

Abstract
The human leukocyte antigen (HLA) complex regulates the adaptive immune response by showcasing the intracellular and extracellular protein content to the immune system, where T cells are able to distinguish between self and foreign. Therefore, a comprehensive map of the entirety of both HLA class I- and class II-presented peptides from different tissues, is a highly sought after resource1, as it enables the investigation of basic immunological questions beyond the exome level. In this work, we describe the HLA Ligand Atlas, a comprehensive collection of matched HLA class I and class II ligandomes from 29 non-malignant tissues and 13 human subjects (208 samples in total), covering 38 HLA class I, and 17 HLA*DRB alleles. Nearly 50% of HLA ligands have not been previously described. The generated data is relevant for basic research in diverse fields such as systems biology, general immunology, and molecular biology. Furthermore, the HLA Ligand Atlas provides essential information for translational applications by supporting the development of effective cancer immunotherapies. The characterization of HLA ligands from benign tissues, in particular, is necessary in informing proteogenomic HLA-dependent target discovery approaches. Thus, this data set provides a basis for novel insights into immune-associated processes in the context of tissue and organ transplantation and represents a valuable tool for researchers exploring autoimmunity. The HLA Ligand Atlas is publicly available as a raw data resource but also in the form of a user-friendly web interface that allows users to quickly formulate complex queries against the data set. Both downloadable data and the query interface are available at www.hla-ligand-atlas.org.
Introduction
Major advances in comprehensive biological analyses include sequencing of the human genome (genomics)2,3, entirely assessing human gene expression (transcriptomics)4 and the study of the human proteome (proteomics)5–7. These discoveries are considered milestones that enable a multi-dimensional understanding of biological processes. In the context of the immune system, another consecutive layer can be defined as the HLA ligandome or the immunopeptidome. Proteins encoded by the human leukocyte antigen (HLA) gene complex present peptides on the cell surface for recognition by T cells, which can distinguish self from foreign8,9. This mechanism plays a crucial role in adaptive immunity. Despite HLA class I ligands originating primarily from intracellular proteins, the correlation with their precursors (mRNA transcripts and proteins) is poor10–12, limiting approaches based on in silico HLA-binding predictions in combination with transcriptomics and proteomics data alone13,14.
Thus, direct evidence of naturally presented HLA ligands is required to prove visibility of target peptides to T cells. This is a challenge, for example, in the context of cancer immunotherapy approaches that aim to identify optimal tumor-specific HLA-presented target antigens15. While their discovery is assisted by the proteogenomic-based exploration of malignancies, a major impediment still resides in the selection of benign tissues as a reference for the definition of tumor specificity12.
Due to the scarce availability of benign human tissue, morphologically normal tissue adjacent to the tumor (NAT) is commonly used as a control in cancer research. However, NATs have been shown to pose unique challenges, since they may be affected by disease and have been suggested to represent a unique intermediate state between healthy and malignant tissues, with a pan-cancer-induced inflammatory response16. Consequently, in this study we employed benign tissues originating from research autopsies of donors that have not been diagnosed with any malignancy and have deceased of other causes, an approach previously described as a surrogate source of normal tissue16,17. Although these donors cannot be referred to as “healthy” since they may be affected by a range of non-cancerous disease processes, we designate them as benign to emphasize morphological normalcy and absence of malignancy. Similarly, the Genotype-Tissue Expression Consortium4,18 provides an ample resource of RNA sequencing data of benign tissues originating from autopsy specimens. In accordance with this approach, we performed a large-scale mass spectrometry (MS)-based characterization of both HLA class I and class II ligands providing data from benign human tissues obtained at autopsy and implemented a user-friendly, web based interface to query and access the data at www.hla-ligand-atlas.org.
The increasing importance of investigating HLA ligandomes in health and disease using MS technologies has been well recognized1, especially to inform precision medicine12,15,19–22 and improve HLA-binding prediction algorithms13,14,23–26. However, technical constraints and limited data availability have hampered progress in this field. In this context, we provide a novel, comprehensive resource comprising HLA class I and II ligandomes of 29 different benign tissue types obtained from 13 human subjects. To our knowledge, this is the first study to map peptides presented by both HLA class I and class II molecules in multiple tissues from the same human subject, allowing the investigation of new immunological questions.
Despite its unprecedented comprehensiveness, we recognize the caveat that only a limited number of human subjects are included in this data set. We envision that the scientific community will extend the knowledge of the HLA ligandome with more subjects and HLA allotypes covering additional tissues and cellular subpopulations. By integrating immunopeptidomics with proteomics and sequencing data, we anticipate a more holistic understanding of immunological processes.
Materials and Methods
Experimental model and subject details
Human tissue samples were obtained post mortem during autopsy performed for medical reasons at the University Hospital Zürich. The study was approved by the Cantonal Ethics Committee Zürich (KEK) (BASEC-Nr. Req-2016-00604). For none of the included patients a refusal of post mortem contribution to medical research was documented and study procedures are in accordance with applicable Swiss law for research on humans (Bundesgesetz über die Forschung am Menschen, Art. 38). In addition, the study protocol was reviewed by the ethics committee at the University of Tübingen and received a favorable assessment without any objections to the study conduct (Project Nr. 364/2017BO2).
None of the subjects included in this study was diagnosed with any malignant disease. Tissue samples were collected during autopsy, which was performed within 72 hours after death. Tissue/organ annotation was performed by a board-certified pathologist. Tissue samples were immediately snap-frozen in liquid nitrogen.
HLA typing
High-resolution HLA typing was performed by next-generation sequencing on a GS Junior Sequencer using the GS GType HLA Primer Sets (both Roche, Basel, Switzerland) at the Department of Transfusion Medicine of the University Hospital of Tübingen. HLA typing was successful for HLA-A,-B, and -C alleles. However, HLA class II typing was only reliable for the HLA-DR locus, and incomplete for the HLA-DP and -DQ loci. The subject characteristics are summarized in Table 1 encompassing information on sex, age, the number of collected tissues and HLA class I and II alleles.
List of human subjects, sex, age at death, number of analyzed tissues, and HLA typing.
HLA immunoaffinity purification
HLA class I and class II molecules were isolated from snap-frozen tissue using standard immunoaffinity chromatography. The antibodies employed were the pan-HLA class I-specific antibody W6/32 27, and the HLA-DR-specific antibody L243 28, produced in house (University of Tübingen, Department of Immunology) from HB-95, and HB-55 cells (ATCC, Manassas, VA) respectively. Furthermore, the pan-HLA class II-specific antibody Tü39 was employed and also produced on house from a hybridoma clone as previously described 29. The antibodies were cross-linked to CNBr-activated sepharose (Sigma-Aldrich, St. Louis, MO) at a ratio of 40 mg sepharose to 1 mg antibody for 1 g tissue with 0.5 M NaCl, 0.1 M NaHCO3 at pH 8.3. Free activated CNBr reaction sites were blocked with 0.2 M glycine.
For the purification of HLA-peptide complexes, tissue was minced with a scalpel and further homogenized with the Potter-Elvehjem instrument (VWR, Darmstadt, Germany). The amount of tissue employed for each purification is documented in Table 2. This information is not available for seven tissues, annotated as n.d. in Table 2. Tissue homogenization was performed in lysis buffer consisting of CHAPS (Panreac AppliChem, Darmstadt, Germany), and one cOmpleteTM protease inhibitor cocktail tablet (Roche) in PBS. Thereafter, the lysate was sonicated and cleared by centrifugation for 45 min at 4,000 rpm, interspaced by 1 h incubation periods on a shaker at 4°C. Lysates were further cleared by sterile filtration employing a 5 µm filter unit (Merck Millipore, Darmstadt, Germany). The first column contained 1 mg of W6/32 antibody coupled to sepharose, whereas the second column contained equal amounts of Tü39 and L243 antibody coupled to sepharose. Finally, the lysates were passed through two columns cyclically overnight at 4°C. Affinity columns were then washed for 30 minutes with PBS and for 1 h with water. Elution of peptides was achieved by incubating four times successively with 100 – 200 µl 0.2% TFA on a shaker. All eluted fractions were subsequently pooled. Peptides were separated from the HLA molecule remnants by ultrafiltration employing 3 kDa and 10 kDa Amicon filter units (Merck Millipore) for HLA class I and HLA class II, respectively. The eluate volume was then reduced to approximately 50 µl by lyophilization or vacuum centrifugation. Finally, the reduced peptide solution was purified five times using ZipTip Pipette Tips with C18 resin and 0.6 µl bed volume (Merck,) and eluted in 32.5% ACN/0.2% TFA. The purified peptide solution was concentrated by vacuum centrifugation and supplemented with 1% ACN/0.05% TFA and stored at −80°C until LC-MS/MS analysis.
Sample input mass for HLA immunoaffinity purification.
Mass spectrometric data acquisition
HLA ligand characterization was performed on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) equipped with a Nanospray FlexTM Ion Source (Thermo Fisher Scientific) coupled to an Ultimate 3000 RSLC Nano UHPLC System (Thermo Fisher Scientific). Peptide samples were loaded with 1% ACN/ 0.05% TFA on a 75 µm x 2 cm Acclaim™ PepMap™ 100 C18 Nanotrap column (Thermo Fisher Scientific) at a flow rate of 4 µl/min for 10 minutes. Separation was performed on a 50 µm x 25 cm PepMap RSLC C18 (Thermo Fisher Scientific) column, with a particle size of 2 µm. Samples were eluted with a linear gradient from 3% to 40% solvent B (80% ACN, 0.15% FA in water) over 90 minutes. The column was subsequently washed by increasing to 95% B within 1 minute, and maintaining the gradient for 5 minutes, followed by reduction to 3% B and equilibration for 23 minutes.
Data acquisition was performed as technical triplicates in data-dependent mode, with customized top speed (3 s) methods for HLA class I- and HLA class II-eluted peptides. HLA class I peptides have a length of 8 - 12 amino acids30,31, therefore, the scan range was restricted to 400 - 650 m/z and charge states of 2 - 3. MS1 and MS2 spectra were detected in the Orbitrap with a resolution of 120,000 and 30,000 respectively. Furthermore, we set the automatic gain control (AGC) targets to 1.5*105 and 7.0*104 and the maximum injection time to 50 ms and 150 ms for MS1 and MS2, respectively. The dynamic exclusion was set to 7 s. Peptides were fragmented with collision-induced dissociation (CID) while the collision energy was set to 35%.
HLA class II peptides have a length of 8 - 25 amino acids32,33, thus the scan range was set to 400 −1,000 m/z and the charge states were restricted to 2 - 5. Readout for both MS1 and MS2 were performed in the Orbitrap with the same resolution and maximum injection times as for HLA class I peptides. The dynamic exclusion was set to 10 s and AGC values employed were 5.0*105 and 7.0*104 for MS1 and MS2, respectively. Higher-energy collisional dissociation (HCD) fragmentation with 30% collision energy was employed for HLA class II peptides.
Database search with MHCquant
MS data obtained from HLA ligand extracts was analyzed using the nf-core34 containerized, computational pipeline MHCquant (revision 1.2.6 - https://www.openms.de/mhcquant/) with default settings. The workflow comprises tools of the open-source software library for LC/MS OpenMS (2.4)35. Identification and post-scoring were performed using the OpenMS adapters to Comet 2016.01 rev. 336 and Percolator 3.1.137 at a local peptide level false discovery rate (FDR) threshold of 1% among replicate sample groups. The human reference proteome (Swiss-Prot, Proteome ID UP000005640, 20,416 protein sequences) extended by a customized list of common contaminants encountered in laboratory settings was used as a database reference. Database search was performed without enzymatic restriction and methionine oxidation as the only variable modification and contaminant peptide hits were excluded subsequently. MHCquant settings for high-resolution instruments involving a precursor mass tolerance of 5 ppm and a fragment bin tolerance of 0.02 Da were applied. The peptide length restriction, digest mass and charge state range were set to 8-12 amino acids, 800-2500 Da and 2-3 for class I and 8-25 amino acids, 800-5000 Da and 2-5 for class II, respectively.
HLA binding prediction
Based on subject-specific HLA allotypes, peptide binding predictions were computed. For HLA class I ligand extracts, NetMHCpan-4.026 both in binding affinity and ligand mode (default) were employed. HLA class II ligand extracts were annotated with NetMHCIIpan-3.238 in binding affinity (default) mode. Peptide-allotype associations were categorized as strong binders if the reported affinity based %Rank score was ≤ 0.5 and as weak binders when the reported %Rank score was ≤ 2. Based on the HLA binding prediction, we defined the purity of HLA ligand extracts as the ratio of predicted binders divided by the total number of peptide identifications of a sample. Technical replicates with purities lower than 50% for HLA class I ligands and lower than 10% for HLA class II ligands were not included in the data set. From the remaining sample replicates, only peptides that were predicted at least weak binders against one of the donor alleles were included in the data set.
Data storage web interface
HLA class I and class II peptides that were predicted to be HLA ligands according to the previously defined criteria, were complemented with their tissue association and stored in an SQL database. A public web server was implemented that allows users to formulate queries against the database, visualizes the results and allows data export for further analysis. The web front-end was implemented in HTML, CSS and JavaScript based on the front-end framework Bootstrap 4. The table plugin DataTables was used to provide rapid browsing and filtering for tabular data. Interactive plots were designed using Bokeh and ApexCharts.
Results
Content and scope of the HLA Ligand Atlas data resource
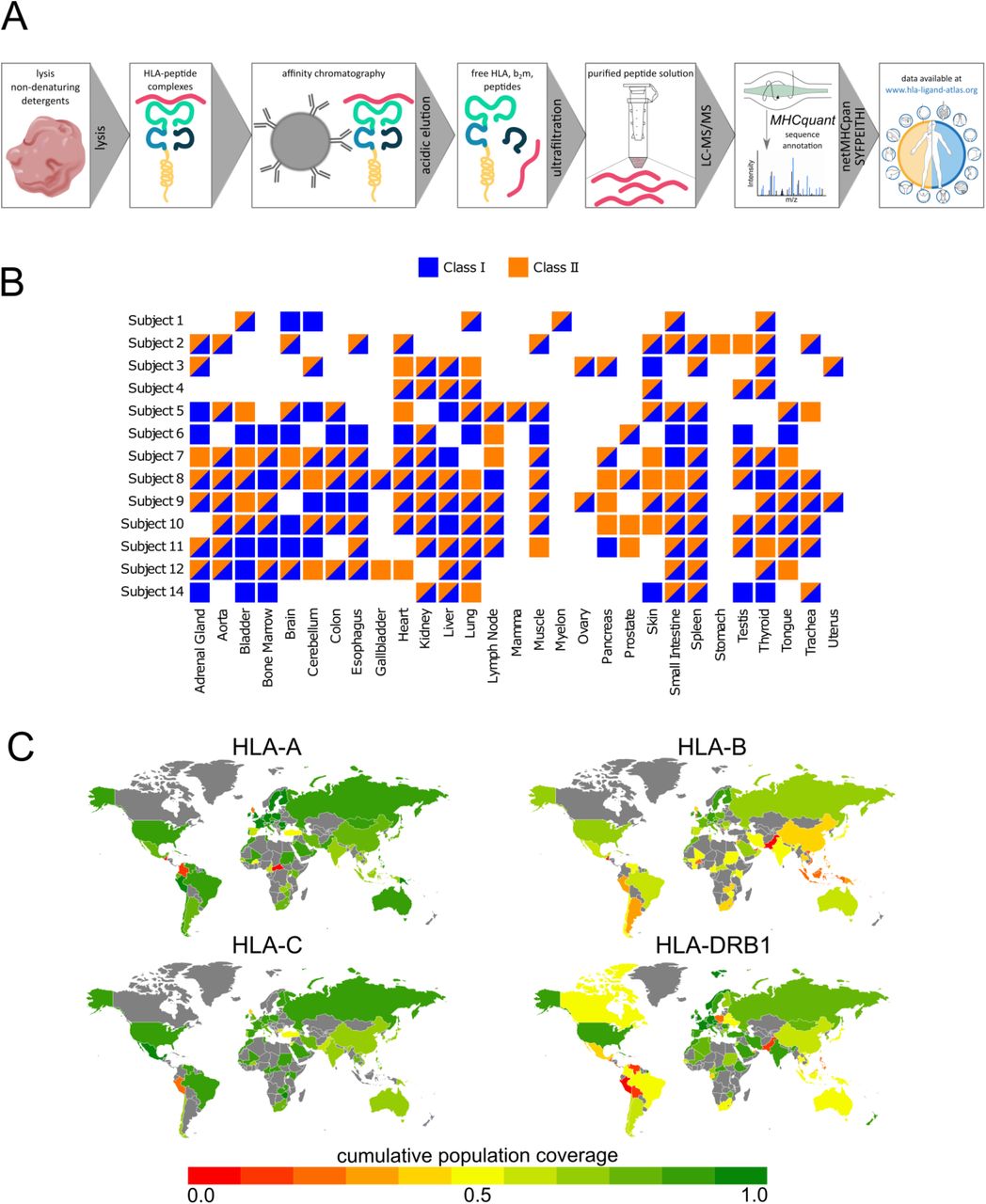
To generate HLA ligandomics data of benign tissues, we employed a well described immunoprecipitation protocol20,39 followed by peptide sequencing via LC-MS/MS. Database search was performed using the open-source, bioinformatics pipeline MHCquant and resulting peptide sequences were filtered according to their binding score and stored in the www.hla-ligand-atlas.org database (Figure 1 A, 2 A).

Workflow schematic and associated experimental design. A) Standard immunoaffinity chromatography was used to isolate and purify HLA ligands followed by peptide sequencing via LC-MS/MS. Resulting HLA ligands are accessible via the www.hla-ligand-atlas.org. B) The HLA Ligand Atlas covers up to 29 different tissues obtained from 13 subjects. The sample matrix illustrates donor-tissue combinations that passed the quality criteria as described in the materials and methods section. C) Cumulative HLA-A, -B, -C, and -DRB1 population coverage. Color-coded population coverages are indicated per country, whereas countries that were not covered by the IEDB data set are marked in grey.
Overall, we have acquired HLA ligandome data from tissues obtained from 13 autopsy subjects, encompassing 29 human tissue types as detailed in Figure 1 B. In total 48,381 HLA class I ligands and 16,146 HLA class II ligands derived from 12,307 source proteins are included in the online resource. Nearly 50% of these peptides have not been described so far. Furthermore, all HLA ligands are linked to the immune epitope database and prediction resource (IEDB; www.iedb.org).
HLA-based vaccine design and diagnostic studies need to take into consideration the HLA polymorphism and their different frequencies in different ethnicities. Thus, we sought to determine an approximation for how frequent the HLA alleles comprised in the HLA Ligand Atlas are represented in different regions of the world (Figure 1 D)15,40. For this purpose, we computed population coverages using the IEDB Analysis Resources (http://tools.iedb.org/population/41). Worldwide, the 12 different HLA-A alleles included in our study achieve a cumulative population coverage of 91.71% with an average match of 1.34 alleles per individual. For HLA-B (16 different alleles) and HLA-C (10 different alleles), the cumulative population coverage is estimated at 63.92% and 85.65% with an average number of 0.78 and 1.19 alleles matching per individual. The 14 different HLA-DRB1 alleles included in the data set cover 86.65% of the world’s population with an average match of 1.23 alleles per individual. Thus, the comprehensive immunopeptidomics data set we provide covers both prevalent and infrequent HLA alleles, which can be easily accessed for HLA-based studies.
The HLA Ligand Atlas web interface
The HLA Ligand Atlas web interface was designed to allow users to conveniently access the data we present here. Users can formulate queries in the form of filters based on peptide sequences, peptide sequence patterns, HLA allotypes, tissues and proteins of origin. These filters can be combined arbitrarily. Additionally, users can submit a file with peptides, either as a plain list or as a FASTA-formatted file. The peptide list is then queried against the database and the resulting hits can again be freely combined with the aforementioned filters. Query results are shown as a list of peptides with plots of the corresponding HLA allotype and tissue distributions. Additionally, detailed views for single peptides and for coverage of proteins are available.
The detailed peptide view features an evidence matrix summarizing the collected HLA allotype - tissue associations for the observed peptide. An example for the peptide sequence MRYVASYL is shown in Figure 2 B. The peptide has been found on eight different subjects and has been annotated to different HLA allotypes, since the binding prediction was always performed against all HLA alleles of the respective donor.

{kind=link}
{kind=link}
A) Data matrix illustrating identification numbers per HLA allotype and tissue. B) Evidence matrix showing the identification of peptide MRYVASYL on all analyzed organs, together with all HLA class I and class II allotypes that were annotated. C) Peptide coverage matrix illustrating positions covered by additional HLA class I or class II ligand identifications within the source protein PRLP2/RLA2_human.
The evidence matrix for each peptide illustrating the HLA-allotype association in the data set can easily be accessed through the HLA Ligand Atlas. Furthermore, it is possible to investigate the source protein of each detected peptide. In case of the provided exemplary peptide MRYVASYL it is the 60S acidic ribosomal protein P2 (RPLP2/RLA2_human). The detailed protein view contains a coverage plot for the protein (Figure 3 C), which illustrates the positions within the respective protein sequence that are covered by identified HLA class I and HLA class II-presented peptides.
Apart from the query interface, the web front-end also displays various aggregate views of the data stored in the database.
Concluding remarks
In this work we provide a comprehensive HLA class I and class II ligandomics data set that can assist in answering multifaceted questions. HLA class I and class II binding prediction algorithms can be substantially improved by increasing the number of subjects with reliable HLA ligandomics data employed for training13,25,44. Further coverage of infrequent and poorly studied HLA allotypes, such as HLA-A*30:01, can increase the accuracy of binding prediction.
Furthermore, the study of non-mutated tumor-associated HLA ligands both from canonical12,19 and non-canonical transcripts45,46, requires a comprehensive map of benign HLA ligandomes as a reference data set20. Thus, direct evidence of HLA ligands by MS is required to prove their presentation and visibility to T cells. Moreover, the investigation of patient- and tissue-specific peptides, hotspots of presentation15,47,48 and the prevalence of their source proteins can be assessed in detail with the data comprised in the HLA Ligand Atlas.
Financial support
funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy - EXC 2180 – 390900677
funded by Deutsche Forschungsgemeinschaft (DFG) SFB 685 „Immunotherapy: Molecular Basis and Clinical Application“
funded by ERC AdG 339842 MUTAEDITING
de.NBI - Deutsches Netzwerk für Bioinformatik
Böhringer Ingelheim Foundation for basic research in medicine
Competing interests
Linus Backert and Daniel J. Kowalewski are employees of Immatics Biotechnologies GmbH. Linus Backert, Daniel J. Kowalewski, Markus W. Löffler, and Stefan Stevanović are inventors of patents owned by Immatics Biotechnologies GmbH. Hans-Georg Rammensee is shareholder of Immatics Biotechnologies GmbH and Curevac AG.
Acknowledgements
We thank Claudia Falkenburger, Ulrich Wulle, Patricia Hrstić, Nicole Bauer, and Beate Pömmerl for excellent technical support.
Footnotes
Abbreviations
- ACN
- Acetonitrile
- AGC
- Automatic gain control
- CID
- Collision-induced dissociation
- Da
- Dalton
- FA
- Formic acid
- FDR
- False discovery rate
- HCD
- Higher-energy collisional dissociation
- HLA
- Human leukocyte antigen
- IEDB
- Immune epitope database and analysis tool
- LC
- Liquid chromatography
- MS
- Mass spectrometry
- MS/MS
- Tandem mass spectrometry
- NAT
- Non-malignant adjacent tissue
- PBS
- Phosphate-buffered saline
- ppm
- Parts per million
- TFA
- Trifluoroacetic acid
- UHPLC
- Ultra-high performance liquid chromatography
References




