

Abstract
Human guanylate binding proteins (GBPs), a family of IFNγ-inducible GTPases, promote cell-intrinsic defence against pathogens and host cell death. We previously identified GBP1 as a mediator of cell death of human macrophages infected with Toxoplasma gondii (Tg) or Salmonella Typhimurium (STm). How GBP1 targets microbes for AIM2 activation during Tg infection and caspase-4 during STm infection remains unclear. Here, using correlative light and electron microscopy and EdU labelling of Tg-DNA, we reveal that GBP1-decorated parasitophorous vacuoles (PVs) lose membrane integrity and release Tg-DNA for detection by AIM2-ASC-CASP8. In contrast, differential staining of cytosolic and vacuolar STm revealed that GBP1 does not contribute to STm escape into the cytosol but decorates almost all cytosolic STm leading to the recruitment of caspase-4. Caspase-5, which can bind LPS and whose expression is upregulated by IFNγ, does not target STm pointing to a key role for caspase-4 in pyroptosis. We also uncover a regulatory mechanism involving the inactivation of GBP1 by its cleavage at Asp192 by caspase-1. Cells expressing non-cleavable GBP1D192E therefore undergo higher caspase-4-driven pyroptosis during STm infection. Taken together, our comparative studies elucidate microbe-specific spatiotemporal roles of GBP1 in inducing cell death by leading to assembly and regulation of divergent caspase signalling platforms.
INTRODUCTION
Most nucleated cells have the ability to defend themselves against infection by viruses, bacteria and eukaryotic parasites in a process called cell-intrinsic immunity. The execution of this process requires the detection of pathogens by membrane-bound and cytosolic sensors and receptors, several of which can also trigger cell death (MacMicking, 2012; Randow et al, 2013; Mostowy & Shenoy, 2015; Jorgensen et al, 2017).The type II interferon, IFNγ, induces expression of up to 2000 interferon-stimulated genes by signalling via the interferon-gamma receptor signalling cascade (MacMicking, 2012). Several newly expressed proteins are involved in killing of or restricting the replication of intracellular pathogens or in inducing host cell death. The guanylate binding protein (GBP) family of cytosolic GTPases are among the most highly expressed proteins in macrophages exposed to IFNγ. The GBP protein family consists of seven members in the human and is involved in defence against bacterial, parasitic and viral intracellular pathogens, regulation of inflammasome signalling and cell death (Shenoy et al, 2012; Man et al, 2017; Santos & Broz, 2018; Gomes et al, 2019). All seven human GBPs possess an N-terminal, globular GTPase domain and a C-terminal helical domain which facilitates membrane interaction. Furthermore, GBP1, GBP2 and GBP5 are isoprenylated at their C-terminal CaaX-box, which can anchor them to membranes. GTPase activity mediates GBP conformational changes and hetero- and homo-oligomerization (Praefcke, 2018). However, recent studies have pointed towards pathogen-specific effects of GBPs and their roles in differentially triggering cell death needs to be better understood (Fisch et al, 2019a).
Starting with a family-wide siRNA screen, we showed that GBP1 is essential in macrophage cell death in response to infection by Toxoplasma gondii (Tg), an apicomplexan parasite, and Salmonella enterica subsp. enterica serovar Typhimurium (STm), a Gram-negative bacterium. GBP1 targeting of both pathogens is necessary for cell death. GBP1 localisation to these divergent pathogens leads to the exposure of pathogen-associated molecular patterns (PAMP), namely Tg-DNA and STm-LPS, which are detected by their cytosolic receptors AIM2 and caspase-4 respectively. As Tg induces the loss of inflammasome proteins, including NLRP3 and caspase-1, human macrophages undergo atypical apoptosis through the assembly of AIM2-ASC-caspase-8 complexes. On the other hand, GBP1 promotes caspase-4 activation and enhances pyroptosis during STm infection (Fisch et al, 2019a). Human macrophages can detect pathogen DNA through either AIM2 or through a newly described NLRP3-mediated pathway in response to pathogens such as Francisella novicida and Modified vaccinia Ankara (Gaidt et al, 2016). Whether human AIM2 is specifically activated through the detection of Tg-DNA remains to be experimentally demonstrated. Similarly, whether caspase-4 is activated by vacuolar or cytosolic STm targeting by GBP1 is not known. As GBP1 targeting of different pathogens elicits divergent host cell death programs, we sought to investigate the mechanistic details of GBP1 during infection by these two unrelated human pathogens. Additionally, GBP1 contains a previously reported caspse-1 cleavage site (Naschberger et al, 2017), thus leading us to hypothesise that GBP1 not only differentially recognises dissimilar microbes to liberate PAMPs, but also provides a functional platform to feedback on the level of caspase activation.
In the current study, we used correlative light and electron microscopy and super-resolution microscopy to delineate the vacuolar compartments targeted by GBP1 and describe how GBP1 promotes the assembly of supramolecular organizing centres (SMOCs) as signalling platforms. Furthermore, we show how cleavage of GBP1 by caspase-1 functions as a negative feedback to balance cell death levels. Overall, we show that GBP1 has unique roles during infection by Tg or STm and thereby provide new insights on its role in host defence.
RESULTS
GBP1 directly contributes to Toxoplasma vacuole opening and infection control
GBP1 is recruited to Tg parasitophorous vacuoles (PV) which results in AIM2 and caspase-8 activation (Fisch et al, 2019a). Like some murine (Yamamoto et al, 2012; Selleck et al, 2013; Degrandi et al, 2013; Kravets et al, 2016), we hypothesized that human GBP1 may promote vacuole opening and cytosolic access to intravacuolar pathogens. To test this, we designed a new assay to quantify Tg vacuole opening. The cytosolic dye CellMask is excluded from PVs but enters the PV once the PV membrane (PVM) is disrupted by detergent-mediated permeabilization (Figure S1A). Increased dye intensity within Tg vacuoles could be reliably quantified using our artificial intelligence-based high-throughput image analysis workflow HRMAn (Fisch et al, 2019b) (Figure 1A and Figure S1B). These analyses revealed an increase in CellMask staining of PVs in IFNγ-primed THP-1 WT cells, indicating their disruption (Figure S1B). We next asked whether GBP1 contributed to Tg vacuole leakiness. Similar assays in IFNγ-primed THP-1 ΔGBP1 cells showed that Tg vacuoles were not disrupted, as seen by the exclusion of CellMask dye (Figure S1B). Doxycycline induced re-expression of GBP1 (THP-1 ΔGBP1+Tet-GBP1 cells) rescued vacuole breakage (Figure S1B); as control, empty vector transduced cells (THP-1 ΔGBP1+Tet-EV) behaved like ΔGBP1 cells (Figure S1B). We used doxycycline-induced expression of mCherry-GBP1 (Fisch et al, 2019a) (THP-1 ΔGBP1+Tet-mCH-GBP1 cells) to allow quantification of GBP1-recruitment to Tg and stratify data on whether PVs that were decorated with mCH-GBP1 lost their integrity. Indeed, a population of GBP1+ PVs were unable to exclude CellMask dye clearly indicating loss of membrane integrity (Figure 1A). Taken together, we concluded that GBP1 is necessary for opening of Tg PVs and GBP1-targeted vacuoles preferentially undergo loss of membrane integrity.
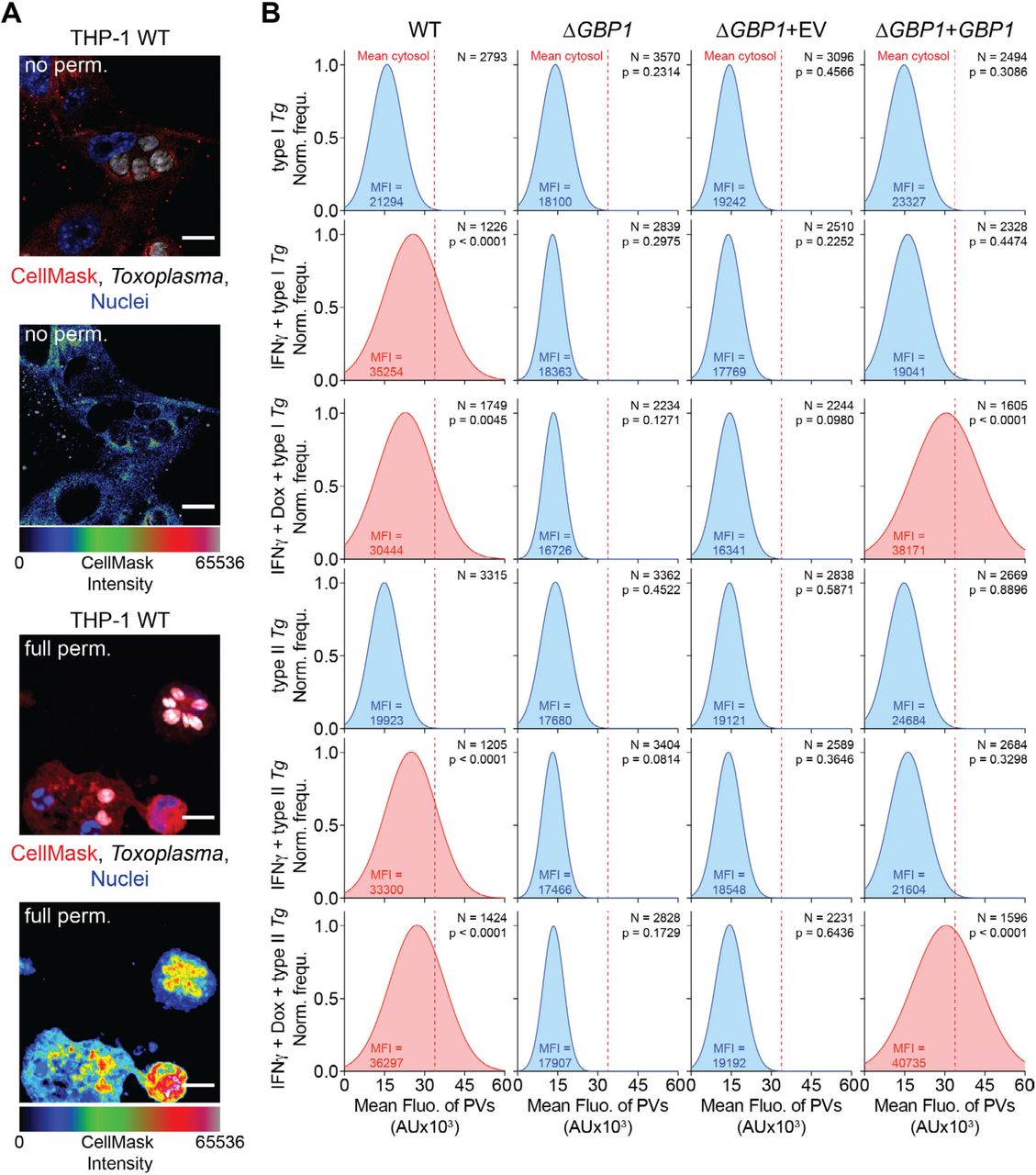
(A) Representative immunofluorescence images from THP-1 WT infected with type I Toxoplasma gondii (Tg) for 18 hours and stained with CellMask but not permeabilized (no perm.; Top) fully permeabilized with saponin (full perm.; Bottom). Rainbow intensity diagram of the CellMask signal from images used for quantification. Red: CellMask; Grey: Tg; Blue: Nuclei. Scale bars 10 µm. (B) Representative normalised frequency plots (Norm. frequ.) of fluorescence intensities of vacuoles in type I or type II Tg-infected THP-1 WT, THP-1 ΔGBP1, THP-1 ΔGBP1+Tet-empty vector (EV) or THP-1 ΔGBP1+Tet-GBP1 cells treated with IFNγ, Doxycycline (Dox) or left untreated and stained with CellMask. Mean fluorescence signal of the cytosol indicated by dashed red line and mean fluorescence intensity of the vacuoles (MFI) displayed in the graphs in Fig 1A. Conditions with increased mean intensity of the vacuole fluorescence are highlighted in red. N = number of vacuoles. Data information: P values in (B) from nested one-way ANOVA comparing means of n = 3 independent experiments from indicated condition to untreated WT cells.

(A) Representative quantification of CellMask fluorescence intensities within vacuoles (PV) of type I or type II Toxoplasma gondii (Tg) infected THP-1 ΔGBP1+Tet-mCH-GBP1 pre-treated with IFNγ and Doxycycline (Dox) to induce GBP1 expression. Plotted depending on whether PVs were decorated with GBP1 (GBP1+) or not (GBP1-). Mean fluorescence signal of CellMask dye in the cytosol is indicated by dashed red line. N = number of vacuoles. (B) Left: Representative immunofluorescence image of type I Tg grown in human foreskin fibroblasts (HFF WT) in the presence of EdU DNA label. Labelled type I Tg were harvested from the HFFs and used to infect (Inf.) THP-1 ΔGBP1+Tet-mCH-GBP1 for 6 hours. THP-1 cells were pre-treated with IFNγ and Dox to induce mCH-GBP1 expression (Right). Parasite DNA released into the cytoplasm was visualised by click-chemistry to label the incorporated EdU. Red: mCH-GBP1; White: EdU-DNA; Green: Tg; Blue: Nuclei. Cells are outlined by the white, dashed line and released Tg-DNA marked by yellow arrowheads. Scale bar 10 µm. (C) Representative images of correlative light and electron microscopy of THP-1 WT or ΔGBP1 cells (flooded with CellMask for fluorescence imaging), pre-treated with IFNγ to induce GBP1 expression and infected with type I (RH) Tg for 6 hours. Parasites indicated within coloured boxes are shown at higher magnifications as indicated (DIC, differential interference contrast; TEM, transmission electron micrograph). Red: CellMask; Green: Tg; Blue: Nuclei. Scale bars as indicated. (D) Representative immunofluorescence images for the endogenous phagosome marker Rab7 in IFNγ-primed THP-1 WT cells infected with type I or type II Tg for 2 hours. Red: Rab7; Grey: Tg; Blue: Nuclei. Scale bar 15 µm. (E) Quantification of Rab7 and GBP1 recruitment to type I and type II Tg vacuoles and Tg-containing phagosomes (Phago.) in IFNγ and Dox treated THP-1 ΔGBP1+Tet-mCH-GBP1. Data are plotted as proportion of GBP1+ parasites at indicated time post-infection. Data information: Graphs in (A+E) from n = 3 independent experiments. P values in (A) from nested t-test comparing GBP1+ to GBP1- PVs following adjustment for multiple comparisons. ** P ≤ 0.01 or *** P ≤ 0.001 in (A) for indicated comparisons of the means of the n = 3 independent experiments.
As Tg infection activates AIM2, we wanted to visualise this process using single-cell assays. To this end, we labelled Tg-DNA with EdU by growing them in human foreskin fibroblasts (HFFs), whose DNA remains unlabelled as they do not replicate due to contact-dependent growth inhibition. Following infection of macrophages with EdU-labelled Tg, we visualised Tg-DNA with Alexa-647 fluorophore using click-chemistry and quantified macrophages containing cytosolic Tg-DNA (Figure 1B). Roughly 35% of infected macrophages contained Tg-DNA in their cytosol 6h p.i. and all cells displaying this phenotype had GBP1+ PVs (Figure 1B). This showed that GBP1 targeting results in the release of Tg-DNA into host cell cytoplasm. We validated CellMask dye assays further by examining the ultrastructure of the vacuole membranes using correlative light-electron microscopy, which revealed ruffled and broken vacuole membranes in cells expressing GBP1 (Figure 1C). In THP-1 ΔGBP1, most PVs analysed by electron microscopy did not show structural defects or loss of membrane integrity (Figure 1C). Taken together, these results point towards an indispensable role for human GBP1 in the rupture of Tg PVs and release of microbial DNA into the cytosol.
We next wanted to address whether GBP1 contributed to phagocytosis of Tg in macrophages. Compartments containing phagocytosed Tg become RAB7+ and the contained parasites are destroyed in lysosomes (Mordue & Sibley, 1997; Sugaya et al, 2011). WT and ΔGBP1 THP-1 cells infected with Tg showed similar recruitment of endogenous RAB7 at 2 h post-infection (p.i.; Figure 1D and Figure S2A) which indicated that GBP1 does not contribute to Tg phagocytosis. By 6 hours p.i, Tg phagosomes had matured and cleared parasites, and only Tg that invaded and formed PVs remained (Figure S2A). To test if GBP1 was also recruited to phagosomes, we quantified the recruitment of mCH-GBP1 (Dox-treated ΔGBP1+Tet-mCH-GBP1 cells) and endogenous RAB7 to Tg (Figure 1E). This revealed an equal distribution of GBP1 on Tg-containing phagosomes (RAB7+) and PVs (RAB7-) at 2 hours p.i. (Figure 1E). At later time points, exclusively PVs remained, of which ∼25% (type I Tg) to 35% (type II Tg) were decorated with GBP1 (Figure 1E). Thus, GBP1 targets both Tg phagosomes and PVs but does not contribute to Tg phagocytosis.

(A) Quantification of Toxoplasma gondii (Tg) phagocytosis by THP-1 WT and THP-1 ΔGBP1 cells pre-treated with IFNγ and infected with type I or type II Tg for the indicated amounts of time. Phagosomes were scored based on the recruitment of Rab7 (Rab7+). (B) Schematic overview of the workflow for Tg plaque assays for parasite viability determination. In brief: Tg was harvested and used to infect THP-1 macrophages treated with IFNγ and Doxycycline (Dox). 18 hours post infection the macrophages were lysed, parasites collected from the cells and the supernatant, diluted and used to infect human foreskin fibroblast (HFF) monolayers and left for 4-5 days until plaques were detected. (C) Representative images of Tg plaques on HFFs in wells of a 24-well plate after 18 hours of culture in THP-1 cells of the indicated genotype and treatment. UT; untreated. (D) Quantification of plaque loss following Tg culture in THP-1 cells for 18 hours. Data presents the reduction in plaque area of the indicated treatment normalised to the untreated (UT) condition. Data information: Graphs in (A+D) show mean ± SEM from n = 3 independent experiments.
Does GBP1 contribute to growth-restriction of Tg? To addressed this, we performed plaque assays to enumerate live Tg after 18 h infection of THP-1 WT, ΔGBP1 and ΔGBP1+Tet-GBP1 reconstituted cells (Figure S2B-D). This revealed that IFNγ-stimulation of macrophages can control Tg-growth in a GBP1-dependent manner as we observed reduced plaque loss with parasites isolated from ΔGBP1 cells (Figure S2D). Reconstitution of GBP1 by addition of Dox reversed the loss of restriction in ΔGBP1 cells. Altogether, we concluded that GBP1 targets Tg PVs, compromises their integrity and parasite viability and releases Tg DNA into the cytosol.
GBP1 does not participate in Salmonella vacuolar escape but targets cytosolic bacteria
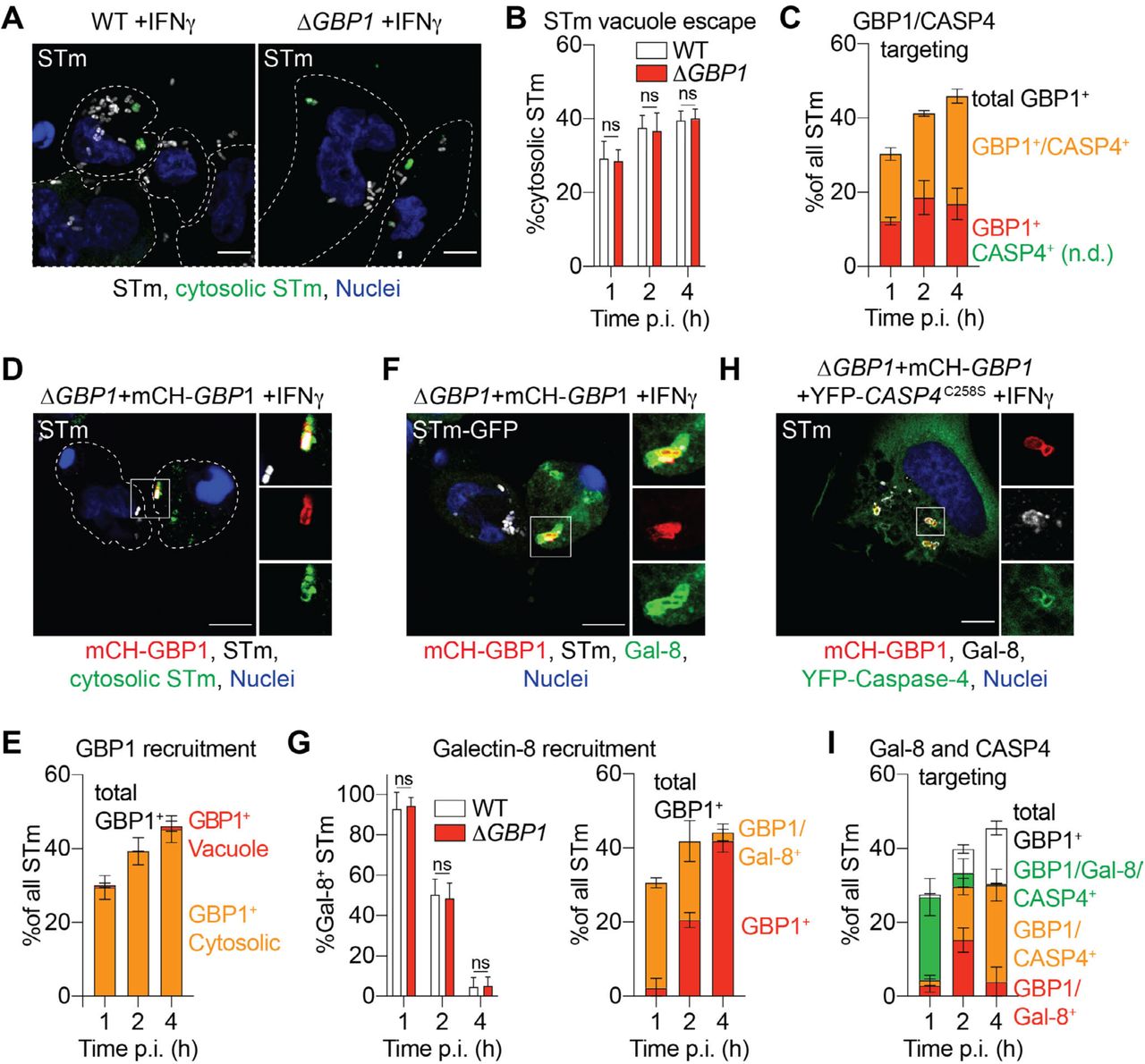
Having established an indispensable role for GBP1 in opening of Tg PVs, we wanted to test if GBP1 also contributed to the escape of STm from Salmonella- containing vacuoles (SCVs). STm secretes effectors that disrupt endocytic trafficking and prevent fusion of degradative lysosomes with SCVs (Spanò & Galán, 2018). We therefore used differential permeabilization (Meunier & Broz, 2015) to determine whether the escape of STm from its vacuole into the cytosol required GBP1. Similar number of cytosolic STm were detected in WT and ΔGBP1 cells, suggesting that GBP1 is dispensable for cytosolic escape of STm (Figure 2A-B). GBP1 recruits caspase-4 to STm and enhances pyroptosis (Fisch et al, 2019a). We performed quantitative immunofluorescence microscopy to determine spatiotemporal differences, if any, in GBP1 recruitment to bacteria over time (Figure 2C). Importantly, differential permeabilization revealed that GBP1 was exclusively recruited to cytosolic STm at all time points (Figure 2D-E). Studies in epithelial cells have shown that the lectin Galectin-8 (Gal-8) is recruited to disrupted SCVs, which promotes bacterial xenophagy and growth-restriction (Thurston et al, 2012). Consistent with a lack of a role for GBP1 in cytosolic escape of STm, similar proportions of STm were decorated with Gal-8 in WT and ΔGBP1 cells (Figure 2G). Immunofluorescence microscopic staining of Gal-8 as a marker for disrupted SCVs further supported our finding that GBP1 is exclusively recruited to broken vacuoles (Figure 2F-G). Temporal studies showed that SCVs were rapidly disrupted (become Gal-8+), but loose this marker over time (Figure 2G), as has been shown before in epithelial cells (Thurston et al, 2012). At later time points as the proportion of Gal-8+ vacuoles decreased, cytosolic STm still retained GBP1 coating (Figure 2G). We therefore hypothesised that bacteria that lose Gal-8 marker become decorated with caspase-4. To assess this possibility, we determined whether Gal-8 and caspase-4 could be recruited to the same bacterium (Figure 2H-I). Remarkably, at 1 hour p.i. STm decorated with Gal-8 and GBP1 were also positive for caspase-4 (Figure 2I) indicating that the same intracellular STm can trigger xenophagy and pyroptosis pathways. Strikingly, at 2 hours p.i. a portion of cytosolic STm were positive for either GBP1-Gal-8 or GBP1-caspase-4 (Figure 2I). Over time the proportion of GBP1-Gal-8+ bacteria decreased, which is consistent with increased GBP1-caspase-4-driven pyroptosis (Figure 2I). Altogether, these single-cell assays revealed that, unlike during Tg infection, GBP1 does not contribute to cytosolic escape of STm, but instead promotes the recruitment of caspase-4 to cytosolic STm, that have not been cleared by xenophagy, to enhance pyroptosis.

(A) Representative immunofluorescence images showing cytosolic Salmonella Typhimurium (STm) and all STm from differentially permeabilised, IFNγ-primed THP-1 WT or ΔGBP1 cells infected with STm SL1344 (MOI = 30) for 2 hours. Cells are outlined by the white, dashed line. Grey: STm; Green: Cytosolic and extracellular STm; Blue: Nuclei. Scale bar 10 µm. (B) Quantification of the proportion of cytosolic STm SL1344 in differentially permeabilised and IFNγ-treated THP-1 WT or ΔGBP1 at the indicated times post infection. (C) Quantification of GBP1 and caspase-4 (CASP4) recruitment to STm SL1344 in IFNγ and Doxycycline (Dox) treated THP-1 ΔGBP1+Tet-mCH-GBP1+YFP-CASP4C258S at the indicated times post-infection. Percentage of all intracellular STm positive for the indicated markers is plotted. n.d. not detected. (D) Representative immunofluorescence images showing the recruitment of GBP1 to cytosolic STm. IFNγ-primed and Dox-treated THP-1 ΔGBP1+Tet-mCH-GBP1 infected with STm SL1344 (MOI = 30) for 2 hours, differentially permeabilised and stained for cytosolic STm and total STm. Cells are outlined by the white, dashed line. Red: mCH-GBP1; Grey: STm; Green: Cytosolic STm; Blue: Nuclei. Scale bar 10 µm. (E) Quantification of GBP1 recruitment to cytosolic and intra-vacuolar STm SL1344 in differentially permeabilised and IFNγ and Dox treated THP-1 ΔGBP1+Tet-mCH-GBP1 at the indicated times post infection (p.i.). (F) Representative immunofluorescence stain for galectin-8 (Gal-8) in IFNγ-primed and Dox-treated THP-1 ΔGBP1+Tet-mCH-GBP1 infected with STm SL1344-GFP (MOI = 30) for 1 hour. Red: mCH-GBP1; Grey: STm; Green: Gal-8; Blue: Nuclei. Scale bar 10 µm. (G) Galectin-8 recruitment to STm is independent of GBP1 and reduces temporally during infection. Left: Quantification of Gal-8 recruitment to STm SL1344-GFP in IFNγ treated THP-1 WT or ΔGBP1 at the indicated times post infection. Right: Quantification of Gal-8 recruitment to STm SL1344-GFP in IFNγ and Dox treated THP-1 ΔGBP1+Tet-mCH-GBP1 at the indicated time post infection. (H) Representative immunofluorescence images showing galectin-8 (Gal-8) recruitment to STm in IFNγ-primed and Dox-treated THP-1 ΔGBP1+Tet-mCH-GBP1+YFP-CASP4C258S. THP-1 cells were infected with STm SL1344 (MOI = 30) for 1 hour. Red: mCH-GBP1; Grey: Galectin-8; Green: YFP-caspase-4; Blue: Nuclei. Scale bar 10 µm. (I) Quantification of Gal-8 and caspase-4 (CASP4) recruitment to STm SL1344 in IFNγ and Dox treated THP-1 ΔGBP1+Tet-mCH-GBP1+YFP-CASP4C258S at the indicated time post infection. Data information: Graphs in (B, C, E, G, I) show mean ± SEM from n = 3 independent experiments. P values for indicated comparisons in (B) and (G) from one-way ANOVA following adjustment for multiple comparisons. ns, not significant.
GBP1 facilitates formation of cytosolic signalling platforms
GBP1-mediated release of Tg-DNA or recruitment of caspase-4 to STm leads to execution of different cell death programs. GBP1 thus promotes the sensing of PAMPs and formation of cytosolic signalling platforms also known as supramolecular organizing centres (SMOCs) (Kagan et al, 2014). We investigated the structure of caspase activation SMOCs promoted by GBP1 actions using structured illumination microscopy (SIM). Upon Tg infection, AIM2 detects DNA and assembles ASC “specks”. Using SIM, we found that these atypical inflammasome complexes appear similar to previously described inflammasomes containing ASC and caspase-1 and caspase-8 (Man et al, 2013, 2014). We found a “donut”-like ASC ring enclosing caspase-8 in the centre (Figure 3A-C and Figure S3A). Furthermore, using THP-1 cells expressing myc-AIM2 (Figure S3B), we detected AIM2 recruitment to ASC specks in Tg-infected macrophages (Figure 3B-C). Similarly, in STm-infected macrophages, we found GBP1 recruited caspase-4 directly to the bacterial surface (Figure 3D). Bacteria were completely covered in GBP1, with a high degree of colocalisation with YFP-caspase-4C258S (Figure 3D). These results clarify the distinct spatial roles of GBP1 during Tg and STm infection.

(A) Representative immunoblots for proCaspase-8, FLAG and β-actin from THP-1 +Tet-CASP8-FLAG cells showing Dox-inducible caspase-8-Flag expression. Cells were treated with Doxycycline (Dox) as indicated or left untreated. * endogenous caspase-8. (B) Representative immunoblots for FLAG, caspase-8 (CASP8), AIM2, myc and β-actin from THP-1 WT, THP-1 +Tet-CASP8-FLAG and THP-1 +Tet-CASP8-FLAG+myc-AIM2 cells showing Dox-inducible caspase-8-Flag expression and constitutive expression of myc-AIM2. Cells were treated with IFNγ, Dox or left untreated as indicated. * endogenous proteins. (C) Representative immunoblots for mCherry, YFP and β-actin from THP-1 ΔGBP1+Tet-mCH-GBP1 cells expressing the indicated mutant of GBP1 and also stably expressing YFP-CASP4C258S. Cells were primed with IFNγ and treated with Dox as indicated. (D) Representative immunoblots for mCherry (mCH), YFP and β-actin from THP-1 ΔGBP1+Tet-mCH-GBP1 +YFP-CASP5C315S. Cells were primed with IFNγ and treated with Dox as indicated. (E) Left: RT-qPCR and Right: representative immunoblot of THP-1 WT cells transfected with non-targeting control (CTRL) siRNA or siRNA against caspase-5 (CASP5). Indicated percentage shows remaining transcript level in siCASP5 transfected cells as compared to CTRL. Data information: Graph in (E) shows mean ± SEM from n = 9 independent experiments.

(A) Left: Representative structured illumination super-resolution immunofluorescence microscopy images from THP-1 WT+Tet-CASP8-FLAG cells treated with IFNγ and Doxycycline (Dox) to induce caspase-8 expression and then infected with type II Toxoplasma gondii (Tg) for 4 hours. Right: 3D reconstruction and slices through the ASC-caspase-8 speck seen in the image on the left. Red: ASC; Grey: Tg; Green: caspase-8; Blue: Nuclei. Scale bar 10 µm. (B) Left: Representative structured illumination super-resolution immunofluorescence microscopy images from THP-1+Tet-CASP8-FLAG+myc-AIM2 cells treated with IFNγ and Dox and infected with type II Tg for 4 hours. Right: 3D reconstruction and slices through the AIM2-caspase-8 speck seen in the image on the left. Cyan: AIM2; Grey: Tg; Green: caspase-8; Blue: Nuclei. Scale bar 10 µm. (C) Ring diameters of the indicated proteins within an inflammasome speck from Tg infected cells shown in (A) and (B). (D) Left: Representative structured illumination super-resolution immunofluorescence microscopy images from THP-1 ΔGBP1+Tet-mCH-GBP1+YFP-CASP4C258S cell treated with IFNγ and Dox and infected with STm SL1344 (MOI = 30) for 2 hours. Right: 3D reconstruction of the GBP1-caspase-4 signalling platform on the cytosolic STm. Red: mCH-GBP1; Grey: STm-LPS; Green: YFP-caspase-4; Blue: Nuclei. Scale bar 10 µm. Data information: Graph in (C) shows quantification from n = 12 inflammasome specks and mean ± SEM. * P ≤ 0.05; **** P ≤ 0.0001 for indicated comparisons in (C) from one-way ANOVA following adjustment for multiple comparisons.
GBP1 specifically promotes caspase-4 activation during STm infection
During STm infection, both caspase-1 and caspase-4 were found to contribute to pyroptosis based on whole-population assays such as LDH release or propidium iodide uptake assays (Fisch et al, 2019a). We asked whether both caspase-1 and caspase-4 were active in the same cell. Indeed, ∼80 % STm-infected cells and cells that deposited GBP1 and caspase-4 on the bacteria also contained an ASC speck (Figure 4A-B). This was indicative of an active canonical inflammasome in response to other bacteria-derived PAMPs. Notably, caspase-4 was not recruited to ASC specks, which is consistent with previous work (Thurston et al, 2016) (Figure 4A). Active canonical inflammasomes were also found in STm-infected cells that lacked GBP1+ STm, but not in uninfected cells (Figure 4B). We therefore concluded that both caspase-1 and caspase-4 can be active in the same infected macrophage.

(A) Representative immunofluorescence images from IFNγ-primed THP-1 ΔGBP1+Tet-mCH-GBP1+YFP-CASP4C258S infected with Salmonella Typhimurium (STm) SL1344 (MOI = 30) for 2 hours stained for ASC. Cells were treated with Doxycycline (Dox) to induce GBP1 expression or left untreated. Yellow arrowheads indicate position of some STm within cells (DNA-staining dye). Red: mCH-GBP1; Cyan: ASC; Green: YFP-caspase-4; Blue: Nuclei. Scale bar 10 µm. (B) Quantification of ASC speck formation and GBP1+caspase-4 recruitment to STm at 2 h post-infection of THP-1 ΔGBP1+Tet-mCH-GBP1+YFP-CASP4C258S cells left untreated of treated with IFNγ or IFNγ + Dox. (C) Representative immunofluorescence images from IFNγ-primed and Dox-treated THP-1 ΔGBP1+Tet-mCH-GBP1+YFP-CASP4C258S cells infected with STm SL1344 (MOI = 30) for 2 hours. Cells expressed the indicated variants of GBP1: GBP1K51A (lacking GTPase activity) or GBP1Δ589-592 or GBP1C589A(which lack the isoprenylation site). Red: mCH-GBP1; Grey: STm; Green: YFP-caspase-4; Blue: Nuclei. Scale bar 10 µm. (D) Quantification of GBP1 and caspase-4 recruitment to STm SL1344 in IFNγ and Dox treated THP-1 ΔGBP1+Tet-mCH-GBP1+YFP-CASP4C258S expressing the indicated version of GBP1 at 2 h post-infection. n.d. not detected. (E) Representative immunofluorescence images from IFNγ and Dox-treated THP-1 ΔGBP1+Tet-mCH-GBP1+YFP-CASP4C258S cells infected with STm SL1344 (MOI = 30) for 2 hours and stained for STm LPS. Red: mCH-GBP1; Grey: STm-LPS; Green: YFP-caspase-4; Blue: Nuclei. Scale bar 10 µm. (F) Quantification of LPS immunofluorescence staining intensity from undecorated, GBP1+ or GBP1+CASP4+ STm in the cytosol of infected THP-1 macrophages. (G) Caspase-5 expression is induced by IFNγ. Representative immunoblots for caspase-5 (CASP5) and β-actin of THP-1 WT treated with IFNγ as indicated. (H) Caspase-5 does not target STm. Representative immunofluorescence images from IFNγ-primed and Dox-treated THP-1 ΔGBP1+Tet-mCH-GBP1+YFP-CASP5C315S infected with STm (MOI = 30) for 2 hours. Red: mCH-GBP1; Grey: STm; Green: YFP-caspase-5; Blue: Nuclei. Scale bar 10 µm. (I) Quantification of GBP1 and caspase-5 recruitment to STm in IFNγ and Dox treated THP-1 ΔGBP1+Tet-mCH-GBP1+YFP-CASP5C315S. n.d. not detected. (J) Caspase-5 is dispensable for STm-induced pyroptosis. Real-time propidium iodide (PI) uptake assay from IFNγ-primed THP-1 ΔGBP1 +Tet-GBP1 cells transfected with indicated siRNA or non-targeting control (CTRL) infected with STm SL1344 (MOI = 30) for 4 h. Plotted as area under the curve (AUC) from real-time assays. Data information: Graph in (B, D, F, I+J) shows mean ± SEM of n = 3 independent experiments. *** P ≤ 0.001 in (F) from nested one-way ANOVA comparing to undecorated vacuoles for the means of the n = 3 independent experiments and in (B+J) from two-way ANOVA following adjustment for multiple comparisons. ns, not significant.
We previously showed that the translocation of GBP1 to STm or Tg requires its GTPase function and isoprenylation (Fisch et al, 2019a). However, as GBP1 was dispensable for bacterial escape from SCVs, we wanted to examine whether lack of GBP1 targeting to STm also affected the recruitment of caspase-4 to bacteria. To address this, we expressed YFP-CASP4C258S in THP-1 ΔGBP1 cells reconstituted with doxycycline-inducible variants of GBP1 that lacked GTPase activity (GBP1K51A) or isoprenylation sites (GBP1C589A or GBP1Δ589-592; Figure S3C). None of these variants localised to STm nor did we observe recruitment of caspase-4 (Figure 4C-D). Along with above described findings on the lack of involvement of GBP1 in STm escape into the cytosol, these results indicated that the inability of GTPase or isoprenylation-deficient GBP1 in enhancing STm-induced pyroptosis was due to their inability to recruit and activate caspase-4 on the surface of cytosolic bacteria (Fisch et al, 2019a). Interestingly, immunofluorescence staining of Salmonella-LPS using a monoclonal antibody revealed that GBP1-CASP4+ bacteria stained not at all or poorly with anti-LPS, which suggested that access to the epitope might be blocked by the thick coat of GBP1 and caspase-4 on the bacterial surface. As LPS is a ligand for caspase-4, this finding is consistent with the possibility that caspase-4 on the bacterial surface precludes antibody-mediated staining of LPS (Figure 4E-F).
Caspase-5 is a human paralogue of caspase-4 and can also detect cytosolic LPS and trigger pyroptosis (Shi et al, 2014). We therefore wanted to assess its role during STm infection. Notably, caspase-5 expression was upregulated upon IFNγ treatment of macrophages (Figure 4G). However, unlike YFP-CASP4C258S, YFP-CASP5C315S was not recruited to STm (Figure S3D, 4H-I). Notably, STm that were negative for caspase-4 but positive for GBP1 could be stained with the anti-LPS antibody (Figure 4H), which suggested that caspase-4 is responsible for the reduced antibody access. Silencing of caspase-5 expression did not affect pyroptosis in STm-infected IFNγ-primed macrophages (Figure 4J, S3E), which further pointed to a unique role for caspase-4 in this scenario. These experiments together established that both caspase-1 and caspase-4 are active within the same cell and GBP1 controls caspase-4 recruitment and activation directly on the surface of cytosolic bacteria, whereas caspase-5 is dispensable for STm-induced pyroptosis.
Caspase-1 controls GBP1-mediated pyroptosis through negative feedback
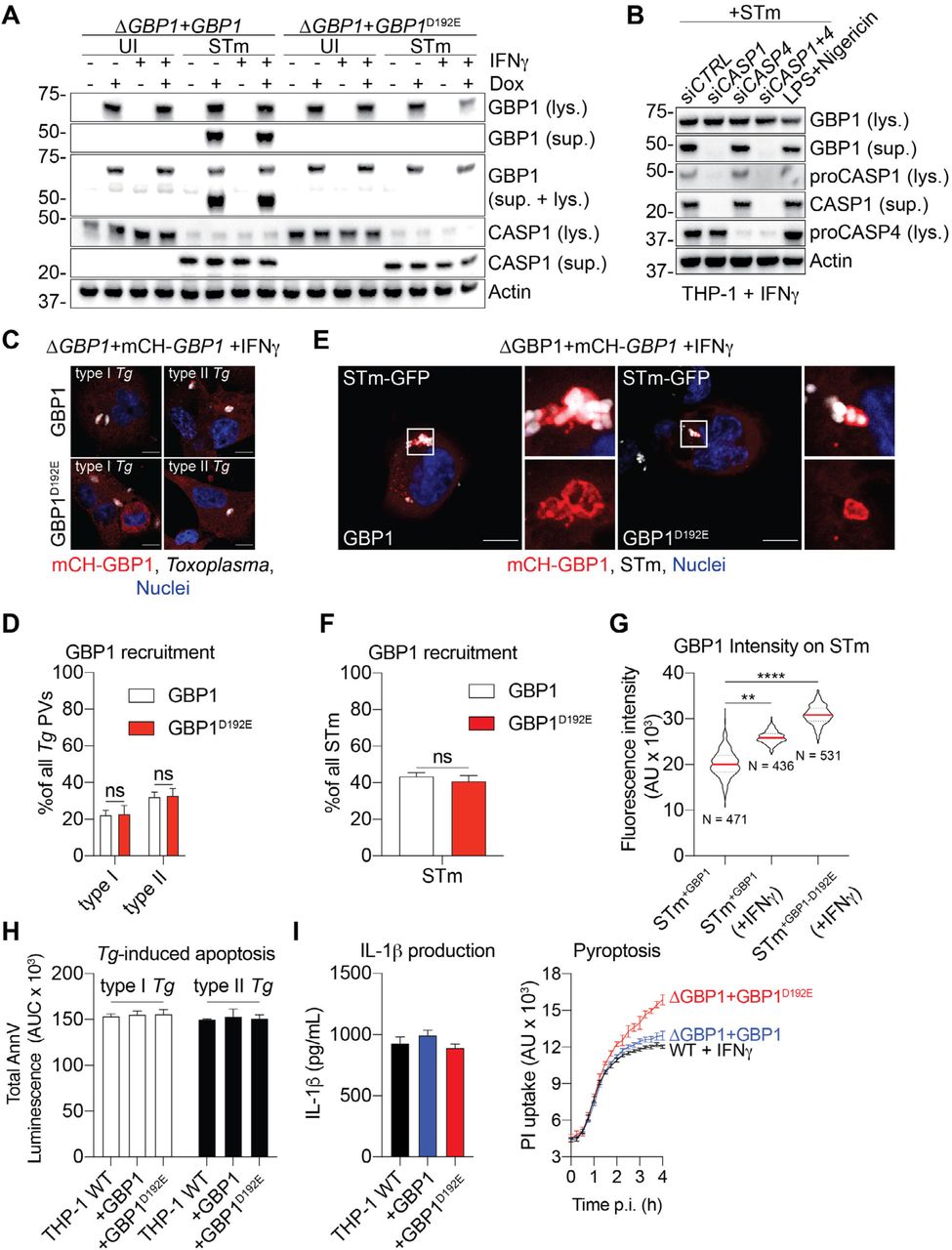
Caspase-1 can cleave GBP1 at Asp192 to generate a stable p47 GBP1 C-terminal fragment in human umbilical vein endothelial cells (HUVECs) (Naschberger et al, 2017). However, the impact of this proteolytic event on GBP1 function during infections has not been examined. To study this further, we created THP-1 ΔGBP1 cells expressing the non-cleavable GBP1D192E without or with an mCherry tag (THP-1 ΔGBP1+Tet-GBP1D192E and THP-1 ΔGBP1+Tet-mCH-GBP1D192E cells; Figure S4A). Aspartate D192 is in a surface-exposed loop within the GTPase domain of GBP1 (Prakash et al, 2000) and therefore likely to be accessible to proteases (Figure S4B). Immunoblotting for GBP1 from STm-infected IFNγ-primed macrophages revealed caspase-1 activation and formation of p47 GBP1 from cells expressing wildtype GBP1 but not GBP1D192E (Figure 5A). Furthermore, silencing caspase-1, but not caspase-4, abolished p47 GBP1 production (Figure 5B), which suggested that caspase-1 plays a dominant role in GBP1 proteolysis. Similarly, chemical activation of caspase-1 by treatment with LPS+Nigericin produced a p47 GBP1 fragment (Figure 5B). To investigate whether trafficking of GBP1D192E is altered, we infected the mCH-GBP1D192E expressing cells with Tg or STm and quantified GBP1 recruitment to PVs or cytosolic STm respectively. Upon infection with Tg we did not observe differences in the proportion of Tg-PVs (Figure 5C-D) or STm (Figure 5E-F) that became GBP1-positive, which indicated that proteolysis of GBP1 is dispensable for its trafficking to microbial vacuoles. However, the mean fluorescence intensity of mCH-GBP1 around decorated STm was markedly higher in cells expressing the GBP1D192E variant (Figure 5G), even though the expression of wildtype GBP1 and GBP1D192E were comparable (Figure S4C-D). We speculated that cleavage of GBP1 may reduce the cellular pool of full-length GBP1, which would also reduce its ability to induce cell death over time. We therefore compared apoptosis and pyroptosis in GBP1 and GBP1D192E expressing cells. Apoptosis induced by Tg infection of THP-1 macrophages expressing GBP1 or GBP1D192E variant were similar, and we reasoned that this is because Tg-infection does not involve activate caspase-1 (Figure 5H). However, during STm infection, GBP1D192E cells underwent higher pyroptosis but released similar levels of IL-1β (Figure 5I). This finding is consistent with a major role for GBP1 in promoting caspase-4-driven pyroptosis, but not canonical caspase-1 activation, which is responsible for IL-1β production (Figure 5I). Taken together, these results suggested that active caspase-1 cleaves GBP1 into an p47 fragment that reduces its recruitment to STm and the recruitment and activation of caspase-4. Because IL-1β maturation was not affected by GBP1D192E mutation, we speculate that this caspase-1-driven feedback mechanism balances cell death and IL-1β secretion during STm infection. Moreover, as caspase-1 does not contribute to cell death during Tg-infection, this feedback regulatory mechanism is pathogen-specific.

(A) Representative immunoblots of GBP1, mCherry (mCH) and β-actin from IFNγ-primed THP-1 ΔGBP1+Tet-GBP1 or GBP1D192E with and without mCh-tag for Doxycycline (Dox)-inducible expression. Cells were treated with Dox as indicated. (B) Crystal structure of human GBP1 (PDB: 1F5N) with GTPase domain highlighted in cyan, C-terminal helical domain in green and aspartate D192 in red. (C) Representative images from THP-1 ΔGBP1+Tet-mCH-GBP1 (Left) or +Tet-GBP1D192E (Right) cells treated with IFNγ and Dox to induce GBP1 expression. Red: mCh-GBP1; Blue: Nuclei. Scale bars 100 µm. (D) Quantification of mean mCH-GBP1 or mCH-GBP1D192E fluorescence per cell from 100 fields of view. Data shows individual values from n = 3 independent experiments and mean (red line) ± SEM. Data information: P values in (D) from t-test comparing means of n = 3 independent experiments. ns, not significant.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) GBP1, but not GBP1D192E, is cleaved during STm infection. Representative immunoblots for GBP1, caspase-1 (CASP1) and β-actin from lysates (lys.) or culture supernatants (sup.) of THP-1 ΔGBP1+Tet-GBP1 WT or D192E cells treated with IFNγ and Dox as indicated and infected with STm SL1344 for 4 hours or left uninfected (UI). (B) Caspase-1 is required for GBP1 cleavage. Representative immunoblots for GBP1, caspase-1 (CASP1), caspase-4 (CASP4) and β-actin from lysates (lys.) or culture supernatants (sup.) of THP-1 WT, transfected with the indicated siRNA and infected with STm SL1344 for 4 hours or treated with LPS and Nigericin for 90 minutes. (C) Representative immunofluorescence images from IFNγ-primed and Doxycycline (Dox)-treated THP-1 ΔGBP1+Tet-mCH-GBP1 or mCH-GBP1D192E cells infected with type I or type II Toxoplasma gondii (Tg) for 6 hours. Red: mCH-GBP1; Grey: Tg; Blue: Nuclei. Scale bar 10 µm. (D) Quantification of recruitment of GBP1 WT or GBP1D192E to type I or type II Tg at 6 hours post infection in THP-1 cells pre-stimulated with IFNγ and Dox to induce GBP1 expression. (E) Representative immunofluorescence images from IFNγ-primed and Dox-treated THP-1 ΔGBP1+Tet-mCH-GBP1 or mCH-GBP1D192E cells infected with STm SL1344-GFP (MOI = 30) for 2 hours. Red: mCH-GBP1; Grey: STm; Blue: Nuclei. Scale bar 10 µm. (F) Quantification of recruitment of GBP1 WT or GBP1D192E at 4 hours post-infection with STm SL1344-GFP in THP-1 cells pre-stimulated with IFNγ and Dox to induce GBP1 expression. (G) Representative fluorescence intensities of the indicated GBP1 variants on the surface of decorated STm SL1344-GFP in THP-1 cells 4 hours post-infection. N depicts the number of measured GBP1+ bacteria in the respective condition. (H) GBP1D192E and WT GBP1 behave similarly in Tg-induced atypical apoptosis. AnnV-Glo assay of THP-1 WT and ΔGBP1 cells stably reconstituted with Tet-GBP1 WT or GBP1D192E mutant as indicated, infected with type I or type II Tg for 18 h. Plotted as area under the curve (AUC) from real-time assays. (I) GBP1D192E promotes increased pyroptosis during STm-infection. Left: IL-1β ELISA from the indicated THP-1 cells primed with IFNγ and Dox infected with STm SL1344 (MOI = 30) at 4 h post-infection. Right: Real-time propidium iodide (PI) uptake assay from IFNγ-primed THP-1 cells of the indicated genotypes and infected with STm SL1344 for 4 h. Data information: Graphs in (D, F, G, H, I) show mean ± SEM from n = 3 independent experiments. ** P ≤ 0.01, *** P ≤ 0.001 **** P ≤ 0.0001 for indicated comparisons in (G) from nested one-way ANOVA comparing means of the n = 3 experiments and in (D, F) from two-way ANOVA following adjustment for multiple comparisons; ns, not significant.
DISCUSSION
IFNγ-inducible GBPs have emerged as important proteins in host defence against a range of pathogens. However, GBPs probably play differential and hence nuanced roles in host defence against different pathogens and these differences remain largely unexplored. Additionally, besides direct attack on pathogens and release of PAMPs, other true mechanistic contribution of GBPs to processes of pathogen restriction and host cell death induction are still lacking. Our studies provide new mechanistic insights on the role of human GBP1 in macrophages infected with two evolutionarily divergent pathogens. We established that GBP1 controlled the fitness of Tg in infected macrophages and promotes lysis of Tg- containing vacuoles, release of Tg-DNA and assembly of the AIM2-ASC-caspase-8 SMOC that induces apoptosis. Tg viability in infected macrophages is reduced through the actions of GBP1. Thus, GBP1 not only removes a replicative niche through apoptotic killing of macrophages, but also controls parasite growth. Unfortunately, due to high levels of pyroptotic loss of macrophages, we could not accurately determine the fate of GBP1-targeted bacteria using gentamicin-protection assays (Miao et al, 2010).
Results obtained are consistent with similar GBP1 recruitment to Tg-containing vacuoles and phagosomes (Qin et al, 2017; Shydlovskyi et al, 2017; Fisch et al, 2019a). Our single-cell assays conclusively reveal that GBP1 decorates two distinct intracellular populations of Tg within the same cell – those formed by host-mediated phagocytosis or those formed by Tg invasion. These compartments are of different origin and have different membrane compositions. GBP1 is recruited to Tg only in IFNγ-primed macrophages, suggesting that other IFNγ-inducible factors also contribute to GBP1 trafficking. In addition, we have previously shown that GBP1 GTPase activity and isoprenylation control its trafficking to Tg (Fisch et al, 2019a). Therefore, GBP1-intrinsic and extrinsic activities control its ability to target Tg, which need to be investigated in the future.
GBP1 targeting of Tg vacuolar membranes was required for their compromise and release of Tg- DNA, which resembled bacterial lysis in the cytosol by murine GBPs (Man et al, 2015; Meunier et al, 2015; Liu et al, 2018). In contrast, STm enters the cytosol independently of GBP1. This yet again indicates that the actions of GBP1 are microbe-specific. GBP1 is a dynamin-like GTPase and thus it may actively alter biological membranes (Huang et al, 2019). Recombinant, farnesylated GBP1 has been shown to bend lipids in the form of giant unilamellar structures in vitro (Shydlovskyi et al, 2017). Membrane disruption has been demonstrated for murine Gbps, as several Gbp family members are responsible for the formation of membrane blebs on Tg vacuoles (Yamamoto et al, 2012; Degrandi et al, 2013; Selleck et al, 2013; Kravets et al, 2016). Results from our correlative light and electron microscopy experiments reveal a similar phenotype, which is GBP1-dependent, in human macrophages. This is the first direct demonstration of a human GBP being capable of disrupting pathogen vacuole membranes. Importantly, using Tg, whose DNA was specifically labelled with EdU, we conclusively show the release of Tg-DNA into the host cytosol for detection by AIM2. Thus, during Tg infection GBP1 acts upstream of cell death execution and facilitates AIM2 signalling.
As caspase-1 and several other inflammasome-associated proteins are absent in Tg- infected cells, AIM2 recruits ASC and caspase-8 (Fisch et al, 2019a). The structure of a caspase-8 containing AIM2 inflammasome, imaged with super-resolution fluorescence microscopy, closely resembles previously published structures of caspase-8 in NLRP3/NLRC4 inflammasomes (Man et al, 2014, 2013). These appear as typical ASC donut-like rings with AIM2 on the inside and caspase-8 in the centre. ASC and AIM2 seem to form a dome under which caspase-8 can oligomerize and self-activate. This resembles death receptor-induced signalling complexes (DISCs)-driven caspase-8 activation during extrinsic apoptosis (Tummers & Green, 2017). It will be interesting to investigate whether the caspase-8 inhibitor c-FLIP (CFLAR) or FADD are recruited to this complex to moderate caspase-8 activity (Wu et al, 2014).
The role of GBP1 during STm infection is to directly recruit caspase-4 to the outer surface of bacteria for LPS-sensing and subsequent pyroptosis. Notably, GBP1 exclusively targeted cytosolic STm and did not contribute to their vacuolar escape. Equally, GBP1 detection of cytosolic STm was independent of recruitment of Gal-8, which belongs to a family of lectins that can detect vacuole rupture when luminal polysaccharides become exposed to the cytoplasm (Coers, 2017). Both GBP1 and Gal-8 were recruited to bacteria early during infection, and it has been suggested that GBP1 can bind endogenous sulphated lipids exposed to the cytoplasm (Bradfield, 2016). Importantly, GBP1 GTPase activity and isoprenylation were essential for GBP1-targeting to STm and subsequent recruitment of caspase-4, pointing further to GBP1-intrinsic and -extrinsic factors in regulating GBP1 trafficking. As STm infection progresses, GBP1-caspase-4 dual recruitment increases, and the proportion of Gal-8-positive bacteria is reduced over time. In epithelial cells, Gal-8 is rapidly recruited to cytosolic STm leading to bacterial xenophagy, during which Gal-8 is lost from bacteria over time (Thurston et al, 2012). In macrophages, we find that GBP1 is present on cytosolic STm even though they have lost the Gal-8 coat. Our findings on GBP1-independent recruitment of Gal-8 to STm are consistent with work with murine GBPs, where galectins-3 and -8 were needed for mGBP recruitment to Gram-negative bacteria (Feeley et al, 2017). We speculate that the cells first try to control the contamination of the cytosol with autophagic bacteria-clearance, but if this fails use GBP1 to induce the more drastic control measure to execute pyroptosis via caspase-4.
GBP1 and caspase-4 formed a dense coat on STm, which reduced bacterial staining with anti-LPS antibody. Whether this was due to reduced antibody access due to the GBP1/caspase-4 coat or blocking of the LPS epitope by caspase-4 cannot be definitively distinguished. As caspase-4 by itself could not recruit to the bacteria, we can hypothesise that GBP1 is involved in exposing parts of the LPS that are buried deeper within the membrane. Data derived from Francisella LPS suggests that tetra-acylated lipid A is recognized by caspase-4, but not murine caspase-11 (Lagrange et al, 2018) and we can therefore assume that GBP1 ‘opens’ the bacterial outer membrane for caspase-4 to gain access to otherwise hidden PAMPs. Interestingly, caspase-5 was not recruited to STm even though previous work has shown that caspase-5 can bind LPS and promote pyroptosis in response to LPS transfection (Shi et al, 2014). These differences between caspase-4 and caspase-5 were unexpected and suggest that like the GBPs, even very similar members of the family members of human caspases have unique roles during infection. In naïve macrophages, the STm SPI2 virulence system, which is expressed late during infection, can subvert NLRP3 and NLRC4 inflammasomes and both caspase-4 and caspase-5 contribute to late pyroptosis (Baker et al, 2015; Bierschenk et al, 2019). As IFNγ-priming results in rapid recruitment of GBP1 and caspase-4 to STm and increased pyroptosis, it is plausible that the late role of caspase-5 in STm-induced cell death in naïve cells is not observed in our experimental conditions.
Taken together, during Tg infection GBP1 releases PAMPs that trigger SMOC formation and during STm infection GBP1 is a part of the SMOC itself. Given the upstream role of GBP1 in cell death, we hypothesised that GBP1 must be part of a regulatory feedback loop to limit its own activity. We found that canonical caspase-1 activation during STm infection, which is absent during Tg infection, limits caspase-4-driven pyroptosis by cleavage of GBP1 at D192. We thus show for the first time that GBP1 proteolysis directly regulates host cell death levels during infection. As caspase-4 does not contribute to IL-1β processing during STm infection, this regulatory feedback does not affect IL-1β production, which is largely driven by the canonical caspase-1 activation pathway. This tightly controlled mechanism is an example of the influence of cell death induction and execution mechanisms controlling not only themselves, but also the amount of inflammation and tissue damage they cause. This contrasts our discovery of the ubiquitin conjugating enzyme UBE2L3 as an indirect target of caspase-1 that specifically control IL-1β production, but not pyroptosis (Eldridge et al, 2017). Studies on cellular targets of caspases may therefore provide new insights on homeostasis and disease (Sanchez-Garrido et al, 2018).
On a whole-organism level, fine tuning inflammation through IL-1β production or pyroptotic cell death is essential to contribute to a balanced immune response. Overall, our studies highlight the GBP1-mediated molecular mechanisms and functions of cell death regulation during infection with evolutionary different microbes.
AUTHOR CONTRIBUTION
DF, ARS and EMF conceived the idea for the study, DF, BC, MCD and TM performed experiments. HB, MY provided essential reagents. LMC provided essential expertise and equipment. DF, ARS and EMF analyzed and interpreted the data and wrote the manuscript. All authors revised the manuscript.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
FUNDING
This work was supported by the Francis Crick Institute, which receives its core funding from Cancer Research UK (FC001076 to EMF, FC001999 to LMC), the UK Medical Research Council (FC001076 to EMF, FC001999 to LMC), and the Wellcome Trust (FC001076 to EMF, FC001999 to LMC). EMF was supported by a Wellcome Trust Career Development Fellowship (091664/B/10/Z). DF was supported by a Boehringer Ingelheim Fonds PhD fellowship. ARS would like to acknowledge support from the MRC (MR/P022138/1) and Wellcome Trust (108246/Z/15/Z). MY was supported by the Research Program on Emerging and Re-emerging Infectious Diseases (JP18fk0108047) and Japanese Initiative for Progress of Research on Infectious Diseases for global Epidemic (JP18fk0108046) from Agency for Medical Research and Development (AMED). HB was supported by Grant-in-Aid for Scientific Research on Innovative Areas (17K15677) from Ministry of Education, Culture, Sports, Science and Technology.
MATERIALS AND METHODS
Cells and parasites
THP-1 (TIB-202, ATCC) were maintained in RPMI with GlutaMAX (Gibco) supplemented with 10% heat-inactivated FBS (Sigma), at 37°C in 5% CO2 atmosphere. THP-1 cells were differentiated with 50 ng/mL phorbol 12-myristate 13-acetate (PMA, P1585, Sigma) for 3 days followed by a rested for 2 days in complete medium without PMA. Cells were not used beyond passage 20. HEK293T and human foreskin fibroblasts (HFF) were maintained in DMEM with GlutaMAX (Gibco) supplemented with 10% FBS at 37°C in 5% CO2 atmosphere. An overview of all cell lines made/ used in this study is provided in Table 1. Tg expressing luciferase/eGFP (RH type I and Prugniaud (Pru) type II) were maintained by serial passage on monolayers of HFF cells. All cell culture was performed without addition of antibiotics unless otherwise indicated. Cell lines were routinely tested for mycoplasma contamination by PCR and agar test.
Cell treatments
Cells were stimulated for 16 h prior to infection in complete medium at 37°C with addition of 50 IU/mL human IFNγ (285-IF, R&D Systems). Induction of GBP1 or caspase-8 expression in the Doxycycline-inducible cells was performed with 200 ng/mL Doxycycline overnight (D9891, Sigma). To chemically activate caspase-1, cells were treated with 10 µM Nigericin (N1495, Invitrogen) and 100 µg/mL LPS-Sm (IAX-100-011, Adipogen).
Toxoplasma gondii infection
Parasite were passaged the day before infection. Tg tachyzoites were harvested from HFFs by scraping and syringe lysing the cells through a 25 G needle. The Tg suspension was cleared by centrifugation at 50 x g for 5 min and then the parasites were pelleted by centrifugation at 550 x g for 7 min from the supernatant, washed with complete medium, centrifuged again and finally re-suspended in fresh medium. Viable parasites were counted with trypan blue and used for infection at a multiplicity of infection (MOI) of 3 for most experiments or 1 for immunofluorescence imaging. Infection was synchronized by centrifugation at 500 x g for 5 min. Two hours after infection, extracellular parasites were removed with three PBS washes.
Plaque assay and quantification
To quantify Tg fitness following infection of macrophages, differentiated THP-1 cells were infected with Tg as described above and 18 hours p.i. supernatant and cells were harvested from the wells of a 12-well plate. Cells were syringe-lysed and obtained parasites from within the cells and the supernatant were diluted 1:10,000 and added to HFFs grown confluent in wells of a 24-well plate. 5 days post infection of the HFFs, cells were fixed with ice-cold methanol and stained with crystal violet. Following 5 washes with PBS, plaques were imaged on a GelCount™ Colony Counter (Oxford Optronix) and cell covered area determined using FIJI ImageJ. Proportion of plaque and plaque loss as compared to Tg grown in untreated THP-1 were calculated subsequently.
Salmonella Typhimurium infection
STm strain SL1344-GFP (with pFPV25.1 plasmid) was maintained under Ampicillin selection (11593027, Gibco) and grown on LB + Ampicillin agar plates. STm SL1344 wildtype strain was maintained in the presence of streptomycin (11860038, Gibco) selection. One day before infection bacteria from a single colony were inoculated into 9 mL LB and grown overnight at 37°C. The overnight culture was diluted 1:50 into LB + 300 mM NaCl (746398, Sigma) and grown shaking in a closed container until an OD600 of 0.9. Bacteria were harvested by centrifugation at 1000 x g for 5 min and washed with serum-free cell culture medium twice and re-suspended in 1 mL medium. Cells were infected with STm at an MOI of 30 and infections were synchronized by centrifugation at 750 x g for 10 min. Infected cells were washed 30 min post-infection three times with warm PBS (806552, Sigma) to remove extracellular bacteria and fresh medium containing 100 μg/mL Gentamicin (15750060, Gibco) was added for 1 h. Medium was then replaced with medium containing 10 μg/mL gentamicin and the infection continued for indicated times. Bacterial MOI used for infections were confirmed by plating on LB agar plates.
Real-time propidium iodide (PI)-uptake assays
To measure live kinetics of cell death, 60,000 cells were seeded per well of a black-wall, clear-bottom 96-well plate (Corning) for differentiation with PMA, treated and infected as described above. Medium was replaced with phenol-red-free RPMI supplemented with 5 μg/mL propidium iodide (P3566, Invitrogen). The plate was sealed with a clear, adhesive optical plate seal (Applied Biosystems) and placed in a plate reader (Fluostar Omega, BMG Labtech) pre-heated to 37°C. PI fluorescence was recorded with top optics every 15 min for times as indicated.
AnnV glow assay
Apoptosis kinetics were analyzed using the RealTime-Glo™ Annexin V Apoptosis Assay (JA1001, Promega) according to the manufacturer’s instruction. 60,000 cells were seeded per well of a white, tissue culture-treated 96-well plate, differentiated, pre-treated and infected as described before. Simultaneously with infection, detection reagent was added. Luminescence was measured using a Fluostar Omega plate reader (BMG Labtech). No-cell, medium-only controls were used for background correction.
IL-1β ELISA
The cell culture supernatant was harvested and cleared by centrifugation at 2000 x g for 5 minutes. Cleared supernatants were then diluted in the buffer provided with the ELISA kit and ELISA was performed according to the manufacturer’s instruction. IL-1β ELISA kit was from Invitrogen (#88-7261, detection range 2 - 150 pg/mL).
Immunoblotting
For immunoblotting, 0.5×106 cells were seeded per well of a 48-well plate, differentiated with PMA, pre-treated and infected. At the end of treatments, cells were washed with ice-cold PBS and lysed for 5 min on ice in 50 μL RIPA buffer supplemented with protease inhibitors (Protease Inhibitor Cocktail set III, EDTA free, Merck) and phosphatase inhibitors (PhosSTOP, Roche). Lysates were cleared by centrifugation at full speed for 15 min at 4°C. BCA assay (Pierce BCA protein assay kit, 23225, Thermo Scientific) was performed to determine protein concentration. 10 μg of total protein per sample were run on 4-12% Bis-Tris gels (Novex, Invitrogen) in MOPS running buffer and transferred on Nitrocellulose membranes using iBlot transfer system (Invitrogen). Depending on the primary antibody used the membranes were blocked with either 5% BSA (A2058, Sigma) or 5% dry-milk (M7409, Sigma) in TBS-T for at least 1 h at room temperature. Incubation with primary antibodies was performed at 4°C overnight (all antibodies used in this study can be found in Table 2). Blots were developed by washing the membranes with TBS-T, probed with 1:5000 diluted secondary antibodies in 5% BSA in TBS-T and washed again. Finally, the membranes were incubated for 2 minutes with ECL (Immobilon Western, WBKLS0500, Millipore) and luminescence was recorded on a ChemiDoc MP imaging system (Biorad).
For immunoblots of culture supernatants, cells were treated in OptiMEM (1105802, Gibco) without serum. Proteins in the supernatants were precipitated with 4x volume cold acetone (V800023, Sigma) overnight at -20°C, and pelleted by centrifugation. Pellets were air dried and re-suspended in 40 μL 2x Laemmli loading dye.
Quantitative RT-PCR (qRT-PCR)
RNA was extracted from 0.25×106 cells using Trizol reagent (15596026, Invitrogen). RNA (1 μg) was reverse transcribed using high-capacity cDNA synthesis kit (4368813, Applied Biosystems). qPCR used PowerUP SYBR green (A25742, Applied Biosystems) kit, 20 ng cDNA in a 20 μL reaction and primers (CASP5-fwd: 5’-GCAAGGAATGGGGCTCACTA-3’; CASP5-rev: 5’-CGTGCTGTCAGAGGACTTGT-3’) at 1 μM final concentration on a QuantStudio 12K Flex Real-Time PCR System (Applied Biosystems). Recorded Ct values were normalized to Ct of human HPRT1 (see Fisch et al, 2019a) and data plotted as ΔCt (Relative expression).
siRNA transfection
Cells were transfected with siRNAs two days prior to infection, at the same time the THP-1 differentiation medium was replaced with medium without PMA. All siRNAs were used at a final concentration of 30 nM. For transfection, a 10x mix was prepared in OptiMEM containing siRNA(s) and TransIT-X2 transfection reagent (MIR 600x, Mirus) in a 1:2 stoichiometry. All siRNAs used in this study were OnTarget Plus smart pool siRNAs (Dharmacon):
Creation of new cell lines
Creation of the Dox-inducible GBP1 and caspase-8 cell lines
THP-1 ΔGBP1+Tet-GBP1 and THP-1 ΔGBP1+Tet-mH-GBP1 have been published before and the THP-1 WT+Tet-CASP8-FLAG cells were created identically using Lentiviral transductions (Fisch et al, 2019a).
To create the caspase-8-3xFLAG expressing Dox-inducible plasmid (pLenti-Tet-CASP8-3xFLAG), the empty vector backbone was digested with BamHI, CASP8 ORF was amplified from pcDNA3-CASP8 by PCR (Addgene #11817, a gift from Guy Salvesen) (Stennicke & Salvesen, 1997), 3xFLAG was amplified from lentiCRISPRv2 (Addgene #52961, a gift from Feng Zhang) (Sanjana et al, 2014) and all fragments assembled with a Gibson assembly. All primers used for cloning PCRs can be found in Table 3.
To make the cells expressing GBP1D192E, the pLenti-Tet-mCH-GBP1 and pLenti-Tet-GBP1 plasmids (Fisch et al, 2019a) were mutated using site-directed mutagenesis and transduced into the THP-1 ΔGBP1+Tet target cells using Lentiviral transduction as described before (Fisch et al, 2019a). Similarly, to make cells expressing YFP-CASP4C258S and mutated GBP1 versions, THP-1 ΔGBP1+Tet-mCH-GBP1K51A, +Tet-mCH-GBP1C589A or +Tet-mCH-GBP1Δ589-592 (Fisch et al, 2019a) were transduced with pMX-CMV-YFP-CASP4C258S (Fisch et al, 2019a) using lentiviral particles
Creation of YFP-CASP5 expressing cell line
To create catalytically-inactive caspase-5C315S, the ORF of pMX-CASP5 (kind gift of Mitchel Pallett) (Munday et al, 1995; Eckhart et al, 2006) was mutated by site-directed mutagenesis and confirmed by Sanger sequencing. Next the CASP5C315S ORF was amplified by PCR and used for Gibson assembly into XhoI digested pMX-CMV-YFP (Eldridge et al, 2017) to create pMX-CMV-YFP-CASP5C315S expression vector.
Caspase-5 encoding plasmids and the packaging plasmids pCMV-MMLV-pack (Shenoy et al, 2012) and pVSVg were transfected into HEK293T cells in serum-free DMEM using Lipofectamine 2000 (11668019, Thermo Scientific) at a ratio of 5:4:1. Medium was replaced 12 h post-transfection with fresh DMEM + 10% FBS to rest the cells. Medium was replaced again after 12 h with complete DMEM containing 5 mM sodium butyrate to boost virus production and left for another 12 h. Medium was replaced with RPMI + 10% FBS and the cells left to produce lentiviral particles for one day. Virus containing supernatant was filtered through a 0.43 μm syringe filter and supplemented with 8 μM polybrene. The THP-1 ΔGBP1+Tet-mCH-GBP1 target cells were then resuspended in 500 μL of the virus containing medium and ‘spinfected’ for 30 mins at 720 x g before being placed back in the incubator. One hour p.i. 1 mL complete medium was added, and the cells left to rest. This infection procedure was repeated for a total of three times before selection with 1 μg/mL Puromycin. Once untransduced control cells had died in the selection medium, the newly created cells were tested by immunoblotting.
Creation of myc-AIM2 expressing cell line
To create a lentiviral vector for constitutive expression of myc-AIM2, the ORF was amplified from pcDNA3-myc-AIM2 (Addgene #73958, a gift from Christian Stehlik) (Khare et al, 2014) and Gibson assembled into BstBI and BsrGI digested pLEX-MCS-ASC-GFP (Addgene #73957, a gift from Christian Stehlik) (de Almeida et al, 2015) to create pLEX-MCS-myc-AIM2. The newly made vector was then transduced into THP-1 +Tet-CASP8-FLAG cells to create THP-1 +Tet-CASP8 + myc-AIM2 cells using Lentiviral transduction as described above.
Fixed immunofluorescence microscopy
For confocal imaging 0.25×106 THP-1 cells were seeded on gelatin-coated (G1890, Sigma) coverslips in 24-well plates. Following differentiation, treatments and infection, cells were washed three times with warm PBS prior to fixation to remove any uninvaded pathogens and then fixed with 4% methanol-free formaldehyde (28906, Thermo Scientific) for 15 min at room temperature. Following fixation, cells were washed again with PBS and kept at 4°C overnight to quench any unreacted formaldehyde. Fixed specimens were permeabilized with PermQuench buffer (0.2% (w/v) BSA and 0.02% (w/v) saponin in PBS) for 30 minutes at room temperature and then stained with primary antibodies for one hour at room temperature. After three washes with PBS, cells were incubated with the appropriated secondary antibody and 1 μg/mL Hoechst 33342 (H3570, Invitrogen) diluted in PermQuench buffer for 1 hour at room temperature. Cells were washed with PBS five times and mounted using 5 μL Mowiol.
Specimens were imaged on a Leica SP5-inverted confocal microscope using 100x magnification and analyzed using LAS-AF software. For structured-illumination super-resolution imaging, specimens were imaged on a GE Healthcare Lifesciences DeltaVision OMX SR imaging system and images reconstructed using the DeltaVision software. All images were further formatted using FIJI software. 3D rendering of image stacks and distance measurements of AIM2-ASC-CASP8 inflammasome specks was performed using Imaris 8.3.1.
Quantification of protein recruitment to pathogen vacuoles and ASC speck formation
Cells seeded as above were infected with Tg MOI = 1 or STm MOI = 30 and fixed at indicated times p.i. Nuclei were stained with Hoechst33342 (H3570, Invitrogen), proteins with antibodies and appropriate secondary antibody and coverslips mounted as described above. Images were acquired using a Ti-E Nikon microscope equipped with LED-illumination and an Orca-Flash4 camera using a 60x magnification. All intracellular parasites/bacteria of 100 fields of view were automatically counted based on whether they show recruitment of the protein of interest using HRMAn high-content image analysis (Fisch et al, 2019b). Further, the analysis pipeline was used to measure the fluorescence intensity of GBP1 on STm vacuoles using the radial intensity measurement implemented in HRMAn.
For quantification of ASC speck formation, 100 Tg-infected cells were manually counted per condition using a Ti-E Nikon microscope equipped with LED-illumination using 60x magnification based on whether they contain an ASC speck and whether STm was decorated with GBP1/CASP4. The experiment was repeated independently three times.
EdU labeling for visualization of Tg-DNA release
Type I (RH) Tg were grown in fully confluent and non-replicating HFFs for 3 days in the presence of 20 µM EdU to incorporate the nucleotide into their DNA. Labelled parasites were then harvested and used for infection as described above. 6 hours p.i. cells were fixed and EdU incorporated into Tg-DNA visualized by staining the specimens using Click-iT™ EdU Cell Proliferation Kit for Imaging, Alexa Fluor™ 647 dye (C10340, Invitrogen) according to the manufacturers instruction. Coverslips were further stained with Hoechst and mounted before imaging on a Ti-E Nikon microscope equipped with LED-illumination and an Orca-Flash4 camera using a 100x magnification.
Correlative light and electron microscopy
1.25×106 THP-1 cells were seeded in a μ-Dish35 mm, high Glass Bottom Grid-500 (81168, ibidi) and differentiated with PMA as described before. Cells were then pre-stimulated with IFNγ overnight and infected with type II (Pru) Tg at an MOI of one for 6 hours. One hour prior to fixation, 1 µg/mL CellMask Deep Red (C10046, Invitrogen) and 20 µM Hoechst 33342 (H3570, Invitrogen) was added to the culture medium to label the cells for detection in fluorescence microscopy. Cells were fixed by adding warm 8% (v/v) formaldehyde (Taab Laboratory Equipment Ltd) in 0.2 M phosphate buffer (PB) pH 7.4 directly to the cell culture medium (1:1) for 15min. The samples were then washed and imaged in PB using a Zeiss AiryScan LSM 880 confocal microscope. Samples were then processed using a Pelco BioWave Pro+ microwave (Ted Pella Inc) and following a protocol adapted from the National Centre for Microscopy and Imaging Research protocol (Deerinck et al, 2010) (See Table 4 for full BioWave program details). Each step was performed in the Biowave, except for the PB and water wash steps, which consisted of two washes followed by two washes in the Biowave without vacuum (at 250 W for 40 s). All chemical incubations were performed in the Biowave for 14 min under vacuum in 2 min cycles alternating with/without 100 W power. The SteadyTemp plate was set to 21°C unless otherwise stated. In brief, the samples were fixed again in 2.5% (v/v) glutaraldehyde (Taab) / 4% (v/v) formaldehyde in 0.1 M PB. The cells were then stained with 2% (v/v) osmium tetroxide (Taab) / 1.5% (v/v) potassium ferricyanide (Sigma), incubated in 1% (w/v) thiocarbohydrazide (Sigma) with SteadyTemp plate set to 40°C, and further stained with 2% osmium tetroxide in ddH2O (w/v). The cells were then incubated in 1% aqueous uranyl acetate (Agar Scientific), and then washed in dH2O with SteadyTemp set to 40°C for both steps. Samples were then stained with Walton’s lead aspartate with SteadyTemp set to 50°C and dehydrated in a graded ethanol series (70%, 90%, and 100%, twice each), at 250 W for 40 s without vacuum. Exchange into Durcupan ACM® resin (Sigma) was performed in 50% resin in ethanol, followed by 4 pure Durcupan steps, at 250 W for 3 min, with vacuum cycling (on/off at 30 s intervals), before embedding at 60°C for 48 h. Blocks were serial sectioned using a UC7 ultramicrotome (Leica Microsystems) and 70 nm sections were picked up on Formvar-coated slot copper grids (Gilder Grids Ltd). Consecutive sections were viewed using a 120 kV Tecnai G2 Spirit transmission electron microscope (Thermo Fischer Scientific) and images were captured using an Orius CCD camera (Gatan Inc). Individual TEM images of ∼25-30 consecutive sections per Tg parasite were converted as Tiff in Digital Micrograph (Gatan Inc.) and aligned using TrakEM2, a plugin of the FIJI framework (Cardona et al, 2012). The stacks were used to check the integrity of the PV and for course alignment with the AiryScan data.
Vacuole breakage assay (HRMAn)
For quantification of Tg vacuole integrity, cells seeded in black-wall 96-well imaging plates were infected and treated as described before. One hour prior to fixation, 1 µg/mL CellMask Deep Red (C10046, Invitrogen) was added to the culture medium to load the cytosol of host cells with this fluorescent dye. Following fixation and staining with Hoechst (H3570, Invitrogen), plates were imaged at 40x magnification on a CellInsight CX7 High-Content Screening (HCS) Platform (Thermo Scientific) and 25 fields of view per well were recorded. Fluorescence of the dye within detected Tg vacuoles was then analyzed using HRMAn (Fisch et al, 2019b). Additionally, HRMAn was used to classify Tg vacuoles based on recruitment of mCH-GBP1 using the implemented neural network and the dataset stratified into decorated and non-decorated vacuoles.
Differential stain for detection of cytosolic STm
To distinguish between STm contained in vacuoles and bacteria that had escaped into the cytosol of infected macrophages, cells were differentially permeabilized using 25 µg/mL digitonin for one minute at room temperature as has been described before (Meunier & Broz, 2015). Cytosolic STm were then stained using anti-Salmonella antibody (ab35156, Abcam) that has been pre-labelled using Alexa Fluor™ 647 Protein Labeling Kit (A20173, Invitrogen) for 15 minutes at 37°C, prior to immediate fixation with 4% paraformaldehyde. Following fixation cells were permeabilized as described above and all STm were stained using the same but unlabeled antibody and corresponding Alexa-488 labelled secondary antibody. Cells were further stained with Hoechst (H3570, Invitrogen) and imaged on a Leica SP5-inverted confocal microscope using 100x magnification. For quantification, 100 fields of view per coverslip (typically >1000 individual STm overall) were acquired using a Ti-E Nikon microscope equipped with LED-illumination and an Orca-Flash4 camera at 60x magnification and analyzed with HRMAn (Fisch et al, 2019b) for colocalization of fluorescent signal of all and cytosolic STm.
Data handling and statistics
Data analysis used nested t-test, one-way ANOVA or two-way ANOVA as groups that were compared are indicated in the figure legends. Benjamini, Krieger and Yekutieli false-discovery rate (Q = 5%) based correction for multiple comparisons as implemented in Prism was used when making more than 3 comparisons. Graphs were plotted using Prism 8.1.1 (GraphPad Inc.) and presented as means of n = 3 experiments (with usually 3 technical repeats within each experiment) with error bars representing SEM, unless stated otherwise.
Supplementary Information
This is supplementary information to the preprint manuscript from Fisch et al. 2019. Supplementary section contains supplementary tables, supplementary figures and figure legends. All further information is available upon request.
Supplementary tables
Abbreviations: CST = Cell Signalling Technologies, IB = immunoblot, IF = immunofluorescence
ACKNOWLEDGEMENTS
We would like to thank Matt Renshaw for help with super-resolution SIM imaging, Julia Sanchez-Garrido for help in optimizing immunoblots and advice on reagents, the Crick High-throughput screening (HTS) STP for help in performing automated imaging experiments, the Crick Genomics and Equipment Park STP for performing sequencing and DNA minipreps for cloning. We thank all members of the Frickel and the Shenoy labs for productive discussion and Crick Core facilities for assistance in the project.
REFERENCES




