

Abstract
Annotating gene structures and functions to genome assemblies is a must to make assembly resources useful for biological inference. Gene Ontology (GO) term assignment is the most pervasively used functional annotation system, and new methods for GO assignment have improved the quality of GO-based function predictions. GOMAP, the Gene Ontology Meta Annotator for Plants (GOMAP) is an optimized, high-throughput, and reproducible pipeline for genome-scale GO annotation for plant genomes. GOMAP’s methods have been shown to expand and improve the number of genes annotated and annotations assigned per gene as well as the quality (based on F-score) of GO assignments in maize. Here we report on the pipeline’s availability and performance for annotating large, repetitive plant genomes and describe how to deploy GOMAP to annotate additional plant genomes. We containerized GOMAP to increase portability and reproducibility, and optimized its performance for HPC environments. GOMAP has been used to annotate multiple maize lines, and is currently being deployed to annotate other species including wheat, rice, barley, cotton, soy, and others. Instructions along with access to the GOMAP Singularity container are freely available online at https://gomap-singularity.readthedocs.io/en/latest/. A list of annotated genomes and links to data is maintained at https://dill-picl.org/projects/gomap/gomap-datasets/.
1 Introduction
Plant genomes are notably repetitive and hard to assemble. As such, long-read sequencing technologies have been quickly and widely adopted (Jiao et al. 2017; Rhoads and Au 2015) to enable high-quality de novo assembly of plant genomes. The number of plant long-read, whole-genome sequencing (WGS) datasets rapidly increasing (See table 1) and would lead to increased number of high-quality plant genome assemblies in near future. In order to make the best use of high-quality assemblies for functional genomics applications, improved computational tools for gene structure and function prediction must also be developed and adopted, just as imroved computational tools for de novo assemblies.
Number of long-read sequencing datasets published in the NCBI SRA database.*
Descriptive summary of input sequences from maize genomes used for GOMAP Annotations.
In 1998, the Gene Ontology (GO) consortium released the first common vocabulary describing gene function across species, thus enabling a genome-wide and comparative approach to functional genomics (Ashburner et al. 2000.) Various tools and approaches were developed to assign GO terms to genes, and a raft of statistical methods to interpret high-throughput experimental results for GO-based gene function implications were developed and released (Grossmann et al. 2007; Jiang et al. 2016; McLean et al. 2010; Radivojac et al. 2013; Young et al. 2010.) More recently, the Critical Assessment of protein Function Annotation (CAFA) competition has enticed research groups to develop tools that improve the accuracy and coverage of gene function prediction (Radivojac et al. 2013.) Unfortunately, methodologies developed through CAFA have not been widely adopted for annotating plant genomes, and existing plant-specific GO annotation pipelines mainly focus on subsets of GO terms rather than the full set of terms available (Amar et al. 2014; Zwaenepoel et al. 2018.)
We sought to assess the performance of some of the best-performing methods produced through CAFA1 for assigning gene function to plant genomes and to produce an improved functional annotation dataset for maize. These efforts were successful, with improvements to prediction outcomes measured in terms of precision, recall, and coverage (Wimalanathan et al. 2018). Obvious next steps are to generalize the developed pipeline and to apply it to additional maize lines and plant species, then to evaluate its performance for annotating gene function to those genomes. The GOMAP (Gene Ontology Meta Annotator for Plants) reported here generalizes the methods used to produce the maize-GAMER datasets, with improvements to computational performance, reproducibility, and portability.
Descriptive summary of input sequences from maize genomes used for GOMAP Annotations.
Steps to run the GOMAP-singularity container
2 Materials and Methods
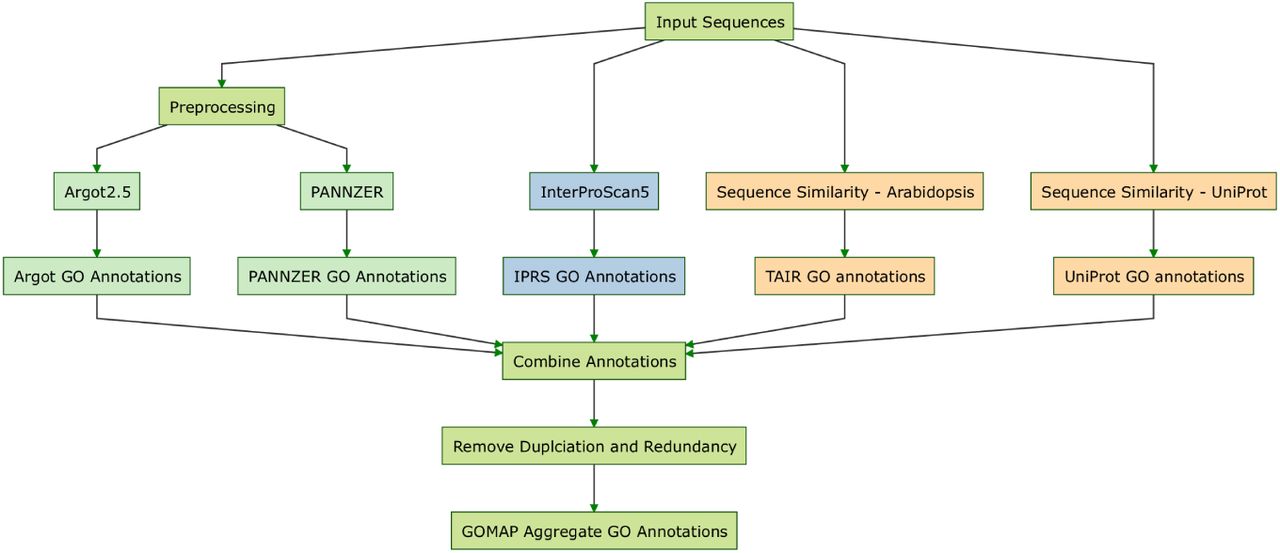
GOMAP uses sequence-similarity, domain-presence, and mixed-method pipelines to annotate GO terms to the plant protein sequences given by the user and produces a single unique and non-redundant GOMAP aggregate dataset (Figure 1). Public datasets and software tools used in GOMAP are outlined below and are kept up-to-date at https://dill-picl.org/projects/gomap/gomap-datasets/ and https://gomap-singularity.readthedocs.io/en/latest/, respectively.

Types of methods and major steps used for the GOMAP pipeline. Sequence-similarity methods are yellow, domain-presence methods are blue, and mixed-methods are green. Outputs from these methods are combined, duplicates and redundancies are removed, and an aggregate dataset is produced.
2.1 Annotation of input sequences
It is recommended that for genes with multiple transcripts, the longest translated protein sequence of individual gene models from a whole plant genome assembly be selected as the representative transcript model for the gene. Sequence similarity searches are performed against two plant datasets, Arabidopsis and UniProt. The Arabidopsis dataset contains protein sequences downloaded from TAIR and curated GO annotations Berardini et al. 2015. The UniProt dataset contains protein sequences from the top plants species that were ranked by number of curated GO annotations available in UniProt Consortium 2015a. The first set of annotations is generated using BLAST-based search to obtain reciprocal-best-hits between input and Arabidopsis sequences, and inheriting curated GO terms from Arabidopsis to the input sequence (Figure 2a; Altschul et al. 1990). The second set of annotations is obtained utilizing a similar approach, but instead of Arabidopsis the search is performed against the datasets of the 10 plant species with the most annotations in the UniProt database (based on counts of genes annotated; Figure 2b). The presence of functional domains in the input sequences (based on 14 types of protein signatures) is assessed by the InterProScan5 pipeline, and GO terms associated with those domains are assigned (Jones et al. 2014) without filtering based on any annotation scoring. Three mixed-method pipelines are additionally deployed by the pipeline to annotate GO terms to input sequences, Argot2.5, FANN-GO and PANNZER (Falda et al. 2012; Clark and Radivojac 2011; Koskinen et al. 2015.) Two of these tools require pre-processed input sequences before they can be used to annotate GO terms (Figure 3). Argot2 requires the BLAST hits of the input sequences to the UniProt database and Pfam hits identified by HMMER search against Pfam domain database (Finn, Bateman, et al. 2014; Finn, Clements, et al. 2011; Consortium 2015b.) PANNZER only requires the BLAST hits to UniProt database for the annotation process. FANN-GO is capable of performing the annotation from the input sequences. The 6 annotation datasets generated by the previous steps are aggregated and any redundancies and duplications (See Defoin-Platel et al. 2011; Wimalanathan et al. 2018 for the definitions and more details about the pipeline itself) introduced by the aggregate step is cleaned to produce a clean annotation dataset.
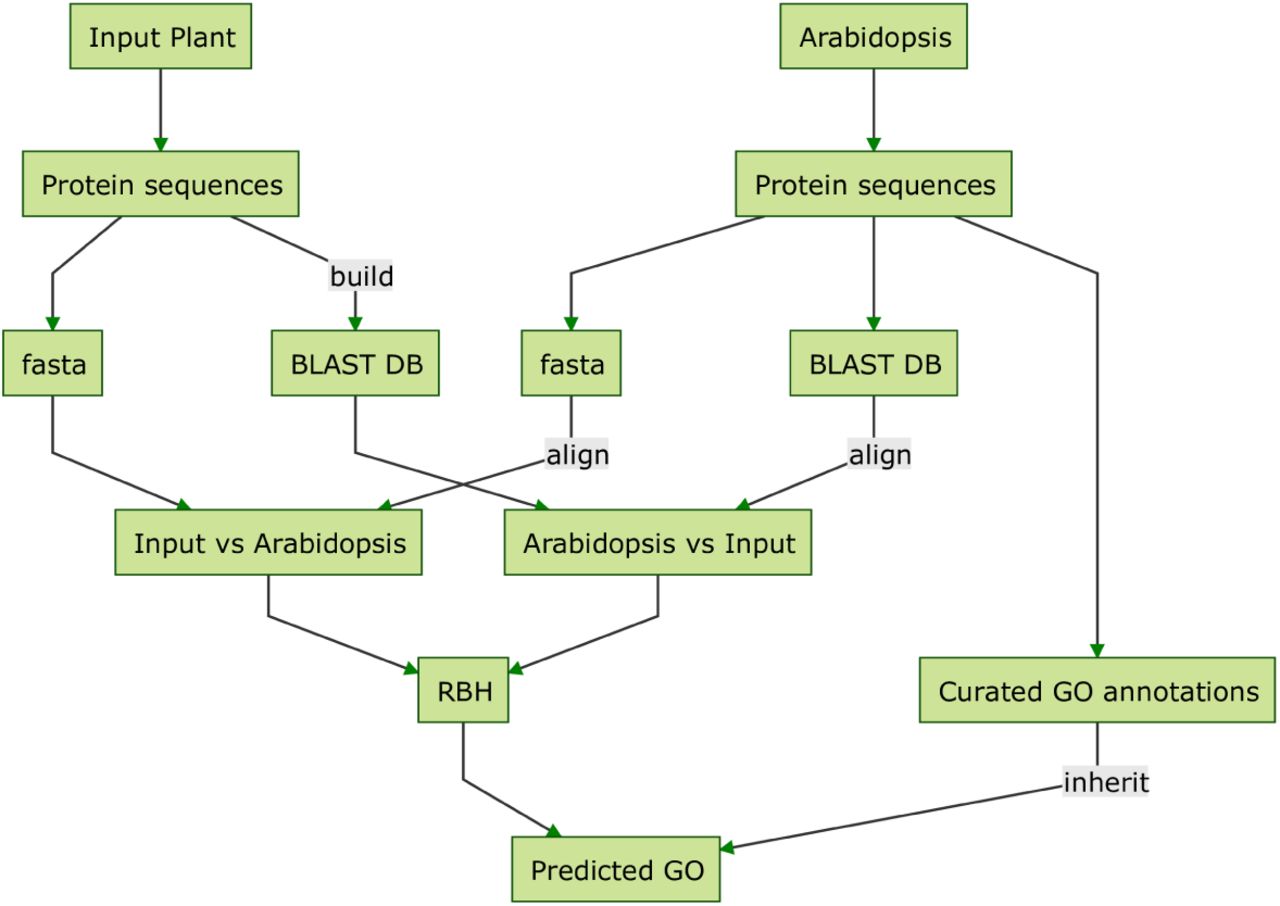
(a) GO annotations using sequence-similarity to Arabidopsis Detailed steps of the approach used to annotate GO terms to the input sequences based on sequence similarity to Arabidopsis gene sequences.

Approached similarly to the Arabidopsis dataset, but repeated independently for each plant species among the top 10 GO annotation counts in UniProt.

Detailed steps of how input sequences are annotated using the mixed-methods Argot2 and PANNZER. Input sequences are preprocessed initially by a BLAST step to UniProt and HMMER step to Pfam database. The output from the preprocessing is used by Argot2 and PANNZER for GO annotation.
2.2 Implementation and Containerization of GOMAP
The GOMAP pipeline was developed by streamlining the code from maize-GAMER and containerizing the resulting code using singularity container (Wimalanathan et al. 2018; Kurtzer et al. 2017). GOMAP is implemented using Python (to run open-source tools for annotating GO terms) and R (to aggregate and clean annotation results). GOMAP was containerized to reduce time spent in installing and configuring dependencies. This improves usability, portability and reproducibility. Singularity containerization was chosen because it works seamlessly in high performance computing (HPC) environments (Kurtzer et al. 2017), and has been widey adopted by differet HPC systems across the United States. Several issues were encountered during the process of containerizing GOMAP; the large size of the pipeline itself, the long runtime on a single machine, and use of MySQL and MATLAB for mixed-method pipelines. These were addressed as follows.
The uncompressed data required for the GOMAP pipeline use about 110 GB of local disk space. This large size is due to the inclusion of external tools and data, which results in a large container that creates issues during the development and distribution of GOMAP via free public resources. Some tools such as PANNZER were dependent on a backend MySQL database, and FANN-GO included MATLAB specific code for the annotation. These two components complicated the containerization and subsequent efforts to run GOMAP on HPC systems. The original PANNZER code was updated to use a sqlite3 database, and the sqlite3 file worked seamlessly in HPC systems without the complications to use MySQL. The FANN-GO code was updated to use open source GNU Octave and the ability to iclue Octave in the container enabled GOMAP to be run on any HPC system and completely enclose all the data and software required for GOMAP. The containers were built and are being shared via Zenodo project and can be used directly as an executable with the correct commands, but using the helper scripts on the GitHub repository is easier.
Run time for GOMAP on a single machine on a single node (via the Iowa State University HPC Condo Cluster; https://www.hpc.iastate.edu/guides/condo-2017) for 40,000 protein sequences takes between 12-14 days. To improve runtime, GOMAP was separated into different steps that run concurrently. In addition, the 2 steps that take the longest, the InterProScan search (1-2 days) and BLAST search against the UniProt sequence database (8-10 days), were parallelized to complete faster. Most HPC environments have a shorter walltime (2-5 days), so parallelizing was necessary to complete these steps within the time limit.
2.3 Results and Conclusions
3 Results
The GOMAP-Singularity container was tested by annotating GO terms to genes derived from three different maize inbred lines: Mo17, W22, and PH207 (Sun et al. 2018; Springer et al. 2018; Hirsch et al. 2016). The Pittsburgh Supercomputing Center (PSC) - Bridges HPC cluster was used to run GOMAP. Each compute node on PSC Bridges is equipped with 28-core processors and 128GB RAM. The analysis was performed on single nodes for the unparallelized steps and was run on 10 nodes for the parallelized steps. The time taken for each step is given in Table 5.5. The number of protein coding gene sequences is as follows: Mo17 had 38,620, W22 had 40,690, and PH207 had 40,557. Each dataset was slightly different from each other in terms of gene length and number of sequences. The differences in time for the pipeline to run each step are largely due to the domain step, and that difference is significant between Mo17 and W22 datasets (Figure 5.4). In addition, the differences in the time taken cannot be accounted for merely by the number of input sequences. For example, although, PH107 and W22 have the same number of input sequences, the runtime is longer for W22. The maize annotation datasets are currently available.

Comparison of runtimes for each step across the three maize genomes annotated here using GOMAP; Mo17, W22, and PH207. Each step of the GOMAP pipeline is colored as per the legend, and steps are given in the order of execution. The domain step for PH207 was calculated by averaging the times for the domain step from the other two inbreds.
We also tested the GOMAP-Singularity container’s performance by annotating GO terms to the genomes of these same three maize inbred lines (Mo17, W22, and PH207) using the Condo Cluster at Iowa State University (regular nodes) and compared that runtime to that of B73, the genome originally annotated in the first annotation created by the first version of the pipeline (reviewed in Wimalanathan et al. 2018). Each genome dataset varied in terms of gene number and assembly length with only negligible differences in runtime ??, though the total walltime taken for all the steps was between 33 and 36 hours. Notably, running steps 1-4 concurrently allows for completion within 24 hours 4. The reproducible methods used and evaluated by the maize-GAMER project have been integrated as the GOMAP pipeline that can annotate GO terms to plant proteins.
3.1 Software and Data Availability
Instructions along with access to the GOMAP Singularity container are freely available online at https://gomap-singularity.readthedocs.io/en/latest/
Genomes annotated using GOMAP are posted to https://dill-picl.org/projects/gomap/gomap-datasets/ as they are completed.
4 Discussion
Each GO annotation method used in GOMAP has its own pros and cons. Sequence similarity methods provide an easy and direct inheritance of GO terms from genes in other species. For an experimentalist, the direct link is invaluable and enables critical evaluation of the GO terms annotated and enables them to generate testable hypotheses. However, sequence-similarity methods annotate lower number of GO terms. Domain-presence methods provide a balanced approach that produce more GO annotations than sequence-similarity and are still based on sequence domains that can be used to find genes in other species. This enables critical evaluation and testable hypothesis for biological experiments. GO annotations produced by domain-presence methods, due to the higher number, are also more suitable for the interpretation of high-throughput experiments. However, both sequence-similarity and domain-presence methods only annotate GO terms to less than half the genes genome-wide. This presents a challenge when interpreting data from high-throughput experiments. State of the art mixed-methods, as such as those developed for the CAFA competition, have better or comparable prediction accuracy to other methods. Perhaps more interestingly, they provide more annotations and annotate more genes than both other methods. The higher number of annotations makes them more suitable for interpreting datasets from large-scale experiments. The utilization of advanced statistical and computational methods makes it difficult to make direct connections to genes from other species.
The tools used in GOMAP were implemented based on the evaluation with an unbiased dataset obtained from MaizeGDB. The mixed-method tools used in GOMAP were the top performing methods in the first iteration of CAFA (Radivojac et al. 2013.) Currently the second and third iteration of the CAFA competition have been completed. More cutting-edge tools have been developed in each iteration (Jiang et al. 2016.) With the use of gold-standard dataset more tools could be evaluated and integrated into GOMAP in the future. Several other pipelines exist for GO annotations, but only a few are plant-specific (Amar et al. 2014; Zwaenepoel et al. 2018.) Other plant-specific GO annotation pipelines available do not focus on predicting genome-scale GO terms, and instead mainly focus on subsets of gene ontology (Zwaenepoel et al. 2018.) GOMAP provides annotations for the all genes and all functions. Containerization of the GOMAP pipeline along with the parallelization of the most time-consuming UniProt BLAST step has enabled the pipeline to annotate new plant genomes in under two days using 10 nodes (demonstrated with the PSC Bridges cluster). A conservative estimate puts the UniProt BLAST step at approximately 7 days on PSC Bridges for approximately 40,000 sequences if the step were run on a single node compared to the 14 hours for 10 nodes. Note: splitting individual GOMAP steps has enabled concurrent execution, which makes real time completion time totals shorter than the totals shown in table ??. Not accounting for the time it takes for steps reliant on the Argot2 web server, a full annotation process should complete in under 24 hours for 40,000 input sequences. Further decreases in runtime may be achieveable if the domain and mixed method steps become parallelized in future releases.
In summary, the reproducible methods used and evaluated by the maize-GAMER project have been integrated as the GOMAP pipeline that can annotate GO terms to plant proteins. GOMAP has been successfully used to annotate GO terms to gene models from other sequenced maize inbred lines and is currently being applied to rice, cotton, wheat, and other crop genomes.
5 Acknowledgements and Funding
Thanks to: R. Walls and D. Campbell for the help generating data DOIs and hosting GOMAP data on CyVerse; N. Weeks for helping adapt FANN-GO to use GNU Octave instead of Matlab, S. Cannon, I.R. Braun, G. Kandoi, A. Jain, H. Vu, P. Joshi, D. Psaroudakis, C.F. Yanarella, and N. Weeks for testing GOMAP pipeline and valuable suggestions. Members of the Dill Plant Informatics and Computational Lab (dill-picl.org) for discussions and helpful suggestions.
This work has been supported by the XSEDE startup allocation awarded to K.W. and C.J.L-D; funding from the Iowa State University Plant Sciences Institute Faculty Scholars Program to C.J.L.D.; and funding from the National Science Foundation [IOS #1027527] to C.J.L-D.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}