

Abstract
Background Dietary composition and antibiotic use are known to have major impacts on the structure and function of the gut microbiome. In turn, the dysbiosis caused by antibiotic treatment or consumption of diets low in microbiota-accessible carbohydrates (MACs) is associated with a number of acute or chronic co-morbities in the host, such as obesity or opportunistic infections. Despite this, little research has been done to explore the role of host diet as a determinant of antibiotic-induced microbiome disruption.
Results Here, we utilize a multi-omic approach to characterize the impact of Western-style diet consumption on ciprofloxacin-induced changes to gut microbiome community structure and transcriptional activity. We found that mice consuming a Western-style diet experienced a greater expansion of Firmicutes following ciprofloxacin treatment than those eating a control diet. At the transcriptional level, we found that ciprofloxacin induced a reduction in the abundance of TCA cycle transcripts on both diets, suggesting that carbon metabolism plays a key role in the response of the gut microbiome to this antibiotic. Despite this shared response, we observed extensive differences in the response of the microbiota to ciprofloxacin on each diet. In particular, at the whole-community level we detected an increase in starch degradation, glycolysis, and pyruvate fermentation following antibiotic treatment in mice on the Western diet, which we did not observe in mice on the control diet. Similarly, we observed diet-specific changes in the transcriptional activity of two important commensal bacteria, Akkermansia muciniphila and Bacteroides thetaiotaomicron, involving diverse cellular processes such as nutrient acquisition, stress responses, and capsular polysaccharide (CPS) biosynthesis.
Conclusions Our findings build on recent work and demonstrates that host diet plays a key role in determining the extent of microbiome disruption induced by antibiotic treatment. Thus, future studies investigating the impact of antibiotics on the microbiota should consider the impact that dietary composition may have on the interpretation of results. In the long term, the relationship between diet and microbiome disruption may help to identify ways to reduce the incidence of dysbiosis following clinical therapy in humans.
Background
The gut microbiome includes the trillions of largely commensal bacteria, archaea, fungi, and viruses and their collective genetic material that inhabit the gastrointestinal tract [1–3]. These communities play an important role in numerous biological processes, such as digestion, neurological development, colonization resistance, and immune function [4–18]. However, the gut microbiome is exquisitely sensitive to perturbation and the disruption of microbial homeostasis, known as dysbiosis, can result in numerous harmful impacts to the host. In particular, broad-spectrum antibiotic use is known to have numerous detrimental impacts on the gut microbiota. Within hours of treatment, antibiotics induce dramatic reductions in both bacterial loads and diversity within the microbiome [19, 20].
While compositional changes are typically transient and recover following the cessation of therapy, oftentimes the structure and diversity of the microbiota never return to their pre-treatment levels. These changes often result in dysbiosis and have numerous acute and chronic impacts on host health. In particular, dysbiosis may increase the risk of infection with opportunistic fungal and bacterial pathogens by reducing colonization resistance [1,5–7,21–25]. Most notably, broad-spectrum antibiotic treatment is known to be a major risk factor in Clostridioides difficile infection, which is responsible for approximately 29,000 deaths worldwide and 15,000 in the United States alone [21,23,26,27]. In addition to short-term complications such as pathogen blooms, persistent dysbiosis is correlated with a number of chronic conditions associated with considerable morbidity and mortality, such as asthma, obesity, and inflammatory bowel disease [5,8–10,13,15,17,18,28].
In addition to eliciting changes in community structure, antibiotic exposure also has a dramatic impact on the functional activity of the gut microbiome by altering the expression of key metabolic genes and the availability of carbohydrates within the gut [20]. Most notably, amoxicillin treatment has been shown to increase the expression of glycoside hydrolases responsible for hydrolysis of polysaccharides, while simultaneously decreasing the abundance of transcripts encoding sugar phosphotransferase systems (PTS) responsible for uptake of simple sugars [20]. Reflecting these changes, amoxicillin also decreases the concentration of glucose within the ceca of mice [20]. Furthermore, dietary intervention experiments have demonstrated that the response of the microbiota to antibiotics can be impacted by nutrient modulation. For example, glucose supplementation reduces the absolute abundance of bacteria, particularly Bacteroides thetaiotaomicron, following amoxicillin treatment in mice [20]. Therefore, it is likely that host diet composition has a major impact on the response of these communities to perturbation.
It is also known that dietary composition has a profound impact on microbiome diversity and overall gut health [29–35]. Diets high in fat and simple sugars, typically referred to as “Western” diets, have been associated with a number of negative health states including obesity, diabetes, allergies, and inflammatory bowel disease [36–46]. Such diets have very low levels of microbiota-accessible carbohydrates (MACs), which are typically found in complex plant polysaccharides and are indigestible and unabsorbable by the host [40,44,47–49]. MACs are typically fermented by the colonic microbiota to produce short-chain fatty acids (SCFAs), which in turn play important roles in regulating energy homeostasis and inflammation within the host [40,45,50–55]. In addition to being associated with increased levels of SCFAs, high-MAC diets have been shown to increase microbial diversity, a classic benchmark for gut microbiota health. Conversely, low-MAC Western diets are known to reduce both microbiome diversity and SCFA production [44,46,49,56]. Due to the absence of MACs, such diets also enrich for muciniphilic microbes that may degrade mucosal lining of the gut, such as Akkermansia muciniphila [40,42,48]. Degradation of the mucosal layer over time is hypothesized to result in compromised gut barrier function that may ultimately lead to increased inflammation and colitis. Lastly, MAC-deficient diets have been shown to exacerbate infection with C. difficile, suggesting that they may negatively impact colonization resistance [26, 27].
Individually, antibiotic usage and the consumption of Western-style diets are known to negatively impact the microbiota, resulting in similar long-term impacts on host health (including obesity, allergies, and inflammatory bowel disease). Despite this, little work has been done to explore how diet impacts the response of the microbiota to antibiotics. Previous work has suggested that dietary composition may play an important role in determining the extent of antibiotic-induced microbiome disruption [20]. In this study, we use a combined metagenomic and metatranscriptomic approach to characterize the impact of a Western-style diet on the taxonomic and functional disruption of the microbiome during ciprofloxacin treatment. Using shotgun metagenomics, we found that ciprofloxacin elicited differential impacts on community composition in mice at both the phylum- and species-level that were diet-dependent. Using metatranscriptomics, we observed that consumption of a Western diet itself induced profound transcriptional changes within the gut microbiomes of mice; furthermore, consumption of this diet modulated the transcriptional response of these communities to antibiotic treatment. In particular, dietary composition had a major impact on the abundance of transcripts encoding key metabolic genes. Notably, we also found that host diet composition had a differential impact on the expression of known virulence factors and antibiotic resistance genes (ARGs) within the microbiome, suggesting that diet may contribute to the expansion of pathogenic bacteria following treatment. Lastly, we were able to detect unique species-specific transcriptional changes in response to both diet and ciprofloxacin treatment in two important commensal bacteria, A. muciniphila and B. thetaiotaomicron. In addition to detecting changes reflective of ciprofloxacin’s primary mechanism of action as an inhibitor of DNA gyrase, we observed that antibiotic treatment impacted a number of diverse cellular processes involving capsular polysaccharide synthesis, stress responses, and nutrient acquisition, amongst others. Together, our findings build on previous literature that demonstrates that antibiotics have a differential impact on the structure and function of the gut microbiome that is dependent on host diet composition.
Results
To determine the impact of dietary composition and antibiotic exposure on the structure and function of the murine gut microbiome, female C57BL/6J mice were fed either a high-fat, high-sugar “Western”-style diet, or a low-fat control diet for seven days. Mice were subsequently treated with a clinically relevant dosage of ciprofloxacin or a vehicle control for 24 hours. This time point was chosen because previously published work demonstrated that 24 hours of ciprofloxacin treatment was sufficient to induce changes in community structure and transcriptional activity [20]. Additionally, this time frame allowed us to profile the acute response of the microbiota to ciprofloxacin exposure, rather than characterizing a post-antibiotic state of equilibrium. Following treatment, the mice were sacrificed to harvest their cecal contents for metagenomic and metatranscriptomic analysis (Figure 1A). Overall, we found that diet and ciprofloxacin treatment had a significant impact on gut microbiome structure (Figure 1B+C).

A. Experimental workflow used in this study. Figure was created with Biorender.com.
B. Alpha diversity of experimental groups as measured by the Shannon Diversity Index. Data are represented as mean ± standard error of the mean (SEM). (**p<0.01, Welch ANOVA with Dunnett T3 test for multiple hypothesis testing).
C. Stacked barplot of the five most abundant bacterial phyla in our dataset. Data are represented as mean + SEM for each phylum.
D. Differentially abundant (Benjamini-Hochberg adjusted p-value < 0.1) bacterial species detected in mice consuming the Western diet (WD). Data are represented as log2 fold change relative to control diet ± standard error. Bar color denotes phylum level taxonomic classification (blue – Verrucomicrobia, red – Firmicutes, green – Bacteroidetes, purple – Proteobacteria, orange – Actinobacteria).
E. Heatmap of the change in abundance of the top 90 bacterial species in response to ciprofloxacin on control (NC) and Western (WD) diets. The Interaction column represents the interaction term generated by DESeq2, denoting the impact of diet on the change in abundance of each species to ciprofloxacin. Cell color denotes log2 fold change of a particular species in response to ciprofloxacin. Heatmap rows were sorted by interaction term value from highest to lowest.
(F-H) Normalized counts of B. thetaiotaomicron (F), A. muciniphila (G), and Blautia sp. YL58 (H) in each experimental group. Data are represented as mean ± SEM. Normalized counts were generated with DESeq2 and subsequently used to perform differential abundance testing. (*p<0.05, ****p<0.0001, Wald test with Benjamini and Hochberg correction).
For all analyses, n ≥ 3. For full DESeq2 results, see Additional File 1.
Mice consuming the Western diet had significantly less diverse gut microbiomes than those fed the control diet (Figure 1B). In general, mice fed a Western diet displayed elevated levels of the phyla Verrucomicrobia and Bacteroidetes, and a reduced abundance of Firmicutes (Figure 1C). At the species level, these shifts appear to be largely driven by an expansion of members of the Bacteroides genus (Figure 1D, Additional File 1). Additionally, the Western diet-fed mice displayed an elevated abundance of several species from the Proteobacteria phylum, which has been previously shown to be suggestive of dysbiosis [57]. Two important bacterial species found in the gut microbiomes of both mice and humans, B. thetaiotaomicron and A. muciniphila, were observed at significantly elevated levels in the mice fed a Western diet [20] (Figure 1F+G, Additional File 1). Notably, both species are known to utilize host-produced mucins; thus, this observation is consistent with earlier studies that have suggested that the consumption of a low-MAC Western diet enriches for muciniphilic bacteria [40,42,48].
Overall, host diet appears to have a major impact on the structure on the microbiome during ciprofloxacin treatment. While ciprofloxacin did not induce a significant reduction in alpha diversity in the timeframe tested, at the phylum level we observed a significant expansion in the relative abundance of Firmicutes following ciprofloxacin treatment on the Western diet (adjusted p-value = 0.0388) but not on the control diet (adjusted p-value = 0.8718) (Figure 1C). To determine which species displayed a differential response to ciprofloxacin on the Western and control diets, we utilized DESeq2 to analyze the interaction between diet and antibiotic treatment [58]. An interaction term describes the direction and magnitude of the differential impact that host diet has on antibiotic perturbation within the microbiome. For example, ciprofloxacin reduces the abundance of Clostridium innocuum on the control diet, while it expands following treatment on the Western diet (Figure 1E, Additional File 1); thus, the interaction term in this case is positive, as it indicates an expansion on the Western diet relative to control following ciprofloxacin exposure.
While most species responded similarly to ciprofloxacin therapy on both diets, there were several notable exceptions. For example, the expansion of several Clostridium species (such as C. innocuum, Clostridium beijerinckii, and Clostridium scindens) following ciprofloxacin tended to be higher on the Western diet than the control (denoted by a positive interaction value in Figure 1E, Additional File 1). Conversely, the reduction of several Bacteroides species following antibiotic treatment tended to be exacerbated on the Western diet (negative interaction values, Figure 1E, Additional File 1). In particular, the reduction of B. thetaiotaomicron in response to ciprofloxacin was enhanced on the Western diet; in contrast, dietary composition had no significant impact on the response of A. muciniphila to ciprofloxacin (Figure 1F+G, Additional File 1). Lastly, Western diet consumption appeared to have the most detrimental impact on Blautia sp. YL58 during ciprofloxacin therapy, as evidenced by its large, negative interaction term (Figure 1H, Additional File 1). While ciprofloxacin did not significantly reduce the abundance of Blautia sp. YL58 in mice consuming the control diet, this bacterium was completely undetectable following treatment on the Western diet. This is particularly notable because this species was present in comparable levels in vehicle-treated animals on both the Western and control diets (Figure 1H, Additional File 1).
Ciprofloxacin Elicits Unique Shifts in Gene Expression on Western and Control Diets
Though many studies have examined the impacts of either diet or antibiotic treatment on the gut microbiome, few have examined their combined effect on the composition or gene expression of these communities. To address this, we first analyzed our metatranscriptomic dataset using the HUMAnN2 pipeline, which normalizes the abundance of RNA transcripts against their corresponding gene abundance in the metagenomic data [59]. Thus, this tool normalizes for differences in community composition between experimental groups and facilitates the comparison of metabolic pathway expression at the whole-community level. A comparison of the transcriptional profile of all experimental groups demonstrates that the microbiota of mice consuming the Western diet display elevated expression of tricarboxylic acid (TCA) cycle and fatty acid degradation pathways in both vehicle and ciprofloxacin treatments, likely reflective of the increased fat and sugar content of this diet (Figure 2A, Additional File 2). Additionally, we found an elevated expression of glycogen degradation genes in the Western diet mice receiving ciprofloxacin, which was not observed in any other group (Figure 2A, Additional File 2).

A. Linear discriminant analysis (LDA) of MetaCyc pathways that were differentially associated with each experimental group.
B. Pairwise LDA of vehicle-treated mice consuming either the Western (WD) or control (NC) diets.
C. Pairwise LDA of vehicle- and ciprofloxacin-treated mice consuming the Western (WD) diet.
D. Pairwise LDA of vehicle- and ciprofloxacin-treated mice consuming the control (NC) diet.
Bar size indicates LDA score and color indicates the experimental group that a MetaCyc pathway was significantly associated with. All LDA scores were generated using LEfSe on unstratified pathway outputs from HUMAnN2. For all analyses, n ≥ 3. For full pathway names and statistics, see Additional File 2.
Conversely, the microbiota of mice consuming the control diet appeared to have elevated expression of amino acid biosynthesis pathways (namely isoleucine, aspartate, asparagine, lysine, and histidine) regardless of antibiotic treatment (Figure 2A, Additional File 2). Interestingly, we also observed elevated levels of several different pathways of nucleotide biosynthesis in the control diet samples receiving vehicle while the Western diet mice displayed elevated levels of adenosine and guanosine nucleotide degradation (Figure 2A, Additional File 2).
A pairwise comparison between the vehicle-treated samples on the Western and control diets reveals that the microbiota exhibits extensive transcriptional changes in response to dietary modulation (Figure 2B, Additional File 2). Notably, the microbiota on the control diet-fed mice displayed elevated expression of nucleotide biosynthesis, glycolysis, gluconeogenesis, starch degradation, and pyruvate fermentation (Figure 2A+B, Additional File 2). We also observed increased expression of the Bifidobacterium shunt, which is known to play a role in SCFA production and may provide mechanistic insight into the the reduced SCFA levels observed on the Western diet in other studies [40, 51] (Figure 2B, Additional File 2).
When we compared the response of the microbiome to ciprofloxacin on each diet, we found key differences in the overall transcriptional profiles (Figure 2C+D, Additional File 2). In mice consuming the Western diet, ciprofloxacin treatment was associated with increased abundance of transcripts from glycogen and starch degradation, glycolysis, and pyruvate fermentation (Figure 2C, Additional File 2). Notably, the expression of glycogen degradation was elevated in vehicle-treated samples on the control diet, suggesting that the utilization of this pathway during ciprofloxacin treatment is diet-dependent (Figure 2D, Additional File 2). On the control diet, we observed an increased abundance of inosine-5-phosphate biosynthesis with ciprofloxacin treatment (Figure 2A+D, Additional File 2). Interestingly, we observed that TCA cycle expression was reduced in ciprofloxacin-treated mice compared to the vehicle treatment in both control and Western diet conditions – the lone commonality between diets (Figure 2C+D, Additional File 2). Previous work has demonstrated that elevated TCA cycle activity increases sensitivity to bactericidal antibiotics, including fluoroquinolones, in vitro [60–64]. Thus, this result suggests that TCA cycle activity may play a key role in the response of the microbiota to ciprofloxacin treatment in vivo, though more work is required to understand its impact.
Ciprofloxacin has a differential impact on the abundance of iron metabolism and mucin degradation transcripts on the Western versus control diets
Due to the potential limitations of the use of a single pipeline, we analyzed our metatranscriptomic dataset with SAMSA2 in parallel with HUMAnN2. While HUMAnN2 normalizes for DNA abundance, SAMSA2 does not have this feature and thus the output is representative of overall transcript levels rather than relative expression. Despite these differences, many of the changes observed using HUMAnN2 were detected using SAMSA2 at the SEED subsystem level. When comparing the vehicle-treated samples on both diets, SAMSA2 detected an increased abundance of transcripts related to respiration in the Western diet, mirroring the increase in TCA cycle expression found with HUMAnN2 (Figure 3A, Additional File 3). The microbiota from the Western diet-fed mice also displayed an increased abundance of transcripts involving fatty acids and iron acquisition, which likely reflect altered nutrient availability (Figure 3A, Additional File 3). Furthermore, transcripts related to virulence and disease were elevated in mice consuming the Western diet, further supporting the hypothesis that consumption of a Western diet may promote dysbiosis and the expansion of enteric pathogens (Figure 3A, Additional File 3). Interestingly, comparatively few subsystems were changed in abundance following ciprofloxacin treatment on either diet (Figure 3B+C, Additional File 3). Most notably, we observed a decrease in transcripts related to dormancy and sporulation in response to ciprofloxacin on both diets (Figure 3B+C, Additional File 3). A similar finding was observed in a recent study in which mice were treated with ciprofloxacin while consuming a non-purified diet, suggesting that these transcripts may play a key role in the response of the microbiota to this antibiotic [20].

A. Differentially expressed (Benjamini-Hochberg adjusted p-value < 0.1) level 1 SEED subsystems in the murine cecal metatranscriptome in vehicle-treated mice consuming the Western (WD) diet. Data are represented as log2 fold change relative to control diet ± standard error. Only features with a base mean ≥ 50 were plotted. See Additional File 3 for full results.
B. Differentially expressed (Benjamini-Hochberg adjusted p-value < 0.1) level 1 SEED subsystems in the murine cecal metatranscriptome in ciprofloxacin-treated mice consuming the control (NC) diet. Data are represented as log2 fold change relative to vehicle control ± standard error. Only features with a base mean ≥ 50 were plotted. See Additional File 3 for full results.
C. Differentially expressed (Benjamini-Hochberg adjusted p-value < 0.1) level 1 SEED subsystems in the murine cecal metatranscriptome in ciprofloxacin-treated mice consuming the Western (WD) diet. Data are represented as log2 fold change relative to vehicle control ± standard error. Only features with a base mean ≥ 50 were plotted. See Additional File 3 for full results.
(D-G) Volcano plots of the metatranscriptomic profile of the murine cecal microbiome in vehicle-treated mice consuming Western diet (D), ciprofloxacin-treated mice on the control diet (E), and ciprofloxacin-treated mice on the Western diet (F). Interaction terms representing the impact of diet on ciprofloxacin-induced changes for each transcript are shown in (G). Data was generated by aligning metatranscriptomic reads to RefSeq using SAMSA2 and analyzing using DESeq2. Points in red represent transcripts for which a statistically significant change in expression was detected (Benjamini-Hochberg adjusted p-value < 0.1). Select genes of interest are labeled. See Additional File 4 for full results.
For all analyses, n = 4.
An additional strength of the SAMSA2 pipeline is that it enables differential abundance testing of individual transcripts in addition to pathway- and subsystem-level analysis [65]. We observed that the microbiota of the mice consuming the Western diet displayed increased abundance of heme b synthase transcripts, which may suggest increased iron acquisition (Figure 3D, Additional File 4). Interestingly, in the untreated mice, we detected large, Western diet-associated increases in the abundance of two different transcripts encoding sialidases, which play a key role in the utilization of host-produced mucins [66] (Figure 3D, Additional File 4). While other studies have shown that the consumption of a Western diet enriches for muciniphilic taxa, this observation demonstrates that such a diet also increases transcriptional activity related to mucin degradation within the microbiome [40, 42]. Furthermore, ciprofloxacin increased the abundance of sialidase transcripts in mice on the control diet, suggesting that this effect may be exacerbated by antibiotic treatment (Figure 3E, Additional File 4).
In addition to the increased abundance of sialidase transcripts, ciprofloxacin induced a number of notable transcriptional changes on each diet. Reflecting the overall reduction in sporulation seen at the subsystem level, we found that the abundance of several sporulation-related transcripts were reduced on the control diet following ciprofloxacin treatment (Figure 3E, Additional File 4). Additionally, we detected an increased abundance of transcripts related to glycoside hydrolase family 98 (GH98; Figure 3E, Additional File 4) on the control but not the Western diet. This family of glycoside hydrolases is known to include endo-beta-galactosidase, an enzyme that cleaves AB blood group surface antigens and has been shown to play a role in the virulence of organisms such as Clostridium perfringens and Streptococcus pneumoniae [67]. On the Western diet, we observed that ciprofloxacin treatment reduced the abundance of transcripts encoding a cluster of sulfate reductases, which may indicate a broader role for these enzymes during antibiotic treatment. Among the transcripts that were increased in abundance on this diet were several that encoded flagellin and related proteins, which can play a role in pathogenicity [68, 69] (Figure 3F, Additional File 4). Additionally, we found increases in transcript levels of several phage-related genes, which has been observed previously in response to ciprofloxacin treatment [20] (Figure 3F, Additional File 4).
Lastly, we examined the interaction of diet and antibiotic treatment on transcript abundance within the microbiome. Notably, we found that the transcript abundance of several sporulation genes following ciprofloxacin treatment was significantly higher on the Western diet than the control (Figure 3G, Additional File 4). Additionally, transcripts encoding phosphotransferase system (PTS) transporters of various substrates (such as cellobiose, fructose, and N-acetylgalactosamine) were also found to be higher on the Western diet following ciprofloxacin treatment (Figure 3G, Additional File 4). Conversely, consumption of the Western diet significantly reduced the change in transcript abundance of both pectate lyase and a hemin receptor following ciprofloxacin therapy. Together, these findings demonstrate that dietary composition significantly impacts the transcriptional response of the microbiome to ciprofloxacin.
Host diet has a major impact on the transcript abundance of antibiotic resistance and virulence factor genes
Previous work has suggested that dietary composition may promote the virulence of several known bacterial pathogens [20,70–73]. Furthermore, the emergence of antibiotic resistance genes (ARGs) during clinical therapy threatens the efficacy of many drugs [74]. Therefore, we sought to profile the impact of diet and ciprofloxacin treatment on the abundance of known ARG and virulence factor transcripts within the microbiome. Overall, diet appeared to have the most dramatic impact on the abundance of ARG transcripts at the class level (Figure 4A, Additional File 5). The microbiota of mice consuming the Western diet had an elevated abundance of ARG transcripts against fosmidomycin, beta-lactam, glycopeptide and peptide antibiotics (Figure 4A, Additional File 5). The observed increase in glycopeptide ARGs appears be driven primarily by increased transcript levels of four genes that confer resistance to vancomycin (vanG, vanN, vanE, and vanL, Additional File 6). Conversely, Western diet-fed mice also exhibited decreased abundances of transcripts encoding ARGs against tetracycline, mupirocin, bacitracin, and phenicol antibiotics (Figure 4A, Additional File 5).

A. Differentially expressed (Benjamini-Hochberg adjusted p-value < 0.1) ARG classes in the murine cecal metatranscriptome in vehicle-treated mice consuming the Western (WD) diet. Data are represented as log2 fold change relative to control diet ± standard error. See Additional File 5
(B-E) Volcano plots of the resistome of the murine cecal microbiome in vehicle-treated mice consuming the Western (WD) diet (B), ciprofloxacin-treated mice on the control diet (C), and ciprofloxacin-treated mice on the Western diet (D). Interaction terms representing the impact of diet on ciprofloxacin-induced changes for each ARG are shown in (E). Data was generated using deepARG-ss and analyzing using DESeq2. Points in red represent ARG transcripts for which a statistically significant change in expression was detected (Benjamini-Hochberg adjusted p-value < 0.1). Select ARGs of interest are labeled. See Additional File 6
For all analyses, n = 4.
Interestingly, ciprofloxacin treatment did not elicit major shifts in class-level ARG transcript abundance on either diet. On the Western diet, we observed statistically significant changes in two ARG classes (an increase in oxazolidinone and a decrease in unclassified ARGs) while ciprofloxacin only changed the abundance of a single class on the control diet (an increase in beta-lactam ARGs). However, ciprofloxacin did elicit numerous changes in the transcript abundance of several individual ARGs on both diets (Additional File 6). On the Western diet, ciprofloxacin increased the abundance of transcripts encoding three tetracycline resistance genes (tetB(60), tetB(46), and otrA) while decreasing expression of several efflux pumps (mefA, mexW, mexB, and oprA). Interestingly, several of the transcripts encoding efflux pumps that were reduced with ciprofloxacin treatment on the Western diet were increased on the control diet (mexW, mefA) along with several other ARGs from this class (mexQ, mexF). Together, these findings suggest that host diet influences the abundance of resistance genes during antibiotic therapy. Additionally, it appears that ciprofloxacin treatment has a comparatively minor impact on the abundance of ARG transcripts within the microbiota relative to host diet. It is important to note that this analytical pipeline does not account for changes in population structure induced by each of the perturbations. As a result, some of the discussed changes in transcript abundance may result from differential bacterial abundances. For example, the Western diet induced an expansion in B. thetaiotaomicron, a bacterium that typically encodes beta-lactamases [20,75–79]. Thus, this may partially explain the higher observed levels of beta-lactam resistance in this condition.
Diet and ciprofloxacin treatment also had a major impact on the abundance of transcripts encoding known virulence factors. In total, we detected 351 virulence factor transcripts with altered abundances on the Western diet (Additional File 7). Supporting our earlier observation (Figure 3D, Additional File 7), we found that the microbiota of the mice consuming the Western diet displayed an increased transcript abundance of numerous enzymes known to hydrolyze host-derived proteins – specifically, hyaluronidase, neuraminidase, sialidase, and exo-alpha-sialidase (Figure 4B, Additional File 7). Additionally, mice consuming a Western diet displayed higher transcript abundances of catalase and superoxide dismutase, which may be indicative of oxidative stress (Figure 4B, Additional File 7). In total, ciprofloxacin treatment altered the abundance of 139 and 90 virulence factor transcripts on the control and Western diets, respectively (Figures 4C+D, Additional File 7). Of note on the control diet, ciprofloxacin increased the abundance of transcripts of sialidase, catalase, a glucose/galactose transporter, and the outer membrane protein OmpA while simultaneously reducing the abundance of transcripts related to urease, iron permease, ferric siderophores, bile salt hydrolase, and twitching motility (Figure 4C, Additional File 7). On the Western diet, ciprofloxacin increased the transcript abundance of several iron-related virulence factors such as an iron-regulated transporter, hemolysin B, and a hemolysin transport protein (Figure 4D, Additional File 7). Lastly, we examined the interaction of both diet and ciprofloxacin treatment on the abundance of transcripts encoding virulence factors (Figure 4E, Additional File 7). Interestingly, we observed that the change in transcript abundance of catalase, superoxide dismutase, hyaluronidase, sialidase, and exo-alpha-sialidase following ciprofloxacin administration were all significantly lower on the Western diet relative to the control (Figure 4E, Additional File 7). However, we previously observed that the baseline levels of these transcripts were significantly elevated in the Western diet before treatment (Figure 4B, Additional File 7). Therefore, it is likely that the reduced expansion of these transcripts following ciprofloxacin treatment on the Western diet is attributable to their high, pre-treatment baseline levels.
Diet and ciprofloxacin alter gene expression within B. thetaiotaomicron and A. muciniphila
All metatranscriptomic analysis conducted thus far has examined the impact of diet and ciprofloxacin treatment on the transcriptional activity of the microbiome at the whole community level. However, we sought to profile how these factors impacted individual species within the microbiota. Thus, we used a previously published pipeline to interrogate the impact of diet and antibiotic treatment on two individual species: B. thetaiotaomicron and A. muciniphila [20, 80]. We focused on these bacteria because they are known human gut commensals, were found in relatively high levels in all samples analyzed, and because they were differentially abundant in a diet-dependent manner.
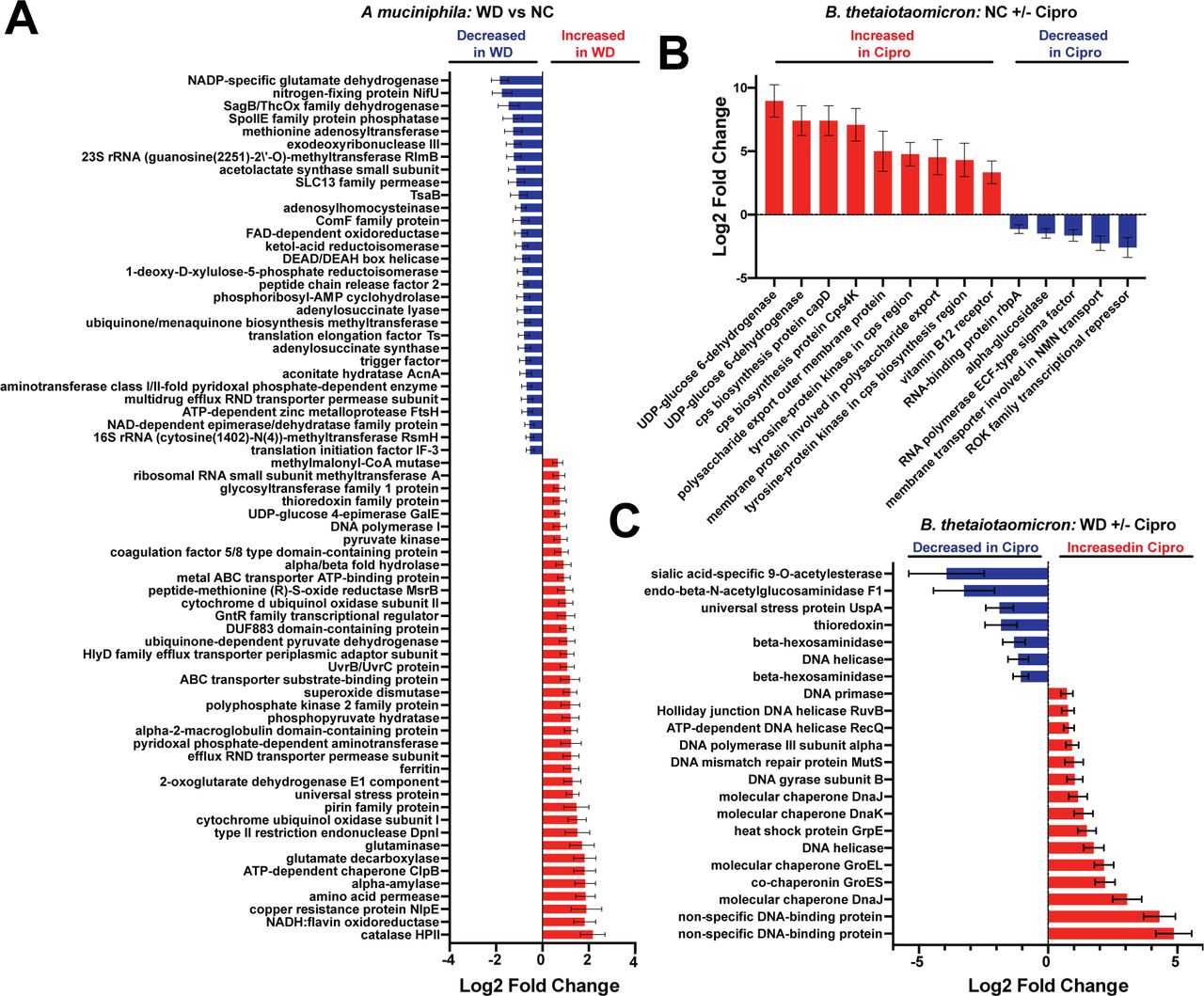
Interestingly, we found that A. muciniphila displayed increased expression of several known stress response genes on the Western diet (Figure 5A, Additional File 8). Specifically, we observed elevated levels of transcripts for catalase HPII (AMUC_RS11055), ATP-dependent chaperone ClpB (AMUC_RS04500), a universal stress protein (AMUC_RS00415), superoxide dismutase (AMUC_RS08510), a UvrB/UvrC protein (AMUC_RS00335), and a thioredoxin family protein (AMUC_RS11695). Together, these changes may indicate that Western diet consumption induces a general stress response in A. muciniphila. Additionally, we observed numerous changes within respiratory and central carbon metabolism, suggesting broad metabolic changes in response to the Western diet (Figure 5A, Additional File 8). We specifically detected increased expression of genes encoding terminal oxidases of the respiratory chain (AMUC_RS09050 - cytochrome ubiquinol oxidase subunit I and AMUC_RS09045 - cytochrome d ubiquinol oxidase subunit II), the TCA cycle (AMUC_RS09040 - 2-oxoglutarate dehydrogenase E1 component), glycolysis (AMUC_RS06320 - phosphopyruvate hydratase, AMUC_RS02385 - pyruvate kinase), and pyruvate metabolism (AMUC_RS01195 - ubiquinone-dependent pyruvate dehydrogenase). Of the genes that were significantly reduced on the Western diet, the most notable were an adenylsuccinate synthase (AMUC_RS11340) and a lyase (AMUC_RS10360), which both play important roles in purine metabolism (Figure 5A, Additional File 8) [81].

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
A. Select differentially expressed (Benjamini-Hochberg adjusted p-value < 0.1) genes of interest in A. muciniphila within the cecum of vehicle-treated mice consuming the Western (WD) diet. Data are represented as log2 fold change relative to control diet ± standard error. See Additional File 7 for full results.
B. Select differentially expressed (Benjamini-Hochberg adjusted p-value < 0.1) genes of interest in B. thetaiotaomicron within the cecum of ciprofloxacin-treated mice consuming the control (NC) diet. Data are represented as log2 fold change relative to control diet ± standard error. See Additional File 8 for full results.
C. Select differentially expressed (Benjamini-Hochberg adjusted p-value < 0.1) genes of interest in B. thetaiotaomicron within the cecum of vehicle-treated mice consuming the Western (WD) diet. Data are represented as log2 fold change relative to control diet ± standard error. See Additional File 8 for full results.
For all analyses, n = 4.
In comparison to diet, ciprofloxacin treatment had a relatively minor impact on A. muciniphila gene expression (Additional File 8). In total, ciprofloxacin significantly altered the expression of 14 and 26 genes on the control and Western diets, respectively (Additional File 8). On the control diet, A. muciniphila increased the expression of the molecular chaperone protein DnaK, which is known to play a role in stress responses [82–84]. Additionally, we observed elevated expression of a MoxR family ATPase following ciprofloxacin treatment on this diet. Though these proteins are poorly characterized, other members of this family have been shown to regulate stress responses in other bacteria [85]. On the Western diet, several genes related to tryptophan biosynthesis and metabolism were elevated following ciprofloxacin treatment (AMUC_RS08210 - tryptophan synthase subunit beta, AMUC_RS08190 - anthranilate synthase component I family protein, AMUC_RS08215 - tryptophan synthase subunit alpha); however, their biological significance is unclear at this time (Additional File 8). Lastly, an examination of the interaction between diet and ciprofloxacin treatment indicated that only six genes (two of which encoded tRNAs) were significantly altered, suggesting that diet does not have a major impact on the response of this bacterium to ciprofloxacin within the microbiome (Additional File 8).
In contrast to A. muciniphila, diet had a relatively minor impact on B. thetaiotaomicron gene expression while ciprofloxacin induced extensive changes. In total, B. thetaiotaomicron altered the expression of 74 genes in response to Western diet consumption (Additional File 9). Of note, this diet increased the expression of an aminoglycoside efflux pump (BT_0305), the universal stress protein UspA (BT_0901), and a hemin receptor (BT_0316). However, more than half of the genes (52.7%) that changed in response to diet are of unknown function and are classified as “hypothetical proteins;” thus, it is difficult to draw conclusions without improved annotation. Overall, ciprofloxacin appeared to induce extensive transcriptional changes within B. thetaiotaomicron regardless of diet. On the control diet, we observed an increased abundance of transcripts encoding a number of proteins involved in capsular polysaccharide (CPS) biosynthesis and export (Figure 5B, Additional File 9). Within B. thetaiotaomicron, CPS production is encoded by a total of 182 genes distributed among eight loci (typically termed cps1-8) [86, 87]. It is hypothesized that an individual bacterium expresses one of these CPS configurations at any given time and that these structures play key roles in processes such as nutrient acquisition and immune evasion [87]. Additionally, the two genes with the greatest increase in expression during ciprofloxacin treatment encoded UDP-glucose 6-dehydrogenase, which plays a key role in the biosynthesis of glycan precursors that are essential for capsule production in other bacteria [88–90]. Together, these findings may suggest a role for CPS state as a determinant of ciprofloxacin susceptibility in vivo.
On the Western diet, ciprofloxacin elicited profound changes in transcriptional activity, altering the expression of 442 different genes (Figure 5C, Additional File 9). Interestingly, B. thetaiotaomicron reduced the expression of a number of genes involved in the utilization of host-derived carbohydrates (sialic acid-specific 9-O-acetylesterase, endo-beta-N-acetylglucosaminidase F1, beta-hexosaminidase) and stress responses (universal stress protein UspA, thioredoxin), mirroring the changes we saw at the whole-community level (Figure 5C, Additional File 9). Conversely, we observed increased expression of several genes that encode molecular chaperones (such as GroEL and GroES) or are involved in DNA replication or damage repair (such as a DNA helicase, DNA gyrase subunit B, DNA mismatch repair protein MutS, DNA polymerase III subunit alpha, Holliday junction DNA helicase RuvB, DNA-binding proteins, and DNA primase) (Figure 5C, Additional File 9). Ciprofloxacin, a fluoroquinolone class antimicrobial, triggers DNA damage via inhibition of DNA gyrase and topoisomerase IV. Thus, these changes in gene expression may be reflective of the primary mechanism of action of this antibiotic and are consistent with previously published data [20]. Furthermore, our ability to detect such changes is an important indication that our analysis is detecting ciprofloxacin-induced transcriptional shifts. Lastly, diet appears to have a significant impact on ciprofloxacin-induced transcriptional changes within B. thetaiotaomicron, modulating the response of 148 genes (Additional File 9). Of note, Western diet consumption in the context of ciprofloxacin treatment had a negative impact on several genes involved in the acquisition of nutrients, such as vitamin B12 and hemin receptors, and transporters of glucose/galactose, hexuronate, arabinose, and Na+ (Additional File 9). Thus, it is likely that the availability of nutrients within the gut plays a role in the response of these bacteria to antibiotics.
Discussion
Previous work has demonstrated that host diet, particularly with respect to sugar and fiber content, plays a major role in the extent of antibiotic-induced microbiome disruption [20]. In Western societies, many people consume a diet high in added sugars and fat, but low in host-indigestible fiber. It is thought that such a composition promotes the development of metabolic syndrome, heart disease, diabetes, and a number of other chronic conditions [36–46]. Furthermore, broad-spectrum antibiotic use and resulting microbiome dysbiosis have been associated with a number of similar co-morbidities along with increased susceptibility to opportunistic infections [1,5–7,21–23,25,26]. Despite this connection, little work has been done examining how host dietary composition impacts the response of the microbiota to antibiotic perturbation. It is known that nutrient availability and metabolic state are a major determinant of antibiotic susceptibility of bacteria in vitro [20,60–63,91–97]. Thus, it is likely that modulating host diet, thus changing the availability of nutrients to the microbiota, would alter the sensitivity of bacteria in these communities to antibiotic therapy.
To address this knowledge gap, we utilized a combined metagenomic and metatranscriptomic approach to profile taxonomic and functional changes in response to both diet and antibiotic treatment. By utilizing these tools in parallel, we are able to link transcriptional changes to observed shifts in community structure on each diet. Using metagenomics, we observed that ciprofloxacin had a differential impact on community composition that was diet-dependent. Specifically, we observed a statistically significant expansion of the Firmicutes phylum following ciprofloxacin treatment on the Western, but not control, diet. Using metatranscriptomics, we observed that ciprofloxacin treatment resulted in a decreased abundance of transcripts from the TCA cycle in both diets, suggesting that this response is diet-independent. Furthermore, this observation is consistent with previous in vitro findings that demonstrate a key role for bacterial respiration as a determinant of susceptibility to fluoroquinolones [60-62,64,91,94]. Conversely, ciprofloxacin had diverging impacts on the abundance of various iron and mucin utilization transcripts on the Western and control diets. Most notably, we found that Western diet consumption (alone and in the presence of ciprofloxacin) influenced the abundance of transcripts encoding known virulence and antibiotic resistance genes, supporting previous literature demonstrating that nutrient availability impacts virulence of enteric pathogens [20,71,98–100]. Lastly, we detected species-specific transcriptional changes in two important commensal bacteria, B. thetaiotaomicron and A. muciniphila. In addition to detecting changes in transcript levels that were reflective of stress responses, we also observed that transcripts involved in diverse cellular processes such as nutrient acquisition, carbon metabolism, and capsular polysaccharide (CPS) biosynthesis were differentially expressed as well.
Despite the advantages of a multi-omic approach, there are a number of drawbacks to these techniques that complicate the interpretation of our results. Most crucially, nearly all analytical pipelines used to analyze microbiome data are reliant on existing databases that are largely incomplete. It is hypothesized that approximately half of all genes within the human gut microbiome have no functional annotation [101]. Thus, the ability to accurately profile the transcriptional activity of these communities is inherently limited by the quality and completeness of the databases utilized. Additionally, elucidating the biological significance of taxonomic of functional changes is often difficult in many microbiome analyses. Due to the complex nature of these communities, it is often difficult to ascertain if the observed transcriptional changes are the result of the direct action of the antibiotic, or the indirect effect of changes in host physiology, nutrient availability, or the disruption of ecological networks within the microbiome. For example, our transcriptional analysis of B. thetaiotaomicron showed that this bacterium differentially expressed receptors for both hemin and vitamin B12, which may suggest that these nutrients play a role in ciprofloxacin toxicity. Alternatively, it is possible that these transcriptional changes are reflective of increased availability of these nutrients due to decreased competition from other members of the microbiota (though these hypotheses are not mutually exclusive). Additionally, it is possible that dietary composition could play a significant role in antibiotic absorption or sequestration in the gut, which in turn would impact the extent of the damage caused to the microbiota.
This study builds on recent work that demonstrates that the availability of metabolites to the bacteria in the host plays an important role in determining the extent of antibiotic-induced microbiome disruption [20]. Taken together, these results demonstrate the need to consider dietary composition in the design and interpretation of experiments focused on understanding the impact of antibiotics on the microbiota. Previous studies have demonstrated that dietary changes induce rapid shifts in gut microbiome composition [32,34,43,56,102–105]. Therefore, in the long-term, dietary modulation could represent an attractive strategy to reduce the collateral damage to commensal bacteria and the resulting complications from dysbiosis caused by clinical therapy. Despite these promising applications, considerable work is required before these findings have direct clinical relevance. In particular, the considerable differences in physiology, microbiome composition, and diet between humans and rodents complicate the direct clinical relevance of these findings. Furthermore, it is unclear which components of diet are responsible for the observed effects and whether short-term dietary modulation has any long-term consequences on either the host or the microbiome. Thus, additional research is warranted to fully elucidate how host diet impacts antibiotic-induced microbiome disruption in humans.
Conclusions
Using a combined metagenomic and metatranscriptomic approach, we demonstrate that murine diet composition has a major impact on the response of the murine gut microbiome to ciprofloxacin therapy. First, we found that the gut microbiome undergoes differential shifts in community structure in response to antibiotic treatment in a diet-dependent manner. At the transcriptional level, we found that ciprofloxacin reduced the abundance of TCA cycle transcripts regardless of diet, suggesting that central carbon metabolism plays a role in the activity of this antibiotic in vivo. Despite this commonality, we observed extensive differences in the transcriptional response of the microbiome to dietary intervention and/or ciprofloxacin treatment. Most notably, we found that diet had a major impact on the abundance of transcripts encoding known virulence and antibiotic resistance genes within the gut microbiome. Mice consuming a Western diet had a high abundance of transcripts encoding proteins known to degrade host-derived polysaccharides such as sialic residues of mucin, suggesting that the consumption of this diet may have detrimental impacts on host physiology. Lastly, we identified species-specific changes in transcript abundance in two key members of the gut microbiome, A. muciniphila and B. thetaiotaomicron. In A. muciniphila, consumption of a Western diet increased the expression of several genes known to play a role in stress responses. In B. thetaiotaomicron, we found that a number of genes involved in CPS biosynthesis were differentially expressed during ciprofloxacin treatment on the control but not Western diet, which may suggest a divergent response of this bacterium to ciprofloxacin that is dependent on nutrient composition. Taken together, these findings demonstrate the important role for host diet as a determinant of antibiotic-induced microbiome perturbations.
Methods
Animal Procedures
All procedures involving animal work were approved by the Institutional Animal Care and Use Committee of Brown University. 4-week-old female C57BL/6 mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and given a 2-week habituation period immediately following arrival at Brown University’s Animal Care Facility. After habituation, mice were switched from standard chow (Laboratory Rodent Diet 5001, St. Louis, MO, USA) to either a Western diet (D12079B, Research Diets Inc., New Brunswick, NJ, USA) or a macronutrient-defined control diet (D122405B, Research Diets Inc., New Brunswick, NJ, USA) for 1 week. Following dietary intervention, mice were given acidified ciprofloxacin (12.5 mg/kg/day), or a pH-adjusted vehicle, via filter-sterilized drinking water ad libitum for 24 hours. Water consumption was monitored to assure equal consumption across cages. Mice were then sacrificed and dissected in order to collect cecal contents. Cecal contents were immediately transferred to ZymoBIOMICS DNA/RNA Miniprep Kit (Zymo Research, Irvine, CA, USA) Collection Tubes containing DNA/RNA Shield. Tubes were processed via vortexing at maximum speed for 5 minutes to homogenize cecal contents, then placed on ice until permanent storage at -80°C.
Nucleic Acid Extraction & Purification
Total nucleic acids (DNA and RNA) were extracted from samples using the ZymoBIOMICS DNA/RNA Miniprep Kit from Zymo Research (R2002, Irvine, CA, USA) using the parallel extraction protocol as per the manufacturer instructions. Total RNA and DNA were eluted in nuclease-free water and quantified using the dsDNA-HS and RNA-HS kits on a Qubit™ 3.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA) before use in library preparations.
Library Preparation
Metagenomic libraries were prepared from DNA (100 ng) using the NEBNext® Ultra II FS DNA Library Prep Kit (New England BioLabs, Ipswich, MA, USA) > 100ng input protocol as per the manufacturer’s instructions. This yielded a pool of 200 – 1000 bp fragments where the average library was 250-500 bp. Metatranscriptomic libraries were prepared from total RNA using the NEBNext® Ultra II Directional RNA Sequencing Prep Kit (New England BioLabs, Ipswich, MA, USA) in conjunction with the NEBNext® rRNA Depletion Kit for Human/Mouse/Rat (New England BioLabs, Ipswich, MA, USA) and the MICROBExpress kit (Invitrogen, Carlsbad, CA, USA). First, up to 1 ug of total RNA was treated with rDNase I and subsequently depleted of bacterial rRNAs using MICROBExpress as per the manufacturer’s instructions. This depleted RNA was then used to prepare libraries with the NEBNext® Ultra II Directional RNA Sequencing Prep & rRNA depletion kits as per the manufacturer’s instructions. This yielded libraries that averaged between 200-450 bp. Once library preparation was complete, both metagenomic and metatranscriptomic libraries were sequenced as paired-end 150 bp reads on an Illumina HiSeq X Ten. We sequenced an average of 2,278,948,631 (± 2,309,494,556) bases per metagenomic sample and 14,751,606,319 (± 3,089,205,166) bases per metatranscriptomic sample. One metagenomic sample from the Western diet + vehicle group had an abnormally low number of bases sequenced (165,000 bp) and was excluded from all subsequent analyses. Following the removal of this sample, we obtained an average of 2,430,867,540 (± 2,306,317,898) bases per metagenomic sample. All reads were deposited in the NCBI Short Read Archive under BioProject number PRJNA563913.
Processing of Raw Reads
Raw metagenomic reads were trimmed and decontaminated using kneaddata utility (version 0.6.1) [106]. In brief, reads were first trimmed to remove low quality bases and Illumina TruSeq3 adapter sequences using trimmomatic (version 0.36) using SLIDINGWINDOW value of 4:20 and ILLUMINACLIP value 2:20:10, respectively [107]. Trimmed reads shorter than 75 bases were discarded. Reads passing quality control were subsequently decontaminated by removing those that mapped to the genome of C57BL/6J mice using bowtie2 (version 2.2) [108]. Additionally, preliminary work by our group detected high levels of reads mapping to two murine retroviruses found in our animal facility: murine mammary tumor virus (MMTV) and murine osteosarcoma viruses (MOV) [20]. Raw metatranscriptomic reads were trimmed and decontaminated using the same parameters. However, in addition to removing reads that mapped to the C57BL/6J, MMTV, and MOV genomes, we also decontaminated sequences that aligned to the SILVA 128 LSU and SSU Parc ribosomal RNA databases [109].
Taxonomic Classification of Metagenomic Reads
Trimmed and decontaminated metagenomic reads were taxonomically classified against a database containing all bacterial and archaeal genomes found in NCBI RefSeq using Kraken2 (version 2.0.7-beta) with a default k-mer length of 35 [110]. Phylum-and species-level abundances were subsequently calculated from Kraken2 reports using Bracken (version 2.0.0) with default settings [111]. The phyloseq package (version 1.28.0) in R (version 3.6.0) was used to calculate alpha diversity using the Shannon diversity index [112]. Metagenomic data was not subsampled prior to analysis.
To perform differential abundance testing, species-level taxonomic output was first filtered to remove taxa that were not observed in >1,000 reads (corresponding to approximately 0.1% of all reads) in at least 20% of all samples using phyloseq in R. Differential abundance testing was subsequently performed on filtered counts using the DESeq2 package (version 1.24.0) using default parameters [58]. All p-values were corrected for multiple hypothesis testing using the Benjamini-Hochberg method [113].
Annotation of Metatranscriptomic Reads Using SAMSA2
Trimmed and decontaminated metatranscriptomic reads were annotated using a modified version of the Simple Annotation of Metatranscriptomes by Sequence Analysis 2 (SAMSA2) pipeline as described previously [20,65,114]. First, the Paired-End Read Merger (PEAR) utility was used to merge forward and reverse reads [115]. Merged reads were then aligned to databases containing entries from the RefSeq, SEED Subsystems, and Virulence Factor (downloaded 04/2019) databases using DIAMOND (version 0.9.12) [116–118]. The resulting alignment counts were subsequently analyzed using DESeq2 (version 1.24.0) using the Benjamini-Hochberg method to perform multiple hypothesis testing correction [20,65,113]. Features with an adjusted p-value of less than 0.1 were considered to be statistically significant.
Analysis of Antibiotic Resistance Gene (ARG) Transcript Abundance
The abundance of transcripts encoding known ARGs was performed using the deepARG pipeline [119]. In short, the fastq-join utility of the ea-utils package (version 1.04.807) was used to join cleaned metatranscriptomic reads [120]. Merged reads were then analyzed for the presence of ARGs using the deepARG-ss algorithm within the deepARG program (version 2.0) using default settings [119]. Count tables at both the individual ARG and ARG class level were then analyzed using DESeq2 (version 1.24.0) using the Benjamini-Hochberg method to correct for multiple hypothesis testing [58, 113]. ARGs with an adjusted p-value of less than 0.01 were considered to be statistically significant.
Metatranscriptomic Analysis using HUMAnN2
To determine the impact of dietary modulation and ciprofloxacin treatment on gene expression within the gut microbiome, we used the HMP Unified Metabolic Analysis Network 2 (HUMAnN2, version 0.11.1) pipeline [59]. First, metagenomic reads were taxonomically annotated using MetaPhlan2 (version 2.6.0) and functionally annotated against the UniRef90 database to generate gene family and MetaCyc pathway level abundances. To ensure consistent assignment between paired samples, the taxonomic profile generated from the metagenomic reads was supplied to the HUMAnN2 algorithm during the analysis of the corresponding metatranscriptomic reads. Metatranscriptomic reads were subsequently annotated as done for metagenomic reads. The resulting gene family and pathway level abundance data from the metatranscriptomic reads was normalized against the metagenomic data from the corresponding sample and smoothed using the Witten-Bell method [121]. Lastly, the resulting RPKM values were unstratified to obtain whole-community level data, converted into relative abundances, and analyzed using LEfSe (version 1) hosted on the Galaxy web server [122].
Transcriptional Analysis of A. muciniphila and B. thetaiotaomicron
A modified version of a previously published pipeline from Deng et al. was utilized to perform transcriptional analysis of individual species within the murine microbiome during dietary modulation and antibiotic treatment [20, 80]. First, Kraken2 (version 2.0.7-beta) was used to identify the fifty most prevalent bacterial species present within the metatranscriptomic samples [110]. Next, the BBSplit utility within the BBMap package (version 37.96) was used to extract reads within our metatranscriptomic dataset that mapped to these fifty most abundant species [123]. Reads from B. thetaiotaomicron and A. muciniphila were subsequently aligned to their corresponding reference genomes using the BWA-MEM algorithm (version 0.7.15) [124]. Lastly, the featureCounts command within the subread program (version 1.6.2) was used to analyze the resulting alignment files to generate a count table for differential expression analysis with DESeq2 [58]. All p-values were corrected for multiple hypothesis testing with the Benjamini-Hochberg method [113]. Features with an adjusted p-value of less than 0.1 were considered to be statistically significant.
List of Abbreviations
- PTS
- phosphotransferase systems
- MACs
- microbiota-accessible carbohydrates
- SCFAs
- short chain fatty acids
- ARGs
- antibiotic resistance genes
- TCA
- tricarboxylic acid
- HUMAnN2
- Human Microbiome Project (HMP) Unified Metabolic Analysis Network 2
- CPS
- capsular polysaccharide
- GH98
- glycoside hydrolase family 98
- SAMSA2
- Simple Annotation of Metatranscriptomes by Sequence Analysis 2
- SRA
- Sequence Read Archive
Declarations
Ethics approval and consent to participate
All animal work was approved by Brown University’s Institutional Animal Care and Use Committee (IACUC) under protocol number 1706000283.
Consent for publication
Not applicable
Availability of data and materials
The datasets generated and analyzed during this study are available from the NCBI Short Read Archive (SRA) under BioProject accession number PRJNA563913 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA563913). Any additional information is available from the corresponding author upon request.
Competing interests
The authors declare that they have no competing interests
Funding
D.J.C, B.J.K, and J.I.W., and S.P. were supported by the Graduate Research Fellowship Program from the National Science Foundation under award number 1644760. P.B. was supported by the National Center for Complementary & Integrative Health of the NIH Award Number R21AT010366, and institutional development awards P20GM121344 and P20GM109035 received from the National Institute of General Medical Sciences within the NIH. The funding agencies had no role in the design of the study or the collection, analysis, and interpretation of data.
Authors’ contributions
DJC planned the study, performed mouse experiments, extracted nucleic acids from cecal samples, conducted analysis of metagenomic and metatranscriptomic data, and was the primary author of the manuscript. JIW assisted with mouse experiments, prepared DNA and RNA into sequencing libraries for metagenomics and metatranscriptomics, assisted in the interpretation of results, and contributed to the writing of the manuscript. BJK performed the analysis of virulence factor and antibiotic resistance gene expression. SP assisted in the interpretation of results. PB conceptualized and planned the study, contributed to the writing of the manuscript, and secured funding. All authors have read and approved of the final manuscript.
Additional Files
Additional File 1: Full DESeq2 results of differential abundance testing of top 90 species detected by shotgun metagenomics
Table S1 – Differential abundance testing of the impact of Western diet (WD) consumption on the abundance of the top 90 bacterial species detected in our dataset. Log2 fold change values were calculated relative to control diet samples.
Table S2 – Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of the top 90 bacterial species in mice consuming the Western diet (WD). Log2 fold change values were calculated relative to vehicle-treated samples on the WD.
Table S3 - Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of the top 90 bacterial species in mice consuming the control diet (NC). Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Table S4 – Interaction term analysis generated by DESeq2 for the impact of host diet consumption on changes in species abundance following ciprofloxacin therapy. Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Additional File 2: Full LEfSe results from the analysis of MetaCyc pathway abundance generated by HUMAnN2. “Class” denotes the experimental group a particular pathway was associated with.
Table S5 – LEfSe analysis of all experimental groups.
Table S6 – Pairwise LEfSe analysis of vehicle-treated samples from mice consuming either the Western (WD) or control (NC) diet.
Table S7– Pairwise LEfSe analysis of ciprofloxacin- and vehicle-treated samples from mice consuming the control diet (NC)
Table S8 – Pairwise LEfSe analysis of ciprofloxacin- and vehicle-treated samples from mice consuming the Western diet (WD)
Additional File 3: Full DESeq2 results of SEED subsystem abundance generated by SAMSA2
Table S9 – Differential abundance testing of the impact of Western diet (WD) consumption on the abundance of SEED subsystems in the murine cecal metatranscriptome. Log2 fold change values were calculated relative to control diet samples.
Table S10 – Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of SEED subsystems in the murine cecal metatranscriptome in animals consuming the Western diet (WD). Log2 fold change values were calculated relative to vehicle-treated samples on the WD.
Table S11 - Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of SEED subsystems in the murine cecal metatranscriptome in animals consuming the control diet (NC). Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Additional File 4: Full DESeq2 results of RefSeq transcript abundance generated by SAMSA2
Table S12 – Differential abundance testing of the impact of Western diet (WD) consumption on the abundance of RefSeq transcripts in the murine cecal metatranscriptome. Log2 fold change values were calculated relative to control diet samples.
Table S13 – Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of RefSeq transcripts in the murine cecal metatranscriptome in animals consuming the Western diet (WD). Log2 fold change values were calculated relative to vehicle-treated samples on the WD.
Table S14 - Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of RefSeq transcripts in the murine cecal metatranscriptome in animals consuming the control diet (NC). Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Table S15 – Interaction term analysis generated by DESeq2 for the impact of host diet consumption on changes in RefSeq transcripts abundance following ciprofloxacin therapy. Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Additional File 5: Full DESeq2 results of ARG class abundance generated by deepARG
Table S16 – Differential abundance testing of the impact of Western diet (WD) consumption on the abundance of ARG classes in the murine cecal metatranscriptome. Log2 fold change values were calculated relative to control diet samples.
Table S17 – Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of ARG classes in the murine cecal metatranscriptome in animals consuming the Western diet (WD). Log2 fold change values were calculated relative to vehicle-treated samples on the WD.
Table S18 - Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of ARG classes in the murine cecal metatranscriptome in animals consuming the control diet (NC). Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Table S19 – Interaction term analysis generated by DESeq2 for the impact of host diet consumption on changes in ARG class abundance following ciprofloxacin therapy. Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Additional File 6: Full DESeq2 results of ARG transcript abundance generated by deepARG
Table S16 – Differential abundance testing of the impact of Western diet (WD) consumption on the abundance of ARG transcripts in the murine cecal metatranscriptome. Log2 fold change values were calculated relative to control diet samples.
Table S17 – Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of ARG transcripts in the murine cecal metatranscriptome in animals consuming the Western diet (WD). Log2 fold change values were calculated relative to vehicle-treated samples on the WD.
Table S18 - Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of ARG transcripts in the murine cecal metatranscriptome in animals consuming the control diet (NC). Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Table S19 – Interaction term analysis generated by DESeq2 for the impact of host diet consumption on changes in ARG transcript abundance following ciprofloxacin therapy. Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Additional File 7: Full DESeq2 results of virulence factor (VF) transcript abundance generated by alignment of reads to the Virulence Factor Database (VFDB) by SAMSA2
Table S20 – Differential abundance testing of the impact of Western diet (WD) consumption on the abundance of VF transcripts in the murine cecal metatranscriptome. Log2 fold change values were calculated relative to control diet samples.
Table S21 – Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of VF transcripts in the murine cecal metatranscriptome in animals consuming the Western diet (WD). Log2 fold change values were calculated relative to vehicle-treated samples on the WD.
Table S22 - Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of VF transcripts in the murine cecal metatranscriptome in animals consuming the control diet (NC). Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Table S23 – Interaction term analysis generated by DESeq2 for the impact of host diet consumption on changes in VF transcript abundance following ciprofloxacin therapy. Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Additional File 8: Full DESeq2 results of transcript abundance analysis of A. muciniphila during dietary intervention and ciprofloxacin treatment
Table S24 – Differential abundance testing of the impact of Western diet (WD) consumption on the abundance of A. muciniphila transcripts within the murine cecal metatranscriptome. Log2 fold change values were calculated relative to control diet samples.
Table S25 – Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of A. muciniphila transcripts within the murine cecal metatranscriptome in animals consuming the Western diet (WD). Log2 fold change values were calculated relative to vehicle-treated samples on the WD.
Table S26 - Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of A. muciniphila transcripts within the murine cecal metatranscriptome in animals consuming the control diet (NC). Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Table S27 – Interaction term analysis generated by DESeq2 for the impact of host diet consumption on changes in A. muciniphila transcript abundance following ciprofloxacin therapy. Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Additional File 9: Full DESeq2 results of transcript abundance analysis of B. thetaiotaomicron during dietary intervention and ciprofloxacin treatment
Table S28 – Differential abundance testing of the impact of Western diet (WD) consumption on the abundance of B. thetaiotaomicron transcripts within the murine cecal metatranscriptome. Log2 fold change values were calculated relative to control diet samples.
Table S29 – Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of B. thetaiotaomicron transcripts within the murine cecal metatranscriptome in animals consuming the Western diet (WD). Log2 fold change values were calculated relative to vehicle-treated samples on the WD.
Table S30 - Differential abundance testing of the impact of ciprofloxacin treatment on the abundance of B. thetaiotaomicron transcripts within the murine cecal metatranscriptome in animals consuming the control diet (NC). Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Table S31 – Interaction term analysis generated by DESeq2 for the impact of host diet consumption on changes in B. thetaiotaomicron transcript abundance following ciprofloxacin therapy. Log2 fold change values were calculated relative to vehicle-treated samples on the NC.
Acknowledgements
Not applicable
Footnotes
References
- 1.↵
- 2.
- 3.↵
- 4.↵
- 5.↵
- 6.
- 7.↵
- 8.↵
- 9.
- 10.↵
- 11.
- 12.
- 13.↵
- 14.
- 15.↵
- 16.
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.
- 23.↵
- 24.
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.
- 31.
- 32.↵
- 33.
- 34.↵
- 35.↵
- 36.↵
- 37.
- 38.
- 39.
- 40.↵
- 41.
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.
- 53.
- 54.
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.
- 73.↵
- 74.↵
- 75.↵
- 76.
- 77.
- 78.
- 79.↵
- 80.↵
- 81.↵
- 82.↵
- 83.
- 84.↵
- 85.↵
- 86.↵
- 87.↵
- 88.↵
- 89.
- 90.↵
- 91.↵
- 92.
- 93.
- 94.↵
- 95.
- 96.
- 97.↵
- 98.↵
- 99.
- 100.↵
- 101.↵
- 102.↵
- 103.
- 104.
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.↵
- 117.
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵




