

Abstract
Host-microbe interactions are crucial for normal physiological and immune system development and are implicated in a wide variety of diseases, including inflammatory bowel disease (IBD), obesity, colorectal cancer (CRC), and type 2 diabetes (T2D). Despite large-scale case-control studies aimed at identifying microbial taxa or specific genes involved in pathogeneses, the mechanisms linking them to disease have thus far remained elusive. To better identify potential mechanisms linking human-associated bacteria with host health, we leveraged publicly-available interspecies protein-protein interaction (PPI) data to identify clusters of homologous microbiome-derived proteins that bind human proteins. By detecting human-interacting bacterial genes in metagenomic case-control microbiome studies and applying a tailored machine learning algorithm, we are able to identify bacterial-human PPIs strongly linked with disease. In 9 independent case studies, we discover the microbiome broadly targets human immune, oncogenic, apoptotic, and endocrine signaling pathways, among others. This host-centric analysis strategy illuminates human pathways targeted by the commensal microbiota, provides a mechanistic hypothesis-generating platform for any metagenomics cohort study, and extensively annotates bacterial proteins with novel host-relevant functions.
Manuscript
Metagenomic case-control studies of the human gut microbiome have implicated bacterial genes in a myriad of diseases. Yet, the sheer diversity of genes within the microbiome and the lack of functional annotations have thwarted efforts to identify the mechanisms by which these bacterial genes impact host health. In the cases where functional annotations exist, they tend to refer to molecular function (e.g. DNA binding, post-translational modification) rather than their role in biological pathways1, and fewer even relate to host cell signaling and homeostasis. Obtaining a clearer idea of the health impacts of each gene has thus far required experimental approaches catered to each gene or gene function2,3.
We hypothesized that host-microbiome protein-protein interactions may underlie health status and could serve to provide additional information, through annotation of human pathways, about the role of bacteria in modulating health. Protein-protein interactions (PPIs) have revealed the mechanisms by which pathogens interact with host tissue through in-depth structural studies of individual proteins3–5, as well as large-scale whole-organism interaction screens6,7. Although there are canonical microbe-associated patterns (MAMPs) that directly trigger host-signaling pathways through pattern recognition receptors present on epithelial and immune tissues8, such as flagellin with Toll-like receptor 5 (TLR5), several recent observations have further underscored a role for commensal-host PPIs in health: An integrase encoded by several Bacteroides species binds human islet-specific glucose-6-phosphatase-catalytic-subunit-related protein (IGRP) thereby protecting against colitis9; a protease secreted by Enterococcus faecalis binds incretin hormone glucagon-like peptide 1 (GLP-1), a therapeutic target for type 2 diabetes (T2D)10; and a slew of ubiquitin mimics encoded by both pathogens11 and gut commensals12 play a role in modulating membrane trafficking.
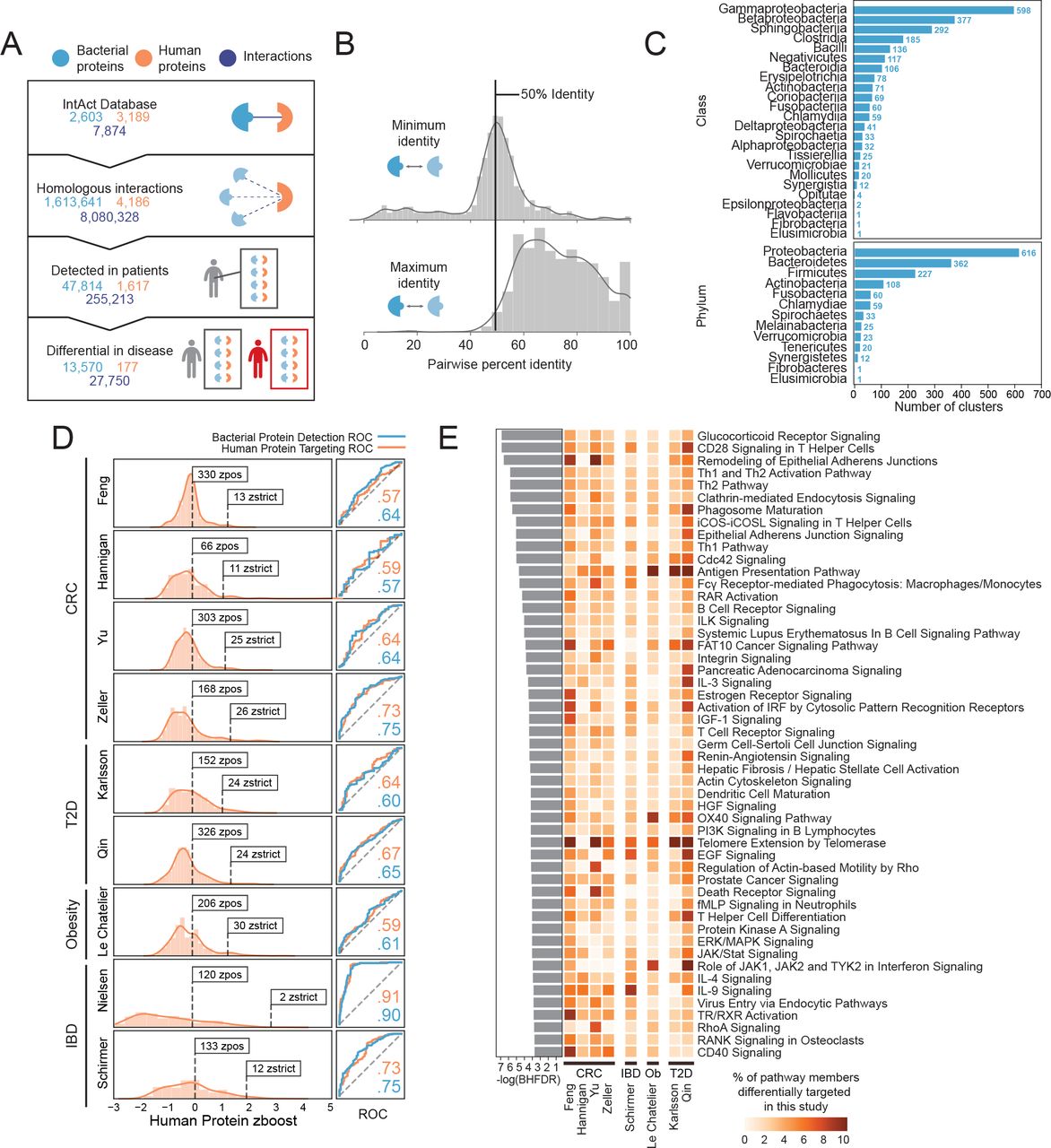
In the absence of experimental data, in silico homology modeling has been used to great effect to inform pathophysiology using inferred host-pathogen PPI networks11,13,14, but such approaches have not yet been applied to the human gut microbiome. Here, we leverage roughly 8,000 experimentally-verified binary inter-species PPIs from the IMEx Consortium members, as curated in the publicly-available IntAct database15 (Figure 1A), to gain insight into host-microbiome interactions. By propagating interactions to all bacterial proteins sharing the same UniRef homology clusters16, we expanded the set of human-microbe PPIs to include over 1.6 million bacterial proteins and 4,186 human proteins, comprising more than 8 million interspecies interactions (Figure 1A, Extended Data Figure 1).

Depiction of the interaction network inference and protein detection pipeline. Note that only bacterial proteins found to be human-interactors through the mapping procedure are used as candidates for detection in metagenomic studies.

(A) The number of interspecies bacterial proteins (blue), human proteins (orange) and interactions (dark blue) in the IntAct database; those inferred using homology clusters (UniRef); those determined to be present in the gut microbiomes from nine metagenomic studies; and those deemed important (zboost greater than zstrict, the magnitude of the minimum zboost) through our comparative metagenomic machine learning approach. If we use the zpos cutoff (zboost greater than zero), we find 40,663 important bacterial proteins (comprising 582 protein homology clusters), 1,156 important human proteins and 149,045 interactions between them. For zstrict, the bacterial proteins comprise 128 protein homology clusters.
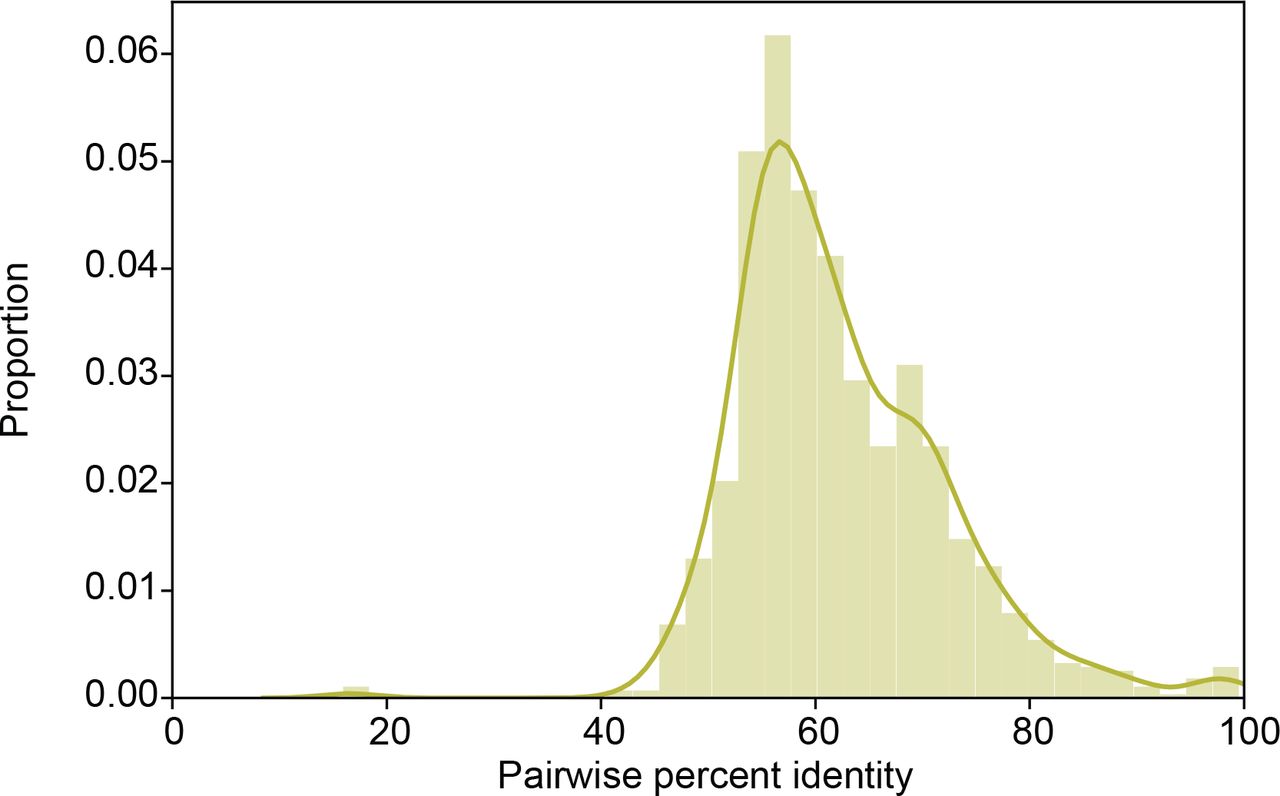
(B) Histograms showing the maximum and minimum percent identity per bacterial cluster between bacterial proteins with experimental verification and proteins detected in human microbiomes. The histograms are annotated with a gaussian kernel density estimate of the distribution.
(C) The number of bacterial clusters that include members from each bacterial phyla and class. Note that most clusters contains proteins from more than one class and phylum.
(D) Distributions of human proteins targeted in the gut microbiomes associated with each study according to their zboost scores (left). Numbers of proteins with zboost scores over zpos and zstrict are noted. Receiver-operator characteristic (ROC) curves for our random forests predictions for each dataset (right) based on bacterial (blue) proteins or their human interactors (orange), along with their corresponding AUC values.
(E) Human cellular pathways overrepresented in the zposhum subset (Benjamini-Hochberg false discovery rate (BHFDR) ≤ 0.05). −log(BHFDR) of each pathway is displayed on the barplot to the left. The heatmap is colored according to the percent of pathway members differentially targeted in each case-control cohort.
Focusing on diseases where abundant information links microbiota with disease phenotypes and where large case-control cohorts exist—namely colorectal cancer (CRC)17–20, T2D21,22, inflammatory bowel disease (IBD)23,24 and obesity25 (Extended Data Table 1)—we then mapped quality-filtered metagenomic sequencing reads from nine case-control study cohorts to our database of bacterial human-protein interactors. Using stringent detection criteria, we find roughly 255,000 potential human-bacterial interactions across the human microbiome. Inferred bacterial interactors found in the human microbiome have strong homology with proteins with experimentally-verified human interaction data (Figure 1B, Extended Data Figure 2). We applied a random forest machine learning algorithm to differentiate between cases and controls in each study based on binary detection vectors of both bacterial protein clusters and targeted human proteins. We calculated a balance-aware forest-based feature importance metric26,27 to rank the disease-association of each bacterial or human protein relative to their detection frequency, hereby called ‘zboost’.

Histogram showing the percent identity between all bacterial proteins with experimental verification and their corresponding detected proteins in human microbiomes. This histogram is annotated with a gaussian kernel density estimate of the distribution.
We noticed that a disproportionate number of bacteria-human PPIs in IntAct were derived from high-throughput screens performed on three intracellular pathogens: Yersinia pestis, Francisella tularensis and Bacillus anthracis6. Nevertheless, we find that patient-detected bacterial clusters are taxonomically diverse, not biased towards the originating classes of these three pathogens—Bacilli or Gammaproteobacteria—and rather, reflect the breadth of taxa typically associated with human gut microbiomes (Figure 1C, Extended Data Figure 3).

Histogram showing the number of species, genera, families, orders, classes and phyla for bacterial clusters with members detected in human microbiomes.
Overall, we are able to reasonably predict disease based on the detection of either bacterial or human interactors (Figure 1D). Interestingly, our approach showed greater predictive capability in some datasets over others, even for the same disease. We suspect this variation may be due to the wide range of etiologies that give rise to these diseases, as is the case for CRC, which can be driven by germ-line mutation, immune status, diet and environmental factors28. Taking these studies together, the variation between the detected human interactors across participants could not be explained purely by the health status of the individual, the specific cohort, or any other available characteristic associated with the samples (Extended Data Figure 5). The only exception was one IBD study where ethnicity correlated with disease status, and was therefore excluded from the remainder of our analyses.
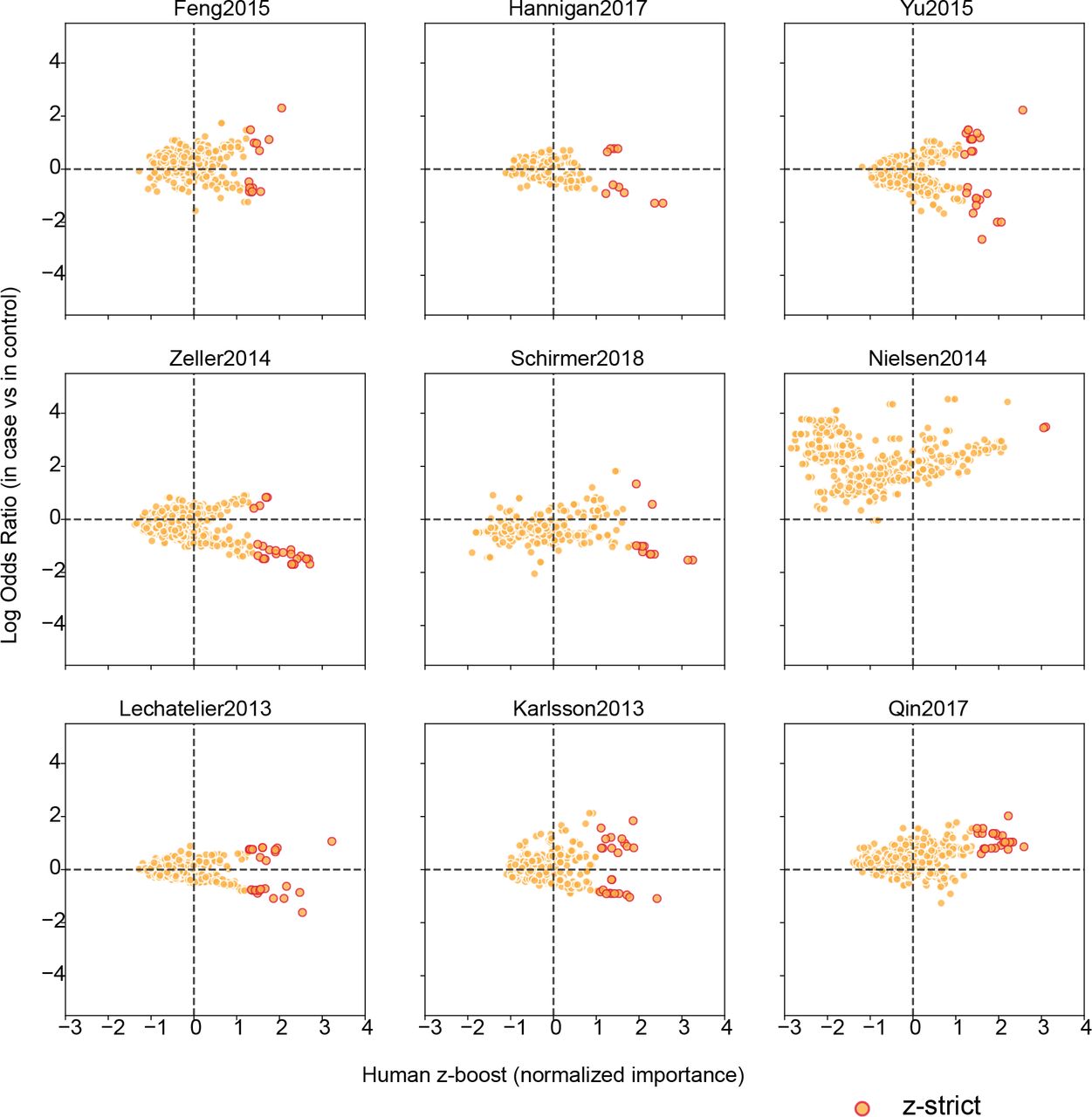
Volcano plots of the human protein interactors present in each study according to their zboost scores and log odds ratios in each case-control cohort study.

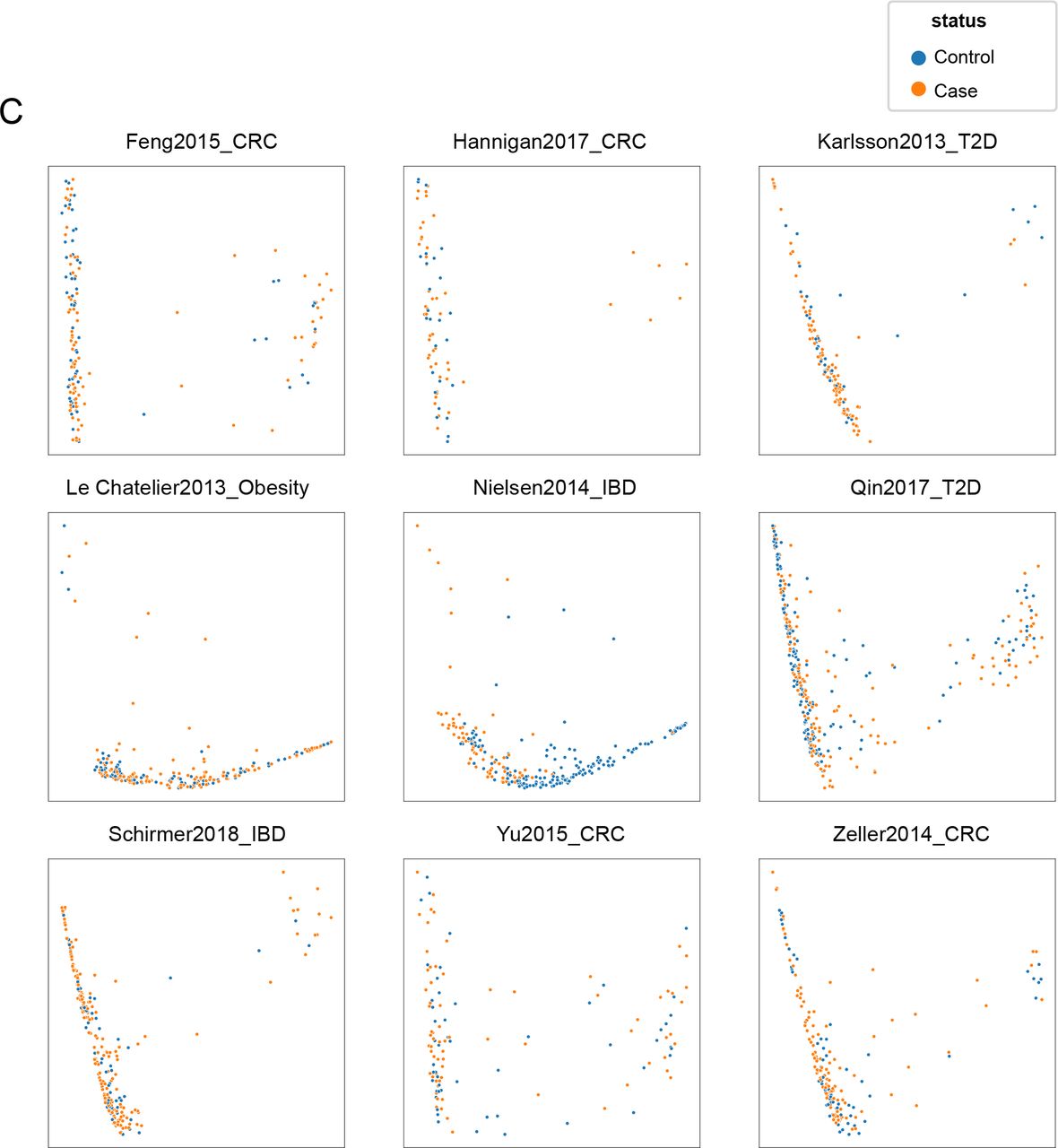
(A) Principal components analysis of detected human protein interactors for samples, according to study.
(B) Principal components analysis of detected human protein interactors for all samples in nine metagenomic studies colored by disease status according to study. Controls are all colored together in blue.
(C) Principal components analysis of detected human protein interactors in each study, separated by controls (blue) and cases (orange).
We identify subsets of important bacterial interactors and their human targets that are predictive of disease (Figure 1D; Extended Data Figure 4). We applied two thresholds to generate protein sets for analysis: those with zboosts greater than 0 (zposbact and zposhum) and those with zboosts greater than the magnitude of the minimum zboost (zstrictbact and zstricthum). Within the larger human subsets (zposhum), we find proteins with established roles in cellular pathways coherent with the pathophysiology of CRC, IBD, obesity and T2D. For example, we find that DNA fragmentation factor subunit alpha (DFFA) is important in T2D (in the Qin et al. cohort), and is involved in death receptor signaling, an important pathway for the destruction of insulin-producing β-cells29. Collagen alpha-1(I) chain (COL1A1) is also a significant target associated with T2D (in the Karlsson et al. cohort), and plays a role in dendritic cell maturation and hepatic fibrosis/hepatic stellate cell activation pathways, capturing known comorbidities between T2D and hepatic steatosis and nonalcoholic steatohepatitis (NASH)30. Proteins important in CRC studies spanned expected bacteria-associated pathways, such as the direct sensing of enterotoxins, e.g. heat-stable enterotoxin receptor GUCY2C (in the Feng et al. and Zeller et al. cohorts); but also classical cancer-associated pathways, such as the maintenance of DNA integrity, e.g. protection of telomeres protein 1 (POT1) (in the Feng et al. and also the Qin et al. cohorts) and X-ray repair cross-complementing protein 6 (XRCC6) (in the Feng et al. and Yu et al. cohorts), the latter of which is required for double-strand DNA break repair. We also find common targeting of human pathways across diseases that speak to their known shared etiologies and symptoms, for instance, actin-related protein 2/3 complex subunit 2 (ARPc2) (in the Yu et al., Schirmer et al. and Karlsson et al. cohorts), a protein involved in remodeling epithelial adherens junctions, a process strongly associated with IBD31, CRC32 and, most recently, T2D33.
These examples are illustrative of a larger trend of disease-associations driven by host-microbiome interactions. A more robust statistical analysis for overall pathway enrichment in the zposhum subset confirms significant enrichment in pathways involving the immune system, apoptosis, oncogenesis, and endocrine signaling, among others (Figure 1E). Although we see significant overlap in the pathways targeted across diseases, which may reflect their associated relative risks33–37, there is some disease-specificity. For example, more human proteins in the antigen presentation pathway are differentially targeted in T2D and obesity cohorts than elsewhere. In the CRC cohorts, more zposhum proteins target the CD40 signaling, RANK signaling in osteoclasts, and TR/RXR activation pathways than other studies.
We next sought to determine whether the human protein interactors that we associated with disease were enriched for relevant previously-reported gene-disease associations (GDA). We find many human targets associated with microbiome-related disorders, such as CRC, diabetes, autoimmune disease, obesity and IBD (Figure 2A). Although none of the cohorts we studied focused on the larger spectrum of autoimmune disease, these disorders are increasingly studied in the context of the gut microbiome38, and therefore we included them in our analysis. Interestingly, our disease annotations were ubiquitous (82.5% of the zstricthum subset had at least one GDA), but were not strictly isolated to the matching metagenomic cohort’s condition (Figure 2A, Extended Data Table 2). Across the larger zposhum subset, GDAs for these microbiome-associated disorders were enriched overall, with the exception of obesity, where annotation is generally scarce (Figure 2B). Surprisingly, in the CRC cohorts were a number of previously identified CRC-associated genetic loci, including well-known cancer genes: tumor protein p53, epidermal growth factor receptor (EGFR), matrix metalloprotease 2 (MMP2), and insulin-like growth factor-binding protein 3 (IGFBP3), among others.
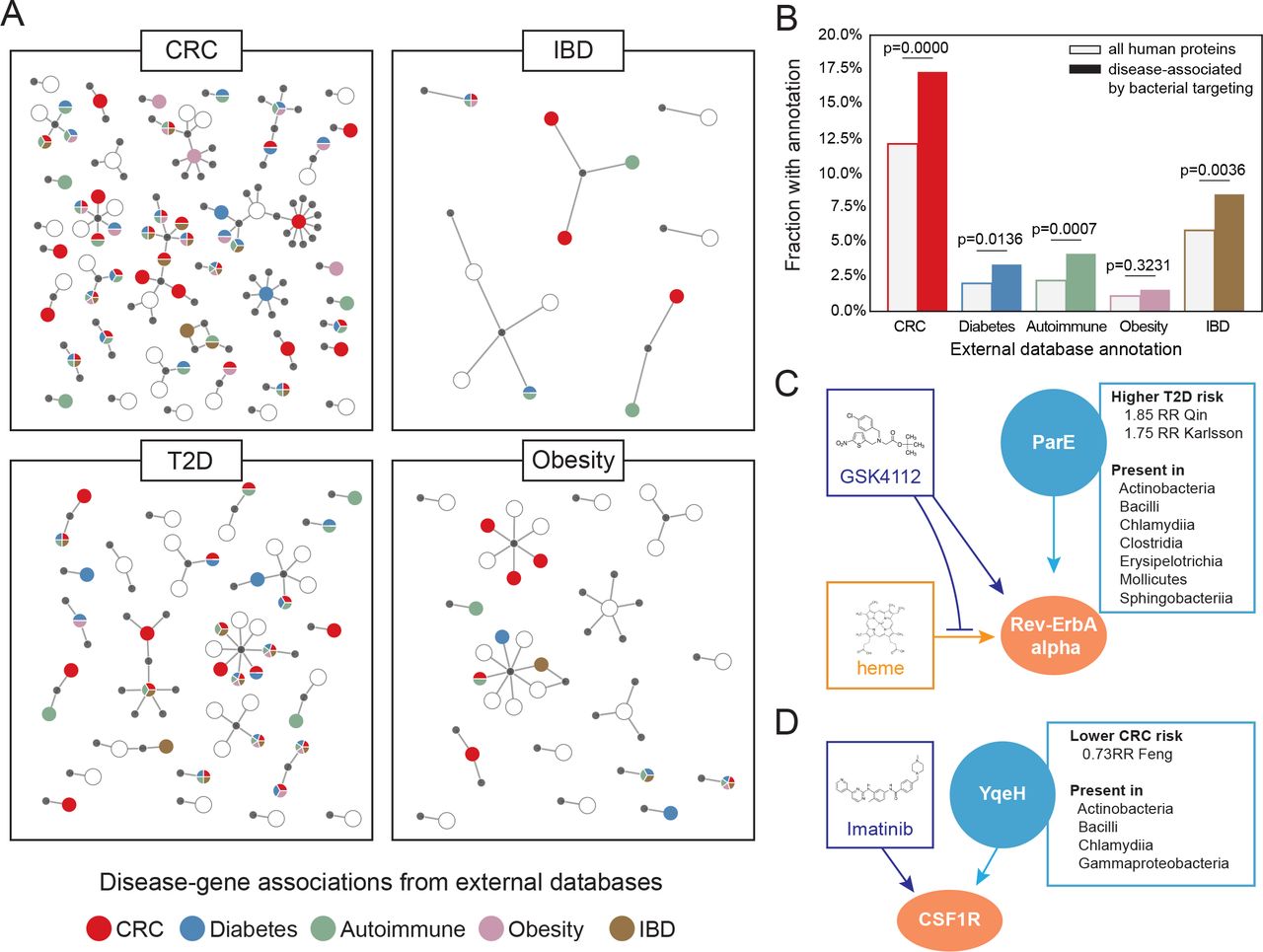
(A) Important human proteins (zstricthum) are plotted with their bacterial partners (gray), according to their disease-gene associations in the DisGeNet database: CRC (red), diabetes (blue), autoimmune disease (green), obesity (mauve) and IBD (brown).
(B) Bar chart comparing the proportions of human proteins with disease-gene associations in important human proteins (zposhum) targeted by microbiomes and all human proteins in DisGeNet.
(C) RevErbA alpha (NR1D1) binds several human proteins (not shown), DNA (not shown) and heme. GSK4112 competitively binds Rev-ErbA alpha, inhibiting binding with heme. ParE is a microbiome protein present in a diverse range of organisms and has a high relative risk associated with T2D.
(D) Macrophage colony stimulating factor 1 receptor (CSF1R) is targeted by imatinib, among other drugs, as well as the uncharacterized bacterial protein YqeH, a protein that has a low relative risk associated with CRC.
Our data suggest many molecular mechanisms that might be regulating human cellular functions through bacterial proteins. In order to better contextualize these mechanisms, we asked whether any of the human proteins in our dataset were already known to be drug targets. Using the Probes & Drugs database39, we find many zstricthum proteins are targeted by drugs (Extended Data Table 3). In many cases, those drugs are known to either treat or affect the pathogenesis of the microbiomes of patients with those diseases. For example, in both T2D cohorts, we found elevated zboost scores associated with human protein Rev-ErbA alpha (NR1D1), the target of the drugs GSK4112, SR9009 and SR9011, which inhibit the binding of Rev-ErbA alpha with its natural ligand, heme (Figure 2C). These drugs have been shown to affect cellular metabolism in vitro and affect hyperglycaemia when given to mouse models of metabolic disorder40,41.
We also find instances where our analysis of a particular disease cohort is consistent not with the therapeutic purpose of a drug targeted by human interactors in that microbiome, but with off-label effects or side effects associated with the drug. For example, we find that imatinib mesylate (brand name: Gleevec), has several human binding partners, including macrophage colony-stimulating factor 1 receptor (M-CSF1R) (Figure 2D), an important target found in CRC (in the Feng et al. cohort), and platelet-derived growth factor receptor-β (PDGFR-B), an important target found in the obesity and T2D (in the Le Chatelier et al. and Qin cohorts, respectively). Literature on imatinib supports these findings: although imatinib is best known as a treatment for leukemia, it has been shown to affect glycemic control in patients with T2D42. Furthermore, imatinib can also halt the proliferation of colonic tumor cells and is involved generally in inflammatory pathways, through its inhibition of TNF-alpha production43.
One of the major advantages of our work is that through homology mapping, we vastly improve our overall ability to annotate host-relevant microbiome functions. When we annotated the microbial pathways using KEGG (Kyoto Encyclopedia of Genes and Genomes)44, we found that 41.2% of the zposbact protein clusters found in human microbiomes lacked any pathway information (Figure 3A, B). Yet, these genes can now be annotated according to the pathways of their human targets, obtaining a putative disease-relevant molecular mechanism (Figure 3A). This host-centric annotation is useful beyond large-scale analysis of metagenomic data, but it broadly enables hypothesis-driven research into how these microbial proteins impact host health.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Paired stacked bar plots showing the disease-associated bacterial cluster pathways annotated by KEGG (left) and their inferred pathways according to the human proteins they target (right), as annotated by WikiPathways51.
(B) Human pathways (annotated using WikiPathways) targeted by disease-associated bacterial clusters. The 75 human pathways with the most previously unannotated bacterial targeters (annotated using KEGG) are shown.
(C) The number of zposbact clusters plotted according to their transmembrane and secretion predictions, i.e. type 3 or type 4 secretion systems (T3SS or T4SS), and/or the presence of eukaryotic-like domains (ELDs).
We examined the means by which bacterial proteins may be interacting with host proteins and found that a majority of bacterial protein clusters (90.2% of zposbact) contain proteins that are transmembrane, are secreted by type 3 or type 4 secretion systems, and/or contain eukaryotic-like domains (Figure 3C), another marker for secretion. Of particular interest were bacterial proteins in this subset that have well-known core functions, e.g. protein chaperones DnaK and GroL, RNA polymerases RpoB and RpoC, and canonical glycolysis enzymes, among others. A number of these proteins have been previously identified as ‘moonlighting’ proteins, which perform secondary functions in addition to their primary role in the cell45. Mycoplasma pneumoniae DnaK and enolase, a protein involved in glycolosis, from a number of pathogens, bind to both human plasminogen and extra-cellular matrix components46,47. Mycobacterium tuberculosis DnaK signals to leukocytes causing the release of the chemokines CCL3-548. Streptococcus pyogenes glyceraldehyde-3-phosphate dehydrogenase (GAPDH), another protein involved in glycosylis, can be shuffled to the cell surface where it plays a role as an adhesin, and can also contribute to human cellular apoptosis49. These examples widely illustrate how bacterial housekeeping proteins are used by pathogens to modulate human health. In this study, we uncover commensal proteins that have ‘interspecies moonlighting’ functions, which are not constrained to pathogenic organisms, but are pervasive throughout our indigenous microbiota.
Here, we reveal for the first time an extensive host-microbiome PPI landscape. This work highlights the myriad host mechanisms targeted by the gut microbiome and the extent to which these mechanisms are targeted across microbiome-related disorders. However, this network is far from complete. Few of the interaction studies on which this interaction network is based were performed on commensal bacteria and therefore, we may be missing interactions specific to our intimately associated bacteria. In addition to large-scale PPI studies involving commensal bacteria and their hosts, further in-depth studies will be needed to fully characterize these mechanisms, such as whether these bacterial proteins activate or inhibit their human protein interactors’ pathways.
This platform enables a high-throughput glimpse into the mechanisms by which microbes impact host tissue, allowing for mechanistic inference and hypothesis generation from any metagenomic dataset. Much as recent studies have uncovered the mechanistic roles of commensal-derived small molecules in disease50, we shed light on a greater role for commensal-derived proteins. By focusing on proteins, our methods connect pharmacology, human genetic variation and microbiome diversity through tangible mechanisms, owing to the large amount of existing data on human proteins. Pinpointing those microbe-derived proteins that interact directly with human proteins will pave the way for novel diagnostics and therapeutics for microbiome-driven diseases, more nuanced definitions of the host-relevant functional differences between bacterial strains, and a deeper understanding of the co-evolution of humans and other organisms with their commensal microbiota.
Extended Data Tables
Extended Data Table 1. Metagenomic studies used in this research. For each study, we list its focus, the labels in the cohort study, the patient count for each of the labels, how we grouped cases and controls, the number of detected bacterial clusters and inferred human interactors, and the number of important bacterial and human proteins, passing each of our thresholds: zpos (zboost greater than zero) and zstrict (zboost greater than the magnitude of the minimum zboost).
Extended Data Table 2. Important human interactors that have known gene-disease associations. Listed are the important zstricthum proteins with gene-disease associations in DisGeNet, along with the study in which they are found to be important.
Extended Data Table 3. Important human interactors that are known drug targets. For each human protein in the zsighum subset, we list the drug interactor and the study in which it was found to be important.
Methods
Building a putative bacteria-human protein-protein interaction (PPI) network
Interactions were downloaded from the IntAct database [August 2018]. Only interactions with evidence codes that indicated binary, experimental determination of the interaction between UniProt identifiers with non-matching taxa were preserved, thereby excluding co-complex associations, small molecule interactions, and predicted interactions. This resulted in a set of 296,103 interspecies PPIs. Interspecies protein interactors were mapped to their UniRef sequence clusters at the 100%, 90%, and 50% identity- to-seed levels, which are publicly available through the UniProt web service. Given two UniRef homology clusters with a known PPI between their members, we map that interaction to all combinations of members from the two clusters. We perform this mapping at all levels of homology (and their combinations). From this large list of putative PPIs, we store only interactions between bacterial proteins and reviewed SwissProt human proteins. The latter step avoids the over-annotation of human isoforms or homologs, or non-verified human proteins. Overall we generate 8,808,328 bacteria-human PPIs involving 1,613,641 bacterial proteins and 4,186 reviewed human proteins. This corresponds to 18,097 interactions between 33,123 UniRef clusters containing bacterial proteins and the aforementioned 4,186 reviewed human proteins.
Detection of human-targeting proteins in metagenomic shotgun sequencing data
Reads from nine metagenomic studies (Extended Data Table 1) were downloaded from the Sequence Read Archive (SRA) using fasterq-dump. Reads belonging to more than one replicate from the same patient were concatenated and treated as a single run. Reads were then dereplicated using prinseq (v0.20.2) and trimmed using trimmomatic (v0.36) with the following parameters:
Dereplication perl prinseq-lite.pl -fastq {1} -fastq2 {2} \ -derep 12345 -out_format 3 -no_qual_header \ -out_good {3} -out_bad {4}; {1,2} Refer to paired read input files {3,4} Refer to output filepaths
Trimming java -Xmx8g -jar trimmomatic-0.36.jar \ PE -threads 5 {1} ILLUMINACLIP:{2}:2:30:10:8:true \ SLIDINGWINDOW:4:15 LEADING:3 TRAILING:3 MINLEN:50 {1} Refer to input files {2} Is the path to a fasta file of Nextera TruSeq adapters
Paired reads were combined into a single file and aligned to a protein library of all 1,613,641 human-interacting bacterial proteins generated above. This read-to-protein alignment was performed using blastx through the diamond command line tool (v0.9.24.125). Read alignments were filtered to only consider results with an identity of at least 90% and no gaps. Bacterial proteins were considered detected with sufficient depth and coverage: more than 10 reads across 95% of the protein sequence, excluding 10 amino acids at each terminus. We assign any bacterial protein detection to its corresponding UniRef homology cluster. Human-interacting bacterial clusters are marked as either ‘detected’ or ‘not detected’ for each patient in each study. For each patient, we also generate a file of human proteins that are targeted by their detected bacterial proteins based on our bacteria-human PPI network.
Prioritization of disease-associated bacterial protein clusters and human targets
Each patient from each study can be represented as either (a) a binary vector of detected bacterial protein clusters or (b) a binary vector of targeted human proteins. We removed human proteins that were considered redundant based on the same exact bacterial protein partners in our database. We used either the bacterial protein clusters or the human interactors to separate case and control cohorts using a random forest machine learning algorithm. Though classically we could simply extract the average Gini coefficient of the trained random forest and use that as a proxy for feature importance, binary labels introduce a complication: the balance of each feature can limit or inflate the Gini coefficients for that feature. In order to avoid for this complication we use an empirical normalization method similar to previous work in the field, that we call zboost.
zboost algorithm
(1) Fit a random forest with 100 estimators (X=protein detection per patient, y=case/control labels), then extract and store the average Gini coefficient for each feature 
(2) For each feature, generate a random binary vector with similar balance, where each protein detection in each patient is a Bernoulli trial where:

(3) Fit a random forest with 300 estimators (X=random protein detection per patient, y=case/control labels), then extract and store the average Gini coefficient for each feature 
(4) Calculate the zboost of each feature as:

(5) Calculate the width of the zboost distribution, given as:

(6) Repeat 1-5 until the width of the zboost distribution does not increase for 200 iterations.
An extra step of filtering is applied to avoid uninformative proteins (too rarely detected, or ubiquitous): any proteins where the minimum value in their expected contingency matrix (case/control vs. detected/not-detected) is less than 5 is removed from consideration. We apply the zboost algorithm to both the bacterial-protein and human-protein binary representations of each patient for all 9 studies. Additionally, we measure our performance on these tasks by training a separate random forest, with 200 estimators, using 5-fold cross-validation.
This work was implemented and applied to our datasets using python (v3.7.3), pandas52 (v0.25.1) and the scikit-learn53 library (v0.21.3). We used two thresholds when conducting analysis on the resulting data: zpos, where zboosts must be greater than zero, and zstrict, where zboost must be greater than the absolute value of the lowest zboost in that learning task.
The high performance in the Nielsen et al. study, along with the lack of significant proteins using zboost (only two passing the zstrict threshold), led to the exclusion of this study from further analysis, as we believe that the signal is driven by the demographic differences between cases and controls in this particular study. No available metadata explained the variation in the other metagenomic studies.
Identity measurements
For each bacterial cluster, we compared the sequence identity between the bacterial proteins with experimentally verified interactions with human proteins and the bacterial proteins detected in human microbiomes from the same UniRef cluster. Original interactors and their homologs were aligned using Smith-Waterman local alignment with a BLOSUM62 matrix via python’s parasail library (v.1.1.17). The identity was calculated as the number of exact matches in the alignment, divided by the total number of alignment columns. Note that this denominator results in an under-estimation of the identity relative to UniRef’s cluster identities.
Pathway enrichment analysis, disease and functional annotations
We performed pathway enrichment analysis using QIAGEN’s Ingenuity Pathway Analysis (IPA)54 tool. All zposhum proteins were uploaded as UniProt identifiers into the interface. Core Enrichment Analysis was conducted on all human tissue and cell lines from all data sources under IPA’s stringent evidence filter. Pathways were considered enriched if they had both a −log(p-value) ≥ 1.3 and a Benjamini-Hochberg False Discovery Rate less or equal to 5%.
Disease annotations were extracted from all of gene-disease associations from DisGeNet (v.6.0). Lacking a simple hierarchy of disease, we binned similar disease terms into the 5 larger categories:
CRC: Adenocarcinoma of large intestine, Hereditary non-polyposis colorectal cancer syndrome, Hereditary nonpolyposis colorectal carcinoma, Malignant neoplasm of colon stage IV, Malignant neoplasm of sigmoid colon, Malignant tumor of colon, Microsatellite instability-high colorectal cancer,
Diabetes: Brittle diabetes, Familial central diabetes insipidus, Fibrocalculous pancreatic diabetes, Gastroparesis due to diabetes mellitus, Insulin resistance in diabetes, Insulin-dependent but ketosis-resistant diabetes, Insulin-dependent diabetes mellitus secretory diarrhea syndrome, Insulin-resistant diabetes mellitus, Insulin-resistant diabetes mellitus at puberty, Latent autoimmune diabetes mellitus in adult, Macroalbuminuric diabetic nephropathy, Maturity onset diabetes mellitus in young, Maturity-onset diabetes of the young, type 10, Maturity-onset diabetes of the young, type 11, Microalbuminuric diabetic nephropathy, Moderate nonproliferative diabetic retinopathy, Monogenic diabetes, Neonatal diabetes mellitus, Neonatal insulin-dependent diabetes mellitus, Non-insulin-dependent diabetes mellitus with unspecified complications, Nonproliferative diabetic retinopathy, Other specified diabetes mellitus, Other specified diabetes mellitus with unspecified complications, Pancreatic disorders (not diabetes), Partial nephrogenic diabetes insipidus, Prediabetes syndrome, Proliferative diabetic retinopathy, Renal cysts and diabetes syndrome, Severe nonproliferative diabetic retinopathy, Transient neonatal diabetes mellitus, Type 2 diabetes mellitus in nonobese, Type 2 diabetes mellitus in obese, Type 2 diabetes mellitus with acanthosis nigricans, Visually threatening diabetic retinopathy, diabetes (mellitus) due to autoimmune process, diabetes (mellitus) due to immune mediated pancreatic islet beta-cell destruction, diabetes mellitus risk, idiopathic diabetes (mellitus), postprocedural diabetes mellitus, secondary diabetes mellitus NEC
Autoimmune: Addison’s disease due to autoimmunity, Adult form of celiac disease, Aneurysm of celiac artery, Ankylosing spondylitis, Ankylosing spondylitis and other inflammatory spondylopathies, Arteriovenous fistulas of celiac and mesenteric vessels, Blood autoimmune disorders, Bullous systemic lupus erythematosus, Chilblain lupus 1, Dianzani autoimmune lymphoproliferative syndrome, Dilatation of celiac artery, Hyperthyroidism, Nonautoimmune, Latent autoimmune diabetes mellitus in adult, Maternal autoimmune disease, Multiple sclerosis in children, Neonatal Systemic lupus erythematosus, Subacute cutaneous lupus, Systemic lupus erythematosus encephalitis, Venous varicosities of celiac and mesenteric vessels, Warm autoimmune hemolytic anemia, diabetes (mellitus) due to autoimmune process, lupus cutaneous, lupus erythematodes
Obesity: Abdominal obesity metabolic syndrome, Adult-onset obesity, Aplasia/Hypoplasia of the earlobes, Childhood-onset truncal obesity, Constitutional obesity, Familial obesity, Generalized obesity, Gross obesity, Hyperplastic obesity, Hypertrophic obesity, Hypoplastic olfactory lobes, Hypothalamic obesity, Moderate obesity, Overweight and obesity, Overweight or obesity, Prominent globes, Simple obesity, Type 2 diabetes mellitus in nonobese, Type 2 diabetes mellitus in obese
IBD: Acute and chronic colitis, Acute colitis, Allergic colitis, Amebic colitis, Chronic colitis, Chronic ulcerative colitis, Crohn Disease, Crohn’s disease of large bowel, Crohn’s disease of the ileum, Cytomegaloviral colitis, Distal colitis, Enterocolitis, Enterocolitis infectious, Eosinophilic colitis, Food-protein induced enterocolitis syndrome, Hemorrhagic colitis, IIeocolitis, Infectious colitis, Left sided colitis, Necrotizing Enterocolitis, Necrotizing enterocolitis in fetus OR newborn, Neonatal necrotizing enterocolitis, Non-specific colitis, Pancolitis, Pediatric Crohn’s disease, Pediatric ulcerative colitis, Perianal Crohn’s disease, Typhlocolitis, Ulcerative colitis in remission, Ulcerative colitis quiescent
We annotated bacterial protein clusters with their corresponding KEGG pathways by blasting all detected bacterial proteins of interest against the KEGG prokaryotes peptide file using blastp. Results had an identity ≥43.9% and e-values below 0.00067.
Human pathway annotation was performed using the mygene python library. Specifically, we queried pathway annotations from Wikipathways.
We submitted our bacterial sequences to EffectiveDB55 in order to obtain predictions for EffectiveT3 (type 3 secretion based on signal peptide), T4SEpre (type 4 secretion based on composition in C-terminus), EffectiveCCBD (type 3 secretion based on chaperone binding sites), and EffectiveELD (predicts secretion based on eukaryotic-like domains). We used the single default cutoffs for T4SEpre, EffectiveCCBD, and EffectiveELD, and chose the ‘sensitive’ cutoff (0.95) rather than the ‘selective’ (0.9999) cutoff for EffectiveT3. Transmembrane proteins or signal peptides were predicted using TMHMM56 (v.2.0c), with a threshold of 19 or more expected number of amino acids in transmembrane helices.
Drug target information was extracted from probes-and-drugs (04.2019 database dump). Bacterial taxonomy information was extracted from NCBI. UniProt identifiers and annotations were downloaded using UniProt SPARQL endpoint.
Statistics
For Figure 2B, p-values for the difference in the proportion of DisGeNet proteins and disease-associated proteins was calculated through a chi squared test (dof=1): The total number of DisGeNet and disease-associated proteins is 17,549 and 767 respectively. For the labels CRC, Diabetes, Autoimmunity, Obesity, and IBD we find {2128, 355, 420, 195, and 1,029} DisGeNet genes against {133, 26, 34, 12, and 65} disease-associated genes respectively. This corresponds to a chi squared statistic of 2.2e-5 (p=0.000022) for CRC, 1.4e-2 (p=0.013619) for Diabetes, 5.9e-4 (p=0.000587) for Autoimmunity, 0.3 (p=323067) for Obesity, and 3.6e-3 (p=0.003627) for IBD.
Footnotes
Conflict of Interest Provisional patents have been filed for both the process described in this paper and therapeutic/diagnostic protein candidates found through this process through Cornell University. Inventors: Ilana Brito and Juan Felipe Beltrán.
References




