

Abstract
Nippostrongylus brasiliensis is a well-defined model of type 2 immunity but the early lung-migrating phase is dominated by innate IL-17A production and neutrophilia. While the importance of IL-17A is well established during microbial infections, its contribution to type 2 immunity is poorly understood. Using N. brasiliensis infection we show that Il17a-KO mice exhibited an impaired type 2 immune response correlating with increased worm burden. Neutrophil depletion and reconstitution studies demonstrated that neutrophils contributed to the subsequent eosinophilia but were not responsible for the ability of IL-17A to promote type 2 cytokine responses. Transcriptional profiling of the lung on day 2 of N. brasiliensis infection revealed an increased IFNγ signature in the Il17a-KO mice confirmed by enhanced IFNγ protein production. Depletion of early IFNγ restored type 2 immune responses in the Il17a-KO mice supporting the hypothesis that the suppression of IFNγ by innate IL-17A sources promotes the protective type 2 immune response. Notably, when IL-17A was blocked later in infection, there was an increased type 2 response. Combined data from both N. brasiliensis and Trichuris muris, a gut-specific pathogen, demonstrated that IL-17A regulation of type 2 immunity was lung-specific. Together our data suggest that IL-17A is a major regulator of type 2 immunity in the lung which supports the development of a protective type 2 immune response but subsequently limits the magnitude of that response.
Introduction
Innate and adaptive sources of interleukin-17A (IL-17A) are responsible for a range of neutrophil-associated inflammatory conditions as well as protection from many bacterial and fungal pathogens [1] [2]. In contrast, type 2 immunity is required for effective control of most helminth infections [3] and is characterized by eosinophilic inflammation and the cytokines IL-4, IL-5 and IL-13. When both type 2 and IL-17 responses are present during helminth infection enhanced pathology is observed, as shown for human schistosomiasis [4], [5] and onchocerciasis [6]. The detrimental relationship between IL-17A and type 2 associated diseases has also been extensively documented in allergic asthma in which the most severe symptoms occur in patients with both high Th2 and Th17 cell responses [7]. Critically, type 2 cytokines can actively suppress IL-17A production which may be an important feedback mechanism to avoid extreme IL-17A-driven pathology [8]–[10]. Despite the evidence for an important relationship between IL-17A and type 2 immune responses during chronic disease, how these responses are connected remains poorly understood.
We and others have demonstrated a prominent role for IL-17A during infection with the lung-migrating nematode Nippostronglyus brasiliensis [8][11], a well-defined pulmonary model of type 2 immunity. After entering the host via the skin, N. brasiliensis larvae migrate through the blood vessels into the lung, causing tissue damage and haemorrhage. IL-17RA-dependent neutrophil recruitment is largely responsible for the lung damage in this model [12]. We previously found that the chitinase-like protein Ym1 induces expansion of IL-17A-producing γδ T cells and Ym1 blockade or IL-17A-deficiency protects mice from peak lung damage [8]. More surprising was our finding that Ym1 neutralisation or IL-17A-deficiency prevents the development of a full type 2 response during N. brasiliensis infection [8], [13].
The notion that IL-17A is required for development of a type 2 response appears counter to the evidence that type 2 cytokines suppress IL-17A production[9], [10]. However, previous studies using murine models of allergic inflammation also show impaired type 2 immunity in the face of IL-17A deficiency [14], [15] or blockade [16]. In an infection or injury context, the specific tissue as well as timing might all play decisive roles in whether IL-17A augments or suppresses type 2 responses. We therefore used N. brasiliensis infection to address the contribution of γδ T cell-derived IL-17A and neutrophils to the development of a subsequent type 2 immune response in the lung. We found that IL-17A suppressed early IFNγ production and that this suppression was essential for the optimal development of a type 2 response. Although neutrophils acted as a major driver of subsequent lung eosinophilia, neutrophils were not responsible for IL-17A-mediated enhancement of type 2 immunity. Once the type 2 response was established, IL-17A acted as a negative regulator, revealing distinct roles during innate and adaptive stages of the response. Notably, Trichuris muris, a nematode restricted to the gastro-intestinal tract also induced a lung type 2 response that was IL-17A-dependent. However, we found no evidence that IL-17A regulated the intestinal type 2 response. Thus, IL-17A serves as a lung-specific regulator of the type 2 immune response.
Materials & Methods
Mice and ethics statement
For experiments using only WT mice, C57BL/6 J mice were obtained from Charles River. C57BL/6 Il17aCreRosa26eYFP mice were originally provided by Dr Brigitta Stockinger[17], [18]. For Il17a-KO experiments C57BL/6 WT mice and C57BL/6 Il17aCreRosa26eYFP homo-zygote mice were bred at the University of Manchester. Mice were age-and sex-matched and all mice were housed in individually ventilated cages. Both males and females were used. Mice were not randomized in cages, but each cage was randomly assigned to a treatment group. Mice were culled by asphyxiation in a rising concentration of CO2. Experiments were performed in accordance with the United Kingdom Animals (Scientific Procedures) Act of 1986.
N. brasiliensis infection
N. brasiliensis was maintained by serial passage through Sprague-Dawley rats, as described [19]. Third-stage larvae (L3) were washed ten times with PBS (Dulbecco’s PBS, Sigma) before infection. On day 0, mice were infected subcutaneously with 250 or 500 larvae (L3). At various time points mice were euthanised, bronchoalveolar lavage (BAL) was performed with PBS containing 1% BSA and lungs were taken for further analysis. For worm counts, the small intestines of infected mice were collected in PBS. Small intestines were then cut longitudinally along the entire length, placed in a 50 ml Falcon and incubated at 37°C for 4h. Settled worms were then counted with the aid of a dissecting microscope.
Flow cytometry
Single-cell suspensions of the lung were prepared by digesting minced lung lobes for 30 min at 37 °C with 0.2 U/ml Liberase TL (Roche) and 80 U/ml DNase (Life Tech) in Hank’s balanced-salt solution before forcing tissue suspensions through a cell strainer (70 µm, Greiner). Red blood cells were lysed using Red Blood Cell Lysing Buffer Hybri Max (Sigma) for 3 min at RT and reaction was stopped by diluting samples in PBS. Total live cells were counted with AO/PI dye on an automated cell counter (Auto2000, Nexcelom). Cells were stained for live/dead (Life Technologies) and then incubated with Fc block (1:500 CD16/CD32 and 1:50 mouse serum) and were then stained with fluorescence-conjugated antibodies. Cells were identified by expression of surface markers as follows: neutrophils Ly6G+CD11b+, eosinophils CD11b+ CD11c− SigF+, CD4 T cells CD4+, TCRβ+CD11b−, γδ Tcells TCRβ−, TCRγδ+, CD11b− and ILC2s Lineage− (CD11b, TCRβ, TCRγδ, Ly6G, F4/80, CD11c, SigF, CD19) CD90.2+KLRG+CD127+. Antibody clones used are listed in Table 1. For staining of intracellular cytokines, cells were stimulated for 4 h at 37 °C with cell stimulation cocktail containing protein transport inhibitor (eBioscience), then stained with live/dead. After surface antibody staining, cells were fixed for 10 min at 4 °C using IC fix (Biolegend) and cells were then incubated in for 20min at RT in Permeabilization buffer (biolegend). Intracellular staining was performed for cytokines using antibodies for IL-5, IL-13, IL-17A and IFNγ as well as for YM1 and Relm-α. Samples were analysed by flow cytometry with LSR Fortessa or LSR II (Becton-Dickinson) and data analysed using FlowJo v10 software.
List of flow cytometry antibodies used.
Quantification of cytokines
Single-cell suspensions of splenocytes, lung-draining lymph nodes or whole lung were stimulated ex vivo with N. brasiliensis excretory secretory product (E/S) antigen [20] (1 µg/ ml) or anti-CD3 (1 µg/ml). Cell supernatants were harvested 72 h later and were stored at −20 °C until further analysis. Mouse IL-13 DuoSet ELISA kit (R&D Systems) was used for measurement of IL-13 levels. Mesenteric lymph node (MLN) cells from T. muris infected or uninfected mice were collected, cultured and restimulated ex vivo for 36 h with E/S as previously described [21]. The concentrations of IL-5, IL-6, IL-9, IL-10, IL-13, IL-17A, TNFα and IFNγ in the MLN culture supernatant were measured by cytokine bead array (CBA, BD Biosciences, UK) as per the manufacturer’s protocol.
Antibody depletion and cell transfer experiments
Neutrophils were depleted via intraperitoneal injection of anti-Ly6G antibody (clone 1A8, BioXcell, West Lebanon, NH, USA) (500 µg/mouse/day) on days −1 and 1 of infection with N. brasiliensis. Control mice were injected with an equal volume of IgG2a isotype control (BioXcell). Neutrophil depletion was confirmed by anti-Gr1 (RB6-8C5) staining for flow cytometry. For neutrophil transfer, neutrophils were purified from bone marrow using Histopaque-based density gradient centrifugation as described before [22]. Collected neutrophils were washed twice with RPMI 1640 and counts and viability was determined (average purity 85% ± 4.42% and 90% ± 1.5% viability). Neutrophils were resuspended in PBS and 0.5 × 106 cells (40µl) transferred intranasally into mice on day 1 post infection with N. brasiliensis. IFNγ was depleted using an anti-IFNγ monoclonal antibody (clone XMG1.2) and injected intraperitoneally (500 µg/mouse/day) on days −1 and 1 of infection with N. brasiliensis. Control mice were injected with an equal amount of corresponding isotype control (GL113). IL-17A was depleted using an anti-IL-17A (17F3) or IgG1 isotype (both Invivo mAB) injected intraperitoneally (100 µg/mouse/day) on days 4, 5 and 6 post-infection with N. brasiliensis.
Histology
For histology, the left lobe of the lung was isolated at different time points following N. brasiliensis infection, inflated and fixed in 10% neutral-buffered formalin (Sigma). Whole left lung lobes were processed using a tissue processor (Leica ASP300S) and were then embedded in paraffin. Paraffin blocks were then sectioned to 5 µm using a microtome (Leica RM2235) and routinely stained with Mayer’s haematoxylin (Merck Millipore HX86014349) and eosin (Sigma) for histological analysis. Slides were imaged using an Olympus slide scanner and high-resolution image files were exported using Pannoramic Viewer software (3DHISTECH). The images were then processed in a KNIME software workflow to obtain 50 random regions of interests (ROIs) across the whole lung section. ROIs that contained lobe boundaries or extensive artefacts were excluded from the analysis. The ROIs were then converted to binary images and lacunarity (Λ) was quantified using the FracLac plugin for ImageJ. The Λ values of all the ROIs were averaged to obtain estimates for the entire lobe.
Extraction of RNA and quantitative real-time PCR
A fragment of the right lung lobe was stored in RNAlater (Ambion) before homogenization of tissue in Qiazol reagent with a TissueLyser (Qiagen). RNA was prepared according to manufacturer’s instructions. RNA was quantified using a ND-1000 Spectrophotometer (NanoDrop Technologies). Reverse transcription of 1 µg of total RNA was performed using Tetro reverse transcriptase (Bioline). For reverse transcription, total RNA was treated with 50 U Tetro reverse transcriptase (Bioline), 40 mM dNTPs (Promega), 0.5 µg PolyT primer for cDNA synthesis (Roche) and RNasin inhibitor (Promega). The abundance of transcripts from the genes of interest was measured by quantitative real-time PCR with the Light Cycler 480 II system (Roche) with a Brilliant III SYBR master mix (Agilent) and specific primer pairs. PCR amplification was analysed by the second-derivative maximum algorithm (Light Cycler 480 Sw 1.5; Roche), and expression of the gene of interest was normalized to that of the housekeeping gene Actb (beta-actin). A list of primer sequences used are shown in Table 2.
List of primer sequences used.
Trichuris muris infection and E/S products
T. muris eggs were prepared from chronically infected stock mice as described previously [23]. Mice were infected by oral gavage with 200 embryonated T. muris eggs suspended in ddH2O. At day 20 and 35 post infection, T. muris burden was assessed by removing the caecum and proximal colon, opening them longitudinally and scraping the contents out with fine forceps. Individual worms were then counted by eye under a binocular dissecting microscope. T. muris adult excretory secretory product antigen (E/S) was prepared as described by [23]. In brief, adult T. muris were cultured ex vivo at 37°C, the culture supernatant was collected and centrifuged to remove eggs and worms. The resultant supernatant was then filter sterilised and stored at −20°C until use for in vitro re-stimulation of MLN cells.
Nanostring RNA Profiling
Samples were run on an Agilent 2200 Tape Station system to ensure high quality lung RNA; samples with a RIN value of < 6.5 were excluded. RNA was diluted to 20ng/μL in RNase free H2O, measured using Qubit™ RNA HS Assay Kit (Thermofisher) and run on a Nanostring nCounter® FLEX system using the Myeloid Innate Immunity v2 panel (XT-CSO-MMII2-12) as per manufacturer’s instructions. Raw data was loaded into nSolver version 4.0 using default settings. non-normalised counts were then exported and subsequent analyses were performed in R (version 3.6) [24] using RStudio Version 1.2.1335 Build 1379 – © 2009-2019 RStudio, Inc. Positive controls were analysed to ensure there was clear resolution at variable expression levels and negative controls were used to set a minimum detection threshold which was applied to all samples. Data were then normalised with EdgeR [25] using the TMM method and differential expression between N. brasiliensis-infected WT and IL-17A-deficient mice was calculated via linear modelling with Empirical Bayes smoothing using the limma R package 2. Heatmaps were then generated from normalized counts of differentially expressed genes using the ComplexHeatmaps R package [26]. The networks and functional analyses of differentially expressed genes were generated with Ingenuity pathway analyser (QIAGEN Inc., https://www.qiagenbio-informatics.com/products/ingenuity-pathway-analysis). Data were then imported into R for visualisation [27].
Statistical analysis
Prism 7.0 (version 7.0c, GraphPad Software) was used for statistical analysis. Differences between experimental groups were assessed by ANOVA (for normally distributed data, tested using Shapiro-Wilk normality test) followed by Sidak’s multiple comparisons test. For gene expression data, values were log2 transformed to achieve normal distribution. Comparisons with a P value of less than 0.05 were considered to be statistically significant.
Results
IL-17A-deficient mice mount a diminished type 2 response
In keeping with the known ability of N. brasiliensis to induce a strong pulmonary type 2 immune response on day 6 post infection (d6pi), we found the BAL and lungs of C57Bl/6 mice to be dominated by eosinophils (Suppl. Fig. 1A, B). This response was accompanied by elevated numbers of CD4+ T cells as well as induction of Group 2 Innate lymphoid cells (ILC2, Suppl. Fig. 1C, D). The establishment of a type 2 response was further confirmed by increased type 2 cytokine expression by CD4+ T cells and gene expression in whole lung (Suppl. Fig. 1E, F). As we and others previously reported [8], [11] infected mice exhibited increased IL-17A production in the first 48 h post infection (Fig. 1A) and consistent with previous reports the main source of IL-17A was γδ T cells [8]. On d2pi the BAL consisted mainly of neutrophils (Suppl. Fig.1A), which, together with N. brasiliensis larvae migration, is known to cause acute lung injury responses [11].

C57BL6/J (WT) and Il17a-KO mice were infected with 250 N. brasiliensis L3’s and cell frequencies and cytokines were measured at different time points post infection. Frequencies of IL-17A producing γδ T cells on d1pi and d2pi and representative flow-plot at d1pi (A). Worm burden in small intestine assessed in WT and Il17a-KO mice on days 2, 4 and 6 post N. brasiliensis infection (B). Absolute count of neutrophils (Ly6G+CD11b+) (C), eosinophils (SiglecF+CD11b+ Cd11c−) in bronchoalveolar lavage (BAL) (D) and lung ILC2 (Lineage-KLRG+CD127+CD90.2+ST2+) as measured via flow cytometry (E). Relative mRNA expression of cytokines Il4, Il5 and Il13 in whole lung as quantified by qRT-PCR (log2 expression relative to actb (β-actin)) (F). IL-13 levels from unstimulated or 72h α-CD3 treated single-suspension lung cells (G). Relative mRNA expression of the mucin genes Muc5ac and Muc5b in whole lung (log2 expression relative to actb) (G). Data are representative (mean ± s.e.m.) of at least 3 individual experiments (A, C, E-F) or pooled of two experiments (D, G, H) with at least 3 mice per group or pooled data from three experiments (B). Data were tested for normality using Shapiro-Wilk test and analysed using One-Way Anova followed by Sidak’s multiple comparisons test for selected groups. NS – not significant. Data in (F, H) were log2 transformed and statistical tests were performed on transformed data *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001
To investigate the role of IL-17A during the development of type 2 immune responses, we infected Il17a-KO mice and corresponding WT C57BL/6J controls with N. brasiliensis L3’s. Larvae leave the lung within 48 h and are expelled from the gut within 6-8 days. Consistent with our previous findings [8] Il17a-KO mice were significantly more susceptible to infection exhibiting an intestinal worm burden almost twice as high as in the WT controls on d4pi and d6pi Fig. 1B). As expected, the early d2 neutrophilia in response to N. brasiliensis infection was muted in the Il17a-KO mice relative to the WT controls (Fig. 1C). Between day 2 and 6 post infection, there was a switch from neutrophilic to eosinophilic responses in the lungs (Suppl. Fig 1A). Whilst increased eosinophil numbers were observed in all infected d6pi mice relative to naïve animals, this increase was less evident for Il17a-KO mice (Fig. 1D). ILC2 displayed a similar pattern, as cell numbers significantly increased with infection in WT but not in Il17a-KO mice (Fig. 1E). Additionally, WT mice exhibited an increased type 2 cytokine expression signature at d6pi, whilst Il17a-KO mice had reduced Il4 expression and a significantly delayed increase in Il5 expression (Fig. 1F). IL-13 protein levels in whole lung samples restimulated with α-CD3 on d6pi were significantly higher in WT mice compared to naïve controls whilst Il17a-KO mice completely failed to upregulate IL-13 levels (Fig. 1G), further demonstrating an impairment of protective type 2 responses. We also measured expression levels of the major mucins in the lung because host mucin production is another feature of anti-helminth protective type 2 responses [28]. N. brasiliensis infection drove an early increase in mucins Muc5ac and Muc5b expression in the lungs of WT mice at day 2 post-infection corresponding to a time when the larvae are transitioning from the lungs. Expression of the mucin Muc5ac was significantly increased in WT mice compared to naïve controls, but reduced in Il17a-KO mice, although not significant (Fig. 1H). A similar pattern was observed for Muc5b expression, which was upregulated in infected WT mice by day 2, but failed to increase in Il17a-KO infected mice (Fig. 1H). By d6pi, when the worms had already migrated to the small intestines, increased mucin expression was no longer observed in infected animals (Fig. 1H). Together these data demonstrate an impairment of type 2 immune responses during helminth infection in the absence of IL-17A.
IL-17A regulates T cell activation and polarization during N. brasiliensis infection
Next we aimed to determine whether the impact of IL-17A-deficiency on type 2 immunity was due to changes in T cell activation or polarisation during N. brasiliensis infection. Using flow cytometry, we observed a reduction in total numbers of CD4+ T cells on d7pi in the lungs of Il17a-KO mice compared to WT controls (Fig. 2A). In contrast, there were no significant differences in CD4+ T cell numbers in the lung-draining lymph nodes (Fig. 2A).

C57BL6/J (WT) and Il17a-KO mice were infected with 250 N. brasiliensis L3 larvae and live, single CD4+ T cells were phenotyped using flow cytometry at d7pi in lung and lung draining lymph nodes (LdLN). Absolute numbers of live CD4+ T cells in lung tissue and LdLN (A). Representative flow-plots showing frequencies of IL-5 and IL-13 production by CD4+ T cells d7pi in lung from WT Il17a-KO mice (B). Absolute numbers of IL-5+ and IL-13+ CD4+ T cells in the lung (C). Expression of CD69, absolute numbers for EGFR, ST2 and PD-1 in lung (D) and LdLNs (E). Data are representative (mean ± s.e.m.) of at least 3 individual experiments with at least n=2 mice in control groups and n=4 mice in infected groups (per experiment). Data was tested for normality using Shapiro-Wilk test and analysed using One-Way Anova followed by Sidak’s multiple comparisons test for selected groups. NS not significant, *P<0.05, **P<0.01.
Not only were there fewer CD4+ T cells in the lungs of Il17a-KO mice, the CD4+ T cells of Il17a-KO mice were producing significantly less IL-13 and IL-5 (Fig. 2B). We also found significantly fewer IL-5+ and IL-13+ CD4+ T cells in the Il17a-KO mice compared to WT controls (Fig. 2C). At the same time point post-infection, we found that expression of the activation marker CD69 was upregulated in the lungs of WT but not Il17a-KO mice (Fig. 2D). However, CD69 did not differ between all tested groups in the lung-draining lymph nodes (Fig. 2E). Recently, Minutti et al. described a role for epidermal growth factor receptor (EGFR) on T cells in protection against N. brasiliensis. EGFR forms a complex with ST2 to allow for IL-33-induced IL-13 expression at the site of infection [29]. We therefore analysed surface expression of EGFR and ST2 on lung and lung-draining lymph node T cells. N. brasiliensis infection increased the number of CD4+ T cells expressing these markers in the lung, but this increase was significantly reduced in Il17a-KO mice. (Fig. 2D). Changes to ST2 and EGFR expression between WT and KO mice were not observed in the lung-draining-lymph nodes (Fig. 2E). We also measured PD-1 expression, an important regulator of T cell function during helminth infection [30], [31]. N. brasiliensis infection in WT mice led to increased numbers of CD4+ T cells expressing PD-1 in the lung (Fig. 2D) with significantly fewer of these cells in Il17a-KO mice (Fig. 2D). Whilst there were also increased numbers of PD-1+CD4+ T cells in the lymph node following infection of WT mice, these were not altered by the absence of IL-17A (Fig. 2D). Overall, these data demonstrate the lung-specific impact of IL-17A-deficiency on CD4+ T cell numbers, type 2 cytokine production and activation status.
Neutrophils regulate eosinophil recruitment but not type 2 responses
IL-17A is a major driver for neutrophil recruitment and activation [32], raising the possibility that impaired neutrophilia in Il17a-KO mice is responsible for diminished type 2 immune responses. This hypothesis was supported by data indicating that neutrophils from N. brasiliensis infected mice can express type 2 cytokines [11]. Therefore, we assessed whether neutrophils contributed to the type 2 immune response by depleting neutrophils during N. brasiliensis infection (Fig. 3A). Injection of anti-Ly6G effectively prevented neutrophil accumulation in the BAL at d2pi compared to isotype control and depletion was still evident in the blood at d6pi (Fig. 3B, Suppl. Fig. 2A). Neutrophilia at day 2 post N. brasiliensis infection is a major driver of the lung injury [11] and here we confirmed these findings (Fig. 3C). Histological sections demonstrated infection-induced injury at d2pi in isotype-treated WT mice, an effect that is almost absent in infected mice depleted of neutrophils. Using lacunarity [33] as an indicator of acute lung injury [34], we show that neutrophil depleted mice show no significant signs of damage (Fig. 3C). Whilst intestinal worm burdens at d2pi and d4pi were not affected by neutrophil depletion, a small but significant increase in parasite numbers at d6pi was observed in mice treated with neutrophil-depleting antibody (Fig. 3D). As expected, eosinophils were prominent in the lung and BAL of isotype-treated mice at d6pi (Fig. 3E). However, depletion of neutrophils in infected mice significantly reduced eosinophils in the lung and BAL compared to the isotype-treated animals. Notably, blood and bone marrow eosinophils were not affected, suggesting a defect in eosinophil recruitment (Supplement Fig. 2B). We therefore assessed chemokines with known eosinophil chemotactic effects (Ccl5, Ccl11, Ccl22), but neutrophil-depletion did not alter mRNA expression of these chemokines in the lung. In addition, we measured Ccl8 as Chen et al. demonstrated that N. brasiliensis-primed neutrophils from d2pi exhibit a notable upregulation of Ccl8 [11]. Expression of Ccl8 was increased in infection, but significantly lower in neutrophil-depleted mice (Suppl. Fig. 2C). As expected, N. brasiliensis infection significantly increased CD4+ T cells in the lung, but the number of CD4+ T cells were significantly reduced following neutrophil depletion (Fig. 3F). Despite effects on both CD4+ T cells and eosinophilia, there were limited changes in type 2 cytokine expression following neutrophil depletion (Fig, 3G, H). Whilst IL-4 levels significantly increased in infected isotype treated mice, there was no significant upregulation of IL-4 expression in whole lung or by CD4+ T cells in neutrophil-depleted mice (Fig. 3G-H). IL-13 and IL-5 were not also altered (Fig. 3G, H, Suppl. Fig. 2D). Thus, neutrophil depletion prevented eosinophil recruitment to the lungs of infected mice, but had a limited effect on type 2 cytokine expression.

C57BL6/J mice were infected with 500 N. brasiliensis L3 larvae and mice were injected with α-Ly6G (1A8) or isotype control on d-1 and d1pi (A). Neutrophil numbers per ml BAL fluid, as well as confirmation of neutrophil depletion via flow cytometry (B). Representative lung sections stained with haematoxylin & eosin and imaged, followed by quantification of lacunarity (Λ) on d2pi and d6pi (C). Gut worm burden after neutrophil depletion d2pi, d4pi and d6pi (D). Eosinophils in lung and BAL (E) as well as CD4+ T cells in lung (F) on d2pi and d6pi as assessed by flow cytometry. Frequencies of intracellular IL-4 and IL-13 from CD4+ T cells in lung (G). Relative expression of Il4 and Il13 in total lung mRNA (log2 expression relative to actb (β-actin)) (H). Il17a-KO mice were intranasally injected with 500,000 neutrophils (+N) d1pi with 250 L3 larvae N. brasiliensis, controls received PBS (I). BAL neutrophils numbers on days 2 and 6 post infection with N. brasiliensis (J). Eosinophil numbers in BAL per gram lung tissue after neutrophil transfer (K). Relative expression of Il4, Il5, and Il13 in whole lung mRNA in WT, Il17a-KO or Il17a-KO mice + neutrophils (+N) (L). Data are expressed as (mean ± s.e.m.) and are representative of at least 3 individual experiments with at least 3 mice per group (B, C, E, J-L) or pooled data from two experiments (D, F). Data were tested for normality using Shapiro-Wilk test and analysed using One-Way Anova followed by Sidak’s multiple comparisons test for selected groups. *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001.
To further address the role of neutrophils, we tested whether transfer of WT neutrophils into Il17a-KO mice could rescue the defect in the development of type 2 immunity. Neutrophils were isolated from the bone marrow of CD45.1+ mice using the histopaque-gradient method [22] and intranasally transferred into Il17a-KO mice (Fig. 3I). Neutrophil transfer restored airway neutrophil frequency in Il17a-KO mice (Fig. 3J). Interestingly, this neutrophilic response was due to increased recipient-derived cells and not the transferred CD45.1+ neutrophils (Suppl. Fig. 2E). One day post-neutrophil transfer (on d2pi), eosinophil numbers were increased in Il17a-KO mice, reaching similar numbers to those observed in infected WT mice (Fig. 3K). Ccl8 levels were also increased following neutrophil transfer (Suppl. Fig. 2F), consistent with the possibility that this chemokine plays a role during eosinophil recruitment to site of infection. At a later timepoint, when eosinophilia was much more dominant and pronounced, the Il17a-KO mice that received neutrophils also displayed an increased lung eosinophilia (Fig. 3L). Despite changes to eosinophil numbers, transfer of neutrophils did not rescue the deficit in type 2 cytokines in Il17a-KO mice (Fig. 3M). Together, this data provides evidence that neutrophils support eosinophil recruitment at the site of infection, but suggests they do not directly contribute to type 2 cytokine responses.
IL-17A leads to a downregulation of early IFNγ during Nippostronglyus infection
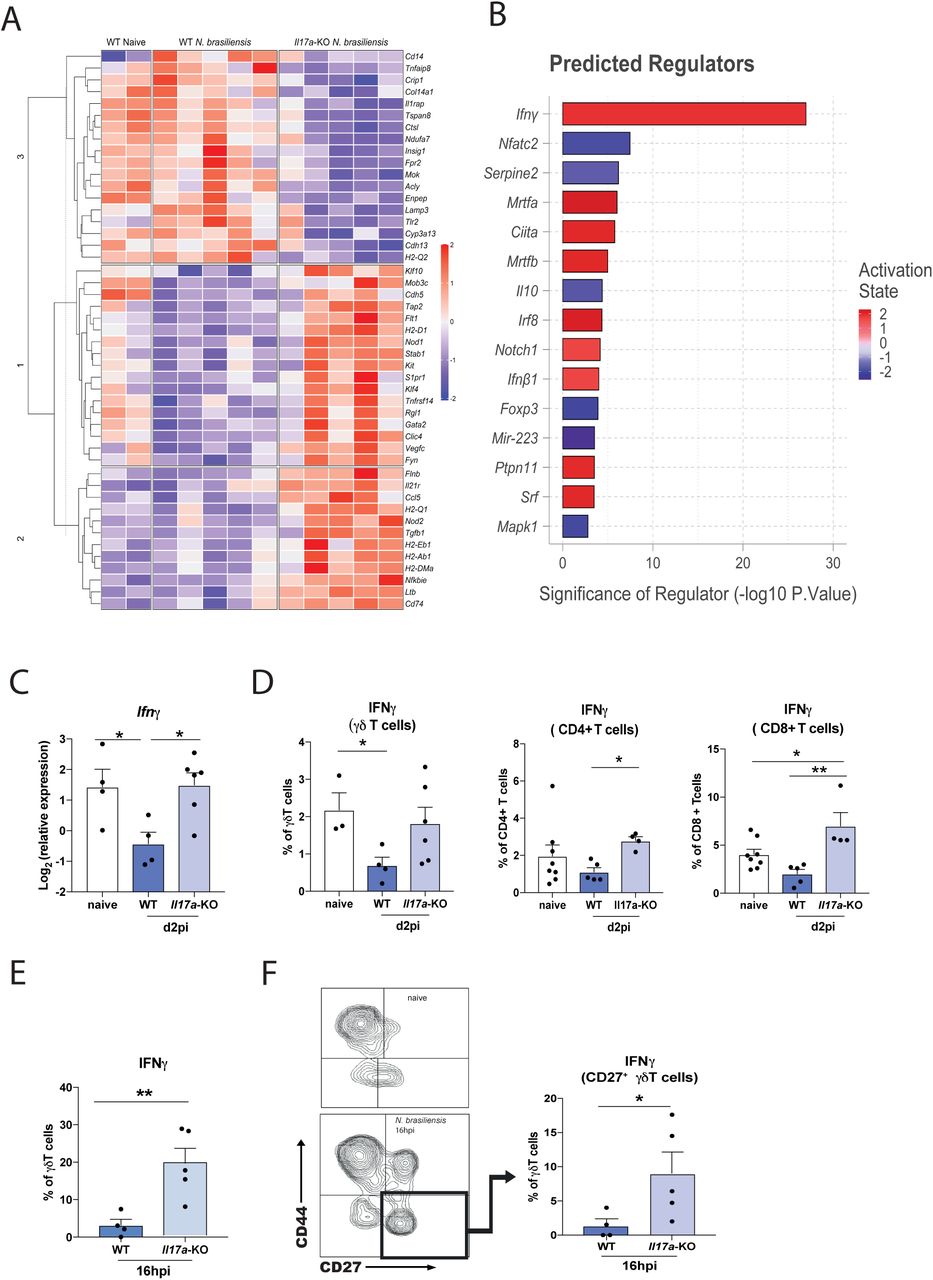
Rapid early IL-17A production is known to be critical for protective immune responses in different settings of lung immunity [1], [35]. To better understand the early events unfolding in the lung during N. brasiliensis infection, we performed a Nanostring gene expression array using a myeloid immunity panel (700 genes). Differentially expressed (DE) genes in whole lung between naïve WT mice and infected WT and Il17a-KO mice at d2pi were assessed in total unamplified RNA (Fig 4A). IL-17A deficiency led to a distinct gene expression profile compared to WT mice in response to N. brasiliensis infection. Notably, when analysing all DE genes (Fig. 4A) using the Ingenuity pathway analyser (Qiagen), IFNγ was predicted as the most significantly increased upstream regulator in N. brasiliensis infected Il17a-KO compared to WT mice (Fig. 4B). This led us to hypothesize that IL-17A may suppress IFNγ, which would facilitate Th2 cell development and explain why mice deficient in IL-17A cannot induce a full type 2 immune response. This hypothesis was also consistent with our unpublished and published [36] finding that Ym1, which induces IL-17A, strongly suppresses IFNγ. To test this possibility, we assessed IFNγ responses in Il17a-KO mice after N. brasiliensis infection. While WT mice exhibited significant suppression of Ifng in whole lung at 2dpi compared to uninfected controls, mice deficient in IL-17A did not show this phenotype (Fig. 4C). By intracellularly staining for IFNγ, we observed that Il17a-KO mice infected with N. brasiliensis failed to exhibit the early downregulation of IFNγ expression seen in infected WT mice (Fig. 4D). Importantly, this failure of suppression was observed across different types of IFNγ-producing cells. Lung CD4+ T cells, CD8+ T cells as well γδ T cells from Il17a-KO mice produced IFNγ at the same proportion as the naïve controls, while WT mice exhibited a downregulation in IFNγ production (Fig. 4D). We further assessed whether early IFNγ was produced by γδ T cell subsets that differ in their expression of CD27 and CD44 [37]. At 16 h post N. brasiliensis infection, IFNγ frequencies were also significantly increased in γδ T cells of Il17a-KO mice compared to WT controls (Fig. 4E). Consistent with expectations [37], the CD27+ γδ T cells were the main producers of IFNγ after infection (Fig. 4F). Overall, our data demonstrated that IL-17A-deficiency enhanced IFNγ production during infection, supporting the hypothesis that IL-17A plays an important role in downregulating IFNγ at the site of infection during the lung migratory phase of N. brasiliensis infection.

Whole lung RNA from C57BL6/J (WT) and Il17a-KO mice on d2pi with N. brasiliensis was analysed by Nanostring. Unsupervised, hierarchically clustered heat map showing differentially expressed genes between infected WT and Il17a-KO mice (A). Top differentially regulated genes between infected WT and Il17a-KO mice were run in Ingenuity pathway analyzer, with top predicted regulators shown in (B). Relative expression of Ifng in whole lung of naïve WT and WT and Il17a-KO mice d2pi with N. brasiliensis (log2 expression relative to actb (β-actin)) (C). Frequency of IFNγ by γδ T cells, CD4+ T cells, CD8+ T cells in WT and Il17a-KO mice d2pi assessed by flow cytometry (D). Frequency of IFNγ+ γδ T cells 16h post N. brasiliensis infection in WT and Il17a-KO mice (E). Representative flow plot showing γδ T cell subsets using CD44 and CD27 in naïve WT mice and Il17a-KO mice 16h post N. brasiliensis infection as well as frequency of IFNγ+ CD27+ γδ T cells 16h post N. brasiliensis infection in WT and Il17a-KO mice (F). Data (C-F) are expressed as (mean ± s.e.m.) and are representative of at least 2 individual experiments with at least 3 mice per infected group. Data were tested for normality using Shapiro-Wilk test and analysed using One-Way Anova followed by Sidak’s multiple comparisons test for selected groups or student’s t-test. *P<0.05, **P<0.01
IFNγ neutralization in Il17a-KO mice rescues the impaired type 2 immune response
To directly test whether active suppression of IFNγ by IL-17A is required for the full development of a type 2 immune response during N. brasiliensis infection, IFNγ was neutralised at day −1 and 1 of infection in Il17a-KO mice and responses examined at day 8 post-infection. (Fig. 5A). The significant defect in eosinophilic responses in Il17a-KO mice compared to WT mice was still evident at day 8 post-infection. However, blocking IFNγ in Il17a-KO mice enhanced eosinophil numbers (Fig 5B). The same pattern was observed for numbers of CD4+ T cells in the lungs (Fig. 5C). To determine whether IFNγ neutralisation altered the type 2 response, we firstly assessed expression of key type 2 cytokines Il4 and Il13 in the lungs of mice. Whilst expression of these genes was significantly reduced in infected Il17a-KO mice compared to WT mice, IFNγ depletion completely recovered expression of these cytokines in Il17a-KO mice compared to isotype-treated animals, and Il4 surpassed the levels seen in WT mice (Fig. 5D). Additionally, we assessed the type 2 marker, Chil3, which also demonstrated enhanced expression in Il17a-KO mice treated with anti-IFNγ compared to similarly treated WT mice (Fig. 5E). Similarly, analysis of numbers of IL-5 and IL-13 producing CD4+ T cells at d8 post infection showed restoration of the type 2 response in Il17a-KO mice that received the neutralising IFNγ antibody (Fig. 5F). Consistent with the ability of IFNγ to regulate type 2 cytokines, IFNγ depletion also restored the activation status of CD4+ T cells in the Il17a-KO mice (Fig. 5G) and increased the numbers of CD4+ T cells expressing type 2 markers EGFR, PD1 and ST2 (Fig. 5H). Again, no effect was observed in these parameters in IFNγ-depleted WT mice. Here, we show that an initial reduction in IFNγ levels during N. brasiliensis infection mediated by IL-17A, allows the subsequent development of type 2 immunity in the lung.

C57BL6/J (WT) and Il17a-KO mice and were treated with α-IFNγ or isotype control on days −1 and 1pi with 250 L3 larvae of N. brasiliensis (A). Absolute cell counts of eosinophils in BAL (B) and CD4+ T cells per gram lung tissue (C) as measured via flow cytometry on d8pi. Relative expression of type 2 cytokines Il4 and Il13 (D) and type 2 marker Chil3 (E) from whole lung RNA. Absolute numbers of IL-5+ and IL-13+ CD4+ T cells (F). Frequency of CD69 expression in CD4+ T cells (G) and numbers of EGFR+, ST2+ and PD1+ CD4+ T cells per gram lung tissue (H). Data (B, D, E, F, G) are representative (mean ± s.e.m.) 2 individual experiments with at least 3 mice per group (per experiment) or pooled data from two experiments (C, F). Data was tested for normality using Shapiro-Wilk test and analysed using One-Way Anova followed by Sidak’s multiple comparisons test for selected groups. Data in (D, E) were log2 transformed and statistical tests were performed on transformed data. *P<0.05.
IL-17A suppresses an established type 2 response in the lung
IFNγ depletion in Il17a-KO mice not only restored the type 2 response, but in some cases exceeded WT levels. Therefore, we hypothesized that IL-17A may suppress the type 2 immune response during later stages of N. brasiliensis infection. To test this hypothesis, we neutralized IL-17A at d4pi, d5pi and d6pi in WT mice and assessed immune responses at d7pi (Fig. 6A). Blocking of IL-17A led to a significant increase in both ILC2 numbers and frequencies in the lung (Fig. 6B), as well as the numbers of ILC2s producing IL-5 and IL-13 (Fig. 6C). However, CD4+ T cell numbers in the lung were comparable between isotype-treated and anti-IL-17A-treated WT mice (Fig. 6D). However, the ability of CD4+ T cells to produce type 2 cytokines may partly rely on IL-17A, as mice administered anti-IL-17A showed a slight increase IL-5 and IL-13 (Fig. 6E). This data demonstrates that IL-17A can have differential effects depending on the time and status of infection. While early IL-17A promotes the type 2 response, later in infection IL-17A acts to suppress and limit excessive the type 2 immunity, particularly in ILC2s.

C57Bl/6 WT mice were treated with α-IL-17A or isotype control on days 3, 4, and 5pi with 250 L3 N. brasiliensis (A). Cells per gram lung tissue and frequency of ILC2 within live lung cells (B). Number of IL-5+ and IL-13+ ILC2 per gram lung tissue (n=5 for naïve, n=11-12 for d7 N. brasiliensis infected groups) (C). Numbers of CD4+ T cells (D) and IL-5+ and IL-13+ CD4+ T cells per gram of lung tissue (n=3 for naïve and n=6 for d7 N. brasiliensis infected groups) (E). Data pooled from two independent experiments (B, C) or are representative (mean ± s.e.m.) of 3 individual experiments with at least 3 mice per group (per experiment) (D, E). Data was tested for normality using Shapiro-Wilk test and analysed using One-Way Anova followed by Sidak’s multiple comparisons test for selected groups or student’s t-test. *P<0.05, **P<0.01.
IL-17A does not regulate type 2 immune responses at the site of T. muris infection
Our data demonstrate an impairment of the type 2 immune response in the lung during infection with the lung-migrating nematode N. brasiliensis. To investigate whether this impairment is lung-specific and if IL-17A also regulates type 2 responses during infection with a gut-specific nematode, we infected WT and Il17a-KO mice with a high dose of 200 Trichuris muris eggs. Infection with T. muris begins with the ingestion of infective eggs that accumulate in the caecum. L1 larvae hatch and penetrate the caecum and proximal colon wall, undergoing moults to L2 (d9-11pi), L3 (d17pi) and L4 (d22 pi). The host type 2 immune response occurs around the time when the parasites moult to L3s at approximately d17pi. Adult worms, if not already expelled, can be found from d32pi onwards [38]. High dose T. muris infection is known to induce a strong type 2 response within the gastrointestinal tract, especially in the caecum and MLNs [21], [39]. Here, worm counts in the caecum of Il17a-KO mice and WT mice were comparable at d19pi and d32pi (Fig. 7A), indicating IL-17A does not alter parasite expulsion rate. Cell numbers in the caecum were analysed and no differences in eosinophil and neutrophil frequency were observed between Il17a-KO mice and WT controls on d19pi and d32pi (Supplement Fig. 3A, B), suggesting an IL-17A-independent recruitment mechanism for both these cell types. CD4+ T cell numbers in the MLN were also comparable on d19pi and d32pi between Il17a-KO mice and WT controls (Suppl. Fig 3C). Although there was an induction of type 2 cytokines in infected mice as measured by intracellular cytokine staining, the numbers of IL-5 or IL-13-producing CD4+ T cells in the MLNs did not significantly differ between the groups (Suppl. Fig 3D). Secreted levels of IL-5, IL-9 and IL-13 in MLN cells also did not show any significant differences between Il17a-KO mice and WT controls (Suppl. Fig 3E). Together, these data failed to provide any evidence that IL-17A was an important regulator of type 2 immunity in the intestine during T. muris infection.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
C57Bl/6 WT and Il17a-KO mice were infected with a high dose of T. muris and immune parameters were investigated at d19 and d32pi. Worms counts in the caecum (A). Frequency of neutrophils (B), eosinophils (C) and CD4+ T cells (D) in the lung at d19pi and d32pi. Absolute counts of IL-5+ and IL-13+ CD4+ T cells per gram of lung tissue (E). Relative expression of cytokines Il4 and Il5 from whole lung RNA (log2 expression relative to actb (β-actin)) (F). Absolute cell counts and frequency of IFNγ by CD4+ T cells per gram of lung tissue (G). Data are expressed as mean ± s.e.m.) and are representative of 3 individual experiments with at least 4 mice per infected group and one mouse per control group. Data was tested for normality using Shapiro-Wilk test and analysed using One-Way Anova followed by Sidak’s multiple comparisons test for selected groups or student’s t-test. *P<0.05, **P<0.01
Previous studies have shown that despite the restriction of the T. muris lifecycle to the gastro-intestinal tract of the mammalian host, evidence of a type 2 immune response can be observed at distant sites, such as the lung [40]. Therefore, the immune response in the lung of T. muris infected WT vs Il17a-KO mice at d19pi and d35pi was assessed. Neutrophils numbers were increased in infected WT animals at d19pi and d35pi but this was significantly reduced in Il17a-KO mice on d19pi (Fig. 7B). No significant changes were observed for eosinophils (Fig. 7C). Whilst lung CD4+ T cell numbers in infected animals did not change compared to naïve controls (Fig. 7D), Il17a-KO mice had significantly fewer IL-5+CD4+ T cells at d19 and d32pi compared to WT controls (Fig. 7E). Although the effect on IL-13+CD4+ T cells was less evident, infected Il17a-KO mice failed to significantly increase numbers of Il-13+CD4+ T cells compared to uninfected controls (Fig. 7E). Supporting the intracellular cytokine staining, qRT-PCR analysis in whole lung tissue showed an impairment of type 2 cytokines in the Il17a-KO mice, with significantly decreased expression of Il4 (d32pi) and Il5 (d19pi) (Fig. 7F).
Similar to infection with N. brasiliensis, we also observed an upregulation of IFNγ in the lung during T. muris infection in Il17a-KO mice. Both the number and the frequency of IFNγ+CD4+ T cells in the lung were significantly increased in Il17a-KO compared to WT infected mice on d19pi (Fig. 7G). This data utilising T. muris infection models suggests that IL-17A-dependent suppression of IFNγ allows promotion of the type 2 immune response specifically in the lungs but not the intestine.
Discussion
IL-17A, the key cytokine of the IL-17 family, is central to barrier immunity, combating fungal infections and inducing antimicrobial proteins as well as neutrophil activating and recruiting chemokines [2]. Here, we explored the relationship between IL-17A and type 2 immunity during N. brasiliensis infection and show that IL-17A, by impinging on IFNγ expression, allows type 2 immunity to fully develop within the lung.
As expected, IL-17A was important for innate neutrophilic responses during N. brasiliensis infection. However, neutrophil depletion and transfer experiments demonstrated that neutrophils themselves only partly contributed to the development of type 2 immunity, mainly through enhancement of the subsequent eosinophilia. Our data highlighted a potential role for the chemokine Ccl8 in neutrophil mediated recruitment of eosinophils. A correlation between CCL8 and eosinophil numbers in allergic airway inflammation has been described previously
[41] and CCL8 was associated with type 2 inflammatory responses in the lung during the protective response to Klebsiella pneumonia [42]. However, neither study demonstrated a direct eosinophil chemotactic function of CCL8. More recently, Puttur et al. revealed that CCL8 is important for the appropriate mobilisation of ILC2s and specifically the localised production of IL-5 by ILCs, possibly explaining why we observed reduced eosinophilia in neutrophil depleted mice [43]
Neutrophils have been implicated in nematode killing [44]–[46]. These studies show the importance of neutrophil influx during the skin stage of nematode infection, while in our model of lung infection, neutrophils seem to play a minor role in worm killing. However, consistent with work from Chen et al, neutrophil depletion did result in reduced lung damage d2pi, confirming early neutrophilia as a main driver of acute lung injury following N. brasiliensis infection [11].
The ability of type 2 cytokines to suppress IL-17 has been well documented [47]–[49], but our study suggests that the development of the type 2 response in the first place can require IL-17A. Our data is supported by a model of murine atopic dermatitis, in which Nakajima et al. show that IL-4 production is reduced by IL-17A deficiency and that in vitro Th2 differentiation from naïve T cells is enhanced with the addition of IL-17A [14]. Notably, the IL-17A-induced IL-4 [14] may subsequently suppress IL-17A production as a feedback mechanism, consistent with the previous observations that type 2 cytokines negatively regulate IL-17A [50]. In allergic airway inflammation models, deficiency of IL-17A also leads to decreased type 2 cytokine production [15] and Th17 cells contribute to Th2-cell-mediated eosinophilic airway inflammation [51]. A combination of type 2 cytokines and IL-17A can be also found as signature for severe disease pathology. For example in a model for severe airway hyperresponsiveness, IL-17A contributes to asthma pathology by enhancing IL-13 activity [52]. Furthermore, during murine and human schistosomiasis and onchocerciasis, a dysregulated balance between IL-17A and type 2 responses can exacerbate the pathology [4]– [6], [53].
In our effort to understand how IL-17A might be required for full type 2 immunity, we identified a role for IL-17A mediated suppression of IFNγ. Evidence that IL-17A can regulate IFNγ exists in the literature, but whether IFNγ is upregulated or downregulated is controversial and likely depends on the setting, timing and location [54]–[56]. During experimental visceral leishmaniasis, Il17a-KO mice have enhanced production of a protective IFNγ response [54], while in Toxoplasma gondii infection, excessive IFNγ in Il17a-KO mice has severe pathological consequences [55]. Evidence also exists in the context of helminth infection, where a lack of IL-17A drives elevated IFNγ during infection with the filarial nematode Litomosoides sigmondontis [57] or Schistosoma japonicum [56] and Schistosoma mansoni [5]. Together these findings support a potentially widespread role for IL-17A in regulating IFNγ.
Our study shows that early in infection, IL-17A suppression of IFNγ in the lung has consequences for the subsequent development of type 2 immune responses. Notably, in Il17a-KO mice the protective type 2 immune response in the small intestine was not impaired as mice were still able to expel N. brasiliensis. To examine whether IL-17A-dependent regulation of IFNγ is site specific, we used high dose infection with the colon dwelling parasite T. muris. The protective type 2 immune response against T. muris in the intestine was intact in mice lacking IL-17A. The surprising finding however was that even though T. muris does not have a lung stage, the concurrent type 2 response in the lung was impaired in Il17a-KO mice. CD4+ T cells in the lung produced less type 2 cytokines and both eosinophils and neutrophils were significantly downregulated in the Il17a-KO mice. Consistent with our findings in N. brasiliensis, CD4+ T cells in Il17a-KO mice produced significantly higher amounts of IFNγ than in their WT counterparts. Together, our data suggest that the impact of IL-17A on type 2 development may be lung restricted. Nonetheless, there may still be a fundamental requirement for IFNγ suppression for type 2 immunity to progress. Artis et al. demonstrated that the type 2 immune response during T. muris requires TSLP, and in very similar experiments to those described here, demonstrated that TSLP functions to suppress IFNγ [58]. Thus, early suppression of IFNγ may be a general pre-requisite for the development of a type 2 environment with a requirement for IL-17A in the lung and TSLP (or other factors) in the gut.
Our data also reveal a potential time dependence on the regulation of type 2 immunity by IL-17A. While the early γδ T cell-derived IL-17A supported the type 2 response, late IL-17A, derived from both Th17 cells and γδ T cells, appeared to be a negative regulator. We observed that depletion of early IFNγ in Il17a-KO mice not only rescued their impaired type 2 immune responses on d8pi, but the type 2 mRNA signature significantly exceeded the WT levels. Thus, removal of IFNγ in IL-17A deficient mice, which allowed normal type 2 progression, revealed a potential role for IL-17A in suppressing type 2 cytokines. To our knowledge, IL-17A suppression of type 2 cytokines has not previously been described in vivo.
We have not yet addressed the full mechanism behind IL-17A-mediated suppression of IFNγ during N. brasiliensis infection but it is notable that IL-17A not only impairs type 2 cytokine production, but also alters the cellular activation status and expression of type 2 markers. Interestingly, in our model, we only observe impairment of type 2 immune responses in the lung itself and not in the Th2 cells from lung-draining lymph nodes. Expression of EGFR and ST2, two markers closely associated with type 2 settings [29], were reduced on the CD4+ T cells of Il17a-KO mice in the lung. EGFR expression on Th2 cells is critical for resistance during GI helminth infection and a signalling complex between EGFR and ST2 can activate Th2 cells to secrete IL-13 in an antigen-dependent manner upon IL-33 exposure. Our data would suggest that this “licensing” of Th2 cells does not occur in the Il17a-KO mice during N. brasiliensis infection, indicating that IL-17A is needed for a proper induction of the adaptive Th2 response in the lung.
Together we demonstrate that the early events in the lung shape the protective type 2 immune response, with IL-17A as a critical regulator through the downregulation of IFNγ. In combination with previous data [58], suppression of IFNγ at barrier sites may be a central paradigm for type 2 immunity. The ability of IL-17A to subsequently suppress type 2 responses reveal an important feedback loop that must go awry during severe asthma and other type 2 conditions, in which IL-17 plays a damaging and pathogenic role.
Disclosures
The authors have no financial conflicts of interest.
Acknowledgments
We thank the Flow Cytometry, Bioimaging, and Biological Services core facilities at the University of Manchester. This work was supported by the Wellcome Trust (106898/A/15/Z to JEA and Z10661/Z/18/Z to RG), the Medical Research Council UK (MR/K01207X/1 to JEA), Medical Research Foundation UK joint funding with Asthma UK (MRFAUK-2015-302 to TES). SC was supported by a Wellcome Trust Studentship (103132/Z/13/Z). We thank Kevin Couper for CD45.1 mice.
Footnotes
The authors declare no conflicts of interest.
References
- [1].↵
- [2].↵
- [3].↵
- [4].↵
- [5].↵
- [6].↵
- [7].↵
- [8].↵
- [9].↵
- [10].↵
- [11].↵
- [12].↵
- [13].↵
- [14].↵
- [15].↵
- [16].↵
- [17].↵
- [18].↵
- [19].↵
- [20].↵
- [21].↵
- [22].↵
- [23].↵
- [24].↵
- [25].↵
- [26].↵
- [27].↵
- [28].↵
- [29].↵
- [30].↵
- [31].↵
- [32].↵
- [33].↵
- [34].↵
- [35].↵
- [36].↵
- [37].↵
- [38].↵
- [39].↵
- [40].↵
- [41].↵
- [42].↵
- [43].↵
- [44].↵
- [45].
- [46].↵
- [47].↵
- [48].
- [49].↵
- [50].↵
- [51].↵
- [52].↵
- [53].↵
- [54].↵
- [55].↵
- [56].↵
- [57].↵
- [58].↵




