

Abstract
Secretory antibody responses (Immunoglobulin A, IgA) against repetitive bacterial surface glycans, such as O-antigens, can protect against intestinal pathogenic bacteria. However, vaccines that rely predominantly on secretory IgA for protection against non-Typhoidal salmonellosis often fail. Here we demonstrate that a major contributor to this failure is rapid immune escape, due to strong selective pressure exerted by high-avidity intestinal IgA. Interestingly, we found that IgA-escape initially occurs via a predictable narrow spectrum of Salmonella O-antigen variants that are fitness-neutral in naïve hosts. This could be attributed both to phase-variation, and to loss-of-function mutations in O-antigen-modifying enzymes. Via a vaccination regimen that simultaneously induced IgA against all observed O-antigen variants, rapid bacterial evolution could be switched from a hindrance into an advantage. Here, IgA generated a selective pressure resulting in fixation of mutations causing loss of polymerized O-antigen. When transmitted into naive hosts, these short O-antigen variants display compromised fitness and virulence, i.e. IgA-mediated pressure generates an evolutionary trade-off. Rational induction of IgA specificities that set “evolutionary traps” could reduce virulent enteropathogen reservoirs, even when sterilizing immunity cannot be achieved. This may become a powerful tool in the management of increasingly drug-resistant enteropathogenic bacteria.
One sentence summary Intestinal IgA responses that recognize all rapidly-evolvable O-antigen variants of a Salmonella Typhimurium strain can drive evolutionary trade-offs for the pathogen.
Main text
While Typhoid fever-causing Salmonella strains can be targeted by either glycoconjugate or live-attenuated vaccines(1, 2), similar approaches have so far failed to generate a licensed vaccine against non-Typhoidal salmonellosis (NTS) in humans(3). Decades of excellent research into live-attenuated vaccines in the mouse model of typhoid fever and invasive NTS have demonstrated protection with complex underlying mechanisms including Immunoglobulin A (IgA (4–6)), T-cell mediated immunity(7, 8), mouse strain-dependent mechanisms(6), and immune-independent niche competition in the gut lumen and tissues(9, 10). However, the ability of attenuated NTS strains to grow rapidly in the gut lumen(11) is associated with prolonged intestinal shedding which may contribute to the poor safety profile described in human vaccine studies(12). In contrast, inactivated oral vaccines and glycoconjugate vaccines have a good safety profile, and can be applied in high-risk groups such as infants, pregnant women and people with immunodeficiencies(13–16). Notably, in the murine streptomycin model of NTS, inactivated oral vaccines protected predominantly via induction of high-avidity O-antigen-specific secretory IgA (17, 18). However, inactivated vaccines are typically associated with weak protection(18, 19). Here we reveal that rapid IgA-driven within-host evolution of NTS strains in the gut lumen is both a contributor and a possible solution to the poor performance of inactivated S.Tm vaccines.
O-Antigen, the long repetitive glycan portion of S-form lipopolysaccharide(20) (LPS), thickly carpets the surface of Salmonella enterica subspecies enterica serovar Typhimurium (S.Tm) (Fig. S1) in the gut lumen. These glycans are sufficiently long to shield non-protruding outer membrane proteins (e.g. most membrane channels(21–23)) from antibody binding(17). Therefore high-affinity intestinal IgA against O-antigen, induced by vaccination or infection(9, 24, 25), is a dominant mechanism driving clumping by enchained growth and agglutination(18). As clumped bacteria are unable to approach the gut wall, this phenomenon provides protection from disease(18, 26, 27).
We initially set out to investigate why IgA-mediated protection fails in non-Typhoidal salmonellosis. Conventional “specific opportunistic pathogen-free” (SOPF; harboring a complex microbiota, 16S amplicon analysis available in (28)) mice were vaccinated with a high-dose peracetic acid inactivated oral S.Tm vaccine (PA-S.Tm). This vaccine is prepared from wild-type S.Tm SL1344 and we have previously demonstrated that it protects the host predominantly via high-avidity O-antigen-binding IgA(18). Four weeks post-vaccination, these mice were antibiotic-treated to generate an open niche in the large intestine, and were subsequently infected with wild-type S.Tm SL1344. Vaccinated mice sporadically developed disease, involving both intestinal inflammation (quantified via fecal lipocalin 2, Fig. 1A) and tissue invasion (mesenteric lymph node colony forming units, CFU), Fig. 1B). Strikingly, disease did not correlatewith IgA titres specific for the wild-type vaccination strain, i.e. occurred despite robust high-titre vaccine-specific secretory IgA induction (Fig. 1C).
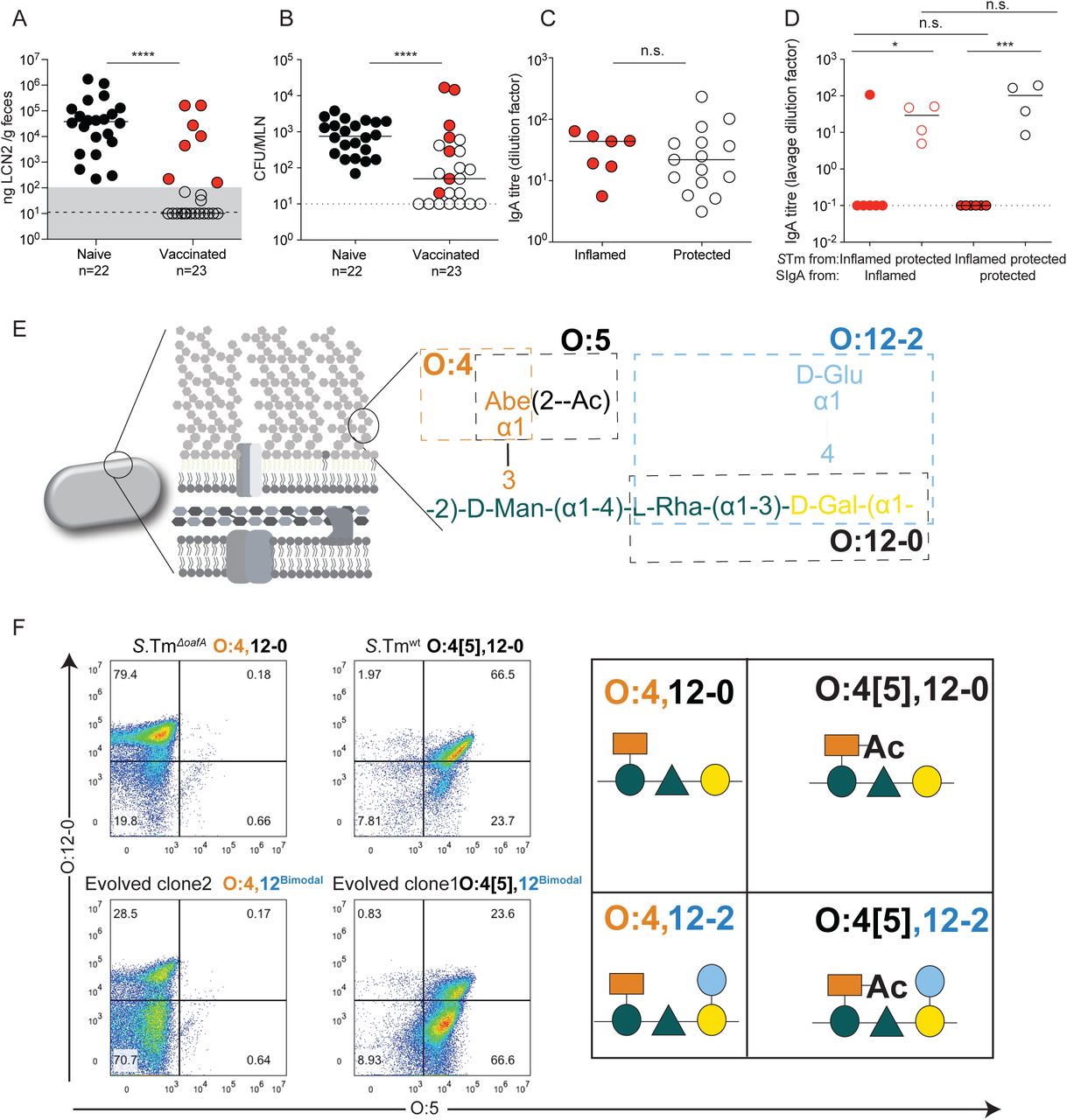
A-C: Naïve (n=22) or PA-S.Tm-vaccinated (Vaccinated, n=23) SOPF mice were streptomycin-pretreated, infected (105 S.Tmwt Colony forming units (CFU) per os) and analyzed 18 h later. A. Fecal Lipocalin 2 (LCN2) to quantify intestinal inflammation, B. Pathogen loads (CFU) in mesenteric lymph nodes (MLN), C. Intestinal IgA titres against S.Tmwt determined by flow cytometry, for vaccinated mice with LCN2 values below (open symbols, protected) and above (filled symbols, inflamed) 100ng/g. p=0.61 by Mann Whitney U test. D. SOPF mice vaccinated and infected as in A-C for 3 days. Titre of intestinal lavage IgA from “inflamed vaccinated” mouse (red borders) or a “protected vaccinated” mouse (black borders) against S.Tm clones re-isolated from the feces of the “inflamed vaccinated” mouse (red filled circles) or “protected vaccinated” mouse (open circles) at day 3 post-infection. 2-way ANOVA with Bonferroni post-tests on log-normalized data. Clones and lavages from n=1 mouse, representative of 9 “vaccinated but inflamed” and 13 “vaccinated protected” mice, summarized in Table S4. E. Schematic of the O-antigen of S.Tm (O:4[5],12), and its common variants (O:4,12_2), coloured to correspond to the “Symbol Nomenclature for Glycans”. F. Overnight cultures of the indicated S.Tm strains and evolved clones arising during infections with S.Tmwt were stained with anti-O:5 and anti-O:12-0 antibodies, followed by fluorescent secondary reagents. Representative flow cytometry analyses of the different O-antigen types, and the “Symbol Nomenclature for Graphical Representations of Glycans”(55) representation of the O-antigen repeat structure present on S.Tm in each quadrant of the flow cytometry plots.
As the IgA response was robust, with higher titres than those observed with live-attenuated vaccines (Fig. S2), we investigated the phenotype of S.Tm clones after growth in the gut lumen of infected mice. Notably in the streptomycin mouse model of NTS, protection is independent of intestinal colonization, i.e. up to 48h post-infection the gut luminal S.Tm population size is similar in both vaccinated protected and vaccinated inflamed mice, with around 109 CFU per gram feces (18). When intestinal IgA from protected or inflamed vaccinated mice was titred against S.Tm clones reisolated from the feces of these mice, a very clear picture emerged. Clones isolated from the feces of protected mice were well-recognised by IgA from both groups, but IgA titres were almost undetectable against S.Tm clones from inflamed mice (Fig. 1D). This suggested the importance of another phenomenon driven by IgA: the presence of IgA exerts a strong selective pressure against S.Tm producing cognate antigens on its surface(18). Combined with the large population size and fast luminal growth of gut luminal pathogen cells(18), these are ideal conditions for rapid evolution of surface structures that are not bound by vaccine-induced IgA.
In order to identify changes in surface antigenicity of S.Tm, we phenotypically and genetically characterized the S.Tm clones from “vaccinated but inflamed” mice. Based on our earlier observations that protection critically depends on the O-antigen (18, 29), we focused on the O-antigen structure. The S.Tm O-antigen is a polymer of -mannose-α-(1→4)-rhamnose-α-(1→3)-galactose-α-(1→2) with an acetylated α-(1→3)-linked abequose at the mannose (Fig. 1E). WT S.Tm strains react strongly to O:12-0-typing antibodies (recognizing the triose backbone, Fig. S3A and B), O:5-typing antisera (recognizing the acetylated abequose) and O:4-typing antisera (recognizing the non-acetylated abequose, present due to chemical deacetylation during growth and non-stoichiometric acetylation(30)). Further, S.Tm has multiple options for rapidly generating O-antigen variants. S.Tm strain SL1344 can lose the O:5 modification, generating an O:4-only phenotype (i.e. from an O-antigen with acetylated abequose, to one with no O-acetylated abequose) by loss of function mutations in oafA, the abequose acetyl transferase (31). It can further shift between O:12-0 (wild-type) and O:12-2 (α- (1→4) glucosylated) serotypes by methylation-dependent expression of a glucosyl transferase operon STM0557-0559 i.e. by phase variation (32, 33). This operon encodes the machinery to add glucose via an α-(1→4) linkage to the backbone galactose(32). Note that S.Tm strain SL1344 lacks a second common operon required for linking glucose via an α-(1→6) linkage to the backbone galactose, generating the O:1 serotype. These and other known variants would need to be taken into account for more diverse strains. It should be noted that both O-acetylation and backbone-glucosylation represent major changes in the hydrophobicity or steric properties of the O-antigen repeat unit, which when extensively polymerized into full-length O-antigen will have major consequences for binding of IgA raised against a different O-antigen variant (34–36).
We applied multiple techniques to determine the O-antigen structure of evolved S.Tm clones. Flow cytometry with serotyping antibodies (Fig. 1F), High Resolution-Magic Angle Spinning (HR-MAS) on intact bacteria (Fig. S4A and B) and 1H-NMR (30) on purified lipopolysaccharide (Fig. S4C) confirmed the complete loss of abequose O-acetylation (O:4[5] to O:4) and non-stoichiometric gain of α-(1→4)-linked glucosylation of galactose (O:12-0 to O:12-2) in clones from vaccinated but inflamed mice.
Live-attenuated vaccine strains of S.Tm colonize the intestine for more than 40 days in the mouse NTS model(9), i.e. sufficiently long for within-host evolution of the vaccine strain to occur. This suggested that one contributor to superior protection offered by live-attenuated vaccines could be the production of additional antibody specificities targeting evolving O-antigen variants. Correspondingly, after day 18 of colonization, when high-avidity IgA is first present in the intestine, we also observed spontaneous emergence of O:4-producing and O:12-2 producing vaccine clones, dependent on the presence of Rag1 and IgA (Fig. S5). Therefore, IgA-dependent selective pressures for O-antigen switching can be generated both by inactivated oral vaccines (Fig. 1) and by immunity arising naturally during colonization with live-attenuated strains (Fig. S5).
We then explored the underlying genetic mechanisms responsible for altered O-antigen structure in the evolved clones. We first determined the stability of the observed O-antigen phenotypes, i.e. whether we would see reversion during cultivation. In vitro serial passages of evolved clones over 5 days revealed that the switch from O:4[5] to O:4 was a stable, unimodal phenotype (Fig. 1F and S6A). Sequencing of O:4[5]-negative evolved clones, observed to emerge spontaneously in 9 of 29 mice vaccinated with an O:4[5]-targeting vaccine (Table S4) revealed a common 7 base-pair contraction of a tandem repeat within the oafA open reading frame, generating a frame-shift and loss of function (Fig. 2A and B). Targeted deletion of oafA (S.TmΔoafA) produced an identical phenotype to the 7 bp deletion (Fig. 2B). The same mutation was detected in 83 of 210 deposited genomes of various Salmonella serovars in the NCBI databank (Fig. S7A)(37)). As there are only two copies of the 7 base-pair motif in the wild-type ORF, the deletion of one 7 base-pair stretch is unlikely to be reversed(38) (Fig. 2A, Fig. S7A). Intriguingly, deposited sequences also indicate variable numbers of 9bp repeats in the promoter region of oafA (Fig. S7B), suggesting a second possible site of microsatellite instability in this gene. oafA also displayed strong evidence for selection-driven inactivation in a recent analysis of genomes of more than 100000 natural Salmonella isolates (39).
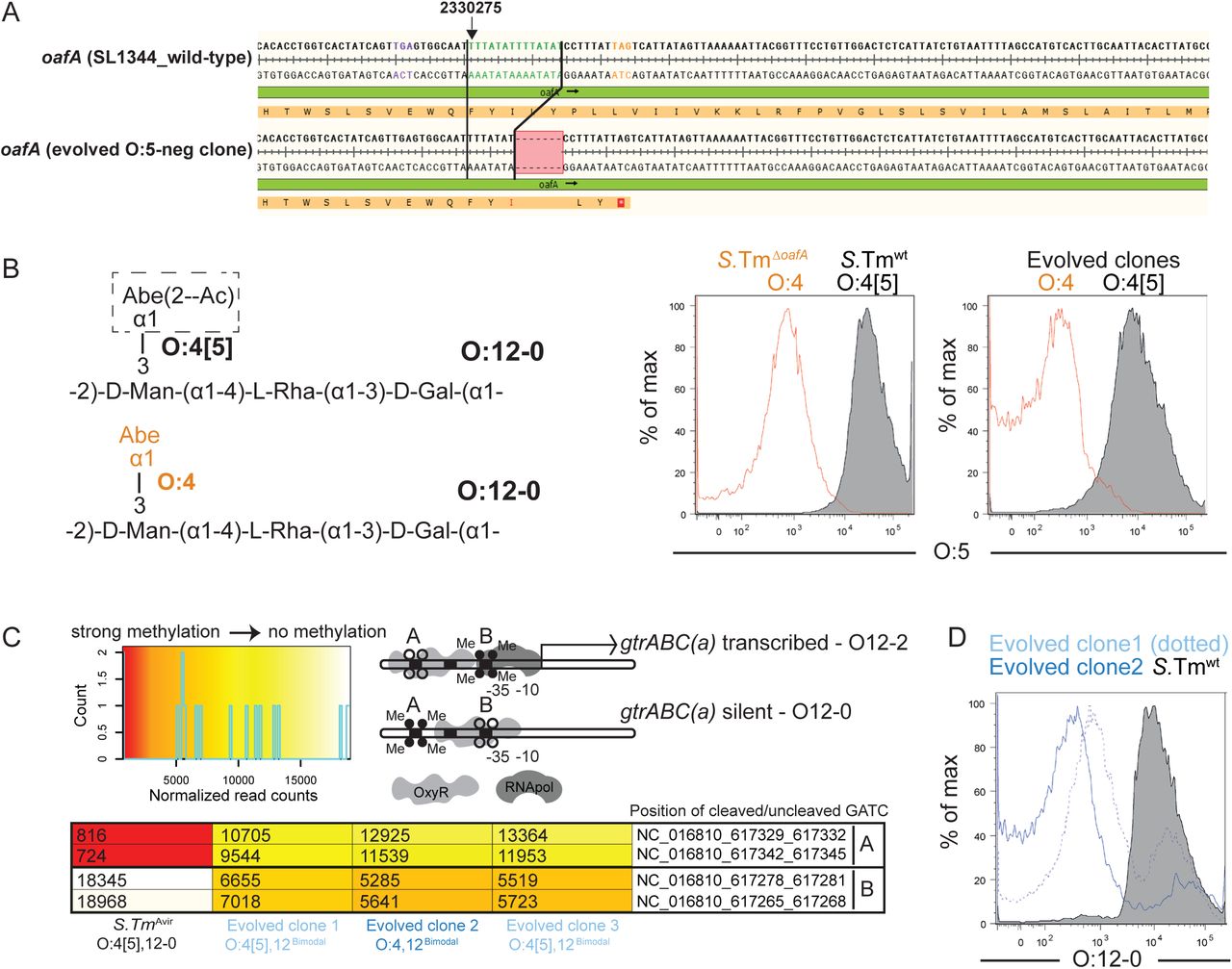
A. Alignment of the oafA sequence from wild type (SL1344_RS11465) and an example O:5-negative evolved clone showing the 7bp contraction leading to premature stop codon (all four re-sequenced O:5-negative strains showed the same deletion). B. Flow cytometry staining of S.Tm, S.TmΔoafA, and two evolved clones differing in O:5 status with anti-O:5 typing sera. C. Methylation status of the gtrABC promoter region in S.Tm, and three O:12Bimodal evolved clones determined by REC-seq. Heat-scale for normalized read-counts, schematic diagram of promoter methylation associated with ON and OFF phenotypes, and normalized methylation read counts for the indicated strains D. Binding of an O-12-specific monoclonal antibody to S.Tm and O:12Bimodal evolved clones, determined by bacterial flow cytometry.
We next assessed the stability of O:12-0 to O:12-2 switching, and its underlying genetic mechanism. In contrast to O:4[5], the loss of O:12-0 staining was reversible during 3 rounds of serial ex vivo passage and both wild-type and evolved clones generated a bimodal staining pattern, consistent with phase variation (Fig. 1F and S6A and B, Supplementary movies A and B, clones referred to henceforth as O:12Bimodal). In line with known epigenetic regulation of the gtrABC operon expression(32), re-sequencing of the O:12Bimodal strains revealed no consistent mutational pattern, although point mutations were observed across the genomes (Table S3). Instead, a semi-quantitative full-genome methylation analysis supported that evolved O:12Bimodal S.Tm clones form mixed populations based on DNA methylation. Populations of evolved clones with up to 80% loss of reactivity with an O12-0-binding monoclonal antibody, presented a high proportion of chromosomes with a methylation pattern typical of the promoter of gtrABC in the ON state(32, 40, 41) and a minor population in the OFF state (Fig. 2C and D): a situation which is reversed in the ancestral strain. Targeted deletion of gtrC (S.TmΔgtrC), the O:12-2 serotype-specific glucosyl transferase of the gtrABC operon, abolished the ability of S.Tm SL1344 to switch to an O:12-bimodal phenotype, even under strong in vivo selection (Fig. S8). Mathematical modeling of O:12-0/O:12-2 population sizes for fixed switching rates (supplementary methods, Fig. S6C-E), and comparison of flow cytometry and a lacZ transcriptional fusion, suggests that in vivo selection of O:12-2-producing clones by IgA is sufficient to explain their relative proportion in the O:12Bimodal population without needing to infer any change in the switching rate (Fig. S6).
Therefore, selective pressure in vaccinated mice, driven by O-antigen-targeting IgA, drives the rapid emergence of S.Tm clones with altered O-antigen structures generated either by mutations or phase-variation. In order to quantify how strongly vaccine-induced IgA can select for O-antigen variants, we designed competition experiments using isogenic mutant pairs carrying targeted deletions in oafA and/or gtrC. This allowed us to study the selective pressure for emergence of each O-antigen variant in isolation.
We first quantified selection for the genetic switch from an O:4[5] to an O:4 serotype. Competitions between S.TmΔoafA ΔgtrC (O:4, O:12-locked) and S.TmΔgtrC (O:4[5], O:12-locked) were carried out in mice vaccinated against either S.TmΔoafA ΔgtrC (O:4) or S.TmΔgtrC (O:4[5]). IgA responses were strongly biased to recognition of the corresponding O:5 or O:4 S.Tm O-antigen epitopes and mediated a substantial selective advantage of expressing the alternative O-antigen variant (up to 107-fold by day 4, Fig. 3A-C). The magnitude of the selective advantage correlated tightly with the magnitude of the specific IgA response against the reactive strain (Fig. 3B-C). Deletion of oafA was fitness-neutral in naïve hosts during 4 days of infection (Fig. 3A). These observations were repeated with identical results in Balb/c mice (Fig. S9). Further, the selective advantage of S.TmΔgtrC over S.TmΔoafA ΔgtrC could also be observed in mice vaccinated with an O:4 variant of a commonly-used live-attenuated strain (S.TmΔaroA ΔoafA ΔgtrC, Fig. S10). However, this was not observed in mice vaccinated with the standard live-attenuated O:4[5] strain, S.TmΔaroA ΔgtrC, that can undergo within-host evolution to the O:4 variant and therefore can prime immunity against both O:5 and O:4 epitopes (Fig. S10). Specific IgA can therefore act as a strong evolutionary pressure selecting for mutations in genes encoding O-antigen-modifying enzymes.
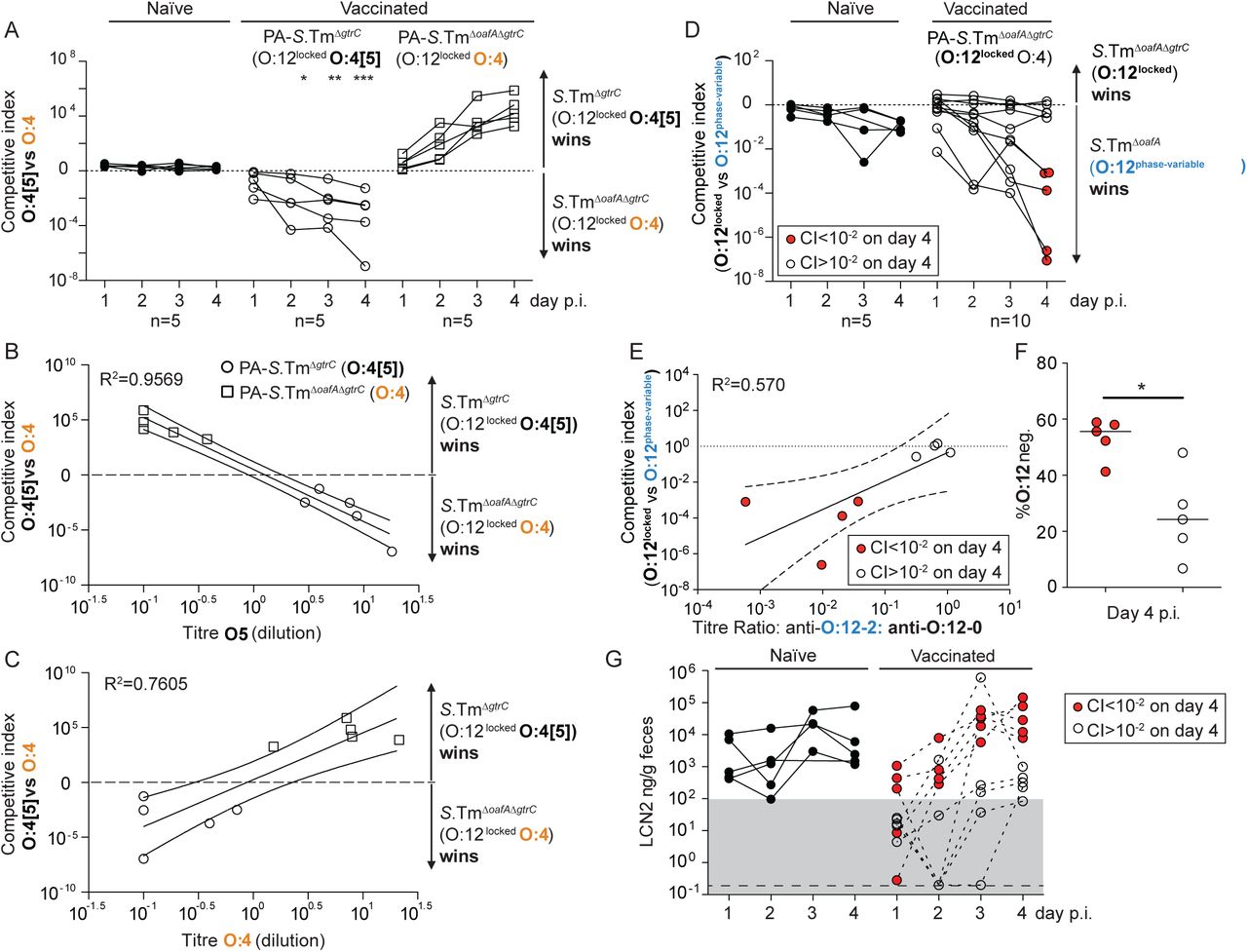
A-C. Naive (closed circles, n=5), PA-S.TmΔgtrC-vaccinated (O:4[5]-vaccinated, open circles, n=5) and PA-S.TmΔgtrCΔoafA-vaccinated (O:4-vaccinated, open squares, n=5) SOPF mice were streptomycin-pretreated, infected (105 CFU, 1:1 ratio of S.TmΔgtrC and S.TmΔgtrC ΔoafA per os). A. Competitive index (CFU S.Tm ΔgtrC/CFU S.TmΔ gtrC ΔoafA) in feces at the indicated time-points. 2-way ANOVA with Bonferroni post-tests on log-normalized values, compared to naive mice. *p<0.05, **p<0.01,***p<0.001, ****p<0.0001. B and C. Correlation of the competitive index with the O:4[5]-specific (B) and O:4-specific (C) intestinal IgA titre, r2 values of the linear regression of log-normalized values. Open circles: Intestinal IgA from O:4[5]-vaccinated mice, Open squares: Intestinal IgA from O:4-vaccinated mice. Lines indicate the best fit with 95% confidence interval D-G. Naive (closed circles, n=5) or PA-S.Tm ΔoafA ΔgtrC -vaccinated (O:4/O:12-vaccinated, open circles and red circles, n=10) SOPF mice were streptomycin-pretreated and infected (105 CFU, 1:1 ratio of S.TmΔoafA (O:12-2 switching) and S.Tm ΔoafA ΔgtrC (O:12-locked) per os). D. Competitive index (CFU S.TmΔoafA ΔgtrC /CFU S.TmΔoafA) in feces at the indicated time-points. Red circles indicate vaccinated mice with a competitive index below 10-2 and are used to identify these animals in panels D-G. Effect of vaccination is not significant by 2-way ANOVA considering vaccination over time. E. Correlation of the competitive index on day 4 with the intestinal IgA titre against an O:12-2-locked S.Tm pgtrABC variant (linear regression of log-normalized values, lines indicate the best fit with 95% confidence interval). F. Enrichment cultures of the fecal S.TmΔoafA population at day 4 were stained for O:12/O:4 and the fraction of O:12-negative S.Tm quantified by flow cytometry. G. Intestinal inflammation quantified by measuring Fecal Lipocalin 2 (LCN2). Note the lines joining the points are to allow individual animals to be tracked over time, and do not imply assumptions about what occurs between the measured time-points.
We next quantified the selective advantage of phase-variation between O:12-0 and O:12-2 using strains with an oafA-mutant background (i.e. O:4-locked, to prevent uncontrolled O:[4]5 to O:4 mutational changes). Mice were mock-vaccinated or vaccinated against S.TmΔoafA ΔgtrC (O:12-locked). Competitive infections were then carried out between S.TmΔoafA (O:12-phase-variable) and S.TmΔoafA ΔgtrC (O:12-locked) strains. In line with published data(32, 42), we observe a very mild fitness benefit of O:12 phase variation in naïve mice. In contrast, phase-variation was a major benefit to S.Tm in a subset of vaccinated animals (Fig. 3D). On closer examination, precisely these animals showed a strong bias of their IgA response towards recognition of O:12-0 only (Fig. 3E). In animals where no out-competition was observed, the IgA response equally recognized strains producing O-antigens with either the O:12-0 or the O:12-2 O-antigen (Fig. 3E). This is likely explained by the stochastic nature of antibody generation towards different epitopes of the O-antigen repeat. Logically, phase-variation is beneficial whenever one phase variant is poorly bound by IgA. Correspondingly, O:12-phase variation, vaccine escape and inflammation were largely observed in mice where IgA bound poorly to the O:12-2 variant (Fig. 3F and G). The mechanistic basis of this selective advantage could be confirmed by complementation of the gtrC gene in trans (Fig. S11). It is interesting to note that gtrABC operons are often found in temperate phage(33, 43), suggesting that the ability of S.Tm to quickly evade IgA mediated immunity may be further promoted by co-option of phage-encoded fitness factors (morons(44)).
Our results led to the conclusion that IgA escapers, i.e. S.Tm mutants or phase variants only weakly recognized by vaccine-induced IgA, are strongly selected for and dominate the luminal S.Tm population within 1-3 days of infection in vaccinated mice. A good correlation was observed between dominance of IgA escapers and initiation of intestinal inflammation, as quantified by fecal Lipocalin 2 (LCN2, Fig. 3G, S12). Thus, both mutation and epigenetic switching processes shape the O-antigen structure of S.Tm SL1344 and can increase the pathogen’s fitness in the intestine of mice immune to specific O-antigen variants. Based on a published analysis of natural Salmonella isolates (39), these changes arise randomly in the natural pathogen populations and appear to be slowly selected against by minor fitness reductions at evolutionary time scales. Correspondingly, they inflict negligible loss of pathogen fitness in naive hosts within the timescales of our experiments (Fig. 3A and D). A major disadvantage of inactivated vaccines over live vaccines is, therefore, their inability to follow this within-host evolution trajectory in order to induce broader IgA responses.
From these observations we hypothesized that a vaccine combining all four observed O-antigen variants (“Evolutionary Trap” vaccine, abbreviated as PA-S.TmET; generated by mixing vaccines containing the O:4[5],12 S.TmΔgtrC,, O:4,12 S.TmΔoafA ΔgtrC, O:4,12-2 S.TmΔoafA pgtrABC, and O:4[5],12-2 S.Tm pgtrABC strains) should generate enhanced disease protection against S.Tm SL1344 by cutting off the observed O-antigen escape pathways. Interestingly although this vaccine induced a broader antibody response (Fig. 4A), we did not reach significance for improved protection as determined by intestinal inflammatory markers, and colonization of mesenteric lymph nodes and spleen (Kruskall-Wallis test on mLN CFU at d3 p.i., Fig. S13). This is likely due to the low-frequency of IgA-escape in this cohort of PA-STmWT-vaccinated C57BL/6 mice, as we do observe a robust improvement in protection in Balb/c mice where the challenge inoculum was a 1:1 mixture of O:4[5] and O:4-producing S.Tm variants (Fig. S9). More striking was that PA-S.TmET vaccination selected for another class of O-antigen variant: mutations generating a single-repeat O-antigen(45). These can be identified by weak binding to typing antisera (Fig. 4B) and by gel electrophoresis of purified LPS (Fig.4C). Sequencing of evolved clones (Table S4) revealed a common large deletion encompassing the wzyB gene (also termed rfc), encoding the O-antigen polymerase(45) (Fig. 4D, Fig. S14 also reported in some “non-typable” S.Tm isolates from broilers(45)). This deletion is mediated by site-specific recombination between direct repeats flanking the wzyB locus, which renders the locus unstable. Short O-antigen-producing clones were detected in 10 of 13 PA-S.TmET vaccinated mice by phenotypic characterization (anti-O5dim flow cytometry staining), and wzyB deletion was confirmed resequencing of n=5 short-O-antigen clones from two different mice (Table S4). Of note, this is not an exhaustive analysis and we cannot currently exclude that other mutations could also give rise to short O-antigen mutants. However, we did not observe mutations in wbaP(45) (complete loss of O-antigen), wzz, fepE (46), or opvAB(47) (dysregulated O-antigen length) in any of the sequenced clones. Intestinal IgA from PA-S.TmΔoafA vaccinated mice showed higher titres against the long O-antigen than the single-repeat O-antigen (Fig. 4E). This weaker binding to the very short O-antigen-producing strains is consistent with lower O-antigen abundance or loss of avidity (Fig. 4B, Fig. S3C).

A and B. S.Tm clones re-isolated from the feces of SOPF mice vaccinated with PBS only, PA-S.TmΔgtrC (n=7) or PA-S.TmET (combined PA-S.TmΔgtrC, PA-S.TmΔoafA ΔgtrC, PA-S.Tm pgtrABC, and PA-S.TmΔoafA pgtrABC, n=8). A. Intestinal IgA titre determined by bacterial flow cytometry against S.TmΔgtrC (O:4[5],12), S.TmΔoafA ΔgtrC (O:4,12), S.TmΔgtrC pgtrABC (O:4[5], 12-2) and S.TmΔoafA ΔgtrC pgtrABC (O:4,12-2). B. Fraction of clones with weak anti-sera staining, as determined by flow cytometry screening, indicative of O-antigen shortening. One point represents one mouse. C. Silver-stained gel of LPS from representative control and evolved S.Tm strains from control and PA-S.TmET vaccinated mice, showing short LPS in clones isolated from 2 different vaccinated PA-S.TmET mice. D. Resequencing of strains with short O-antigen revealed a large genomic deletion between inverted repeats, comprising the wzyB gene (O-antigen polymerase) (n=5 clones, isolated from 2 different mice). E. Intestinal IgA titre from PA-S.TmΔoafA ΔgtrC -vaccinated SOPF mice specific for S.Tm ΔoafA ΔgtrC (long O-antigen) and S.TmΔoafA ΔgtrC ΔwzyB (short O-antigen). F, G, H, Single 24h infections in streptomycin pretreated naïve mice (n=14, short O-antigen, n=9 long O-antigen). Evolved and synthetic wzyB mutants have reduced ability to colonize the gut (F, CFU/g feces) and to spread systemically (G, CFU per mesenteric lymph node (MLN)). This translates into diminished propensity to trigger intestinal inflammation in comparison to wzyB wild type strains (H, fecal Lipocalin 2 (LCN2)). I. Mock-vaccinated wild type (C57BL/6, n=10), PA-STmΔoafA ΔgtrC -vaccinated JH-/- mice (JH-/-n=6), PA-STmΔoafAΔgtrC -vaccinated wild type (C57BL/6, n=16) and PA-STmΔoafA ΔgtrC -vaccinated JH+/- littermate controls (JH+/-, n=5 mice were streptomycin pre-treated and infected with 105 CFU of a 1:1 ratio S.Tm ΔoafA ΔgtrC ΔwzyB and S.TmΔoafA ΔgtrC i.e. serotype-locked, short and long O-antigen-producing strains. Competitive index of S.Tm in feces on the indicated days. J. Feces from the indicated mice (grey-filled circles) from panel I were transferred into streptomycin-pretreated naive mice (one fecal pellet per mouse, n=5) and competitive index in feces calculated at day 2 post-infection. I: 2-way ANOVA and J: Paired t-test results are shown. K and L. Fecal Lipocalin 2 (LCN2) corresponding to panels I and J respectively. A, I, K. 2-way ANOVA on log-Normalized data. Bonferroni post-test statistics are shown. In panel I, competitive index in vaccinated mice is significantly higher than 1 at all time-points by Wilcoxon signed rank tests. D, F, G, H: Mann-Whitney U 2-tailed tests.
Of note, we have previously published that IgA responses against the surface of rough Salmonella are identically induced by vaccination with either rough or wild-type Salmonella oral vaccines(17). Correspondingly, including the wzyB mutant into our PA-S.TmET mix does not further improve IgA titres specific for S.TmΔoafA ΔgtrC ΔwzyB (Fig. S13), and does not improve the protective power of PA-S.TmET (Fig. S13). Rather we suggest that the single-repeat O-antigen represents a fitness optimum for S.Tm SL1344 in orally vaccinated mice, where the decreased abundance of O-antigen repeats, and/or loss of binding by IgA specificities requiring more than one repeat, provides an advantage.
Single infections revealed that, in comparison to isogenic wild type counterparts, wzyB-deficient mutants (synthetic or evolved) are significantly less efficient at colonizing the gut of streptomycin pretreated naïve mice (Fig. 4F), disseminating systemically (Fig. 4G) and triggering inflammation (Fig. 4H). This attenuation can be attributed to compromised outer membrane integrity(48) and also manifests as an increased sensitivity to complement-mediated lysis, bile acids and weak detergents (Fig S15).
To verify that IgA can select for short-O-antigen-producing strains, we then tested whether IgA-mediated selection could drive outgrowth of synthetic wzyB deletion mutants. Competitions between S.TmΔoafA ΔgtrC (O:4,12-locked, long O-antigen) and S.TmΔoafA ΔgtrC ΔwzyB (O:4,12-locked, single repeat O-antigen) mutants in the intestine of vaccinated and mock-vaccinated or antibody-deficient mice revealed a substantial fitness loss due to the wzyB deletion in naive animals, as observed in earlier studies(45, 49) (Fig. 4I). However, in the gut of vaccinated mice, the fitness cost of decreased outer-membrane integrity in wzyB mutants was clearly outweighed by the benefit of avoiding O-antigen specific IgA binding (Fig. 4I). Vaccinated IgA-/- mice were indistinguishable from naive mice in these experiments, i.e. IgA and not any other effect of the vaccine was responsible for the phenotype. PA-S.TmET-elicited IgA can therefore select for S.Tm SL1344 mutants with a fitness cost in naïve hosts.
To demonstrate that vaccine-induced IgA, and not further genetic change in S.Tm, drives this out-competition, we carried out fecal transfer experiments into naïve, streptomycin-treated recipients. Full fecal pellets from PA-STmET vaccinated mice that had been infected for 4 days with the short/long O-antigen mixture were delivered to streptomycin-treated naïve hosts. S.TmΔoafA ΔgtrC ΔwzyB (short O-antigen) dominated the population in the donor feces. However, on transfer to the naive environment the wzyB mutant was rapidly out-competed by the S.TmΔoafA ΔgtrC (full length O-antigen) (Fig. 4J-L). Thus, outgrowth in vaccinated mice is not due to compensatory mutations in the wzyB mutants, but to antibody-mediated selection.
IgA-mediated evolutionary traps work on the premise that S.Tm experiences two levels of selection: within-host and between-host. By dramatically altering the within-host selective landscape, we fix mutations that are detrimental under between-host selective pressures. Between-host selection, for example between farm animals, includes a period of exposure to environmental stresses, as well as transit through the hostile upper gastrointestinal tract, and invasion into a limited intestinal niche. Both published data (50), and our own analyses of susceptibility of short-O-antigen strains to membrane stress (Fig. S15) indicate that these strains are less likely to survive in the environment, or in the presence of high concentrations of bile acids. As the streptomycin mouse model involves a very stable intestinal niche for S.Tm, requiring only a single virulent S.Tm to be transferred in order to generate full-blown disease in the naïve host(51), we instead investigated the effect of wzyB mutations on S.Tm infection and transmission in the western diet model(28). Fecal pellets were recovered from mice pre-treated with streptomycin and infected with S.Tm with either a complete or single-repeat O-antigen. Recipient mice fed western diet for 24h prior to exposure received a complete fecal pellet from an infected donor and S.Tm counts in feces, mesenteric lymph nodes and spleen were tracked over 4 days of infection. Exposure of western diet-fed mice to fecal pellets from streptomycin-pretreated mice only resulted in disease when the donor mouse was infected with S.TmWT (Fig. S16), i.e. emergence of wzyB-mutants could block the disease transmission chain under these conditions.
As mouse models of transmission are necessarily rather contrived, we additionally generated a mathematical model of Salmonella transmission in mice and scaled to the more relevant setting of a commercial pig farm. This model is based on literature-derived parameters and quantitatively investigates the effect of two consequences of vaccination on transmission: 1) Successful vaccination decreases the duration of shedding of viable Salmonella from the infected donor animals(18), and 2) fixation of wzyB mutations increases the death/clearance rate of Salmonella between shedding from the infected donor and arrival in the lower gastrointestinal tract of a new host (Fig. S15 and 16). Both of these factors were shown to have strong effects on the predicted R0 of a Salmonella outbreak where the required infectious dose is large and the period of microbiota disturbance in the recipient animals is limited (Fig. S17). Correspondingly, a weak effect of vaccination on transmission is expected in the streptomycin mouse model of infection, but a strong effect (sufficient to keep R0 below 1) would be predicted on a pig farm (see supplementary methods for detailed model description).
In conclusion, high-avidity intestinal IgA targeting the bacterial surface exerts a strong negative selective pressure on S.Tm. Combined with rapid growth and a large population size in the gut lumen, this selects for phase-variants or fixation of mutations that alter major cell surface antigens, and therefore facilitates S.Tm immune escape. Via a tailored cocktail of IgA specificities, it is possible to turn this rapid immune escape into a tool to force an evolutionary trade-off, i.e. driving outgrowth of S.Tm mutants producing very short O-antigens. Short O-antigen-bearing mutants have a fitness disadvantage in the environment and on transmission into naive hosts(50, 52), with important implications for disease spread. While we stress that these observations have been made only with the SL1344 strain of Salmonella Typhimurium, the concept of tracking the evolutionary trajectories of bacterial pathogens in vaccinated animals to design IgA “evolutionary traps” should extend to more diverse strains. This concept converts the rapid within-host evolution of intestinal bacteria from a major barrier, into a major benefit for such vaccination strategies. Of note, the evolutionary trap principle should also be relevant for the design of recombinant or synthetic glycoconjugate vaccines, and even O-antigen-targeting phage therapy. While other immune mechanisms, such as cellular immunity and surface protein-targeting antibodies can clearly improve protection (7, 53), the ability of tailored IgA responses to drive O-antigen evolution in the gut lumen will be a powerful addition to the vaccination toolbox. Evolutionary trap strategies can therefore be applied in the race to contain increasingly antibiotic resistant Enterobacteriaceae(54) in both humans and farm animals.
Funding
MD is supported by a SNF professorship (PP00PP_176954). ES acknowledges the support of the Swiss National Science Foundation (40B2-0_180953, 310030_185128, NCCR Microbiomes) and Gebert Rüf Microbials (GR073_17). MD and ES acknowledge the Botnar Center for Child Health Multi-Invesitigator Project 2020. BMS acknowledges the support of R01 AI041239/AI/NIAID NIH HHS/United States. WDH acknowledges support by grants from the Swiss National Science Foundation (SNF; 310030B-173338, 310030_192567, NCCR Microbiomes), the Promedica Foundation, Chur, the Gebert Rüf foundation (Displacing ESBL, with AE) and the Helmut Horten Foundation. EB is supported by a Boehringer Ingelheim Fonds PhD fellowship. BM acknowledges support by the Swiss National Science Foundation (200020_159707).
Author contributions
MD, WDH and ES designed the project and wrote the paper. MD and ES designed and carried out experiments. MvdW, BHM, RM contributed to experimental design / data interpretation. EB, DH, VL FB, KSM, AH, JA, PV, LV, DW, FB, AE, GZ, OH, MA, CL carried out analyses shown in Fig1-4 and S1-S17. ND produced strains 891 and 931. LP, AL and BMS generated novel antibody reagents. All authors critically reviewed the manuscript.
Conflict of Interest
The authors declare that Evolutionary Trap Vaccines are covered by European patent application EP19177251.
Data and materials availability
All data and materials are in the manuscript or will be provided on request to the corresponding authors.
List of Supplementary Materials
Materials and Methods
Supplementary Figures S1-11
Supplementary Table S1-5
Supplementary Movies 1 and 2
Supplementary .fcs files used to generate presented data
Supplementary Materials
Materials and methods
Ethics statement
All animal experiments were approved by the legal authorities (licenses 223/2010, 222/2013 and 193/2016; Kantonales Veterinäramt Zürich, Switzerland) and performed according to the legal and ethical requirements.
Mice
Unless otherwise stated, all experiments used specific opportunistic pathogen-free (SOPF) C57BL/6 mice. 129S1/SvImJ, IgA−/− (56), Balb/c, JH-/- (57), Rag1-/- (58) (all C57BL/6 background) and 129.SJL mice, were re-derived into a specific opportunistic pathogen-free (SOPF) foster colony to normalize the microbiota and bred under full barrier conditions in individually ventilated cages in the ETH Phenomics center (EPIC, RCHCI), ETH Zürich and were fed a standard chow diet. Low complex microbiota (LCM) mice (C57BL/6) are ex-germfree mice, which were colonized with a naturally diversified Altered Schaedler flora in 2007(9) and were bred in individually ventilated cages or flexible-film isolators at this facility, and received identical diet. Vaccinations were started between 5 and 6 weeks of age, and males and females were randomized between groups to obtain identical ratios wherever possible. As strong phenotypes were expected, we adhered to standard practice of analysing at least 5 mice per group. Western diet without fibre (BioServ, S3282; 60% kcal fat; irradiated; per mass: 36% fat, 20.5% protein, 35.7% carbohydrates, 0% fibre) was fed ad libitum where indicated. Researchers were not blinded to group allocation.
Strains and plasmids
All strains and plasmids used in this study are listed Table S1.
For cultivation of bacteria, we used lysogeny broth (LB) containing appropriate antibiotics (i.e., 50 µg/ml streptomycin (AppliChem); 6 µg/ml chloramphenicol (AppliChem); 50 µg/ml kanamycin (AppliChem); 100 µg/ml ampicillin (AppliChem)). Dilutions were prepared in Phosphate Buffer Saline (PBS, Difco) In-frame deletion mutants (e.g. gtrC::cat) were performed by λ red recombination as described in(59). When needed, antibiotic resistance cassettes were removed using the temperature-inducible FLP recombinase encoded on pCP20(59). Mutations coupled with antibiotic resistance cassettes were transferred into the relevant genetic background by generalized transduction with bacteriophage P22 HT105/1 int-201(60). Primers used for genetic manipulations and verifications of the constructions are listed Table S2. Deletions of gtrA and gtrC originated from in-frame deletions made in S.Tm 14028S, kind gifts from Prof. Michael McClelland (University of California, Irvine), and were transduced into the SB300 genetic background.
The gtrABC operon (STM0557-0559) was cloned into the pSC101 derivative plasmid pM965(61) for constitutive expression. The operon gtrABC was amplified from the chromosome of SB300 using the Phusion Polymerase (ThermoFisher Scientific) and primers listed Table S2. The PCR product and pM965 were digested with PstI-HF and EcoRV-HF (NEB) before kit purification (SV Gel and PCR Clean up System, Promega) and ligation in presence of T4 ligase (NEB) following manufacturer recommendations. The ligation product was transferred by electro-transformation in competent SB300 cells.
Targeted sequencing
Targeted re-sequencing by the Sanger method (Microsynth AG) was performed on kit purified PCR products (Promega) from chromosomal DNA or expression vector templates using pre-mixed sequencing primers listed Table S2.
Whole-genome re-sequencing of O:12Bimodal isolates
The genomes of S.Tm and evolved derivatives were fully sequenced by the Miseq system (2×300bp reads, Illumina, San Diego, CA) operated at the Functional Genomic Center in Zürich. The sequence of S.Tm SL1344 (NC_016810.1) was used as reference. Quality check, reads trimming, alignments, SNPs and indels calling were performed using the bioinformatics software CLC Workbench (Qiagen).
Whole-genome sequencing of S.Tm isolates from “Evolutionary Trap” vaccinated mice and variant calling
Nextera XT libraries were prepared for each of the samples. The barcoded libraries were pooled into equimolar concentrations following manufacturer’s guidelines (Illumina, San Diego, CA) using the Mid-Output Kit for paired-end sequencing (2×150 bp) on an Illumina NextSeq500 sequencing platform. Raw data (mean virtual coverage 361x) was demultiplexed and subsequently clipped of adapters using Trimmomatic v0.38 with default parameters(62). Quality control passing read-pairs were aligned against reference genome/plasmids (Accession numbers: NC_016810.1, NC_017718.1, NC_017719.1, NC_017720.1) with bwa v0.7.17(63). Genomic variant were called using Pilon v1.23(64). with the following parameters: (i) minimum coverage 10x; (ii) minimum quality score = 20; (iii) minimum read mapping quality = 10. SnpEff v4.3 was used to annotate variants according to NCBI and predict their effect on genes(65).
PA-STm vaccinations
Peracetic acid killed vaccines were produced as previously described(17). Briefly, bacteria were grown overnight to late stationary phase, harvested by centrifugation and re-suspended to a density of 109-1010 per ml in sterile PBS. Peracetic acid (Sigma-Aldrich) was added to a final concentration of 0.4% v/v. The suspension was mixed thoroughly and incubated for 60 min at room temperature. Bacteria were washed once in 40 ml of sterile 10x PBS and subsequently three times in 50 ml sterile 1x PBS. The final pellet was re-suspended to yield a density of 1011 particles per ml in sterile PBS (determined by OD600) and stored at 4°C for up to three weeks. As a quality control, each batch of vaccine was tested before use by inoculating 100 µl of the killed vaccine (one vaccine dose) into 300 ml LB and incubating over night at 37 °C with aeration.
Vaccine lots were released for use only when a negative enrichment culture had been confirmed. For all vaccination, 1010 particles, suspended in 100µl PBS were delivered by oral gavage, once weekly for 4 weeks. Where multiple strains were combined, the total number of vaccine particles remained constant, and was roughly equally divided between the constituent strains. Unless otherwise stated, PA-STm vaccinated mice were challenged orally on d28 after the first vaccination.
Vaccination by attenuated strains of non-typhoidal Salmonella
6-week-old C57Bl/6 or 129 Sv/Ev mice were orally pretreated 24 h before infection with 25 mg streptomycin. ΔgtrC and ΔoafA ΔgtrC derivatives of the ΔaroA mutant (Table S1, (66)) were cultivated overnight separately in LB containing streptomycin. Subcultures were prepared before infections by diluting overnight cultures 1:20 in fresh LB without antibiotics and incubation for 4 h at 37°C. The cells were washed in PBS and 50 µl of resuspended pellets were used to infect mice per os (5×107 CFU).
Feces were sampled at day 1, 9 and 42 post-infection, homogenized in 1 ml PBS by bead beating (3mm steel ball, 25 Hz for 1 minute in a TissueLyser (Qiagen)), and S.Tm strains were enumerated by selective plating on MacConkey agar supplemented with streptomycin. Samples for lipocalin-2 measurements were kept homogenized in PBS at -20 °C.
Non-typhoidal Salmonella challenge infections
Infections were carried out as described(67). Unless otherwise stated (i.e. where western diet was used for microbiota disruption), in order to allow reproducible gut colonization, 8-12 week-old C57Bl/6 or 129 Sv/Ev mice, naïve or vaccinated, were orally pretreated 24 h before infection with 25 mg streptomycin or 20 mg of ampicillin. Strains were cultivated overnight separately in LB containing the appropriate antibiotics. Subcultures were prepared before infections by diluting overnight cultures 1:20 in fresh LB without antibiotics and incubation for 4 h at 37°C. The cells were washed in PBS and 50 µl of resuspended pellets were used to infect mice per os (5×105 CFU). Competitions were performed by inoculating 1:1 mixtures of each competitor strain.
Feces were sampled daily, homogenized in 1 ml PBS by bead beating (3 mm steel ball, 25 Hz for 1 min in a TissueLyser (Qiagen)), and S.Tm strains were enumerated by selective plating on MacConkey agar supplemented with the relevant antibiotics. Samples for lipocalin-2 measurements were kept homogenized in PBS at -20°C. At endpoint, intestinal lavages were harvested by flushing the ileum content with 2 ml of PBS using a cannula. The mesenteric lymph nodes, were collected, homogenized in PBS Tergitol 0.05% v/v at 25 Hz for 2 min, and bacteria were enumerated by selective plating.
Competitive indexes were calculated as the ratio of relative population sizes of competitors at a given time point, normalized for the ratio in the inoculum.
Where western diet was used to disrupt the microbiota, mice were switched from standard chow to western diet (BioServ, S3282) 24 h prior to infection and were maintained on western diet throughout the infection.
Non-typhoidal Salmonella transmission
Donor mice were vaccinated with PA-S.TmΔoafA ΔgtrC once per week for 5 weeks, streptomycin pretreated (25 mg streptomycin per os), and gavaged 24 h later with 105 CFU of a 1:1 mixture of S. TmΔoafAΔgtrCwzyB::cat (CmR) and S. TmΔoafAΔgtrC Kan (KanR). On day 4 post infection, the donor mice were euthanized, organs were harvested, and fecal pellets were collected, weighed and homogenized in 1 ml of PBS. The re-suspended feces (centrifuged for 10 s to discard large debris) were immediately used to gavage (as a 50 µl volume containing the bacteria from on fecal pellet) recipient naïve mice (pretreated with 25 mg streptomycin 24 hours before infection). Recipient mice were euthanized and organs were collected on day 2 post transmission. In both donor and recipient mice, fecal pellets were collected daily and selective plating was used to enumerate Salmonella and determine the relative proportions (and consequently the competitive index) of both competing bacterial strains.
Quantification of fecal Lipocalin2
Fecal pellets collected at the indicated time-points were homogenized in PBS by bead-beating at 25 Hz, 1min. Large particles were sedimented by centrifugation at 300 g, 1 min. The resulting supernatant was then analysed in serial dilution using the mouse Lipocalin2 ELISA duoset (R&D) according to the manufacturer’s instructions.
Analysis of IgA-coating, and O:5/O:12-0 expression on S.Tm in cecal content
Fresh cecal content or feces was re-suspended in sterile PBS by bead-beating at 25 Hz, 1 min (previously demonstrated to disrupt IgA cross-linked clumps(18)). An aliquot estimated to contain not more than 106 S.Tm was directly stained with a monoclonal human IgG-anti-O:12-0 (STA5(18)) and biotin-conjugated anti-mouse IgA clone RMA-1 (Biolegend), and/or Rabbit-anti-Salmonella O:5 (Difco). After washing, secondary reagents Alex647-anti-human IgG (Jackson Immunoresearch), Pacific Blue-conjugated streptavidin (Molecular Probes), Phycoerythrin-conjugated streptavadin (Molecular Probes) and/or Brilliant violet 421-anti-Rabbit IgG (Biolegend) were added. After a final washing step, samples were analysed on a BD LSRII flow cytometer, or a Beckman Coulter Cytoflex S, with settings adapted for optimal detection of bacterial-sized particles. The median fluorescence intensity of IgA staining on S.Tm was determined by “gating” on bacterial sized particles and calculating the appropriate median fluorescence corresponding to O:12-0 or O:5 staining FlowJo (Treestar, USA). Gates used to calculate the % of “ON” and “OFF” cells were calculated by gating on samples with known ON or OFF phenotypes.
Analysis of specific antibody titers by bacterial flow cytometry
Specific antibody titers in mouse intestinal washes were measured by flow cytometry as described(18, 68). Briefly, intestinal washes were collected by flushing the small intestine with 5 ml PBS, centrifuged at 16000 g for 30 min and aliquots of the supernatants were stored at -20°C until analysis. Bacterial targets (antigen against which antibodies are to be titered) were grown to late stationary phase or the required OD, then gently pelleted for 2 min at 3000 g. The pellet was washed with sterile-filtered 1% BSA/PBS before re-suspending at a density of approximately 107 bacteria per ml. After thawing, intestinal washes were centrifuged again at 16000 g for 10 min. Supernatants were used to perform serial dilutions. 25 μl of the dilutions were incubated with 25 μl bacterial suspension at 4°C for 1 h. Bacteria were washed twice with 200 μl 1% BSA/PBS before resuspending in 25 μl 1% BSA/PBS containing monoclonal FITC-anti-mouse IgA (BD Pharmingen, 10 µg/ml) or Brilliant violet 421-anti-IgA (BD Pharmingen). After 1 h of incubation, bacteria were washed once with 1% BSA/PBS and resuspended in 300 μl 1% BSA/PBS for acquisition on LSRII or Beckman Coulter Cytoflex S using FSC and SSC parameters in logarithmic mode. Data were analysed using FloJo (Treestar). After gating on bacterial particles, log-median fluorescence intensities (MFI) were plotted against antibody concentrations for each sample and 4-parameter logistic curves were fitted using Prism (Graphpad, USA). Titers were calculated from these curves as the inverse of the antibody concentration giving an above-background signal.
Flow cytometry for analysis of O:5, O:4 and O:12-0 epitope abundance on Salmonella in cecal content, enrichment cultures and clonal cultures
1 µl of overnight cultures, or 1µl of fresh feces or cecal content suspension (as above) was stained with STA5 (human recombinant monoclonal IgG2 anti-O:12(18)), Rabbit anti-Salmonella O:5 or Rabbit anti-Salmonella O:4. After incubation at 4°C for 30 min, bacteria were washed once with PBS/1% BSA and resuspended in appropriate secondary reagents (Alexa 647-anti-human IgG, Jackson Immunoresearch, Brilliant Violet 421-anti-Rabbit IgG, Biolegend). This was incubated for 10-60 min before cells were washed and resuspended for acquisition on a BD LSRII or Beckman Coulter Cytoflex S.
Live-cell immunofluorescence
200 uL of an overnight culture was centrifuged and resuspended in 200 μL PBS containing 1 μg recombinant murine IgA clone STA121-AlexaFluor568. The cells and antibodies were co-incubated for 20 min at room temperature in the dark and then washed twice in 1 mL Lysogeny broth (LB). Antibody-labeled cells were pipetted into an in-house fabricated microfluidic device(69). Cells in the microfluidic device were continuously fed S.Tm-conditioned LB(69) containing STA121-AlexaFluor568 (1 μg/mL). Media was flowed through the device at a flow rate of 0.2 mL/h using syringe pumps (NE-300, NewEra PumpSystems). Cells in the microfluidic device were imaged on an automated Olympus IX81 microscope enclosed in an incubation chamber heated to 37°C. At least 10 unique positions were monitored in parallel per experiment. Phase contrast and fluorescence images were acquired every 3 min. Images were deconvoluted in MatLab(70). Videos are compressed to 7 fps, i.e. 1 s = 21 mins.
HR-MAS NMR
R. Typhimurium cells were grown overnight (∼18 h) a to late stationary phase. The equivalent of 11–15 OD600 was pelleted by centrifugation for 10 min 4 °C and 3750 g. The pellet was resuspended in 10% NaN3 in potassium phosphate buffer (PPB; 10 mM pH 7.4) in D2O and incubated at room temperature for at least 90 min. The cells were then washed twice with PPB and resuspended in PPB to a final concentration of 0.2 OD600/μl in PPB containing acetone (final concentration 0.1% (v/v) as internal reference). The samples were kept on ice until the NMR measurements were performed - i.e. for between 1 and 8 h. The HR-MAS NMR spectra were recorded in two batches, as follows: S.TmWT, S.TmwbaP, S.TmEvolved_1, S.TmEvolved_2 were measured on 16.12.2016, S.TmOafA was measured on 26.7.2017.
NMR experiments on intact cells were carried out on a Bruker Biospin AVANCE III spectrometer operating at 600 MHz 1H Larmor frequency using a 4 mm HR-MAS Bruker probe with 50 µl restricted-volume rotors. Spectra were collected at a temperature of 27 °C and a spinning frequency of 3 kHz except for the sample of OafA (25°C, 2 kHz). The 1H experiments were performed with a 24 ms Carr–Purcell– Meiboom–Gill (CPMG) pulse-sequence with rotor synchronous refocusing pulses every two rotor periods before acquisition of the last echo signal to remove broad lines due to solid-like material(30). The 90° pulse was set to 6.5 μs, the acquisition time was 1.36 s, the spectral width to 20 ppm. The signal of HDO was attenuated using water pre-saturation for 2 s. 400 scans were recorded in a total experimental time of about 30 minutes.
O-Antigen purification and 1H-NMR
The LPS was isolated applying the hot phenol-water method(71), followed by dialysis against distilled water until the phenol scent was gone. Then samples were treated with DNase (1mg/100 mg LPS) plus RNase (2 mg/100 mg LPS) at 37°C for 2 h, followed by Proteinase K treatment (1 mg/100 mg LPS) at 60°C for 1 h [all enzymes from Serva, Germany]. Subsequently, samples were dialyzed again for 2 more days, then freeze dried. Such LPS samples were then hydrolyzed with 1% aqueous acetic acid (100°C, 90 min) and ultra-centrifuged for 16 h at 4°C and 150,000 g. Resulting supernatants (the O-antigens) were dissolved in water and freeze-dried. For further purification, the crude O-antigen samples were chromatographed on TSK HW-40 eluted with pyridine/acetic acid/water (10/4/1000, by vol.), then lyophilized. On these samples, 1D and 2 D (COSY, TOCSY, HSQC, HMBC) 1H- and 13C-NMR spectra were recorded with a Bruker DRX Avance 700 MHz spectrometer (1H: 700.75 MHz; 13C: 176.2 MHz) as described(72).
Atomic force microscopy
The indicated S.Tm strains were grown to late-log phase, pelleted, washed once with distilled water to remove salt. A 20 µl of bacterial solution was deposited onto freshly cleaved mica, adsorbed for 1 min and dried under a clean airstream. The surface of bacteria was probed using a Dimension FastScan Bio microscope (Bruker) with Bruker AFM cantilevers in tapping mode under ambient conditions. The microscope was covered with an acoustic hood to minimized vibrational noise. AFM images were analyzed using the Nanoscope Analysis 1.5 software.
Methylation analysis of S.Tm clones
For REC-Seq (restriction enzyme cleavage–sequencing) we followed the same procedure described by Ardissone et al, 2016(73). In brief, 1 µg of genomic DNA from each S.Tm was cleaved with MboI, a blocked (5’biotinylated) specific adaptor was ligated to the ends and the ligated fragments were then sheared to an average size of 150-400 bp (Fasteris SA, Geneva, CH). Illumina adaptors were then ligated to the sheared ends followed by deep-sequencing using a HiSeq Illumina sequencer, the 50 bp single end reads were quality controlled with FastQC (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). To remove contaminating sequences, the reads were split according to the MboI consensus motif (5’-^GATC-3’) considered as a barcode sequence using fastx_toolkit (http://hannonlab.cshl.edu/fastx_toolkit/) (fastx_barcode_splitter.pl --bcfile barcodelist.txt --bol --exact). A large part of the reads (60%) were rejected and 40% kept for remapping to the reference genomes with bwa mem(63) and samtools(74) to generate a sorted bam file. The bam file was further filtered to remove low mapping quality reads (keeping AS >= 45) and split by orientation (alignmentFlag 0 or 16) with bamtools(75). The reads were counted at 5’ positions using Bedtools(76) (bedtools genomecov -d -5). Both orientation count files were combined into a bed file at each identified 5’-GATC-3’ motif using a home-made PERL script. The MboI positions in the bed file were associated with the closest gene using Bedtools closest(76) and the gff3 file of the reference genomes(77). The final bed file was converted to an MS Excel sheet with a homemade script. The counts were loaded in RStudio 1.1.442(78) with R version 3.4.4(79) and analysed with the DESeq2 1.18.1 package(80) comparing the reference strain with the 3 evolved strains considered as replicates. The counts are analysed by genome position rather than by gene. The positions are considered significantly differentially methylated upon an adjusted p-value < 0.05. Of the 2607 GATC positions, only 4 were found significantly differentially methylated and they are all located in the promoter of the gtrABC operon.
The first step in the reads filtering was to remove contaminant reads missing the GATC consensus motif (MboI) at the beginning of the sequence. These contaminant reads are due to random fragmentation of the genomic DNA and not to cuts of the MboI restriction enzyme. Using fastx_barcode_splitter.pl about 60% of the reads were rejected because they did not start with GATC. The rest (40%) was analyzed further. Random DNA shearing and blunt-ended ligation of adaptors, combined with sequencing noise at the beginning of reads likely generates this high fraction of reads missing at GTAC sequence.
gtrABC expression analysis by blue/white screening and flow cytometry
About 200 colonies of S.TmgtrABC-lacZ (strain background 4/74, (32)) were grown from an overnight culture on LB agar supplemented with X-gal (0.2 mg/ml, Sigma) in order to select for gtrABC ON (blue) and OFF clones (white). These colonies were then picked to start pure overnight cultures. These cultures were diluted and plated on fresh LB agar X-gal plate in order to enumerate the proportion of gtrABC ON and OFF siblings. The proportion of O:12/O:12-2 cells was analyzed by flow cytometry.
In vitro growth and competitions to determine wzyB-associated fitness costs
Single or 1:1 mixed LB subcultures were diluted 1000 times in 200 µl of media distributed in 96 well black side microplates (Costar). Where appropriate, wild type S.Tm carried a plasmid for constitutive expression of GFP. To measure growth and competitions in stressful conditions that specifically destabilize the outer membrane of S.Tm, a mixture of Tris and EDTA (Sigma) was diluted to final concentration (4 mM Tris, 0.4 mM EDTA) in LB; Sodium cholate (Sigma) and Sodium Dodecyl Sulfate (SDS) (Sigma) were used at 2% and 0.05% final concentration respectively. The lid-closed microplates were incubated at 37°C with fast and continuous shaking in a microplate reader (Synergy H4, BioTek Instruments). The optical density was measured at 600 nm and the green fluorescence using 491 nm excitation and 512 nm emission filter wavelengths every 10 minutes for 18 h. Growth in presence of SDS is causing aggregation when cell density reaches OD=0.3-0.4, therefore, it is only possible to compare the growth curves for about 250 minutes. The outcome of competitions was determined by calculating mean OD and fluorescence intensity measured during the last 100 min of incubation. OD and fluorescence values were corrected for the baseline value measured at time 0.
Serum resistance
Overnight LB cultures were washed three times in PBS, OD adjusted to 0.5 and incubated with pooled human serum obtained from Unispital Basel (3 vol of culture for 1 vol of serum) at 37°C for 1 h. Heat inactivated (56°C, 30 min) serum was used as control treatment. Surviving bacteria were enumerated by plating on non-selective LB agar plates. For this, dilutions were prepared in PBS immediately after incubation.
Modeling antigen switching between O12 and O12-2
The aim of this modeling approach is to test whether a constant switching rate between an O12 and an O12-2 antigen expression state can explain the experimentally observed bimodal populations.
To this end, we formulated a deterministic model of population dynamics of the two phenotypic states as:

 where O12 and O12-2 denote the population sizes of the respective antigen variants, μ denotes the growth rate, which is assumed to be identical for the two variants, K the carrying capacity, and s→12-2 and s→12 the respective switching rates from O12 to O12-2 and from O12-2 to O12.Growth, as well as the antigen switching rates, are scaled with population size in a logistic way, so that all processes come to a halt when carrying capacity is reached.
where O12 and O12-2 denote the population sizes of the respective antigen variants, μ denotes the growth rate, which is assumed to be identical for the two variants, K the carrying capacity, and s→12-2 and s→12 the respective switching rates from O12 to O12-2 and from O12-2 to O12.Growth, as well as the antigen switching rates, are scaled with population size in a logistic way, so that all processes come to a halt when carrying capacity is reached.
We use the model to predict the composition of a population after growth in LB overnight, and therefore set the specific growth rate to μ = 2.05h-1, which corresponds to a doubling time of roughly 20min. The carrying capacity is set to K = 109 cells. We ran parameter scans for the switching rates s→12 and s→12-2, with population compositions that start either with 100% or 0% O12, and measure the composition of the population after 16h of growth (Fig. S6C). The initial population size is set to 109 cells Experimentally, we observe that when starting a culture with an O12 colony, after overnight growth the culture is composed of around 90% O12 and 10% O12-2 cells, whereas starting the culture with O12 cells yields around 50% O12 and 50% O12-2 cells after overnight growth (Fig. S6B). To explain this observation without a change in switching rates, we would need a combination of values in s→12 and s→12-2 that yield the correct population composition for both scenarios. In Fig. S6D, we plot the values of s→12 and s→12-2 that yield values of 10% O12-2 (starting with 0% O12-2, green dots) and 50% O12-2 (starting with 100% O12-2, orange dots). The point clusters intersect at s→12 = 0.144h-1 and s→12-2 = 0.037h-1 (as determined by a local linear regression at the intersection point).
We then used the thus determined switching rates to produce a population growth curve in a in a deterministic simulation, using the above equations for a cultures starting with 100% O12-2, (Fig. S6E, Left-hand graph) and for a culture starting with 0% O12-2 (Fig. S4E, right-hand graph).
These switching rates are consistent with published values (31). Our results show that the observed phenotype distributions can be explained without a change in the rate of switching between the phenotypes.
Modeling Realistic Transmission
Section 1: Central concepts in non-Typhoidal Salmonella transmission
Salmonella spread in a population when contaminated matter from an infected host is ingested by other susceptible hosts, which may then develop a symptomatic Salmonella infection. Successful vaccination itself will affect the duration of infection and the concentration of Salmonella shed in the feces during the infection. The Evolutionary Trap vaccine will further affect the concentration of S.Tm arriving in the lower GIT of a new host, as short-O-antigen mutants have a higher loss in the environment and during transit of the intestine of the next host (Fig. S17A). In section 2 we consider the effect of infection duration on transmission in a host population. In section 3 we calculate the probability of successful infection from a one-time exposure. In section 4 we discuss realistic parameters. In section 5 we show the expected impact of vaccination per se and evolutionary trap-driven loss-of-fitness on Salmonella transmission in a host population.
Section 2: Duration of infection with non-Typhoidal Salmonella
If the infected host is vaccinated, then the host is protected from inflammation even if an open niche in the lower gastrointestinal tract (GIT) allows Salmonella colonization(18). As Salmonella competes poorly with the microbiota in the absence of inflammation(9), the microbiota will regrow and ultimately outcompete. Thus, the duration of Salmonella shedding is reduced in successfully vaccinated animals. In the limit of a large and well-mixed population, if the duration of infection is reduced k-fold, then R0, the average number of hosts successfully infected by one infected host at the beginning of an outbreak, is also reduced by k-fold. If the population is small or not well-mixed, such that the same host may eat contaminated matter during several distinct periods of susceptibility, then the reduction will be less than k-fold.
Section 3: The probability of infection with non-Typhoidal Salmonella, given a one-time exposure
3.1 : Concepts
A mass m of feces, containing Salmonella at concentration sd, are ingested by the recipient. The number of viable Salmonella ingested with this fecal material will be determined by bacterial death in the environment, prior to ingestion, and death during transit of the upper GIT of the recipient, before reaching the replicative niche in the lower GIT. We denote p the overall probability of survival for each bacterium between leaving the infected animal and arriving in the lower GIT of the recipient. We define s0 as the initial concentration of growing Salmonella present in the lower-GIT of the recipient as:
s0 = msd p/M
with M the mass of the contents of the lower GIT. Successful vaccination per se reduces sd, the concentration of Salmonella shed by the infected donor. The short O-antigen Salmonella have a reduced sd as well as a reduced p, i.e. probability to survive the period of time between shedding and arrival in a new replicative niche.
For Salmonella to successfully cause inflammatory disease, which is required for Salmonella to outcompete the microbiota and generate a large stable nice for Salmonella in the gut lumen, its population has to reach a certain threshold of Salmonella concentration T in the lower GIT(79). If the microbiota is undisturbed, Salmonella cannot grow and reach the threshold. In reality, the exact nature of this colonization resistance is highly complex, but for the purpose of a workable model, let us denote τ the duration of a microbiota perturbation, during which the microbiota is in a low enough concentration that the growth rate of Salmonella will be close to its maximal growth rate. Let us define rs the maximal growth rate and cs the loss rate of Salmonella. If Salmonella does not get to T before the end of τ, then the gut will remain uninflamed, the microbiota will rapidly outcompete Salmonella and the host animal will remain healthy. The lower the initial concentration of Salmonella, s0, the more time it takes to reach the threshold T, and thus the less likely it is that Salmonella can compete with regrowth of the microbiota. Let us denote tw the time window during which transmission leads to a successful Salmonella infection of a recipient host under a given set of conditions. If the recipient host is at most exposed once during τ, then a k-fold reduction in the window of possible infection tw (for example because the short O-antigen Salmonella transmitted from a vaccinated host has a smaller survival probability p, leading to a shorter tw), will also reduce R0 by k-fold. Note that if a recipient host is ingesting contaminated matter more than once during τ, then the reduction will be less than k-fold.
In what follows, we calculate the window of possible infection, as a function of the other parameters.
3.2 Calculations
3.2.1 : In the absence of inflammation, the equations for the Salmonella concentration S(t) are given by (with K the carrying capacity, and F the effective concentration of the microbiota which competes for the same resources as Salmonella):
 We assume that T «K and that during τ, F «K, thus defining reff = rs - cs the effective net growth rate:
We assume that T «K and that during τ, F «K, thus defining reff = rs - cs the effective net growth rate:

3.2.2 : Window of successful salmonella infection
tw, the time window during which successful transmission can occur, can be defined as the time interval required (between t = 0 (time of start of microbiota perturbation in the recipient host) and the end of the microbiota perturbation τ) for Salmonella growth to increase the population concentration from s0 to T. This translates into the following equation
 And thus
And thus

4. Parameters
Duration of Salmonella shedding
In a vaccinated infected donor, the concentration of Salmonella in the lower GIT (and therefore the density of Salmonella shed in the feces) drops more rapidly that in naïve infected donors. Salmonella with a short O-antigen induce less inflammation (Fig. 4H) at 24 h post-infection, even in naïve, streptomycin-pre-treated hosts. As R0 is roughly proportional to this duration of shedding in the population, there is a strong dependence on these observations.
Net growth rate (reff): The wild-type Salmonella maximum net growth rate in the gut lumen reff, is experimentally found to be around 1.3 h−1. (18). From competitive infections of short- and wild-type-O-antigen strains, as shown in Fig. 4 I and J, we see that the relative proportion of the short-O-antigen strain decreases steadily over the 4 days of infection. The decrease in the competitive index is of the order of 100.5-fold a day (Fig. 4I and J), thus we can estimate the difference in net growth rate is of the order of 0.05h−1.
Period of microbiota disturbance (τ): The duration of the period τ during which the microbiota is disturbed enough that Salmonella can grow at reff depends on the type of perturbation present.
• Streptomycin pretreatment (1g/kg) results in a perturbation in the order of 3 days.
• Western diet exposure in mice has a τ in the order of 12 h (49).
The threshold T above which Salmonella induces inflammation in mice is around 5.107/g (79). As similar densities of Salmonella colonization can cause disease in pigs, we make the bold assumption that this value is similar in both species.
The initial concentration of Salmonella s0 in the wild-type case with no vaccination. Mice are naturally coprophagic. In non-Typhoidal salmonellosis, the relevant gut compartment for Salmonella growth is the cecum, which hold a typical mass of M = 1 g contents. A contaminated fecal pellet (mass of typically m = 10 mg), which at a concentration of c = 109 bacteria/g, with a limited death of bacteria (let’s assume p = 0.3 for instance) will mean s0 = mcp/M = 3×106/g Salmonella will be present at the start of the infection.
In swine in natural settings, while c and p will likely be of the same order of magnitude, m/M is different, and will depend strongly on the conditions. The amount of contaminated matter ingested is not well characterized, and likely highly variable. The highest level will be reached for deliberate ingestion of feces. Young piglets eat daily about 20 g of feces (80), have about 24h digestion time, and total mass of their gut content if of the order of 130 g(81), which leads to m/M 0.15, instead of the 0.01 discussed previously for mice. Thus s0 = 5×107/g is an upper bound in piglets.
The reduction in the initial concentration of Salmonella due to successful vaccination or successful vaccination and dominance of short O-antigen clones in the donor, s!0/s0
To simplify this case, we focus on a transmission from the donor occurring during the first 3 days after it becomes infected. During this time, Salmonella loads in the gut are comparable between vaccinated and unvaccinated donor mice, and reduction in transmission will be predominantly due to loss-of-fitness of the shed Salmonella i.e.:.
As vaccination does not change fecal mass likely to be ingested (m) or the mass of lower GIT content before infection (M), m/M will remain the same. Vaccination by itself do not change c, at least in the first days; however for short O-antigen Salmonella, based on Fig. 4F, c’/c will be approximately 10-0.5, giving a lowed net growth in the recipient host. The vaccination itself is not a priori changed by the vaccination itself, however for the short-O-antigen strain, p’/p will be below 1, as this strain is more susceptible to both environmental and host-derived stress. A conservative estimate of s0’/s0 =0.1 is reasonable, and the actual difference may be much larger, as the viability of the short O-antigen Salmonella may be strongly affected (Fig. S15F).
5. Results and Discussion
In Fig. S17B, we compare the relative change in R0 in different scenarios. We take the parameters for mice, and for swine as listed above, and vary two critical parameters (shedding duration of the infected donor, and concentration of Salmonella arriving in the host lower gastrointestinal tract) over a large range of values.
We simulated the effect on the relative R0 as a function of the relative duration of active Salmonella shedding (i.e. the relative length of the infectious period for a vaccinated versus and a naïve donor). Fig. S17B shows, as expected, that the duration of high-density Salmonella shedding is an important parameter, as R0 will scale with this parameter. This duration can be affected both by the vaccination preventing inflammation, and by the presence of short O-antigen strains that are less effective colonizers of the GIT.
Another hypothesis arising from a study of the model parameters is that s0, i.e. the number of viable Salmonella arriving in the lower GIT of a recipient host, will be lower in the case of a short-O-antigen strain, that for a wild-type O-antigen strain due to weakened resistance to environmental and host stressors. Fig. S17B (y axes) shows that the long, robust microbiota perturbation in the mouse non-Typhoidal salmonellosis (streptomycin) model, makes the effect of initial dose on R0 very small. In contrast, in a more natural system with a shorter microbiota perturbation, this parameter has more importance (Swine model).
Significance of this model for Salmonella transmission
An estimate for Salmonella transmission, based on a Danish pig farm outbreak suggests that the R0 in typical conditions maybe around 2 (although with a large uncertainty in the estimate). In order for disease introductions to never result in a major outbreak, R0 needs to be below 1. Therefore, achieving and R0’/R0 of 0.5 or lower will generate effective protection. In this model, outbreak prevention in a pig population can be achieved by decreasing the duration of shedding of the infected animal by 50%; or by 25%, if combined with a decrease in viable Salmonella reaching the lower GIT of the recipient of around 100-fold. The combination of faster decay of Salmonella fecal concentrations in vaccinated animals, and the poorer survival in the environment and during transit through the upper GIT can therefore both contribute to effective prophylaxis in realistic settings.
5.3 Sensitivity to the parameters
As we see on Fig. S17B, R’0/R0 has a strong dependence on the duration of shedding, and a more moderate dependence on s’0/s0. What about the other parameters? Let us use the parameters of Fig. S17B and take s’0/s0=0.1 and a 2-fold reduction of shedding duration. The main difference between the mice and swine case is the duration τ of microbiota perturbation. If the microbiota perturbation was only 6 h instead of 12 h for the swine, then R’0/R0 would be further reduced from 0.41 to 0.33. The dependence on reff is small. A reduction of reff by 20% decreases R’0/R0 from 0.41 to 0.38 in the swine case, and from 0.47 to 0.46 in the mice case. We have also assumed that the reff of the short-O-antigen Salmonella is 0.05 h-1 smaller than the wild-type Salmonella, but this is not the main effect, as removing this assumption increases R’0/R0 by only about 4 % for both the mice and the swine cases. The dependence on s0 and the threshold T is very weak. Decreasing s0 10-fold or increasing T 10-fold only change R’0/R0 from 0.41 to 0.39; and in the mouse, the dependence is even weaker, a similar change reduces R’0/R0 by 0.2% only.
Caveats of the model
This is a highly simplified model and cannot begin to represent the full complexity of this system. Several critical assumptions have needed to be made: 1) that infected animals with high Salmonella shedding remain in the herd. Daily health checks may in fact remove actively infected animals from the herd into quarantine, decreasing this period of shedding and thus R0, also in the unvaccinated case. 2) we have modelled a highly uniform system with a single deterministic equation. In reality, infection spread is highly stochastic. 3) we have not explicitly modeled a time-dependence of s0 in new recipients relative to the time of the first infection, although this will vary over time. 3) the housing situation on a typical Danish pig farm may differ significantly to conditions in pig farms around the world and the necessary reduction in R0 required to protect these farms would require a more detailed assessment of pen sizes, exposure to fecal material, cleaning regimens, etc. 4) We assume that the interaction between short O-antigen-producing Salmonella and vaccination is additive and not that one effect always supersedes the other. In reality, this may be more complex, as for example the evolutionary trap vaccines should better prevent outgrowth of fitness-neutral escape mutations.
Supplementary figure legends

A-C. Atomic force microscopy phase images of S.Tmwt, S.TmΔwzyB (single-repeat O-antigen), and S.TmΔwbaP (rough mutant - no O-antigen) at low magnification (A) and high magnification (B and C). Invaginations in the surface of S.TmΔwbaP (dark colour, B) show a geometry and size consistent with outer membrane pores(84). These are already less clearly visible on the surface of S.TmΔwzyB with a single-repeat O-antigen, and become very difficult to discern in S.Tmwt. C. Fast-Fourier transform of images shown in “B” demonstrating clear regularity on the surface of S.TmΔwbaP, which is progressively lost when short and long O-antigen is present.

Intestinal lavage IgA from PA-STm-vaccinated mice at d28 after the first vaccination (n=5) or S.TmΔsseD-chronically infected mice at day 40 post-infection (n=9) were titred against wild-type S.Tm by bacterial flow cytometry. Dilution factor of lavages givine IgA MFI=500 is shown. Mann Whitney U test result shown.

A. Recombinant monoclonal STA5 human IgG1 was used to surface stain overnight cultures of the indicated Salmonella enterica serovars. Bacterial surface binding was detected with a Dylight-633-anti human IgG secondary antibody and analysed by flow cytometry. STA5 binds to all serovars that include the O:12 epitope B. Overnight cultures of the indicated S.Tm synthetic mutants were stained with STA5 and analysed as in (A). Over-expression of gtrABC producing O:12-2-modified O-antigen results in loss of STA5 staining. C. Overnight cultures of the indicated S.Tm synthetic mutants were stained with STA5 and analysed as in (A), or with a and a recombinant murine monoclonal dimeric IgA STA121, detected with Brilliant violet 421-anti-mouse IgA. Note that STA5 binds equally well to S.Tm with short or long O-antigen, but STA121 cannot bind to the single-repeat O-antigen. wzyB mutations are therefore associated, at least, with escape from vaccine-induced antibodies with similar specificities to STA121.

A. Schematic diagram of expected NMR peaks for each molecular species B. HR-MAS 1H-NMR spectra. Spectra show predicted peak positions, and observed spectra for C1 protons of the O-antigen sugars. C. 1H NMR of purified LPS from the indicated strains. Note that non-acetylated abequose can be observed in wild-type strains due to spontaneous deacetylation at low pH in late stationary phase cultures(30). A gtrA mutant strain is used here to over-represent the O:12-2 O-antigen variant due to loss of regulation(40).

IgA-/- and Rag1-/- and heterozygote littermate controls were pre-treated with streptomycin and infected with S.TmΔsseD orally. Feces were collected at the indicated time-points, enriched overnight in LB plus kanamycin, stained for O:5 and O:12-0 and analysed by flow cytometry. The fraction of the population that lost O:5 and O:12-0 antisera staining is shown over time. Note that lines joining the points are to permit tracking of individual animals through the data set, and may not be representative of what occurs between the measured time-points.

A. Wild type and evolved S.Tm clones were picked from LB plates, cultured overnight, phenotypically characterized by O:12-0 (left panel) and O:5 staining (right panel), plated and re-picked. This process was repeated over 3 cycles with lines showing the descendants of each clone. B. Comparison of fractions of O:12-0-positive and O:12-0-negative bacteria (in fact O:12-2) determined by flow cytometry staining with typing sera and by blue-white colony counts using a gtrABC-lacZ reporter strain. C-E. Results of a mathematical model simulating bacterial growth and antigen switching (see supplementary methods). C. Switching rates from O12-0 to O12-2 and from O12-2 to O12-0 were varied computationally, and the fraction of O12-2 was plotted after 16 h of growth. Left-hand plot depicts the results of the deterministic model when starting with 100% O12-2, right-hand plot depicts the results when starting with 100% O12-0. D. depicts only the switching rates that comply with the experimentally observed antigen ratios after overnight growth (90% O12-0 when starting with O12-0, and 50% O12-0 when starting with O12-2). Right-hand plot is a zoomed version showing values for switching rates between 0 – 0.1 h-1 (marked by a grey rectangle in D). left-hand plot. Dashed lines are linear regressions on the values in this range, and their intersection marks the switching rates used for the stochastic simulation in (E). E. Simulation results of bacterial population growth, when starting with only O12-2 (left-hand plot) or only O12-0 (right-hand plot). was kept constant in all simulations; switching rates were varied in steps of in (C and D), and kept constant at and in (E); the starting populations were always individuals of the indicated phenotype; carrying capacity was always 109 cells.

Aligned fractions of the oafA ORF from a natural isolate (from chicken) presenting the same 7 bp deletion detected in mutants of S.Tm SL1344 emerging in vaccinated mice. S.Tm SL1344 was used a reference(85). B. Aligned oafA promoter sequences from three natural isolates of human origin (stool or cerebrospinal fluid(86)) showing variations in the number of 9 bp direct repeats.

Wild type 129Sv mice were mock-vaccinated or were vaccinated with PA-S.TmΔoafA ΔgtrC as in Fig. 1A. On d28, all mice were pre-treated with streptomycin, and infected with the indicated strain. A. Feces recovered at day 10 post-infection, was enriched overnight by culture in streptomycin, and stained for O:12. Fraction O:12-low S.Tm was determined by flow cytometry. Percentage of S.Tm that are O:12-negative was quantifed over 10 days and is plotted in panel B.

A-C. Naive (closed circles), PA-S.TmΔgtrC-vaccinated (open circles) and PA-S.TmET-vaccinated (crossed-circles) SPF Balb/c mice were streptomycin-pretreated, infected (105 CFU, 1:1 ratio of S.TmΔgtrC and S.TmΔgtrC ΔoafA per os). Note that naïve Balb/c mice were euthanized on day 3 due to severe disease. A. Secretory IgA titres (intestinal lavage dilution) against O:4[5], 12-0, and an O:4, 12-0 S.Tm. B. Competitive index (CFU S.Tm ΔgtrC/CFU S.TmΔ gtrC ΔoafA) in feces at the indicated time-points. 2-way ANOVA with Bonferroni post-tests on log-normalized values, compared to naive mice. ****p<0.0001. C and D. Correlation of the competitive index with the O:4-specific (C) and O:4[5]-specific (D) intestinal IgA titre, r2 values of the linear regression of log-normalized values. Open circles: Intestinal IgA from PA-S.TmΔgtrC -vaccinated mice, crossed circles: Intestinal IgA from PA-S.TmET - vaccinated mice. Lines indicate the best fit with 95% confidence interval. E. CFU of S.TmΔgtrC (black symbols) and S.TmΔgtrC ΔoafA (orange symbols) per gram feces at the indicated time-points. F and G. CFU of S.TmΔgtrC (black symbols) and S.TmΔgtrC ΔoafA (orange symbols) per organ and day 4 post-infection (vaccinated) and day 3 post-infection (naïve). H. Fecal Lipocalin 2 as a marker of inflammation in the indicated groups. 2-way repeat-measures ANOVA on log-normalized data. ***p<0.001,****p<0.0001 I and J. Correlation between fecal lipocalin 2 on d3 post-infection and O:4 and O:4[5]-specific intestinal IgA titres. r2 values of the linear regression of log-normalized values. Lines indicate the best fit with 95% confidence interval. Note that lines joining the points are to permit tracking of individual animals through the data set, and may not be representative of what occurs between the measured time-points.

For challenge, S.TmΔaroA ΔgtrC-exposed (O:4[5]-vaccinated, closed circles) and S.TmΔaroA ΔgtrC ΔoafA-exposed (O:4-vaccinated, closed squares) SOPF mice (C57BL/6, black symbols; 129 Sv/Ev, blue symbols) were ampicillin-pretreated, infected (107 CFU, 1:1 ratio of ampicillin-resistant S.TmΔgtrC and S.TmΔgtrC ΔoafA per os). A. Competitive index (CFU S.Tm ΔgtrC/CFU S.TmΔgtrC ΔoafA) in feces at the indicated time-points. Mean significantly different from 0, One-sample t test *p<0.05, **p<0.01,***p<0.001. B. Fecal Lipocalin 2 (LCN2). The timeline is divided between days post-infection by attenuated aroA strains and days post-challenge with competing virulent strains. C. Vaccine strains shedding at day 1, 9 and 42 post-infection. The resident E. coli population eventually excluded aroA Salmonella mutants in the 129 Sv/Ev mice (Blue symboles) D and E. Correlation of the competitive index with the O:4[5]-specific (D) and O:4-specific (E) intestinal IgA titre, r2 values of the linear regression of log-normalized values. Closed circles: Intestinal IgA from O:4[5]-vaccinated mice, Closed squares: Intestinal IgA from O:4-vaccinated mice. Lines indicate the best fit with 95% confidence interval
Mice were vaccinated and pre-treated as in Fig. 3. The inoculum contained a 1:1 ratio of S.TmΔoafA and S.TmΔoafA ΔgtrC pgtrABC. Competitive index in feces was determined by differential selective plating over 4 days post-infection.

Fig. 3A. Fecal Lipocalin 2 (LCN2) over days 1-4 plotted against the competitive index of infection for animals from Fig. 3A, vaccinated either against O:5-producing S.Tm (A), or O:4-producing S.Tm (B). Symbols indicate different days post-infection as indicated in the legend.
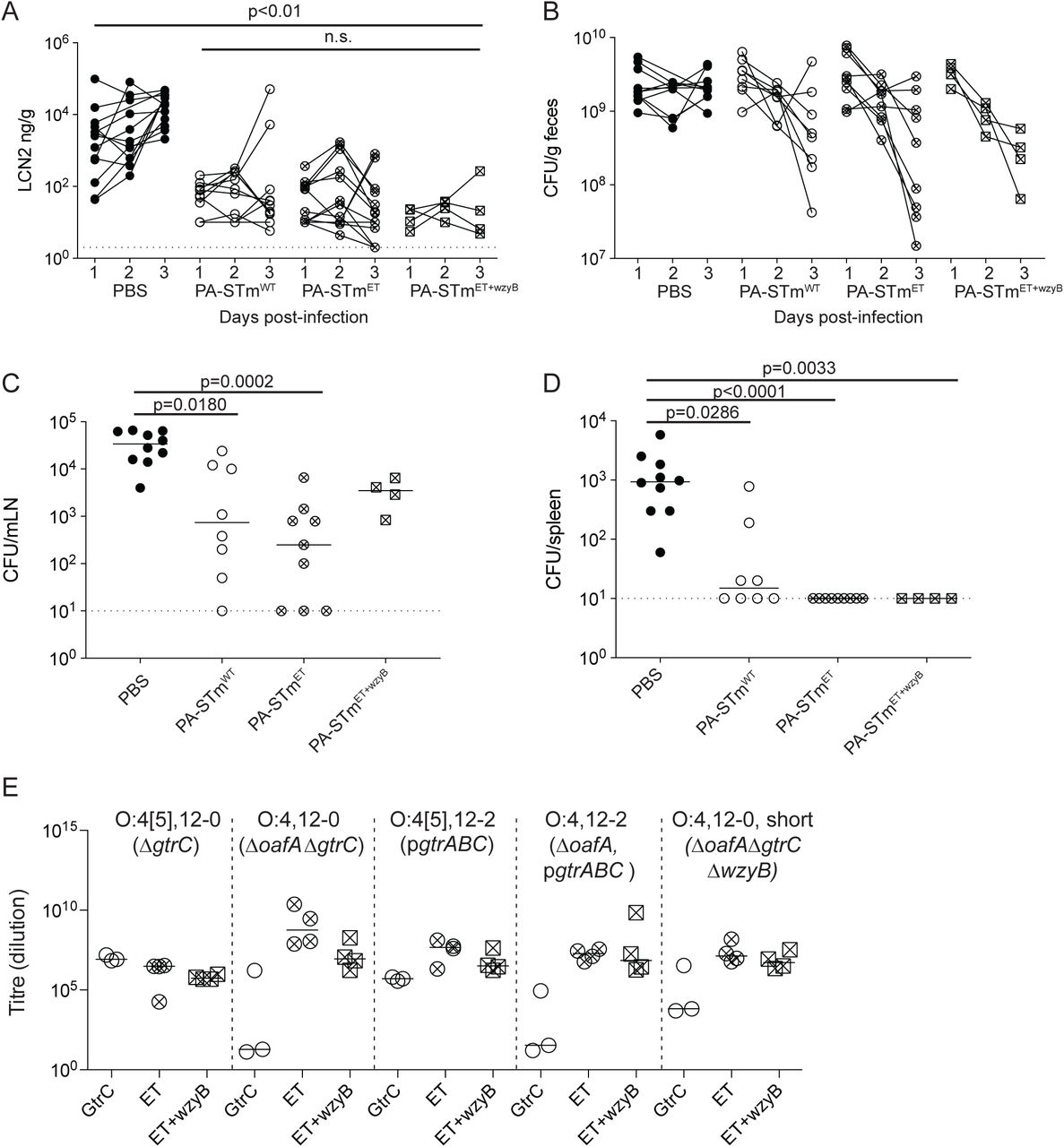
Mice were vaccinated with vehicle only (Naïve, n=10), PA-S.Tmwt (n=8), PA-STmET (combined PA-S.TmΔgtrC, PA-S.TmΔoafA ΔgtrC, PA- S.Tm pgtrABC, and PA-S.TmΔoafA pgtrABC, n=9) or PA-STmET+wzyB (combined PA-S.TmΔgtrC, PA-S.TmΔoafA ΔgtrC, PA-S.Tm pgtrABC, PA-S.TmΔoafA pgtrABC and PA-S.TmΔoafA ΔgtrC ΔwzyB, n=4). On day 28 after the first vaccination, mice were streptomycin pre-treated and challenged with 105 S.Tmwt orally. Fecal Lipocalin-2 (LCN2) at day 1-3 post-infection (A) and CFU S.Tmwt per gram feces on day 1-3 post-infection (B), CFU S.Tmwt per mesenteric lymph node (MLN) at day 3 post-infection (C), and CFU S.Tmwt per spleen at day 3 post-infection (D). A and B, repeat-measures ANOVA reveals no significant difference between the vaccinated groups at any time-point. C and D: Kruskal-Wallis analyses were carried out for significance. Exact p values displayed. E. IgA titres of a final experiment not included in Fig. 4A), and additionally showing the group PA-S.TmΔoafA ΔgtrC ΔwzyB. 2-way ANOVA on log-normalized data. Note that lines joining the points are to permit tracking of individual animals through the data set, and may not be representative of what occurs between the measured time-points.


The deletion of wzyB does not affect the growth of S.Tm or S.TmΔoafA ΔgtrC in LB (No stress) (A) but impairs growth in presence of Tris-EDTA (B), 2% cholate (C) and 0.05% SDS (D). Dashed lines represent the range of variations between experiments. This was in line with the outcome of competitions between S.Tm expressing constitutive Green Fluorescent Protein (S.TmGFP) and S.Tm ΔoafA ΔgtrC ΔwzyB or a wzyB mutant isolated from an Evoltrap vaccinated mouse (E). The level of GFP corrected for the optical density (OD) served as readout to quantify the S.TmGFP population at the end of the overnight growth, in presence of S.TmΔoafA ΔgtrC, S.Tm ΔoafA ΔgtrC ΔwzyB or an evolved S.TmΔwzyB, in LB with or without Tris-EDTA. Values above 1 (dashed line) indicates that relatively more GFP was detected in presence of Tris-EDTA than without, which resulted from a competitive advantage of S.TmGFP in presence of stress. F. The deletion of wzyB makes Salmonella sensitive to human serum complement. Values below 100 (dashed line) indicates that the number of colony-forming units (CFU) detected after incubation in human serum was lower than after incubation in heat inactivated human serum.

A-C. SOPF C57BL/6J mice were switched onto a western diet 24 h before infection with 108 S.TmΔoafA ΔgtrC (long O-antigen, red diamonds, n=6) or 108 S.TmΔoafAΔgtrC ΔwzyB (Short O-antigen, black diamonds, n=7) p.o. A. S.Tm CFU per gram feces on day 1-3 post infection. B. S.Tm CFU per mesenteric lymph node (mLN) at d 3 post-infection, C. S.Tm CFU per spleen at d3 post-infection. A. Repeat-measures ANOVA on log-normalized data with Bonferroni post-tests. B and C. Mann-Whitney U tests. C. and E. A donor cohort of mice were pre-treated with streptomycin and infected with either 105 S.TmΔoafA ΔgtrC (long O-antigen, n=6) or 105 S.TmΔoafA ΔgtrC ΔwzyB (Short O-antigen, n=6) p.o. The recipient mice were placed onto western diet. At 24 h post-infection (donors), and 24 h after starting western diet feeding (recipients), fecal pellets were collected from the donors, homogenized, plated to determine the donor CFU/g feces (D), and then concentrated into 100µl. S.Tm from one fecal pellet was used to transmit the infection into western-diet-fed recipients. E. S.Tm CFU per gram feces in recipient mice at the indicated time-points.
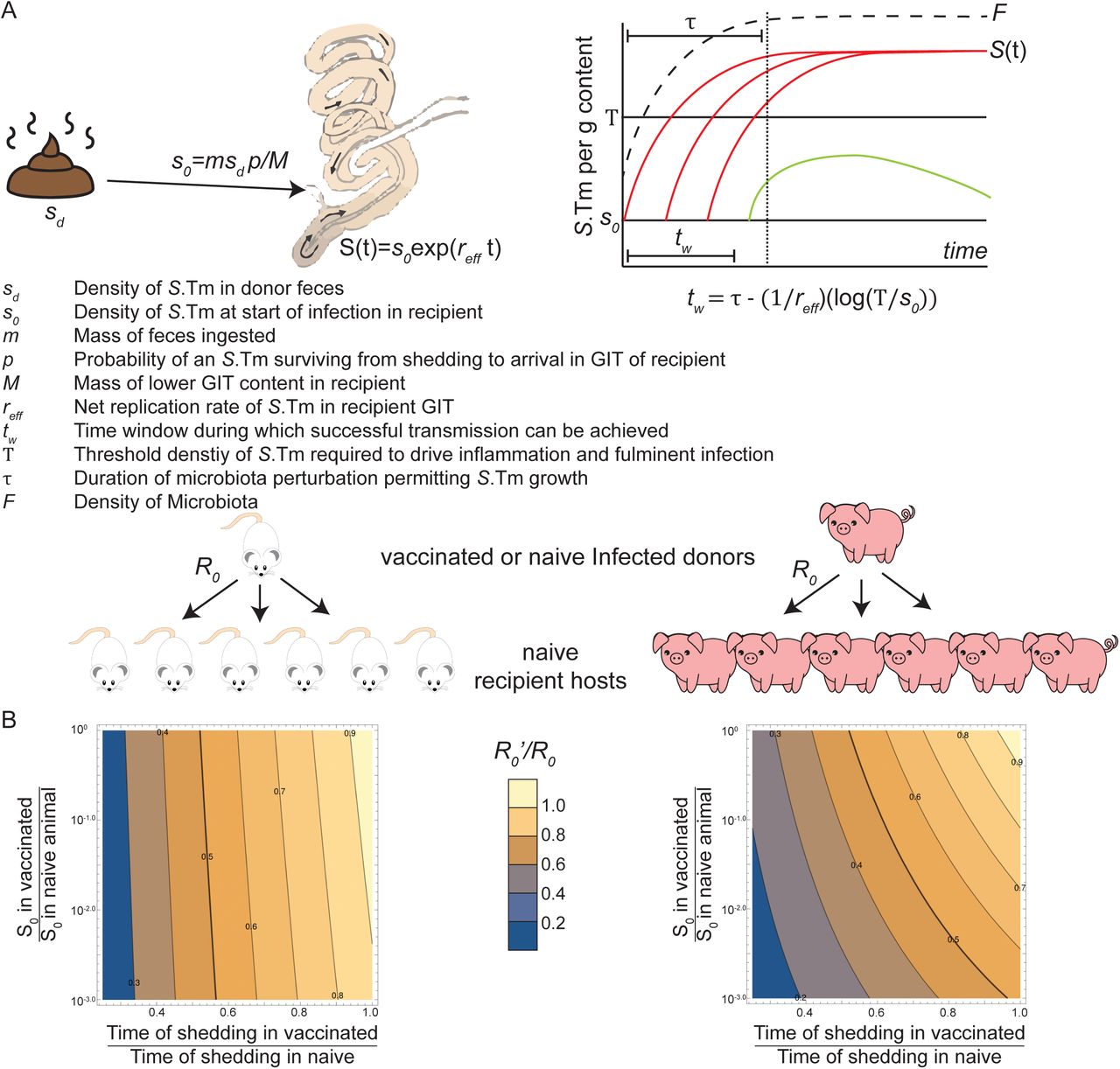
A. Schematic of the model in which a vaccinated or naïve donor transmits infection into a naïve population. A new host becomes infected by ingesting a certain mass of feces containing an original Salmonella density of sd Individual Salmonella shed from the donor have a probability p of arriving alive in the lower gastrointestinal tract (GIT) of the new host, generating the seeding population at density s0. The nature of the perturbation present in the new host will determine the duration τ during which these Salmonella will be able to grow. If the Salmonella population size reaches a threshold, T, before the microbiota recovers then inflammation is triggered, and a fulminant infection develops. If s0 is low, or the infection event occurs too late during the microbiota perturbation, then T is not reached, and the host remains healthy (see supplementary methods for more detail). B. Here the equations in A are used to investigate the effect of a shorter infectious period and lower seeding densities (s0), due vaccination on the relative value of R0 (R0’/R0 colour scale). We explore two possible scenarios. T = 5×107 CFU/g for both mice and pigs. In mice pre-treated with streptomycin, s0 = 3×106 /g and τ = 72 h, in swine s0 = 5×107 /g and τ = 12 h. As the R0 for Salmonella spread in a standard Danish pig farm is estimated to be in the order of 2, a 50% reduction in R0 will result in extinction of the outbreak.
Supplementary Movies A and B
Visualization of O:12 phase variation using live-cell immunofluorescence. Cells expressing GFP (green) pre-stained with fluorescently-labeled recombinant murine IgA STA121 specific for the O:12-0epitope (red) were loaded into a microfluidic chip for time-lapse microscopy. Cells were fed continuously S.Tm-conditioned LB containing fluorescently-labeled recombinant murine IgA STA121 specific for the O:12-0epitope.
(A) Loss and (B) gain of antibody reactivity (red staining) was observed, indicative of O:12 phase variation.
Supplementary Tables
Table S1: Salmonella strains and genotypes
Table S2: Details of primers used in strain construction, testing and sequencing
Table S3: Details of mutations found in resequenced O12-0 or O12 bimodal evolved clones studied by REC-Seq as shown in Fig. 1F and 2C-D. Numbers indicate the position of the mutation, numbers in brackets indicates the percentage of reads were the mutation was detected.
Table S4: Details of experiments in which Salmonella O-antigen shifts were observed

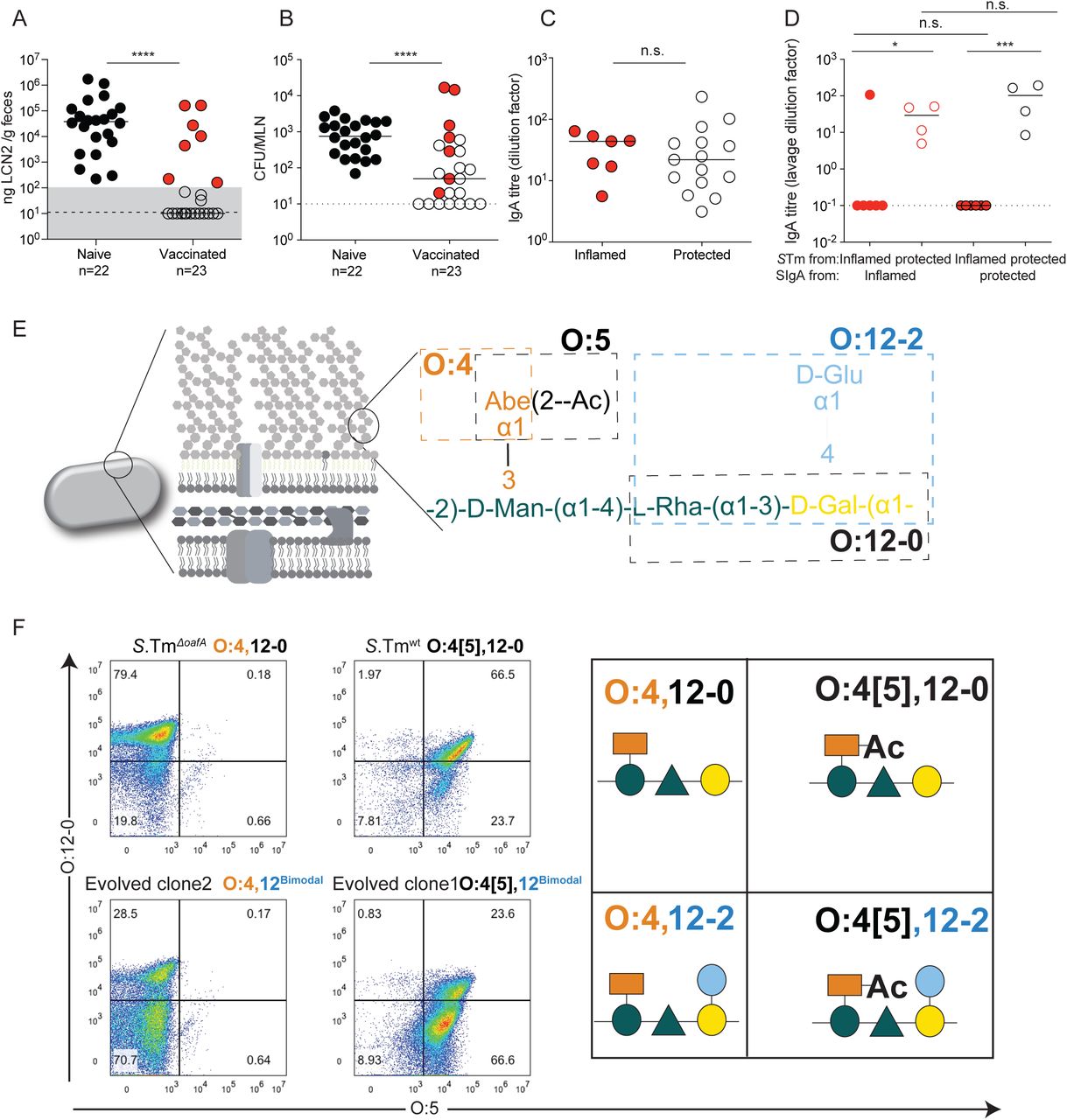{kind=link}
{kind=link}
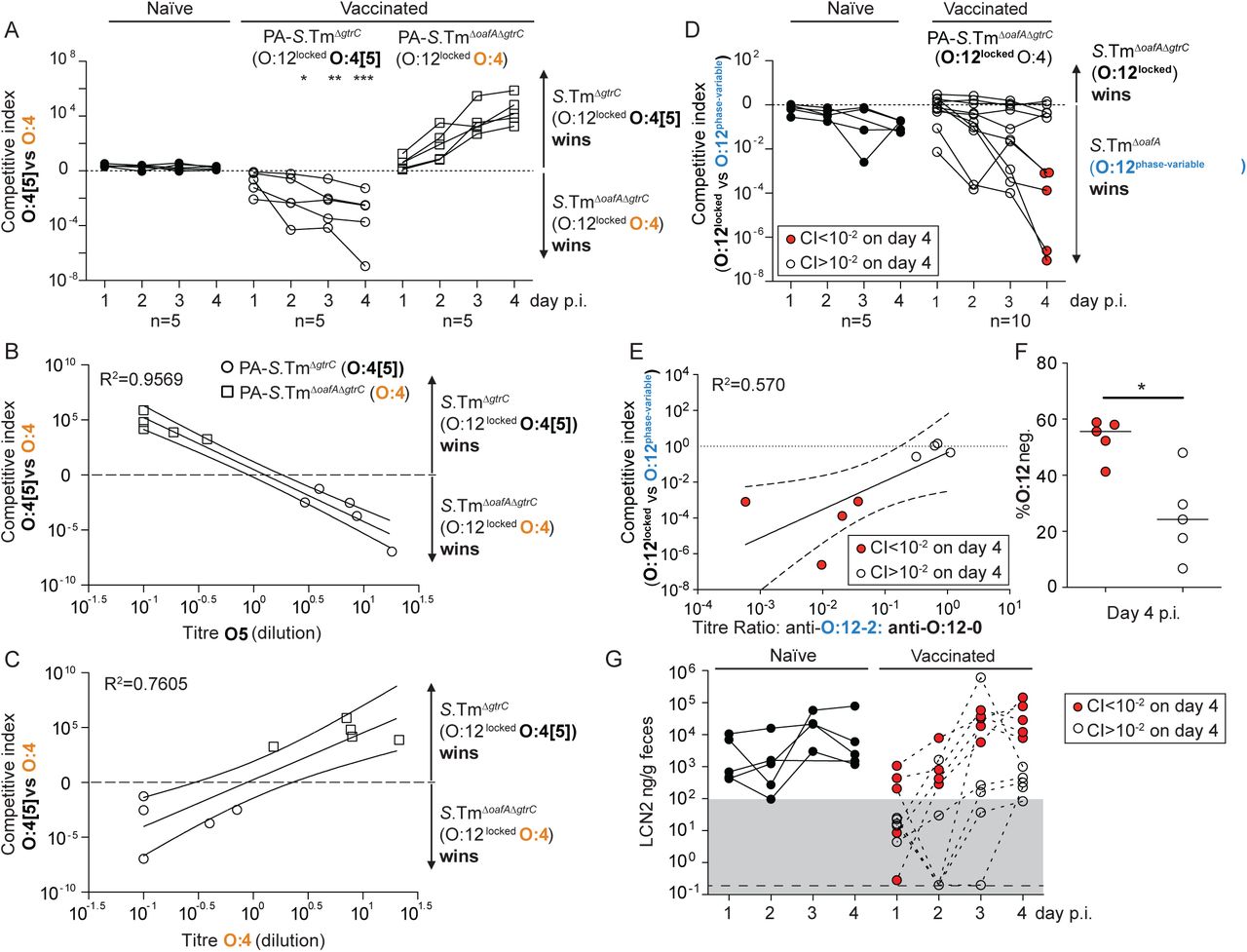{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
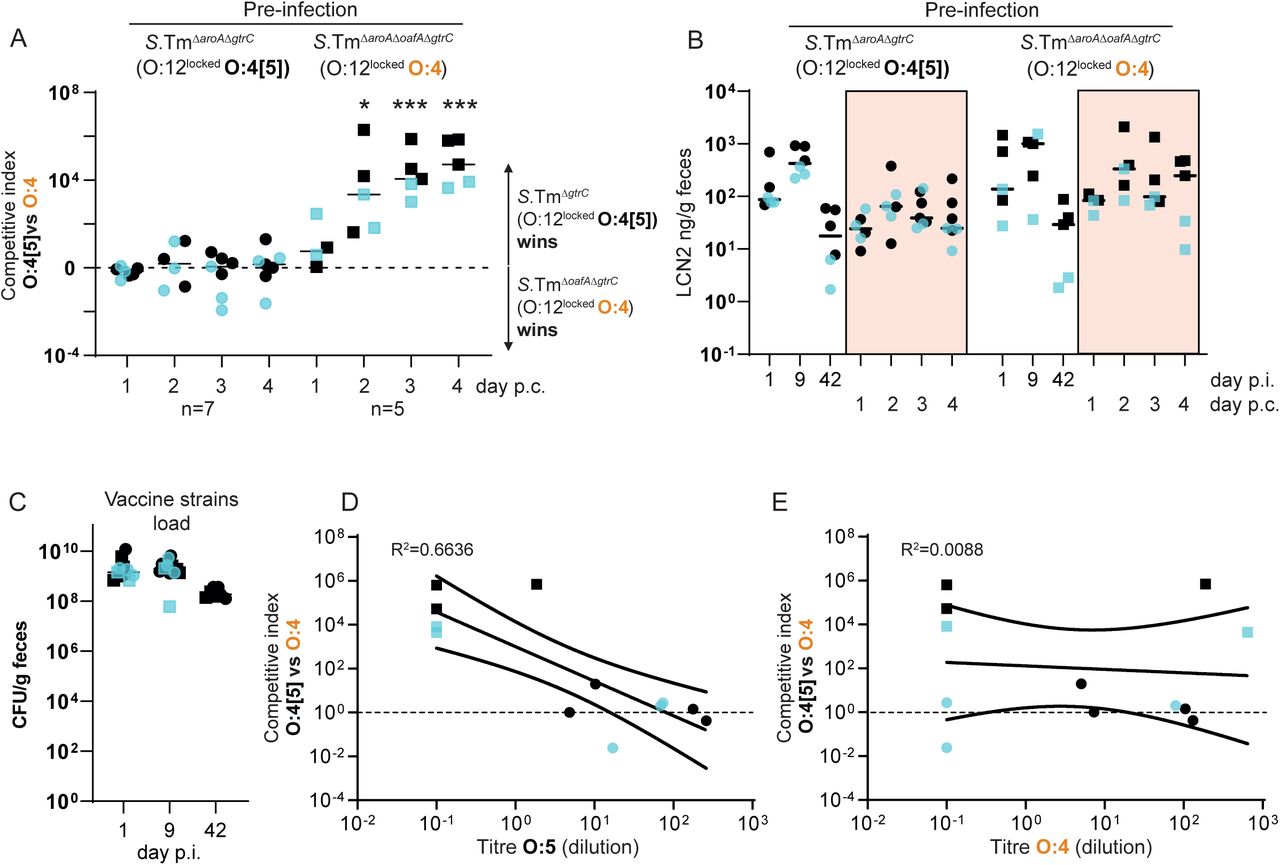{kind=link}
{kind=link}
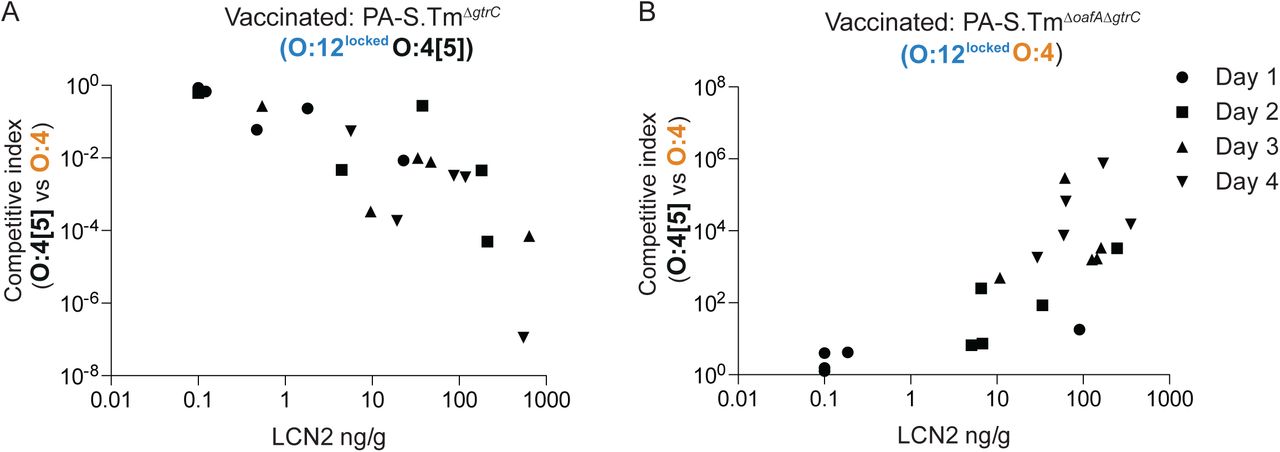{kind=link}
{kind=link}
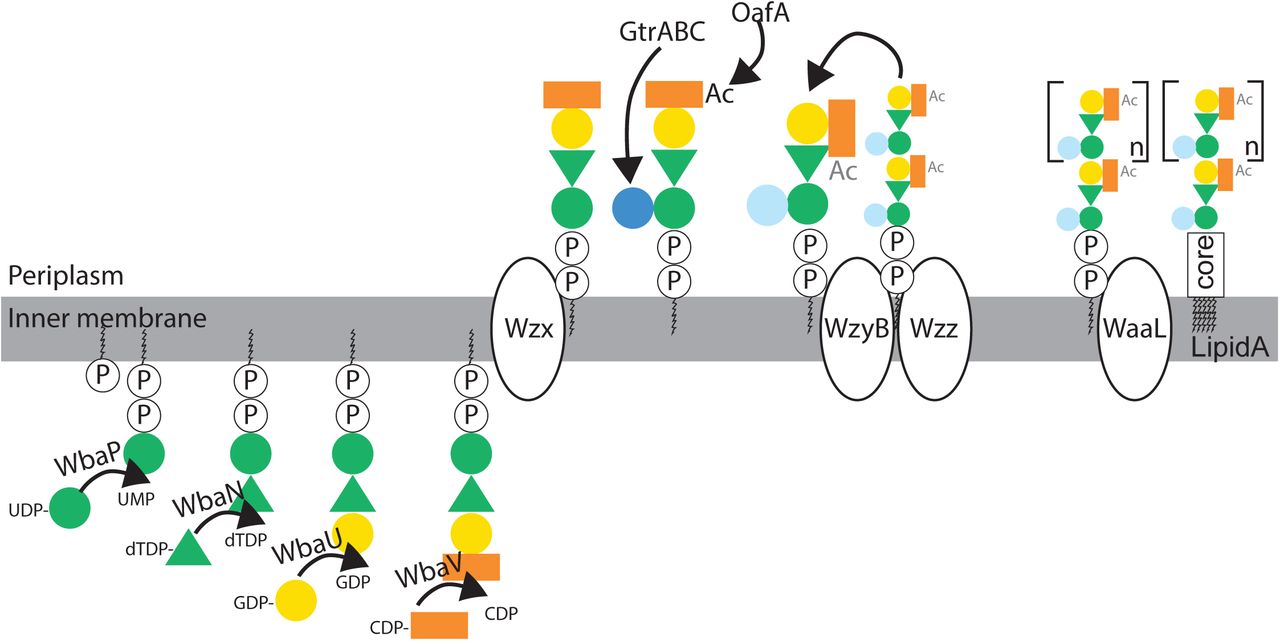{kind=link}
{kind=link}
{kind=link}
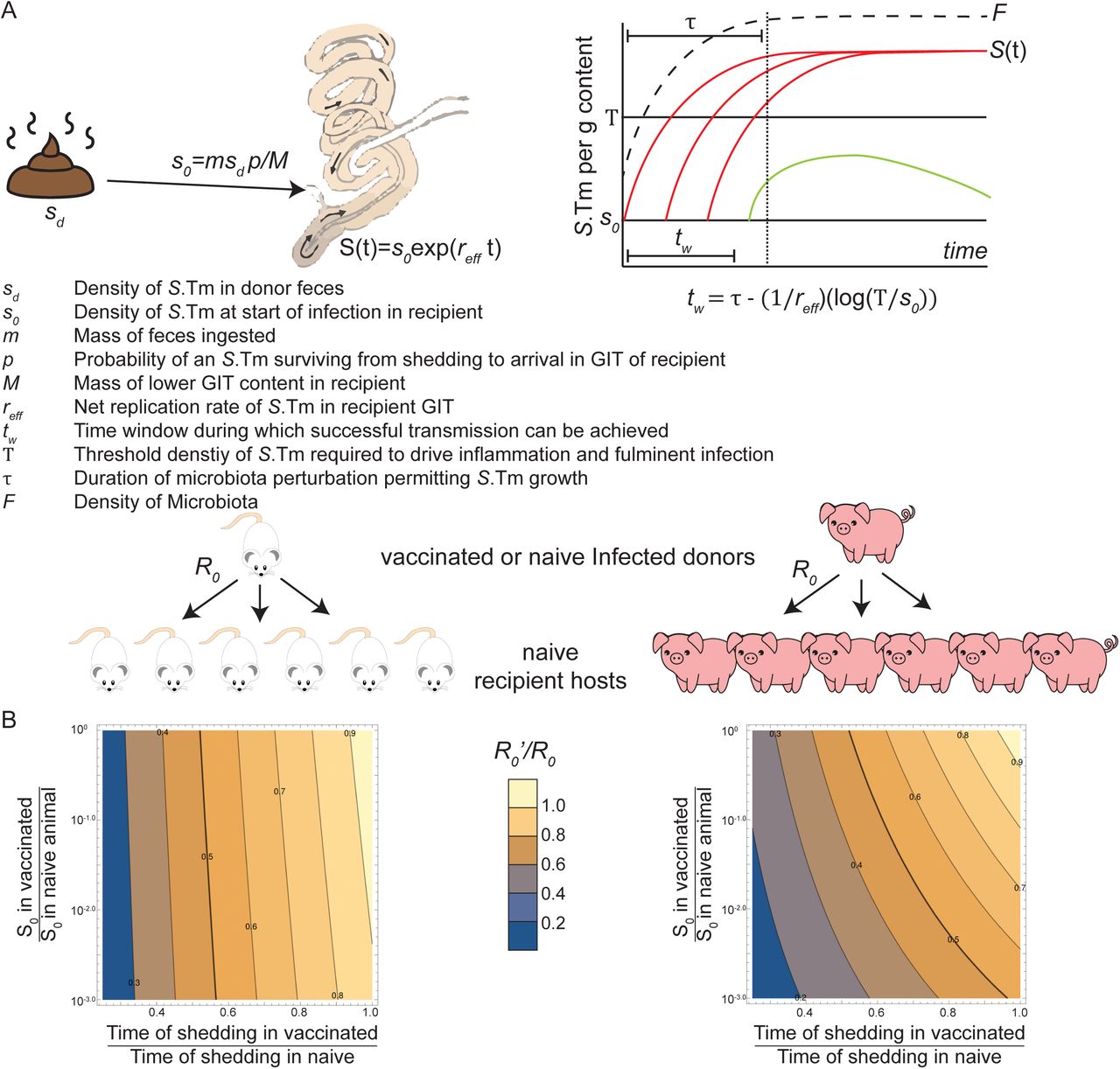{kind=link}
{kind=link}
Acknowledgements
OH acknowledges Heiko Käßner for recording NMR spectra, Regina Engel for GLC-MS, and Katharina Jakob and Sylvia Düpow for technical support. We want to thank Magdalena Schneider, Christine Kiessling, Elisabeth Schultheiss, Rosa-Maria Vesco and Clarisse Straub for the DNA extraction, library preparations and sequencing of the bacterial isolates. MD acknowledges Delphine Cornillet and the group of Prof. Dirk Bumann for serum resistance measurements.
ES, WDH and MD acknowledge Prof. Markus Aebi and Dr. Joshua Cherry for their helpful discussion and insight, as well as members of the Hardt, Slack, and Diard groups for their comments.
Footnotes
Extensive modifications of the text to clarify aims of the work. Corrections to mis-aligned data in figure 1 Re-drawing of sub-figures to allow individual animals to be tracked over time. Additional data points added to sub-panels Additional supplementary figures added to demonstrate influence of short O-antigen on disease transmission Concept repeated in an additional mouse line (Balb/c)
References
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.
- 6.↵
- 7.↵
- 8.↵
- 9.↵
- 10.↵
- 11.↵
- 12.↵
- 13.↵
- 14.
- 15.
- 16.↵
- 17.↵
- 18.↵
- 19.↵
- 20.↵
- 21.↵
- 22.
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.↵
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.
- 36.↵
- 37.↵
- 38.↵
- 39.↵
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.↵
- 47.↵
- 48.↵
- 49.↵
- 50.↵
- 51.↵
- 52.↵
- 53.↵
- 54.↵
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.↵
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.↵
- 75.↵
- 76.↵
- 77.↵
- 78.↵
- 79.↵
- 80.↵
- 81.↵
- 82.
- 83.
- 84.↵
- 85.↵
- 86.↵
- 87.↵




