aligned around indels (GATK version 3.6) 58. Tumor variants (SNVs and indels) were called from unambiguously mapped reads using VarScan2 (version 2.4) 59 and annotated using Variant Effect Predictor from Ensembl 60. Variants found in pooled normal controls (uninvolved skin from TB and TN mice) or listed in NCBI dbSNP build 150 were excluded.
Analysis of SNVs and CNAs
SNV burden (variants per Mb) was calculated as a function of the total capture region. Discovered SNVs mapping to murine genes with an identified human ortholog were used to determine the mutational overlap between mouse and human tumors. Genes altered in ≥15% of human tumors were identified using provisional TCGA data 61. Oncoprints were made with ComplexHeatmap version 2.0.0 62 and enriched gene ontologies identified using Ingenuity Pathway Analysis (QIAGEN Inc.) 63. To calculate the overlap with human tumors, CNAs were identified using CNVkit 64. Reported CNAs passed a log2 segmentation threshold of 0.2 with support from at least five bins. Genome fraction containing a CNA was determined by computing the footprint of segments surpassing the copy number threshold and dividing this by the total footprint of all segments.
Mutational spectrum analysis
The total burden and relative contribution of each mutation type to No UV-, UVA- and UVB-induced melanomas was determined using the “mut_type occurences” algorithm in the R package for MutationalPatterns 65. Differences in the absolute number of mutations were assessed using a Mann-Whitney U test. Differences in frequency of each SNV type between UVA or UVB samples versus controls (No UV) were determined using t-tests with Holm’s adjustment for multiple comparisons (p< 0.05 considered significant).
Lego plots were generated in Matlab after SNVs in each tumor were classified into one of the 96 possible trinucleotide context substitutions (6 substitution classes X 4 possible flanking 5’ bases X 4 possible flanking 3’ bases), averaged across samples from the same experimental group, and normalized to the relative rate at which each respective trinucleotide repeat occurs within the mm10 exome.
MutationalPatterns was used to examine strand bias, identify de novo mutational signatures and compare mutational signatures with those appearing in COSMIC version 3 33. De novo signatures were selected using non-negative matrix factorization, and a rank of 3 was selected based on maximization of variance explained and a cophenetic score. To assess the enrichment of our de novo or COSMIC v3 signatures, we used a modified version of the “fit_to_signatures” function in the MutationalPatterns package that allowed for the evaluation of strand sensitive mutational signatures. COSMIC v3 signatures were attained from Alexandrov et al. 33. Patients were binned into the signature-present group if the contribution of a given signature was non-zero. We used a Fischer Exact Test to determine if the proportion of patients showing the presence or absence of a signature was significantly different between disease groups.
Immunoblotting
Flash-frozen tumors were homogenized in PBS with Halt phosphatase inhibitor (Thermo Scientific) and protease inhibitor (Sigma) using the Precellys Evolution Homogenizer with Cryolys (Bertin Instruments). The preset elastic setting was used for homogenizing. Tumor homogenates were centrifuged to remove PBS and resuspended in RIPA buffer with phosphatase and protease inhibitors. Lysates were sonicated 2 × 10 seconds using a Branson digital sonifier at 10% amplitude. Samples were centrifuged at 15,000 rpm for 5 min at 4°C and supernatants collected and quantified by Bradford assay (BioRad). Samples (35 µg total) were blotted for BRAF (Santa Cruz sc-5284; 1:500) and β-Actin (Cell Signaling #3700; 1:1000) and imaged using a LI-COR Odyssey CLx system. Bands were quantified using Image Studio Version 5.2 software (LI-COR Biosciences).
Footnotes
Financial support: This work was supported by the Melanoma Research Alliance (309669 to C.E.B.), Damon Runyon Foundation (38-16 to C.E.B and 22-17 to R.L.B.), Pelotonia (R.C.H., E.R.C.) and The National Institutes of Health (F31CA236418 to B.M.M. and P30 CA016058 to The Ohio State University Comprehensive Cancer Center).
Conflict of interest statement: R.L.L. is on the supervisory board of Qiagen, is a scientific advisor to Imago, C4 Therapeutics and Isoplexis which include equity interest. He is a former SAB member of Loxo which was acquired by Eli Lilly. He receives research support from and consulted for Celgene and Roche and has consulted for Lilly, Janssen, Astellas, Morphosys and Novartis. He has received honoraria from Roche, Lilly and Amgen for invited lectures and from Gilead for grant reviews.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}