

SUMMARY
Spatial gene expression heterogeneity plays an essential role in a range of biological, physiological and pathological processes but it remains a scientific challenge to conduct high-spatial-resolution, genome-wide, unbiased biomolecular profiling over a large tissue area. Herein, we present a fundamentally new approach – microfluidic Deterministic Barcoding in Tissue for spatial omics sequencing (DBiT-seq). Parallel microfluidic channels (10μm, 25μm, or 50μm in width) are used to deliver molecular barcodes to the surface of a formaldehyde fixed tissue slide in a spatially confined manner. Crossflow of two sets of barcodes A1-A50 and B1-B50 followed by ligation in situ yields a 2D mosaic of tissue pixels, each containing a unique combination of full barcode AiBj (i=1-50, j=1-50). It permits simultaneous barcoding of mRNAs, proteins, or even other omics on a fixed tissue slide, enabling the construction of a high-spatial-resolution multi-omics atlas by NGS sequencing. Applying it to mouse embryo tissues revealed all major tissue types in early organogenesis, distinguished brain microvascular networks, discovered new developmental patterning in forebrain, and demonstrated the ability to detect a single-cell-layer of melanocytes lining an optical vesicle and asymmetric expression of Rorb and Aldh1a1 within it, presumably associated with the onset of retina and lens, respectively. Automated feature identification using spatial differential expression further identified dozens of developmental features. DBiT-seq is a highly versatile technology that may become a universal method for spatial barcoding and sequencing of a range of molecular information at a high resolution and the genome scale. It can be readily adopted by biologists with no experience in microfluidics or advanced imaging, and could be quickly disseminated for broader impacts in a variety of fields including developmental biology, cancer biology, neuroscience, and clinical pathology.
INTRODUCTION
In multicellular systems, cells do not function in isolation but are strongly influenced by spatial location and surroundings(Knipple et al., 1985; Scadden, 2014; van Vliet et al., 2018). Spatial gene expression heterogeneity plays an essential role in a range of biological, physiological and pathological processes(de Bruin et al., 2014; Fuchs et al., 2004; Yudushkin et al., 2007). For example, how stem cells differentiate and give rise to diverse tissue types is a spatially regulated process which controls the development of different tissue types and organs(Ivanovs et al., 2017; Slack, 2008). Mouse embryonic organogenesis begins during the end of the first week right after gastrulation and continues through birth(Mitiku and Baker, 2007). When and how exactly different organs emerge in an early stage embryo is still inadequately understood due to dynamic heterogeneity of tissues and cells during a rapid developmental process. An embryonic organ at this stage could differ substantially in anatomical and molecular definitions as compared to their adult counterparts. In order to dissect the initiation of early organogenesis in the whole embryo context, it is highly desirable to not only identify genome-wide molecular profiles to define emerging cell types but also interrogate their spatial organization in the tissue at a high resolution.
Despite the latest advent of massively parallel single-cell RNA-sequencing (scRNA-seq)(Klein et al., 2015; Macosko et al., 2015) that revealed astonishing cellular heterogeneity in many tissue types, including the dissection of all major cell types in developing mouse embryos from E9 to E14(Cao et al., 2019; Pijuan-Sala et al., 2019), the spatial information in the tissue context is missing in scRNA-seq data. The field of spatial transcriptomics emerged to address this challenge. Early attempts were all based on multiplexed single-molecule fluorescent in situ hybridization (smFISH) via spectral barcoding and sequential imaging(Pichon et al., 2018; Trcek et al., 2017). It evolved rapidly over the past years from detecting a handful of genes to hundreds or thousands (e.g., seqFISH, MERFISH)(Chen et al., 2015; Lubeck et al., 2014), and recently to the whole transcriptome level (e.g, SeqFISH+)(Eng et al., 2019). However, these methods are technically demanding, requiring high-sensitivity optical imaging systems, sophisticated image analysis process, and a lengthy repeated imaging workflow to achieve high multiplexing(Perkel, 2019). Moreover, they are all based upon a finite panel of probes that hybridize to known mRNA sequences, limiting their potential to discover new sequences and variants. Fluorescent in situ sequencing methods (e.g., FISSEQ, STARmap)(Lee et al., 2015; Wang et al., 2018) were additionally reported but the number of detectable genes is limited, and their workflow resembles sequential FISH, again requiring a lengthy, repeated, and technically demanding imaging process.
It is highly desirable to develop new methods for high-spatial-resolution, unbiased, genome-scale molecular mapping in intact tissues, which does not require sophisticated imaging but can instead capitalize on the power of high-throughput Next Generation Sequencing (NGS). This year, a method called Slide-seq was reported that utilizes a self-assembled monolayer of DNA-barcoded beads on a glass slide to capture mRNAs released from a tissue section placed on top. It demonstrated spatial transcriptome sequencing at a 10µm resolution(Rodriques et al., 2019). A similar method, called HDST, used 2µm beads in a microwell array chip to further increase the nominal resolution(Vickovic et al., 2019). However, these emergent NGS-based methods have the following limitations: (a) The way to decode the array of DNA-barcoded beads is through manual sequential hybridization or SOLiD sequencing, similar to seqFISH, again requiring a lengthy and repeated imaging process. (b) The number of detected genes from the 10µm resolution Slide-seq data is very low (∼150 genes/pixel) and thus, it can hardly visualize the spatial expression of individual genes in a meaningful way even if the collective gene sets can locate major cell types. (c) These methods, including a previously reported low-spatial-resolution (∼150um) approach(Stahl et al., 2016), are all based upon the same mechanism – “barcoded solid-phase RNA capture”(Salmen et al., 2018). They require newly sectioned tissues to be carefully transferred to the bead or spot array and lysed to release mRNAs. Although the mRNAs are presumably captured only by the beads right underneath, the lateral diffusion of free mRNAs is unavoidable. (d) All these genome-scale methods are all technically demanding and difficult to use in most biology laboratories. Finally, it is not obvious how these methods can be extended to other omics measurements and how easy researchers from other fields can adopt them. Therefore, high-spatial-resolution omics is still a scientific challenge but also an opportunity that, if fully realized and democratized, will shift the paradigm of research in many fields of biology and medicine. Current methods are either technically impractical or fundamentally limited by the approaches themselves for enabling wide-spread adoption.
Inspired by how molecular barcoding of individual cells in isolated droplets or microwells served as a universal sample preparation method(Dura et al., 2019; Klein et al., 2015; Macosko et al., 2015) to barcode single cells for massively parallel sequencing of mRNAs, DNAs, or chromatin states, we sought to develop a universal method to spatially barcode tissues, forming a large number of barcoded tissue pixels each containing a distinct molecular barcode. Similarly, the barcoded mRNAs or proteins in the tissue pixels can be retrieved, pooled, and amplified for NGS sequencing but, in this case, to generate a spatial omics atlas. We have previously developed microfluidic channel-guided deposition and patterning of DNAs or antibodies on a substrate for multiplexed protein assay(Lu et al., 2013; Lu et al., 2015). Thus, we hypothesized that a microfluidic channel-guided delivery of molecular barcodes could be similarly utilized to perform spatial barcoding.
Herein, we report on a fundamentally new technology for spatial omics – microfluidic Deterministic Barcoding in Tissue for spatial omics sequencing (DBiT-seq). A microfluidic chip with parallel channels (10, 25 or 50μm in width) is placed directly against a fixed tissue slide to introduce oligo-dT tagged DNA barcodes A1-A50 that bind mRNAs and initiate in situ reverse transcription. This step results in stripes of barcoded cDNAs in the tissue section. Afterwards, the first chip is removed and another microfluidic chip is placed perpendicular to the first flow direction to introduce a second set of DNA barcodes B1-B50, which are ligated at the intersection to form a 2D mosaic of tissue pixels, each of which has a distinct combination of barcodes Ai and Bj (i=1-50, j=1-50). Then, the tissue is lysed and spatially barcoded cDNAs are retrieved, pooled, template-switched, amplified by PCR, and tagmentated to prepare a library for NGS sequencing. Proteins can be co-measured by applying a cocktail of antibody-derived DNA tags (ADTs) to the fixed tissue slide prior to flow barcoding, similar to Ab-seq or CITE-seq(Shahi et al., 2017; Stoeckius et al., 2017). Using DBiT-seq, we demonstrated high-spatial-resolution co-mapping of whole transcriptome and a panel of 22 proteins in mouse embryos. It faithfully detected all major tissue types in early organogenesis. The spatial gene expression and protein atlas further identifies a differential pattern in embryonic forebrain development and microvascular networks. The 10μm-pixel resolution can detect a single-cell-layer of melanocytes lining around an optical vesicle and discovered asymmetric gene expression within it, which has not been observed previously. DBiT-seq does not require any DNA spot microarray or decoded DNA-barcoded bead array but only a set of reagents. It works for an existing fixed tissue slide, not requiring newly prepared tissue sections that are necessary for other methods(Rodriques et al., 2019; Stahl et al., 2016). It is highly versatile allowing for the combining of different reagents for multiple omics measurements to yield a spatial multi-omics atlas. We envision that this may become a universal approach to spatially barcode a range of molecular information including DNAs, epigenetic states, non-coding RNAs, protein modifications, or combined. The microfluidic chip is directly clamped onto the tissue slide and the barcode flow step requires no experience in microfluidic control. Reagent dispensing is similar to pipetting into a microliter plate. Thus, DBiT-seq is potentially a platform technology that can be readily adopted by researchers from a wide range of biological and biomedical research fields.
RESULTS
DBiT-seq workflow
The workflow of DBiT-seq is described in Figure 1A. It does not require a newly microtomed tissue section to start with as a standard tissue slide that has already been fixed and banked is compatible with our approach. If a frozen tissue section were the starting material, it can be transferred to a poly-L-lysine coated slide, fixed with formaldehyde, and stored in −80°C until use. A polydimethylsiloxane (PDMS) microfluidic chip containing parallel microchannels (down to 10μm in width) is placed on the tissue slide to introduce a set of DNA barcode solutions (Figure S1). Each barcode is composed of an oligo-dT sequence for binding mRNAs and a distinct barcode Ai (i=1 to 50). Reverse transcription is conducted during the first flow for in situ synthesis of cDNAs that immediately incorporate barcodes A1-A50. Then, this PDMS chip is removed and another PDMS chip is placed on the same tissue with the microchannels perpendicular to those in the first flow barcoding. Next, a second set of barcodes Bj (j=1 to 50) are flowed in to initiate in situ ligation that occurs only at the intersections, resulting in a mosaic of tissue pixels, each of which has a distinct combination of barcodes Ai and Bj (i=1 to 50 and j= 1 to 50). The tissue slide being processed is imaged during each flow as well as after both flows such that the exact tissue region comprising each pixel can be identified unambiguously. To perform multi-omic measurements of proteins and mRNAs, the tissue slide is first stained with a cocktail of antibody-derived tags (ADTs)(Stoeckius et al., 2017) prior to microfluidic flow barcoding. The ADTs have a polyadenylated tail that allows for detecting proteins using a workflow similar to detecting mRNAs. After forming a spatially barcoded tissue mosaic, cDNAs are collected, template-switched, and PCR amplified to make a sequencing library. Using 100×100 pair-ended NGS sequencing, we can detect spatial barcodes (AiBj, i=1-50, j=1-50) of all pixels and the corresponding transcripts and proteins to computationally reconstruct a spatial expression atlas. It is worth noting that unlike other methods, DBiT permits the same tissue slide being imaged with microfluidic channels to precisely locate the pixels and perform correlative analysis of tissue morphology and omics at high resolution and high accuracy.

(A) Detailed microfluidic device design (left panel) and barcoding chemistry protocol (right panel). Left panel: fresh frozen tissue sections were first allowed to warm to room temperature for 10 minutes. Then, 4% Formaldehyde was added, and tissue was fixed for 20 minutes at room temperature. After fixation, a cocktail of 22 antibody-DNA tags (ADTs) were added and incubated at 4°C for 30 minutes. After washing three times with PBS, 1st PDMS chip was attached to the glass slide. Barcode A (A1-A50) along with reverse transcription mixture was flowed through each channel. After reverse transcription, the 1st PDMS chip was removed and a 2nd PDMS was attached. Ligation solution along with Barcode B (B1-B50) was flowed into each channel. When finished, the 2nd PDMS chip was removed and a PDMS gasket was attached to the glass slide. Lysis solution was added into the gasket and the lysate was collected. cDNA and ADT derived cDNA were extracted using streptavidin coated magnetic beads. Template switch and PCR were then performed. The sequencing library was finally built with standard tagmentation. Right panel: DNA barcode A consists of a poly T region, a barcode region and a ligation region. The poly T region will recognize the poly A tail of mRNA and ADTs. DNA Barcode B consists of a ligation region, a barcode region, a UMI region and a PCR primer handle region. During ligation process, the ligation region will be ligated to the ligation region of barcode A. The cDNA product will then be template-switched. The final product is further amplified by PCR.
(B) DAPI and ethidium homodimer-1 (both are nuclei staining dyes) staining of mouse embryo (E.10) tissue section. After attaching the 1st PDMS chip, DAPI (purple color) solution was flowed through each channel and incubated for 20 minutes at room temperature. 1st PDMS was removed and a 2nd PDMS was attached. Ethidium homodimer-1 (Red) was then flowed through. The resulted tissue will form a mosaic like structure, with the intersections to be stained by both nuclei dyes.

(A) Schematic workflow. A standard fixed tissue slide is used as the starting material, which and incubated with a cocktail of antibody-DNA tags that recognize a panel of proteins of interest. A custom-designed PDMS microfluidic chip with 50 parallel microchannels in the center is aligned and placed on the tissue slide to introduce the 1st set of barcodes A1-A50. Each barcode is tethered with a ligation linker and a poly-T trail for binding mRNAs or the DNA tag for protein detection. Then, reverse transcription (RT) is conducted in situ to yield cDNAs that are covalently linked to barcodes A1-A50. Afterwards, this microfluidic chip is removed and another microfluidic chip with 50 parallel microchannels perpendicular to those in the first microfluidic chip is placed on the tissue slide to introduce the 2nd set of DNA barcodes B1-B50. These barcodes contain a ligation linker, a unique molecular identifier (UMI) and a PCR handle. After introducing the barcodes B1-B50 and the universal complementary ligation linker through the second microfluidic chip, the barcodes A and B are joined through ligation in situ and then the intersection region of the two microfluidic channels defines a distinct pixel with a unique combination of A and B, giving rise to a 2D array of spatially addressable barcodes AiBj (i=1-50, j=1-50). Afterwards, the second PDMS chip is removed and the tissue remains intact while spatially barcoded for all mRNAs and the proteins of interest. The barcoded tissue is imaged under an optical or fluorescence microscope to visualize individual pixels. Finally, cDNAs are extracted from the tissue slide, template switched to add PCR handle to the other side, and amplified by PCR for making the sequencing via tagmentation. A pair-ended high-throughput sequencing is performed to read the spatial barcodes (AiBj) and cDNA sequences from either mRNAs or antibody DNA tags. In silicon reconstruction of a spatial mRNA or protein expression map is realized by matching the spatial barcodes AiBj to the corresponding cDNA reads. The reconstructed spatial omics atlas can be correlated to the tissue image collected after DBiT to identify the exact spatial location of individual pixels.
(B) Schematic of the biochemistry protocol to add spatial barcodes to a tissue slide. Proteins of interest are labeled with antibody DNA tags (ADTs), each of which consists of a unique antibody barcode (15mer, see Table S1) and a poly-A tail. Barcode A1-A50 contains a ligation linker(15mer), a unique spatial barcode Ai (i=1-50, 8mer, see Table S2), and a poly-T sequence(16mer), which detects mRNAs and proteins through binding to poly-A tails. After introducing barcodes A1-A50 to the tissue slide, reverse transcription is conducted in situ to generate cDNAs from mRNAs as well as antibody barcodes. Barcode B1-B50 consists of a ligation linker(15mer), a unique spatial barcode Bj(j=1-50, 8mer, see Table S2), a unique molecular identifier (UMI)(10mer), and a PCR handle (22mer) terminally functionalized with biotin, which facilitates the purification in the later steps using streptavidin-coated magnetic beads. When the barcodes B1-B50 are introduced to the tissue sample that is already barcoded with A1-A50 using an orthogonal microfluidic delivery, a complementary ligation linker is also introduced and initiates the covalent ligation of barcodes A and B, giving rise to a 2D array of spatially distinct barcodes AiBj (i=1-50 and j=1-50).
Barcode design and chemistry
The key elements of DNA barcodes and the chemistry to perform DBiT is described in Figure 1B. To detect proteins of interest, the tissue is firstly labeled with ADTs, each of which consists of a unique antibody barcode (15mer, see Table S1) and a poly-A tail. Barcode A contains a 15mer ligation linker, a unique spatial barcode Ai (i=1-50, 8mer, see Table S2), and a 16mer poly-T sequence, which binds mRNAs and ADTs through binding to poly-A tail. After permeabilization, DNA barcodes A1-A50 are flowed in along with a reverse transcriptase mixture and reverse transcription is conducted in situ to generate cDNAs as well as incorporate barcode A in the tissue stripes within individual microchannels. Barcode B consists of a 15mer ligation linker, a unique spatial barcode Bj(j=1-50, 8mer, see Table S2), a 10mer unique molecular identifier (UMI), and a 22mer PCR handle terminally functionalized with biotin, which is used later to perform cDNA purification with streptavidin-coated magnetic beads. During the second flow to introduce barcodes B1-B50, a complementary ligation linker and the T4 ligase are also introduced to initiate in situ ligation of barcodes A and B only at the intersections of two flow, which completes the deterministic barcoding of a tissue slide and yields a mosaic of tissue pixels with distinct barcodes in each of the 50×50 = 2,500 pixels. This chemistry is versatile and can be readily expanded to a larger array (e.g., 100×100 = 10,000) of pixels or extended to other omics measurement by changing the binding chemistry from poly-T to, for example, spicing-site specific sequences.
Microfluidic device for the DBiT process
The PDMS microfluidic chip design (Figure S2) is simple, consisting of 50 parallel microchannels in the center which are connected to the same number of inlet and outlets on two sides of the PDMS slab. It is made of silicone rubber, which is sticky to the glass slide surface and can be placed on the tissue slide to introduce solution without noticeable leakage if no positive pressure is applied. To further assist the assembly, a simple clamp is used to hold the PDMS firmly against the slide at the tissue specimen region (Figure 2A). The inset (inlet) holes which are ∼2mm in diameter and 4mm in depth allow the ∼5μL of barcode reagents to be directly pipetted with no need for any microfluidic handling setup. The outlet holes are enclosed (or roofed?) with a global cover connected to a house vacuum to pull the reagents from the insets (inlets) into the tissue region. It takes several seconds to pull the solution from inlets through outlets for a 50μm microfluidic chip and up to 3min for a 10μm microfluidic chip. After flow barcoding, the microfluidic chip is sonicated and rinsed with 0.5M NaOH solution and DI water for reuse. Thus, this device requires no sophistic microfluidic control systems, can be readily assembled by a scientist with no experience in microfluidics, and the workflow is readily adoptable in a conventional biology laboratory.

(A) A photograph of PDMS chip (1st direction) with fifty 10µm-width channels in the center. Holes were punched with a 2mm diameter puncher.
(B) AutoCAD design of PDMS chip with 10µm width channels.
(C) PDMS chip with 25 µm width channels. (D) PDMS chip with 50 µm width channels.

(A) Microfluidic device used in DBiT-seq. A series of microfluidic chips were fabricated with 50 parallel microfluidic channels in the center that are 50μm, 25μm, and 10μm in width, respectively. The PDMS chip containing 50 parallel channels is placed directly on a tissue slide and, if needed, the center region is clamped using two acrylic plates and screws. All 50 inlets are open holes ∼2mm in diameter and capable of holding ∼13μL of solution. Different barcode solutions are pipetted to these inlets and drawn into the microchannels via a global vacuum applied to the outlets situated on the opposite side of the PDMS chip. Basically, this device is a universal approach to realize spatially defined delivery of DNA barcodes to the tissue surface at a resolution of down to 10μm to even better.
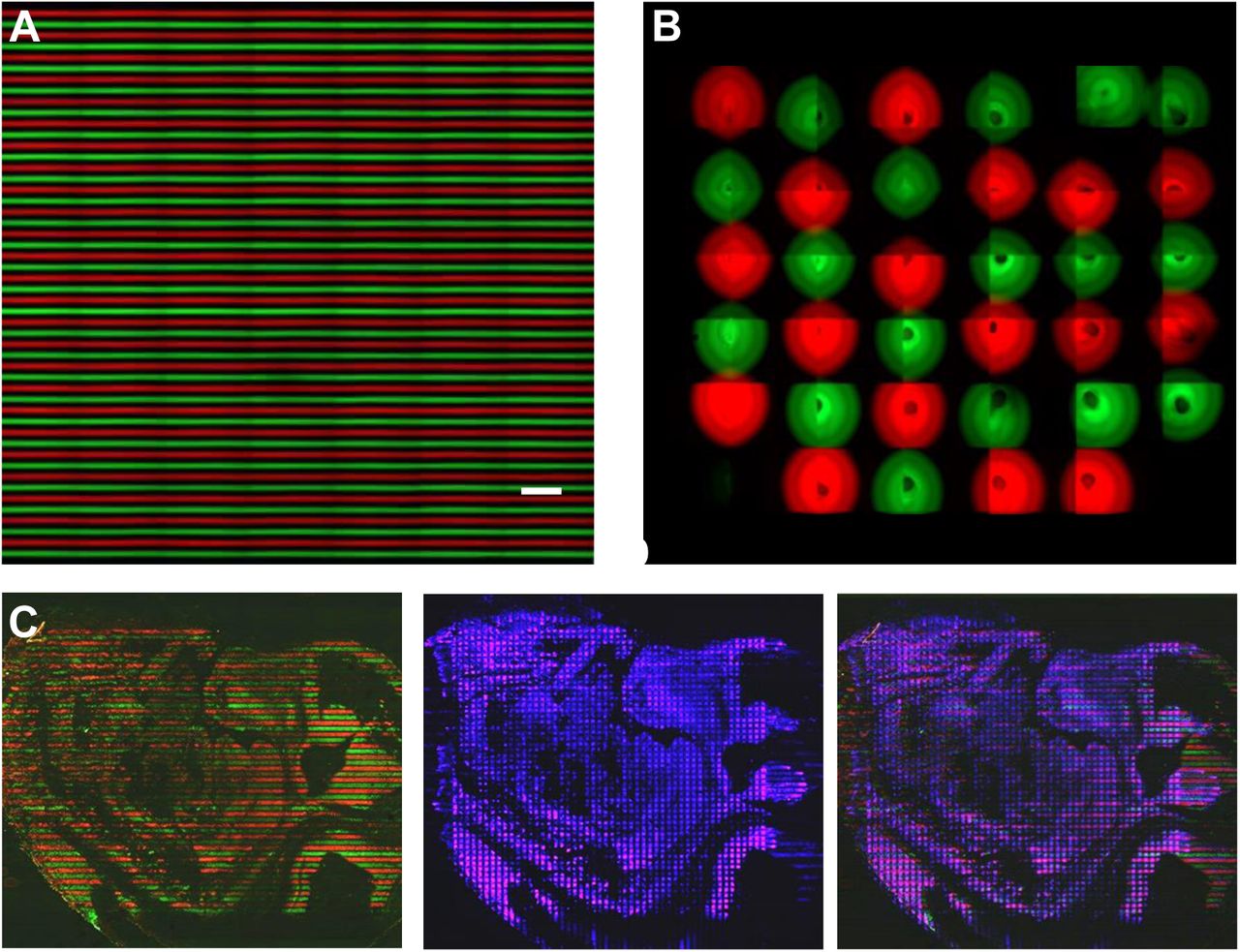
(B) Validation of spatial barcoding using fluorescent DNA probes. The images show parallel lines of Cy3-labelled barcode A (red, left panel) on the tissue slide defined by the first flow, the square pixels of FITC-labeled barcode B (green, right panel) defined as the intersection of the first and the second flows, and the overlay of both fluorescence colors (middle). Because barcode B is ligated to the immobilized barcode A in an orthogonal direction, it occurs only at the inter-section of the first and second flows. Microfluidic channel width = 50μm.
(C) Validation of leak free flow barcoding. This fluorescence image shows an E14 mouse embryo tissue slide stained with two barcode A reagents pre-conjugated with Cy3 (red) and FITC (green), respectively, delivered to the tissue surface using the aforementioned microfluidic chip. It shows well separated red and green fluorescence with no sign of leakage across a large distance and over complex tissue topography. Microfluidic channel width = 50μm.
(D) Validation of spatial barcoding for 10μm pixels. A tissue slide was subjected to spatial barcoding or DBiT and the resultant pixels were visualized by optical imaging (upper left) of the tissue post-DBiT and fluorescent imaging (upper right) of the same tissue sample using FITC-labeled barcode B. Pressing microfluidic channels against the tissue slide resulted in a small plastic deformation of the tissue matrix, which enabled us to directly visualize all individual pixels created by DBiT, which turned out to in excellent agreement with the fluorescent image of all pixels using FITC-labelled barcode B. Enlarged views (low panels) further show discrete pixels down to a 10-μm level.
(E) Line profile of fluorescence intensity. It allows for quantitative measurement of the pan-mRNA-FISH signal within and between pixels. The half-peak-height-spreading of fluorescence intensity is theoretically associated with the diffusion distance of DNA barcodes through the tissue matrix underneath the microfluidic channel walls.
(F) Qualification of the diffusion “leak-out” distance, the measured size of pixels, and the number of cells per pixel. Quantitative analysis of the line profile revealed the barcode diffusion through the dense tissue matrix is small (0.9μm, obtained with 10μm microchannels fixed by clamps). The measured pixel size agreed with the microchannel size. Using a cell nuclear staining DAPI, the number of cells in a pixel can be visualized. The average cell number is 1.7 per a 10μm pixel and 25.1 per a 50μm pixel. Therefore, the pixel size is approaching the single cell level.
(G) Sequencing data quality assessment and comparison. A high-resolution spatial transcriptome sequencing data was obtained using a 10μm microfluidic barcoding approach. The number of genes detected per pixel is >2000. In contrast, Slide-seq only detected ∼150 genes per pixel (10μm), which is insufficient for spatial visualization of individual genes in a meaningful way. The low-resolution ST method yielded a similar number of genes per pixel but the pixel size is ∼100-150μm, which is almost 100 times larger in area.
Evaluation of DBiT using fluorescent in situ hybridization (FISH)
Although no noticeable leakage was observed between microchannels during the vacuum driven flow barcoding, it is unclear if the DNA barcode solutions could diffuse through the tissue matrix and result in cross-contamination. The diffusion distance in an aqueous solution decreases substantially with the increase of molecular size, which was utilized to perform diffusion-limited reagent exchange in microfluidics for multiple chemistry reactions. We hypothesize that the diffusion through a dense matrix is even more restricted. A validation experiment was designed to monitor our workflow step by step using fluorescent probes and to evaluate the effect of diffusion underneath the microchannel walls (Figure 2B&C, Figure S1B). We conjugated barcodes A(1-50) with fluorophore Cy3 and barcodes B(1-50) with fluorophore FITC, and then imaged the tissue during and after DBiT at a 50μm pixel resolution. The first flow is supposed to yield stripes of Cy3 signal (red) corresponding to barcodes A hybridized in situ to tissue mRNAs. We observed distinct stripe pattern with no visually noticeable diffusion between stripes. The second flow adds barcodes B only to the intersections, yielding isolated squares of FITC signal (green), which is exactly our observation(Figure 2B). Due to autofluorescence of tissue excited by blue light (488nm), the faint green fluorescence appears in between squares but the average intensity is an order of magnitude lower. Next, two sets of fluorescent barcodes A were synthesized with Cy3 and FITC, respectively, and flowed through alternately over a large area of a mouse E14 embryo tissue section, giving rise to alternating green and red lines with no cross-contamination (Figure 2C, Figures S3&4). These images were taken when using a 50μm device without clamp. To evaluate the feasibility of reducing to 10μm flow barcoding, we performed a full DBiT using fluorescent barcodes B with FITC and observed a clean pattern of green fluorescence pixels (Figure 2D and Figure S5). Interestingly, this pan-mRNA FISH signal in each tissue pixel is not uniform but can reflect the underlying cell morphology. As mentioned above, our approach allows for the imaging of the same tissue slide during and after flow barcoding. We found that the clamping step compressed the tissue underneath the microchannel walls and led to localized plastic deformation. As a result, the light field optical image of the tissue region processed by cross-flow barcoding show imprinted topological patterns with readily distinguishable tissue pixels, which can be used to assist the correlation of tissue histology with spatial omics sequencing data. The compressed tissue region underneath the microchannel walls has a higher matrix density and may further reduce the diffusion distance. We used the fluorescence intensity line profile (Figure 2E) to calculate the half-peak-width-intensity increase, which represents a quantitative measure of the “diffusion” distance between microchannels. It turned out to only 0.9±0.2μm for 10μm flow channels operated with clamp and 4.5±1μm for 50μm flow channels without clamp (Figure 2F). Thus, we speculate the theoretical limit of DBiT spatial resolution can be as good as ∼2μm.

These pictures are from a PDMS chip (50 µm channel width) covered mouse embryo (E.14) tissue slides.
(A) Whole embryonic tissue;
(B) Enlarged view of front head and a section of the lower body.
(C) Enlarged view of a region at the front head.

(A) Test possible crosstalk between two neighboring channels by flowing alternatively a FITC (Green) labelled Barcode A and a Cy3 (Red) labelled Barcode A. Scale bar = 400 µm.
(B) Analyze possible leakage at the injection area. Each circle represents an inset hole. Green colored circles are loaded with FITC labelled Barcode A and red colored circles are loaded with Cy3 labelled Barcode A.
(C) Staining mouse embryo (E.14) mRNAs with FITC (Green) labelled Barcode A and Cy3 (Red) labelled Barcode A (left panel). Further staining with Cy5 labelled Barcode B (Middle panel). Overlay of two previous panels (right panel).

(A) Measure possible cross talk between two neighboring channels by flowing alternatively a FITC (Green) labelled Barcode A and a Cy3 (Red) labelled Barcode A. Scale bar = 100 µm.
(B) Staining part of mouse embryo (E.14) mRNAs with FITC (Green) labelled Barcode A and Cy3 (Red) labelled Barcode A (figure not show). Further staining with Cy5 labelled Barcode B.
(C) Overlay of three fluorescent channels.
Evaluation of DBiT-seq data quality
The PCR amplicons were analyzed for cDNA size distribution, which peaks at 900-1100bp for a sample fixed right after preparation(Figure S6). A frozen tissue section slide left at room temperature for 24 hours or longer led to significant degradation and the shift of the main peak to ∼350bp. However, after fixation and flow barcoding, it still resulted in usable sequencing data for quantification of gene expression. A HiSeq pair-ended (100×100) sequencing was conducted to identify spatial barcodes and the expression of proteins and mRNAs on each pixel. The alignment was done using DropSeq tools(Macosko et al., 2015) to extract UMI, Barcode A and Barcode B, from Read 2. The processed read was trimmed, mapped against the mouse genome(GRCh38), demultiplexed annotation(Gencode release M11) using the Spatial Transcriptomics pipeline reported previously (Navarro et al., 2017). With that, similar to scRNA-seq quality evaluation, we calculated the total number of transcript reads (UMIs) per pixel and the total number of genes detected (Figure 2G and Figure S7). Compared with the literature data from Slide-seq(Rodriques et al., 2019) and the low resolution Spatial Transcriptomics (ST) sequencing data(Stahl et al., 2016), our data from a 10μm DBiT-seq experiment was able to detect 22,969 genes in total and 2,068 genes per pixel. In contrast, Slide-seq, which has the same pixel size (10μm), detected ∼ 150 genes per pixel(spot). It is worth pointing out that this significant improvement in data quality allows DBiT-seq to directly visualize the expression pattern of individual genes but Slide-seq could not do that in a meaningful way due to data sparsity. The number of UMIs or genes per pixel detected by low-resolution ST method is similar to our approach but the size of the pixel in ST is ∼100-150μm, which is ∼100× larger in area. This marked increase in data quality is presumably attributed to the uniqueness in flow barcoding method that does not require a tissue lysis step to release mRNAs and avoids the loss of released mRNAs because of their lateral diffusion into the solution phase. Although it has long been recognized that retrieving mRNAs from fixed tissue specimens for NGS sequencing has decreased yield due to degradation, recent studies showed that the quality of mRNAs in tissue remains largely intact but rather, it is the tissue lysis and RNA retrieval step that leads to the degradation and the consequential poor recovery.

(A) Fresh frozen tissue sample tested with DBiT-seq immediately.
(B) Fresh frozen sample left at room temperature for 24 hours before testing with DBiT-seq. Due to degradation, shorter cDNA was observed when left at room temperature for extended time.

Our sequencing data was obtained using a 10μm microfluidic barcoding approach. The number of reads per pixel is close to 5,000. Slide-seq only have ∼200 reads per pixel (10μm and the low-resolution ST method yielded a similar number of reads per pixel, but the pixel size is ∼100-150 μm, about 100 times larger than ours.
Whole mouse embryo spatial multi-omics atlas mapping
The dynamics of embryonic development, in particular, the formation of different organs (organogenesis) at the early stages, is intricately controlled spatiotemporally. The results from a large number of laboratories around the world and obtained using a range of techniques such as FISH, immunohistochemistry(IHC), and RNAseq, have been integrated to generate a relatively complete mouse embryo gene expression database such as eMouseAtlas(Armit et al., 2017). Thus, the developing mouse embryos are well suited for validation of a new spatial omics technology by providing known reference data for comparison. We applied DBiT-seq to a E.10 whole mouse embryo tissue slide at a pixel size of 50μm to computationally construct a spatial multi-omics atlas. The tissue histology image from an adjacent section was stained for H&E (Haemotoxylin and Eosin) (Figure 3A left). The read counts of mRNA transcripts in individual pixels, equivalent to pan-mRNA detection, are shown as a spatial heatmap (Figure 3A middle), which is found to correlate well with tissue density and H&E morphology. The total read counts from a panel of 22 protein markers (see Table S1) combined in each pixel appear to be more uniform and less dependent on tissue density and morphology (Figure 3A right). The quality of sequencing data is excellent with an average of ∼4500 genes detected per pixel, which is higher than that in the 10μm-pixel DBiT-seq data (Figure 2G), due in part to the larger pixel size and subsequent increased cell type diversity per pixel. Unsupervised clustering of all pixels based on mRNA transcriptomes reveals eleven major clusters (Figure 3B) as shown in a tSNE plot that, once mapped back to the spatial atlas, are found to correlate with the major tissue types at this stage including telencephalon (forebrain), mesencephalon (midbrain), rhombencephalon (hindbrain), branchial arches, spinal neural tube, heart, limb bud, and ventral and dorsal side of main body for early internal organ development. We anticipate more tissue subtypes to be identified using higher resolution DBiT-seq. Based upon literature database and the classical Kaufman’s Atlas of Mouse Development(Baldock and Armit, 2017), we performed anatomical annotations of 13 major tissue types(Figure 3C), among which 9 were identified by unsupervised clustering. Interestingly, even at this resolution (pixel size = 50μm), some fine features identified by clustering – such as the small clusters in the middle of the brain and a distinct stripe of pixels between the dorsal and ventral layers of the body – are not readily distinguishable in H&E. The former is indicative of early eye and ear development and the latter is less clear but may correlate with the dorsal aorta.

(A) Pan-mRNA and pan-protein-panel spatial expression maps (pixel size 50μm) reconstructed from DBiT-seq, together with the H&E image of an adjacent tissue section. Whole transcriptome signal correlated with anatomic tissue morphology and density.
(B) Unsupervised clustering of all pixels and mapping back to the spatial atlas. The results are shown in a tSNE plot and mapped back to spatial map, which is further compared to the anatomical structure via overlay with the H&E image.
(C) Anatomic annotation of tissue (sub)types. To facilitate the examination of tissue morphology in correlation with the spatial gene expression atlas, an adjacent tissue section was stained for H&E and overlaid with the unsupervised clustering map. The clustering analysis revealed 11 major tissue (sub)types, which are in general agreement with anatomically annotated tissue regions (1-13).
(D) Spatial expression of four individual proteins and cognate mRNA transcripts in a whole mouse embryo. These are Notch1(Notch1), CD63(Cd63), Pan-Endothelial-Cell Antigen(Plvap), and EpCAM(Epcam). Multi-omic DBiT-seq allows for head-to-head comparison of a panel of proteins and the expression of cognate genes. It shows consistence as well as discordance between mRNA and protein for selected pairs, but the spatial resolution is adequate to resolve the fine structures in specific organs.
(E) Correlation between mRNAs and proteins in anatomically annotated tissue regions (see subpanel (C)). The average expression levels of individual mRNAs and cognate proteins in each of the thirteen anatomically annotated tissue regions are compared.
(F) Comparison to immunofluorescence tissue staining. Pan-endothelial antigen (PECA), which marks the formation of embryonic vasculature, is expressed extensively at this stage (E.10), consistent with the protein and mRNA expression revealed by DBiT-seq. EpCAM, an epithelial marker, already show up but in several highly localized regions, which were also identified by DBiT-seq (both mRNA and protein). P2RY12 is a marker for microglia in CNS, which depicts the spatial distribution of the neural system.
(G) Comparison to single-cell RNA-seq data. Integrating the global transcriptome profiles of four E.10 embryo samples sequenced using DBiT-seq conform well into the correct stage (E.10) of a single-cell RNA-seq data UMAP generated from mouse embryos ranging from E9.5-E13.5(Cao et al., 2019).
Correlation between proteins and mRNAs in spatial expression patterns
While single-cell RNA/protein co-sequencing such as CITE-seq can directly compare the expression level of individual proteins to cognate mRNAs in a cell, the correlation between their spatial expression patterns in the tissue context are missing. Herein, high quality spatial multi-omics data allows for head-to-head comparison between individual proteins and mRNA transcripts pixel-by-pixel in a tissue. As such, all 22 proteins analyzed are compared with their corresponding mRNAs (see Figure S8). Selected mRNA/protein pairs are discussed below (Figure 3D). Notch signaling plays a crucial role in regulating a vast array of embryonic developmental processes. Notch1 protein is found to be highly expressed throughout the whole embryo, which is consistent with the observation of extensive Notch1 mRNA expression although it appears to mirror the tissue density. CD63 is an essential player in controlling cell development, growth, proliferation, and motility. Its mRNA transcript is indeed expressed extensively in the whole embryo with a higher expression in hindbrain and heart. Pan-EC-Antigen(PECA) or MECA-32, as a pan-endothelial marker, is expressed in many tissue regions, but the spatial pattern is difficult to identify at this resolution. The expression of EpCAM, a pan-epithelial marker, is highly localized in terms of both mRNA and protein, the expression patterns of which are also highly consistent. Several other genes are discussed as below. Integrin subunit alpha 4 (ITGA4), known to be critical in epicardial development, is indeed highly expressed in embryonic epicardium but also observed in many other tissue regions. Its protein expression is seen throughout the whole embryo. Many genes show strong discordance between mRNA and protein such as NPR1. A pan-leukocyte protein marker CD45 is seen extensively but apparently enriched in the dorsal aorta region and brain, although the expression level of its cognate mRNA Ptprc is low. We further generate a comprehensive chart of tissue region-specific mRNAs and proteins by calculating the average expression in each of 13 anatomically annotated tissue regions (Figure 3E). Next, to validate the DBiT-seq data, immunofluorescence was performed using antibodies to staining for P2RY12 (microglia in central nerve system) PECA (endothelium), and EpCAM (epithelium). We observed a highly consistent pattern of EpCAM between immunostaining and DBiT-seq (Figure 3F). Spatial transcriptome sequencing (without ADTs) was repeated with a separate E.10 embryo tissue slide and the results are consistent (Figure S9). Finally, a “bulk” transcriptional profile could be derived from spatial DBiT-seq data and compared to scRNA-seq of mouse embryos E9.5 – E13.5, which revealed that our data are correctly positioned in the UMap when compared to literature data(Cao et al., 2019) (Figure 3G).

Protein and mRNA pairs as shown in Figure 3D.

(A) Left panel: bright field microscope images of E.10 mouse embryo after removal of the 2nd PDMS chip. Plastic deformation of the tissue forms identifiable 50µm x 50µm “pixels”. Middle panel: Matching pan-mRNA data with microscope images. Right panel: clustering of pixels based on expression levels.
(B) Clustering of pixels by whole transcriptome data. Four clusters were identified and with Gene Ontology (GO) analysis, they are assigned to brain, heart, neuron and blood vessel.
(C) Four representatives of differentially expressed genes that matches well with whole transcriptome clustering.
(D) Additional differentially expressed genes that show clustering of whole embryo.
Higher resolution mapping of an embryonic brain
The data in Figure 3 indicates non-uniform expression of genes such as CD63, PECA, and EpCAM, but the resolution can hardly resolve their spatial patterns. We conducted a higher resolution (pixel size = 25μm) DBiT-seq to analyze the brain region of an E.10 mouse embryo tissue sample. Pan-mRNA map (Figure 4A) clearly resolves many more fine structures in brain as compared to Figure 3A. Pan-protein-panel map begins to show resolvable fine structures and both can be correlated with the H&E histology (Figure 4A). We surveyed all 22 individual proteins (Figure S10) and observed distinct expression patterns in at least 12 proteins in the brain region. Four selected proteins are discussed below (Figure 4B). CD63 is expressed extensively except in forebrain with a clear spatial pattern. Surprisingly, PECA, a pan-endothelial marker, explicitly delineates the brain microvascular network using DBiT-seq whereas the tissue histology can barely distinguish the location of microvasculature. EpCAM is found in extremely localized and highly defined regions as thin as a single line of pixels (∼25μm) with an ultrahigh signal-to-noise ratio. A distinct differential expression pattern of MAdCAM is seen in a part of the forebrain, which has never been identified previously. Overlay of CD63 and MAdCAM1 reveals a strong anticorrelation relationship not only in forebrain but also other regions including hindbrain and neck. A highly organized spatial expression pattern is also seen among MAdCAM1, PECA and EpCAM. To validate these observations, we performed immunofluorescence imaging using an adjacent tissue section to detect EpCAM and PECA. Spatial expression maps obtained by DBiT-seq and immunofluorescence staining images are superimposed on a H&E image (Figure 4C), which show surprisingly perfect correlations at a near-single-pixel level, providing a strong validation of DBiT-seq.

Protein expression of all 22 individual proteins in embryonic mouse brain (E.10, 25 µm pixel size).

(A) Pan-mRNA and pan-protein-panel spatial expression maps of the brain region of a mouse embryo (E.10) obtained at a higher resolution (pixel size 25μm) in comparison with the H&E image taken from an adjacent tissue section. The spatial pattern of whole transcriptome signal correlates with cell density and tissue morphology.
(B) Visualization of individual proteins CD63, pan-endothelial-cell antigen (PECA), EpCAM, MAdCAM1. Left two columns: spatial protein expression heatmaps explicitly showing brain region-specific expression and the network structure of brain microvasculature. Right column: overlay of MAdCAM1/CD63 or PECA/MAdCAM1/EpCAM reveals spatial correlation or anti-correlation between these protein markers.
(C) Validation by immunofluorescence. Spatial expression of EpCAM and PECA reconstructed from DBiT-seq and the immunofluorescence staining of the same proteins are superposed on the H&E image for comparison. A highly-localized expression pattern of EpCAM is in near-perfect correlation with immunostaining. The network of microvasculature revealed by PECA in DBiT-seq is strongly correlated with the immunostaining signal.
(D) Volcano plot showing spatial proteomics-guided differential mRNA expression analysis to search for region-specific transcriptomic signature. The targeted protein panel DBiT-seq yielded a high spatial resolution protein expression atlas and highly defined tissue features presumably due to the higher depth per protein as compared to mRNAs in DBiT-seq. Here, we used the protein MAdCAM1 expression to extract all the pixels of interest in the forebrain and perform differential mRNA gene expression analysis to search for search for other differentially expressed genes.
(E) Visualization of two transcripts (Pax6 and Lhx3) identified by the analysis in (D). Both are enriched in the MAdCAM+ region of the forehead. Also perform is pathway analysis of MAdCAM1+ forebrain region.
(F) Examples of individual genes preferentially expressed in the MAdCAM-regions. Myh6 is expressed mainly in the heart. Fbn2 is extensively expressed in the brain but exhibits a strong anti-correlation with MAdCAM. Trpm1 is highly localized around the optical vesicle. Stmn2 is expressed in multiple regions of the brain but enriched in the neck region.
Spatial proteomics-guided differential gene expression analysis for mechanistic discovery
To further discern the genes associated with MAdCAM1 patterning in forebrain and discover the underlying mechanisms, we took advantage of our spatial multi-omics data atlas to use high-quality protein data to define the tissue region of interest and then extract the mRNA transcriptomes from the pixels in this region, which are subsequently subjected to differential gene expression analysis and/or submitted to Gene Ontology (GO) pathway analysis to infer mechanisms. The volcano plot (Figure 4D) shows the result of differential gene expression analysis between MAdCAM1+ forebrain region and MAdCAM1-region, and revealed Pax6 and Lhx2 are enriched in the MAdCAM1+ forebrain. A panel of top ranked genes differentially expressed are also visualized in the spatial atlas (Figure S11) and we found that Pax6 and Lhx2 are indeed enriched in this specific region (Figure 4E upper panel). Pathway analysis reveals the top 10 identified gene regulatory pathways (Figure 4E lower panel) are broadly implicated in telencephalon(forebrain) development, but it suggests pallium and cerebral cortex development at this stage. The GO analysis was also performed on MAdCAM-negative tissue pixels and a wide range of pathways were identified. Four top ranked genes are visualized (Figure 4F). Myosin heavy chain 6 (Myh6) is strongly localized in a specific feature, which appears to be the heart. Interestingly, fibrillin-2 (Fbn2) expression – known to be important in directing the assembly of elastic fibers to form microfibrils that support the development of nerves, muscles, lens during embryonic development –is also observed extensively throughout the brain but not in the MAdCAM+ region of the forebrain. Stathmin-2 (Stmn2), which controls microtubule dynamics, is found not expressed in midbrain and sparsely seen in forebrain, but is expressed extensively in the periphery of hindbrain and neck. All these shed new light on the spatial organization of gene expression patterns in embryonic brain development.
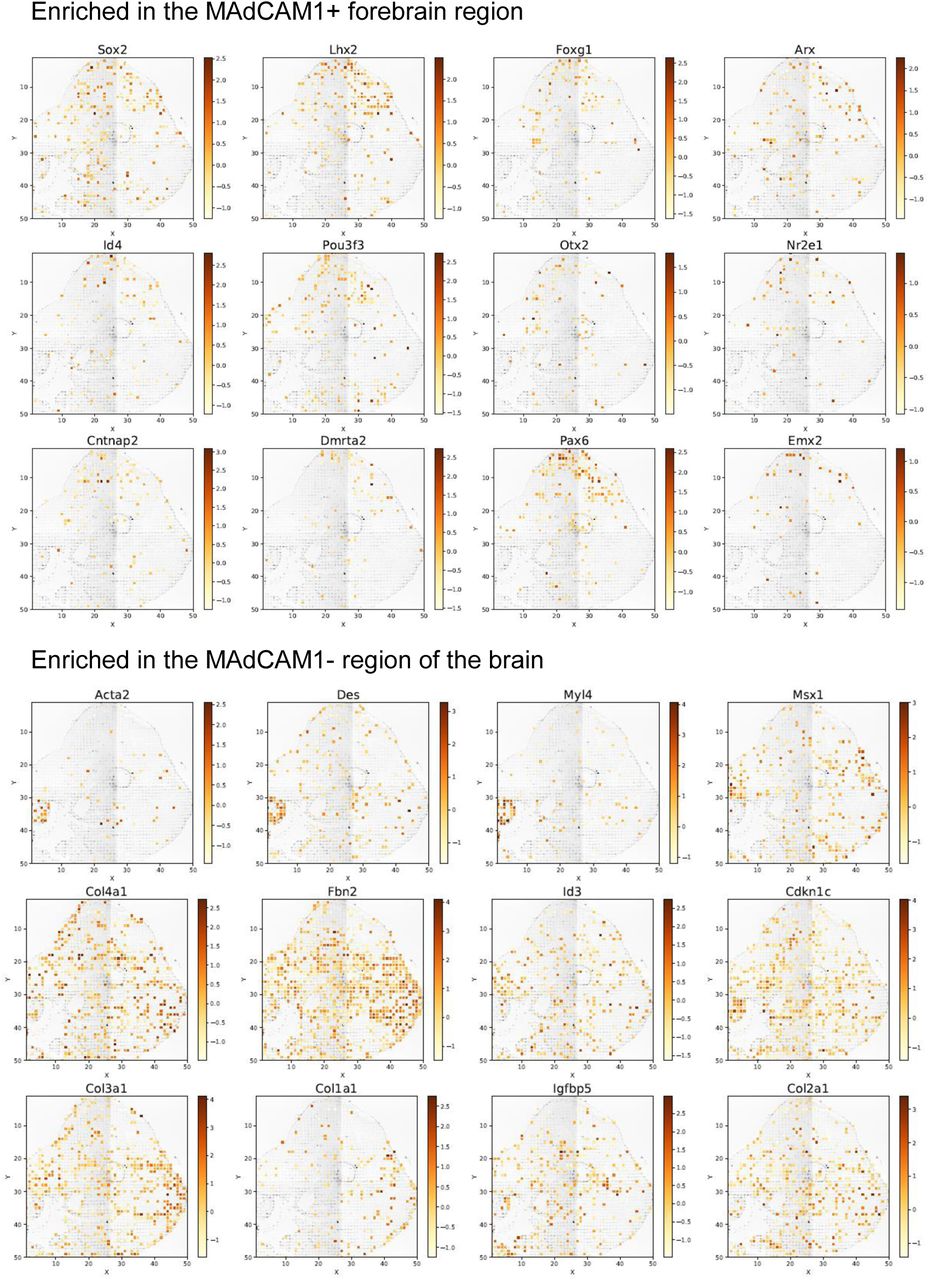
Spatial expression of mRNAs enriched in MAdCAM1 protein + or – regions identified by spatial proteomics-guided differential gene expression analysis.
Resolving a single-cell layer of melanoblasts in eye development
We further conducted the mapping of spatial transcriptome of a brain region using 10μm microfluidic channels. Again, it is performed on a whole mouse E10 embryo tissue slide and the pan-mRNA signal is superimposed in the region of tissue barcoded and analyzed by DBiT-seq (Figure 5A upper panel). An enlarged view of the tissue region processed with flow barcoding shows the imprinted morphology for differentiating all individual pixels (Figure 5A mid panel). An adjacent tissue section was stained for H&E (Figure 5A lower panel) for comparison. It turns out that the most noticeable gene set identified in the sequencing data is associated with eye development. Four of the top ranked genes are visualized (Figure 5B). At this stage (E.10), the eye development likely reaches a late optical vesicle stage. Pax6, a gene broadly implicated in embryonic organ and tissue development, is found to be strongly enriched within the optical vesicle(Heavner and Pevny, 2012; Smith et al., 2009). Trpm1, observed sparsely in Figure 4F, perfectly lines around the optical vesicle. Six6, a gene known for specification and proliferation of the eye field in vertebrate embryos, is expressed within the optical vesicle as well as other regions in this tissue region(Heavner and Pevny, 2012). Pmel, a pigment cell-specific gene(Kwon et al., 1991) involved in developing fibrillar sheets, also lines perfectly around the optical vesicle. It turns out this is a single-cell layer of pigment cells or melanoblasts developed around the optical vesicle as evident in H&E and even a light field image because of the dark appearance of these cells. This result demonstrates the ability for DBiT-seq to resolve a single-cell-layer structure at high precision. Overlay of these images reveals additional features(Figure 5C). Pax6 is not only expressed within the optical vesicle but also co-expressed partially in the lining of melanoblasts and the optical nerve bundle (to the left of the optical vesicle). Six6 is located well within the optical vesicle with no overlap with the Pmel+ cell lining. We further performed GO analysis to identify major pathways and signature genes (Figure 5D). Eye development and melanin emerge as the two major categories. Visualizing all these top ranked genes (Figure S12) reveals additional fine features that have not been observed previously. Thanks to the unique advantage of our technology to precisely match pixel-by-pixel the spatial omics sequencing data to the tissue image, the spatial relationship between different genes can be examined at high resolution and high precision (Figure 5E). For example, Trpm1 perfectly overlaps with the dark circle of pigment cells. Rorb, a gene encoding retinoic acid-related orphan receptor beta, is expressed but relatively enriched at the back side, whereas Aldh1a1, a gene encoding Aldehyde Dehydrogenase 1, is strongly localized at the surface ectoderm side of the optical vesicle, suggesting their differential role in eye development. The “global” pathway analysis indicates Rorb is implicated in photoreceptor cell differentiation, which may contribute to retina development, whereas Aldh1a1, implicated in embryonic camera-type development, may be associated with early development of lens. The differential patterning of these genes at this stage have not been observed previously and this result highlights the utility of high-spatial-resolution omics atlas for the discovery of new mechanisms in embryonic organogenesis at an unprecedented resolution. Such high-resolution spatial transcriptome data allows for the selection of specific regions such as left vs right side of the optical vesicle to perform genome-wide differential gene expression analysis (Figure 5F). The result reveals the most distinct gene expressed on the right side is Aldh1a1, but a large number of genes are differentially expressed on the left side of the optical vesicle, which warrants further investigations.

Spatial expression patterns of top ranked genes identified in the eye development region (10 µm pixel).

(A) Images showing a select region of the brain analyzed by high-resolution DBiT-seq (pixel size 10μm). The upper image shows the location of this region in relation to the whole mouse embryo (E.10), which is superposed with pan-mRNA signal from sequencing. In this case, all 50×50 pixels are within the brain tissue with nearly homogeneous tissue density and thus pan-mRNA signal can barely differentiate tissue morphology. The middle panel is an optical image of the tissue slide post-DBiT, which clearly shows the pattern of pixels imprinted by microfluidic channels. The lower panel is the corresponding H&E image from an adjacent tissue section. The most obvious feature is the optical vesicle lined by a single-cell-layer of dark pigment cells (melanoblasts).
(B) Visualization of selected genes Pax6, Pmel, Trpm1, and Six6. They are all associated with early stage embryonic eye development, but mark different fine structures. Pmel and Trpm1 are expressed in a layer of melanoblast cells lining the optical vesicle. Pax6 and Six6 are mainly expressed inside the optical vesicle but also seen in other regions mapped in this data.
(C) Overlay of selected genes reveals the relative spatial relationship at ultra-high resolution. For example, Pax6 is expressed in whole optical vesicle including the single-cell layer of melanoblasts defined by Pmel and the optical nerve fiber bundle on the left. Six6 is expressed in the eye vesicle but does not overlap with the melanoblast layer even though they are in close spatial proximity to each other.
(D) Pathway analysis and differentially expressed genes. The most profound pathways identified are eye development including the associated sensory nerves and melanin.
(E) Enlarged view of Trpm1, Rorb, and Aldh1a1 spatial expression superimposed on the darkfield image of the imprinted tissue (pixel size 10μm). The expression of Trpm1 matches perfectly the location of pigment cells in the optical image. It also reveals a differential expression of Rorb and Aldh1a1. These genes appear to already show asymmetric expression within the optical vesicle before the initiation of eye cup formation; it shows a distinct and localized expression of Rorb at the anterior end, presumably associated with lens development, and the enrichment of Aldh1a1 toward the posterior side, presumably associated with the development of photoreceptor cells later on. This is in agreement with the particularly anti-correlated pathways in (D) between Rorb and Aldh1a1.
(F) Volcano plot showing whole transcriptome-wide differential gene expression between left and right side of the optical vesicle. Aldh1a1 emerges as the most distinct gene expressed at the right side while a large number of genes show up in the left side of optical vesicle.
Automated feature identification via spatial differential gene expression analysis
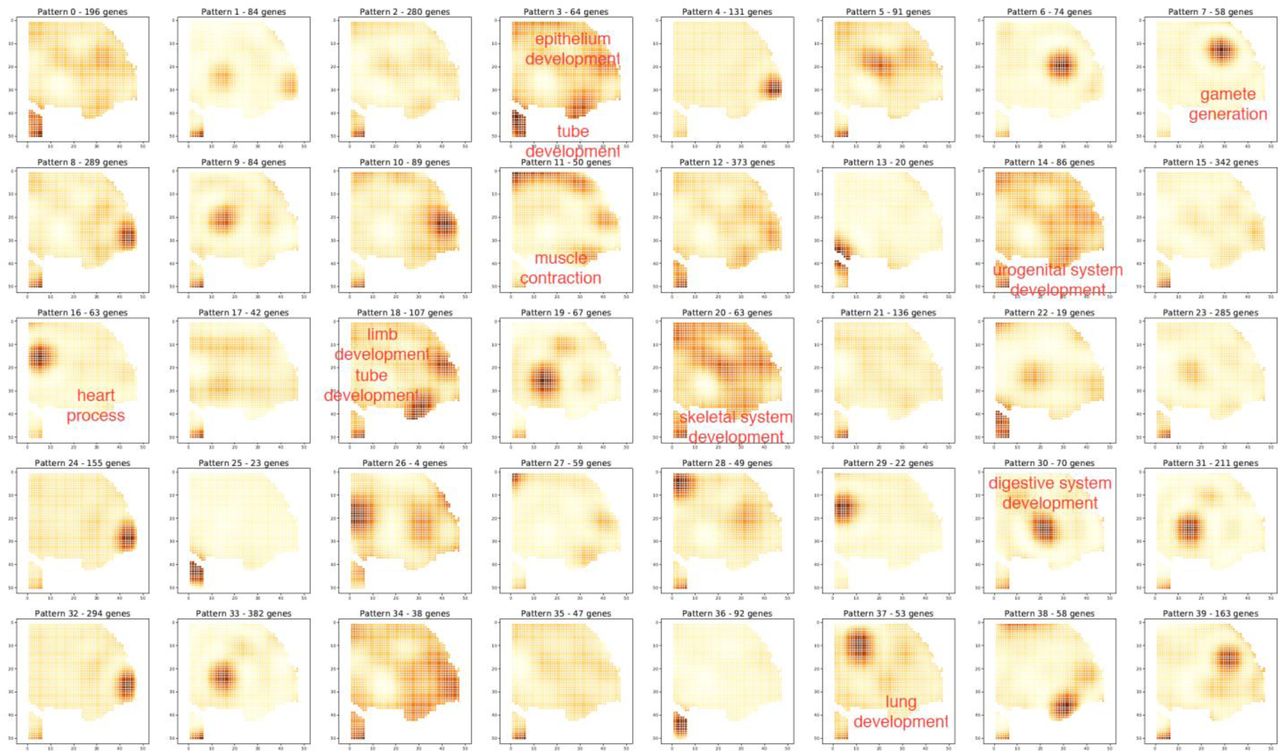
The expressions of Pax6 and Six6 are also seen in other regions of Figure 5B, suggesting the presence of additional tissue types. However, if only a small area of tissue image is available, it is technically challenging to annotate different anatomic regions. In pathological tissue specimens, the anatomical morphology may alter substantially, causing difficulty to precisely annotate small tissue regions. Therefore, an automated feature identification method based solely upon spatial omics data is useful. We applied a spatial differential expression(spatialDE) pipeline(Svensson et al., 2018a) developed for low-resolution spatial transcriptomics to our data as shown in Figure 5 and further identified 20 features (see Figure S13 and Figure 6A) including eye, ear, muscle, forebrain, and epithelium. Some of these features are almost undistinguishable using the corresponding tissue image such as ear (presumably due to too early stage in the developmental process) and forebrain (barely covered in the field of view).

Features identified in region around the eye field from the spatial transcriptome data (10 µm pixel).

(A) Major features identified in the ultra-high-spatial-resolution data described in Figure 5. It revealed not only eye but numerous other tissue subtypes. The right panel is a combined view of 5 selected features.
(B) Major features identified in the lower body of a later-stage mouse embryo (E.12), which showed a wide variety of tissue types developed. The right panel is a combined view of 5 selected features.
Many internal organs have not begun to develop in an E.10 mouse embryo. To further evaluate the ability for spatialDE to detect more distinct tissue types, an E.12 mouse embryo was analyzed using DBiT-seq. Interestingly, in only 1/3 of the whole embryo tissue section we were able to identify 40 distinct features including several internal organs such as heart, lung, urogenital system, digestive system, and male gonad (testis) (see Figure S14 and Figure 6B). Many of these features are still too early to identify based on tissue histology. We revisited the E.10 whole mouse embryo spatial transcriptome data (Figure 3) and identified ∼20 distinct features (Figure S15), which is much less than that from a fraction of the E.12 embryo, indicating that the features identified are indeed associated with the progress in development and the emergence of new organ structures. Herein we demonstrated the utility of automated feature identification using spatial DGE in high-spatial-resolution transcriptomics data and this success is also due in part to the quality of our DBiT-seq data, which can detect more than ∼1500 genes per 10μm pixel. This analysis may find applications in mechanistic discovery and pathological samples.

Feature identified in an E.12 mouse embryo tissue slide using SpatialDE.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Features identified in the whole mouse embryo (E.10) tissue slide using SpatialDE.
DISCUSSION
We developed a completely new technology for high-resolution (∼10um) spatial omics sequencing. All early attempts towards spatial transcriptomics were all based on multiplexed fluorescent in situ hybridization(Chen et al., 2015; Eng et al., 2019; Lubeck et al., 2014; Perkel, 2019). Recently, a major breakthrough in the field arises from the use of high throughput NGS sequencing to reconstruct spatial transcriptome maps(Rodriques et al., 2019; Stahl et al., 2016), which is unbiased, genome-wide, and presumably easier to adopt by a wider range of biological and biomedical research community. The core mechanism of these NGS-based methods to achieve spatial transcriptomics is through a method called “barcoded solid-phase RNA capture”(Trcek et al., 2017), which uses a DNA barcode spot array such as ST seq (Stahl et al., 2016) or a barcoded bead array such as Slide-seq (Rodriques et al., 2019) to capture mRNAs from a freshly sectioned tissue slice placed on top and lysed to release mRNAs. These approaches are still technically demanding, requiring a lengthy and sophisticated step to decode the beads, while the mRNA capture efficiency and the number of dateable genes per pixel at the 10μm size level is markedly below optimal. Additionally, it is not obvious how they can be extended for other omics measurements. Herein, spatial DBiT-seq is a fundamentally different approach. It does not need to lyse tissue to release mRNAs and is compatible with existing formaldehyde-fixed tissue slides. It is highly versatile and easy to operate. It requires only a simple microchannel device and a set of reagents. It does not need to conduct sophisticated sequential hybridization or SOLiD sequencing to decode beads before experiments. This standalone device is highly intuitive to use with no need for any microfluidic handling system and thus can be readily adopted by biologists who have no microfluidics training.
With this technology, we conducted the spatial multi-omics atlas (proteins and mRNAs) sequencing of whole mouse embryos and generated numerous new insights. Major tissue types in a mouse embryo could be identified during early organogenesis stages. Spatial protein and gene expression atlas revealed a differential pattern in embryonic forebrain defined by MAdCAM1 expression. Reconstructed spatial protein expression map can readily resolve brain microvascular networks, which are barely distinguishable in tissue histology images. We further demonstrated the ability for DBiT-seq to resolve a single-cell layer of melanocytes lining around the optical vesicle and discovered an asymmetric gene expression pattern between Rorb and Aldh1a1 within the optical vesicle that may contribute to the subsequent development of retina and lens, respectively. DBiT-seq demonstrated not only high spatial resolution but also high quality of sequencing data with a much higher genome coverage and a greater number of genes detected per 10μm pixel when compared to Slide-seq. This improvement enabled us to visualize the spatial expression of individual genes whereas the Slide-seq data are too sparse to query individual genes in a meaningful way.
Thanks to the versatility of our technology, we can readily combine multiple omics on the same pixel. As demonstrated in this work, we simultaneously measured whole mRNA transcriptome and a panel of 22 protein markers, allowing for comparing individual proteins and mRNAs for their spatial expression patterns. We demonstrated the use of high-quality spatial protein expression data to guide the tissue region-specific transcriptome analysis for differential gene expression and pathway analyses, leading to a new approach for mechanistic discovery that one type of omics data cannot readily provide. Moreover, DBiT has the capability to become a universal sample preparation step to enable high-spatial-resolution mapping of many other molecular information. For example, it can be applied to barcoding DNA sequences for high-spatial-resolution Assay for Transposase-Accessible Chromatin (ATAC)(Chen et al., 2016) and potentially for detecting chromatin modifications via in-tissue Cut-Run(Skene and Henikoff, 2017) followed by DBiT.
This spatial barcoding approach is not limited to tissue specimens but also applicable to single cells dispensed on a substrate to perform deterministic barcoding for massively parallel transcriptome, proteome, or epigenome sequencing. In this way, a variety of cellular assays such as cell migration, morphology, signal transduction, drug responses, etc. can be done before hand and linked to the omics data, enabling direct correlation of single-cell omics to live cell functions in every single cell. This may further address a long-standing problem in the field of single-cell RNA sequencing – the unavoidable perturbation of cellular states including protein and mRNA expression during trypsinization and single-cell suspension preparation.
Like any other emerging technologies, DBiT-seq has limitations. First, although it is close to single-cell level spatial mapping, it does not directly resolve single cells. However, due to the unique capability of DBiT-seq to obtain precisely matched tissue image from the same tissue slide, we believe molecular imaging such as IHC or FISH can be performed to outline the boundaries of individual cells, which could be used to identify the number and the type of cells in each pixel. A large database of IHC or FISH on a similar type of tissue can be used to train a machine learning (ML) neural network to predict the spatial expression in individual cells based on tissue histology. Then, the trained neural network can be applied to DBiT-seq data and matched histology images to computationally reconstruct single-cell spatial gene or protein expression atlas. Second, there is a theoretical resolution limit. Based on our validation data, this limit is ∼2μm, which is challenging to do using microfluidic DBiT. However, we are optimistic to push it down to ∼5μm, in which most pixels contain 1 or less than one cell. Third, current DBiT-seq approach relies on a 50×50 orthogonal barcoding array, which yields a ∼1×1mm2 mappable area at the 10μm pixel size. But this can be readily expanded to a larger area by increasing the number of barcode reagents to 100×100 or even 200×200. Third, current DBiT device requires the tissue section being placed relatively in the center of the slide (in a 10mmx10mm region). Many banked tissue slides contain tissue sections on different locations of the slide. To solve this problem, a microfluidic device with a large-sized reagent delivery handle chip bond onto a small flow barcoding chip can be fabricated such that the footprint required to attach the microfluidic flow barcoding region to the slide is small and can be aligned to the tissue section anywhere on the slide.
In summary, we report on an enabling and versatile technology called microfluidic deterministic barcoding in tissue for omics sequencing (DBiT-seq) to perform high-resolution spatial barcoding for simultaneous measurement of mRNA transcriptome and a panel of proteins on a fixed tissue slide at high spatial resolution (10μm), in an unbiased manner, and at the genome scale. DBiT-seq is a fundamentally different approach for spatial omics and has the potential to become a universal method for mapping a range of molecular information (proteins, transcriptome, and epigenome). The potential impact is broad and far-reaching in many different fields of basic and translational research including embryology, neuroscience, cancer and clinical pathology.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCE TABLE
LEAD CONTACT AND MATERIALS AVAILABILITY
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
METHOD DETAILS
Microfluidic device fabrication and assembly
DNA barcodes and other key reagents
Tissue procurement and handling
Tissue slides, fixation, and storage
Tissue histology and H&E staining
Immunofluorescence staining
Application of DNA-antibody conjugates to the tissue slide
Adding the first set of barcodes and reverse transcription
Adding the second set of barcodes and ligation
cDNA collection and purification
Template switch and PCR amplification
Amplicon purification, sequencing library preparation and quality assessment
QUANTIFICATION AND STATISTICAL ANALYSIS
Sequence alignment and generation of gene expression matrix
Clustering and spatial differential gene expression analysis
DATA AND CODE AVAILABILITY
SUPPLEMENTAL INFORMATION
Supplementary Information can be found online at [to be inserted, SI is also provided as part of the manuscript submission].
AUTHOR CONTRIBUTIONS
Conceptualization: R.F.; Methodology, Y.L., M.Y., Y.D., and G.S.; Experimental investigation, Y.L., Y.D., G.S., C.C.G., D.K., Z.B., Y.X., and D.Z.; Data Analysis, M.Y., Y.L., Y.D. and R.F.; Resources, D.K., Z.B., and Y.X.; Writing – Original Draft, R.F., Y.L., and M.Y.; Writing – Review and Editing, R.F., Y.L., M.Y., G.S., and C.C.G.
DECLARATION OF INTEREST
R.F., Y.L. and Y.D. are inventors of and have submitted a patent on the deterministic spatial barcoding technology. R.F. is co-founder and scientific advisor of, and hold equity in, IsoPlexis and Singleron Biotechnologies. These companies have no relationship with this technology. The interests of R.F. were reviewed and are managed by Yale University Provost’s Office in accordance with the University’s conflict of interest policies.
STAR METHODS
KEY RESOURCES TABLE

LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents may be directed to the corresponding author Rong Fan (rong.fan{at}yale.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
Mouse: C57BL/6NCrl (Charles River Laboratories)
This mouse is purchased from Charles River Laboratories, and the embryo was collected the day the mouse was received. For detailed information about mouse maintenance and care, refer to the manufacturer’s information sheet.
METHOD DETAILS
Microfluidic device fabrication and assembly
The microfluidic device was fabricated with polydimethylsiloxane(PDMS) using soft lithography. The chrome photomasks with 10 µm resolution were ordered from front range photomasks (Lake Havasu City, AZ). The molds were fabricated using SU-8 negative photoresist according to the following microfabrication process. A thin layer of SU-8 resist (SU-8 2010, SU-8 2025 and SU-8 2050, Microchem) was spin-coated on a clean silicon wafer following manufacturer’s guidelines. The thickness of the resistant was ∼50 µm for the 50-µm-wide microfluidic channel device, ∼28 µm for 25-µm-wide device, and ∼20 µm for 10-µm-wide device. A protocol to perform SU-8 photo lithography, development, and hard baking was followed based on the manufacturer (MicroChem)’s recommendations to yield the silicon molds for PDMS replication.
PDMS microfluidic chips were then fabricated via a replication molding process. The PDMS precursor was prepared by combining GE RTV PDMS part A and part B at a 10:1 ratio. After stir mixing, degassing, this mixture was poured to the mold described above, degassed again for 30min, and cured at 75 °C for ∼2 hours or overnight. The solidified PDMS slab was cut out, peeled off, and the inlet and outlet holes punched to complete the fabrication. The inlet holes were ∼2 mm in diameter, which can hold up to 5 µL of solution. A pair of microfluidic chips with the same location of inlets and outlets but orthogonal microfluidic channels in the center were fabricated as a complete set of devices for flow barcoding a tissue slide. To do that, the PDMS slab was attached to the tissue section glass slides and a custom-designed acrylic clamp was used to firmly hold the PDMS against the tissue specimen to prevent leakage across microfluidic channels without the need for harsh bonding processed such as thermal bonding or plasma bonding(Temiz et al., 2015).
DNA barcodes and other key reagents
Oligos used were listed in Table S1 Antibody-Oligo sequences and Table S2 DNA oligos and DNA barcodes. All other key reagents used were listed as Table S3.
Tissue Handling
Formaldehyde fixed tissue or frozen tissue slides were obtained from a commercial source Zyagen (San Diego, CA). The protocol Zyagen used to prepare the embryonic tissue slides is the following. The pregnant mice (C57BL/6NCrl) were bred and maintained by Charles River Laboratories. More information can be found in the information sheet. The time-pregnant mice (day 10 or day 12) were shipped to Zyagen (San Diego, CA) the same day. The mice were sacrificed at the day of arrival for embryos collection. The embryo sagittal frozen sections were prepared by Zyagen (San Diego, CA) as following: the freshly dissected embryos were immersed into OCT and snapped frozen with liquid nitrogen. Before sectioning, the frozen tissue block was warmed to the temperature of cryotome cryostat (−20°C). Tissue block was then sectioned into thickness of <7 µm and placed in the center of a poly-L-lysine coated glass slide (CatLog no. 63478-AS, electron microscopy sciences). The frozen slides were then fixed with 4% formaldehyde or directly kept at −80 °C if a long-time storage is needed.
Tissue slides and fixation
To thaw the tissue slides, they were taken out of the freezer, placed on a bench at room temperature for 20 minutes, and then cleaned with 1X phosphate buffer saline (PBS) supplemented with RNase inhibitor (0.05U/μL, Enzymatics). If the tissue slides were frozen sections, they were first fixed by immersing in 4% formaldehyde (Sigma) for 20 minutes. Afterwards, the tissue slides were dried with forced nitrogen air and then ready to use for spatial barcoding.
Tissue histology and H&E staining
An adjacent tissue section was also requested from the same commercial resource which could be used to perform tissue histology examination using H&E staining. Basically, the fixed tissue slide was first cleaned by DI water, and the nuclei were stained with the alum hematoxylin (Sigma) for 2 minutes. Afterwards, the slides were cleaned in DI water again and incubated in a bluing reagent resolution (0.3% acid alcohol, Sigma) for 45 seconds at room temperature. Finally, the slides were stained with eosin for 2 more minutes. The stained embryo slide was examined immediately or stored at −80 °C fridge for future analysis.
Immunofluorescence staining
Immunofluorescence staining was performed either on the same tissue slide or an adjacent slide to yield validation data. Three fluorescent-labelled antibodies listed below were used for visualizing the expression of three target proteins: Alexa Fluor® 647 anti-mouse CD326 (Ep-CAM) Antibody, Alexa Fluor® 488 anti-mouse Panendothelial Cell Antigen Antibody, PE anti-P2RY12 Antibody. The procedure to stain the mouse embryo tissue slide is as follows. (1) Fix the fresh frozen tissue sections with 4% Formaldehyde for 10 mins, wash three times with PBS. (2) Add 1% bovine serum albumin (BSA) in PBS to block the tissue and incubate for 30 mins at RT. (3) Wash the tissue with PBS for three times. (4) Add the mixture of three antibodies (final concentration 25 µg/mL in 1% BSA, PBS) to the tissue, need around 50 µL. Incubate for 1 hour in dark at RT. (5) Wash the tissue with PBS for three times, with 5 mins washing each time. (6) Dip the tissue in water shortly and air dry the tissue. (7) Imaging the tissue using EVOS (Thermo Fisher EVOS fl), with a resolution of 10 x. Filters used are Cy5, RFP and GFP.
Application of DNA-antibody conjugates to the tissue slide
In order to obtain spatial proteomic information, we incubate the fixed tissue slide with a cocktail of DNA-antibody conjugates prior to microfluidic spatial barcoding. The cocktail was prepared by combining 0.1 µg of each DNA-antibody conjugates (see Key Resource Table). The tissue slide was first blocked with 1% BSA/PBS plus RNase inhibitor, and then incubated with the cocktail for 30 minutes at 4°C. Afterwards, the tissue slide was washed 3 times with a washing buffer containing 1% BSA + 0.01% Tween 20 in 1X PBS and one time with DI water prior to attaching the first PDMS microfluidic chip.
Adding the first set of barcodes and reverse transcription
To perform spatial barcoding of mRNAs for transcriptomic mapping, the slides were blocked by 1% BSA plus RNase inhibitor (0.05U/μL, Enzymatics) for 30 minutes at room temperature. After cleaning with 1x PBS and quickly with DI water, the first PDMS microfluidic chip was roughly aligned and placed on the tissue glass slide such that the center of the flow barcoding region covered the tissue of interest. This tissue section was then permeabilized by loading 0.5% Trition X-100 in PBS into each of the 50 channels followed by incubation for 10 minutes and finally were cleaned thoroughly by flowing through 20μL of 1X PBS. A vial of RT mix was made of 50 μL of RT buffer (5X, Maxima H Minus kit), 32.8 µL of RNase free water, 1.6 µL of RNase Inhibitor (Enzymatics), 3.1 µL of SuperaseIn RNase Inhibitor (Ambion), 12.5 µL of dNTPs (10 mM, Thermo Fisher), 25 µL of Reverse Transcriptase (Thermo Fisher), 100 µL of 0.5X PBS with Inhibitor (0.05U/μL, Enzymatics). To perform the 1st microfluidic flow barcoding, we added to each inset 5 µL of solution containing 4.5 µL of the RT mix described and 0.5 µL of one of the 50 DNA barcodes (A1-A50) solution (25 µM), and then pulled in using a house vacuum for 5-10 minutes depending on channel width. Afterwards, the binding of DNA oligomers to mRNAs fixed in tissue was allowed to occur at room temperature for 30 minutes and then incubated at 42 °C for 1.5 hours for in situ reverse transcription. To prevent the evaporation of solution inside the channels, the whole device was kept inside a sealed wet chamber(Gervais and Delamarche, 2009). Finally, the channels were rinsed by flowing NEB buffer 3.1(1X, New England Biolabs) supplemented with 1% RNase inhibitor (Enzymatics) continuously for 10 minutes. During the flow barcoding step, optical images could be taken to record the exact positions of these microfluidic channels in relation to the tissue section subjected to spatial barcoding. It was done using an EVOS microscope (Thermo Fisher EVOS fl) in a light or dark field mode. Then the clamp was removed and the PDMS chip was detached from the tissue slide, which was subsequently dipped into a 50 mL Eppendorf tube containing RNase free water to rinse off remaining salts.
Adding the second set of barcodes and ligation
After drying the tissue slides, the second PDMS chip with the microfluidic channels perpendicular to the direction of the first PDMS chip in the tissue barcoding region was carefully aligned and attached to the tissue slide such that the microfluidic channels cover the tissue region of interest. The ligation mix was prepared as the following. It contains 69.5 µL of RNase free water, 27 µL of T4 DNA ligase buffer (10X, New England Biolabs), 11 µL T4 DNA ligase (400 U/µL, New England Biolabs), 2.2 µL RNase inhibitor (40 U/µL, Enzymatics), 0.7 µL SuperaseIn RNase Inhibitor (20 U/µL, Ambion), 5.4 µL of Triton X-100 (5%). To perform the second flow barcoding, we added to each channel a total of 5 µL of solution consisting of 2 µL of the aforementioned ligation mix, 2 µL of NEB buffer 3.1(1X, New England Biolabs) and 1 µL of DNA barcode B (25 µM). Reaction was allowed to occur at 37 °C for 30 minutes and then the microfluidic channels were washed by flowing 1X PBS supplemented with 0.1% Triton X-100 and 0.25% SUPERase In RNase Inhibitor for 10 minutes. Again, the images showing the location of the microfluidic channels on the tissue slide could be taken during the flow step under the light or dark field optical microscope (Thermo Fisher EVOS fl) before peeling off the second PDMS chip.
cDNA collection and purification
We devised a square well PDMS gasket, which could be aligned and placed on the tissue slide, creating an open reservoir to load lysis buffer specifically to the flow barcoded tissue region to collect cDNAs of interest. Depending on the area of this region, the typical amount of buffer is 10 - 100 µL of Proteinase K lysis solution, which contains 2 mg/mL proteinase K (Thermo Fisher), 10 mM Tris (pH = 8.0), 200 mM NaCl, 50 mM EDTA and 2% SDS. Lysis was carried out at 55 °C for 2 hours. The lysate was then collected and stored at −80 °C prior to use. The cDNAs in the lysate were purified using streptavidin beads (Dynabeads MyOne Streptavidin C1 beads, Thermo Fisher). The beads (40 µL) were first washed three times with 1X B&W buffer (Ref to manufacturer’s manual) with 0.05% Tween-20, and then stored in 100 µL of 2X B&W buffer (with 2 μL of SUPERase In Rnase Inhibitor). To perform purification from stored tissue lysate, it was allowed to thaw, and the volume was brought up to 100 µL by RNase free water. Then, 5 µL of PMSF (100 µM, Sigma) was added to the lysate and incubated for 10 minutes at room temperature to inhibit the activity of Proteinase K. Next, 100 µL of the cleaned streptavidin bead suspension was added to the lysate and incubated for 60 minutes with gentle rotating. The beads with cDNA were further cleaned with 1X B&W buffer for two times and then with 1X Tris buffer (with 0.1% Tween-20) once.
Template switch and PCR amplification
The cDNAs bound to beads were cleaned and resuspended into the template switch solution. The template switch reaction mix contains 44 μL of 5X Maxima RT buffer (Thermo Fisher), 44 μL of 20% Ficoll PM-400 solution (Sigma), 22 μL of 10 mM dNTPs each (Thermo Fisher), 5.5 μL of RNase Inhibitor (Enzymatics), 11 μL of Maxima H Minus Reverse Transcriptase (Thermo Fisher), and 5.5 μL of a template switch primer (100 μM). The reaction was conducted at room temperature for 30 minutes followed by an additional incubation at 42 °C for 90 minutes. The beads were rinsed once with a buffer containing 10 mM Tris and 0.1% Tween-20 and then rinsed again with RNase free water using a magnetic separation process. PCR was conducted following these two steps. In the first step, a mixture of 110 µL Kapa HiFi HotStart Master Mix (Kapa Biosystems), 8.8 μL of 10 μM stocks of primers 1 and 2, and 92.4 μL of water was added to the cleaned beads. If the protein detection was conducted in conjunction using a process similar to CITE-seq, a primer 3 solution (1.1 μL, 10 μM) was also added at this step. PCR reaction was then done using the following conditions: first incubate at 95°C for 3 mins, then cycle five times at 98°C for 20 seconds, 65°C for 45 seconds, 72°C for 3 minutes and then the beads were removed from the solution by magnet. Evagreen (20X, Biotium) was added to the supernatant with 1:20 ratio, and a vial of the resultant solution was loaded into a qPCR machine (BioRad) to perform a second PCR step with an initial incubation at 95°C for 3 minutes, then cycled at 98°C for 20 seconds, 65°C for 20 seconds, and finally 72°C for 3 minutes. The reaction was stopped when the fluorescence signal just reached the plateau.
Amplicon purification, sequencing library preparation and quality assessment
The PCR product was then purified by Ampure XP beads (Beckman Coulter) at 0.6×ratio. The mRNA-derived cDNAs (>300 bp) were then collected from the beads. If the cDNAs were less than 300 bp, they remained in in the supernatant fraction. If the protein detection was conducted similar to CITE-seq, this faction(fraction?) was actually used instead. For sequencing antibody-DNA conjugate-derived cDNAs, we further purified the supernatant using 2X Ampure XP beads. The purified cDNA was then amplified using a PCR reaction mix containing 45 µL purified cDNA fraction, 50 µL 2x KAPA Hifi PCR Master Mix(Kapa Biosystems), 2.5 µl P7 primer of 10 µM and 2.5 µL P5 cite primer at 10 µM. PCR was performed in the following conditions: first incubated at 95°C for 3 minutes, then cycled at 95°C for 20 seconds, 60°C for 30 seconds and 72 °C for 20 seconds, for 10 cycles, lastly 72 °C for 5 minutes. The PCR product was further purified by 1.6X Ampure XP beads. For sequencing mRNA-derived cDNAs, the quality of amplicon was analyzed firstly using Qubit (Life Technologies) and then using an Agilent Bioanalyzer High Sensitivity Chip. The sequencing library was then built with a Nextera XT kit (Illumina) and sequenced using a HiSeq 4000 sequencer using a pair-end 100×100 mode. To conduct joint profiling of proteins and mRNAs, the DNA-antibody conjugate-derived sequencing library was combined with mRNA-derived cDNA library at a 1:9 ratio, which is sufficient to detect the finite set of proteins and minimally affects the sequencing depth required for mRNAs.
QUANTIFICATION AND STATISTICAL ANALYSIS
Sequence alignment and generation of gene expression matrix
For obtaining transcriptomics data, the Read 2 was processed by extracting the UMI, Barcode A and Barcode B. The processed read was trimmed, mapped against the mouse genome(GRCh38), demultiplexed and annotated(Gencode release M11) using the ST pipeline v1.7.2 (Navarro et al., 2017), which generated the gene expression matrix for down-stream analysis. Spatially variable genes were identified by SpatialDE(Svensson et al., 2018b). The resulting DGE list was submitted to ToppGene(Chen et al., 2009) for GO and Pathway analysis.
For obtaining proteomics data, the Read 2 was processed by extracting the antibody-derived barcode, spatial Barcode A and Barcode B. The processed read was trimmed, demultiplexed using the ST pipeline v1.7.2 (Navarro et al., 2017), which generated the gene protein matrix for down-stream analysis.
Clustering and spatial differential expression analysis
Spatially variable genes generated by SpatialDE was used to conduct the clustering analysis, Non-negative matrix factorization(NMF) was performed using the NNLM pacakges in R, after the raw values were log-transformed, we chose k of 11 for the mouse embryo DBiT-seq transcriptome data obtained at a 50μm pixel size. For each pixel, the largest factor loading from NMF was used to assign cluster membership. NMF clustering of pixels was plotted by tSNE using the package “Rtsne” in R.
DATA AND CODE AVAILABILITY
Data availability
The accession number for the sequencing data reported in this paper is GEO: GSExxxx (to be inserted).
Code availability
Code for sequencing data analysis is available (to be inserted).
ACKNOWLEDGEMENTS
We thank Drs. Haifan Lin, Andre Levchenko, Dianqing Wu, Jun Lu, and Stephanie Halene for helpful discussions. R.F. dedicates this work to his senior colleagues/mentors Drs. W. Mark Saltzman and Jay Humphrey on the occasions of their 60th birthday celebration. This research was supported by Packard Fellowship for Science and Engineering (to R.F.), Stand-Up-to-Cancer (SU2C) Convergence 2.0 Award (to R.F.), and Yale Stem Cell Center Chen Innovation Award (to R.F.). Y.L. was supported by the Society for ImmunoTherapy of Cancer (SITC) Postdoctoral Fellowship. The molds for microfluidic chips were fabricated at the Yale University School of Engineering and Applied Science (SEAS) Nanofabrication Center. We used the service provided by the Genomics Core of Yale Cooperative Center of Excellence in Hematology (U54DK106857). This sequencing service was conducted at Yale Stem Cell Center Genomics Core Facility which was supported by the Connecticut Regenerative Medicine Research Fund and the Li Ka Shing Foundation. It was also conducted using the sequencing facility at the Yale Center for Genomic Analysis (YCGA).
Footnotes
↵4 Lead contact
Methods Section References
REFERENCES




