

ABSTRACT
The immediate-early gene Arc is a master regulator of synaptic function and a critical determinant of memory consolidation. Arc protein is localized to excitatory synapses, where it controls AMPA receptor endocytosis, and to the nucleus, where it associates with Tip60, a subunit of a chromatin modifying complex. Here we show that Arc interacts with dynamic chromatin loops and associates with histone markers for active enhancers and transcription in cultured hippocampal neurons. When Arc induction by pharmacological network activation was prevented using a short hairpin RNA, the expression profile was altered for over 1900 genes. Many gene families were affected by the absence of Arc, most notably those associated with synaptic function, neuronal plasticity, intrinsic excitability (channels, receptors, transporters), and signaling pathways (transcription factors/regulators). Interestingly, about 100 genes whose activity-dependent expression level depends on Arc are associated with the pathophysiology of Alzheimer’s disease, suggesting a critical role for Arc in the development of neurodegenerative disorders. When endogenous Arc expression was induced in a non-neuronal cell line (HEK293T), the transcription of many neuronal genes was increased, suggesting Arc can control expression in the absence of activated signaling pathways. Taken together, these data establish Arc as a master regulator of neuronal activity-dependent gene expression and a significant factor underlying the pathophysiology Alzheimer’s disease.
INTRODUCTION
The neuronal immediate-early gene Arc1,2 plays a critical role in memory consolidation3–6. Arc expression is rapidly and transiently induced by novel behavioural and sensory experiences7–11, while its mRNA is enriched in dendrites and targeted to recently activated synapses, where it is locally translated12,13. Arc protein resides in excitatory synapses, where it controls AMPA receptor endocytosis14, allowing it to act as a master regulator of synaptic function and plasticity15,16 that implements homeostatic synaptic scaling at the neuronal network level17–19. While the synaptic role of Arc has been well documented, the observed failure to convert early-to late-LTP in Arc knockout mice cannot be explained by an AMPA receptor endocytosis deficit4. This suggests that Arc may have additional functions. Interestingly, Arc protein can also be localized in the nucleus, where it binds to a beta-spectrin IV isoform and associates with PML bodies20–22, sites of epigenetic regulation of gene tran-scription23. Nuclear Arc has been reported to regulate transcription of the GluA1 AMPA receptor24. Recently, another nuclear function for Arc has been demonstrated: Arc interacts with the histone-acetyltransferase Tip6025, a subunit of a chromatin modifying complex26–28. Arc expression level correlates with the acetylation status of one of Tip60’s substrate: lysine 12 of histone 4 (H4K12)25, a memory-associated histone mark which declines with age29. These newly discovered nuclear functions may point to an epigenetic role for Arc in memory consolidation. We have therefore investigated Arc’s interaction with chromatin and its association with histone marks in cultured hippocampal and cortical neurons. Fluorescent microscopy experiments demonstrated a highly dynamic interaction between chromatin and Arc, as well as a tight association between Arc and histone marks for active enhancers and active transcription. RNA-Sequencing (RNA-Seq) experiments in which activity-dependent Arc expression was prevented using a short hairpin RNA showed that Arc regulates the transcription of over nineteen hundred genes controlling memory, cognition, synaptic function, neuronal plasticity, intrinsic excitability and intracellular signaling. Interestingly, Arc also controls the expression of susceptibility genes for Alzheimer’s disease, as well as many genes implicated in the pathophysiology of this disorder. A Gene Ontology (GO) analysis identified downstream signaling pathways and diseases associated with the observed changes in mRNA levels, while an Ingenuity Pathway Analysis (IPA) revealed upstream regulators predicted by the change in gene expression profile caused by Arc knockdown. Finally, we induced expression of Arc in human embryonic kidney 293T (HEK-293T) cells, using CRISPR-Cas9, which resulted in the increased transcription of many neuronal genes. Taken together, our data demonstrate that Arc controls neuronal activity-dependent expression of many genes underlying higher brain functions and may be involved in the development of Alzheimer’s disease (AD) and other neurodegenerative disorders.
RESULTS
Arc is a neuronal activity-dependent immediate-early gene1,2, whose expression is induced by exposure to a novel environment or a new sensory experience7,8,11. Knockdown of Arc expression abrogates long-term memory without affecting short-term memory, indicating a critical role for Arc in memory consolidation3–6. Arc protein localizes to dendritic spines, where it regulates AMPA receptor endocytosis14, and to the nucleus20,24,30,31, where its function is less understood. In this study we have used cultured hippocampal and cortical neurons to study the role of Arc in the nucleus. Arc expression can be induced by increasing network activity in neuronal cultures, using a combination of 4-aminopyridine (4AP), bicuculline and forskolin (4BF), a form of pharmacological long-term potentiation (LTP)21,22,32,33. Figure 1 shows that this form of network activation strongly induces the expression of Arc in a subset of neurons. In this in vitro paradigm, Arc localizes predominantly to the nucleus four hours after network activity-dependent induction of its expression.

Hippocampal neurons (DIV19-21) were treated with 4BF, which pharmacologically stimulates network activity and induces long-term potentiation of excitatory synapses. After 4 hours of enhanced network activity, neurons were fixed and stained for Arc (red) and the neuronal marker Map2 (green). Under vehicle (DMSO) treatment, very little Arc staining could be detected (left panel, Vehicle), whereas the increase in network activity induced strong nuclear Arc expression in approximately half of the neurons: 49 ± 8 % (n=3) (right panel, 4BF).
Memory consolidation requires de novo gene expression34,35, which is induced by activation of signaling cascades that originate in the synaptic connections potentiated during learning36–40. This synapse-to-nucleus signaling results in post-translational modifications of chromatin, including acetylation, methylation, phosphorylation, and sumoylation of histones and methylation of DNA41,42. Chromatin modification alters its nanostructure, which controls accessibility of gene promoters to the transcription machinery43,44. These synaptic activity-induced epigenetic processes can alter gene expression and have been shown to be critical for learning and memory45–52. We therefore characterized the structure and dynamics of chromatin in cultured hippocampal neurons, evaluated how pharmacological LTP (4BF treatment) affected chromatin structure, and compared chromatin properties of neurons expressing Arc protein with control neurons that do not.
Chromatin reorganization in Arc-positive neurons
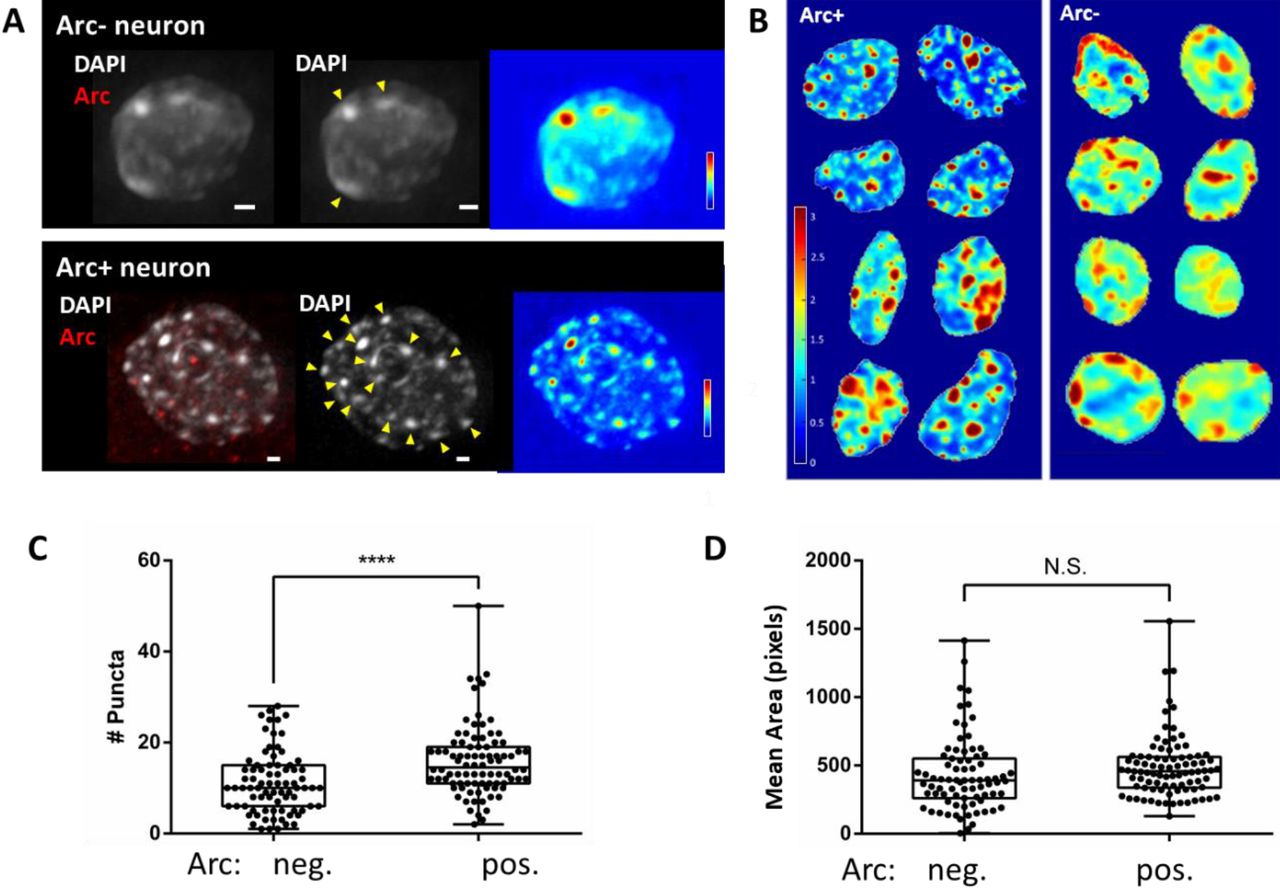
Induction of Arc protein expression by pharmacological network activation (Fig. 1) is relatively slow and reaches a maximum level between 4 and 8 hours and Arc is only expressed in a subset of neurons. As shown in Figure 2, chromatin organization is different between neurons that are positive and negative for Arc. Chromatin was visualized by labeling DNA with the fluorescent dye 4’,6-diamidino-2-phenylindole (DAPI). Whereas chromatin in Arc-negative neurons is relatively homogenous, the nuclei of Arc-positive neurons contain many bright puncta, representing chromocenters with densely packed chromatin, in which genes are likely silenced (Fig. 2A and 2B). The puncta are interpersed with domains of highly open chromatin, which is more supportive of efficient gene transcription. The number of puncta increased from 11.1±0.8 puncta in Arc-negative nuclei to 15.9±0.8 puncta in Arc-positive nuclei (Fig. 2C). However, the mean area of the puncta was not significantly different between Arc-positive and Arc-negative neurons (Fig. 2D).

Arc expression was induced in a subset of cultured hippocampal neurons by a 4-hour treatment with 4BF. Cells were fixed and stained for Arc (C7 antibody, Santa Cruz). DNA was labeled using DAPI. Z-stacks of DAPI images were obtained for neuronal nuclei that were positive and negative for Arc expression. Out-of-focus fluorescence was removed using 3D deconvolution (AutoQuant). (A) Max-projection images of a representative nucleus from an Arc-negative (top) and Arc-positive neuron (bottom). The white bar indicates a scale bar of 1 μm. DNA, labeled by DAPI, is shown in white while Arc expression is shown in red. Yellow arrowheads indicate DNA puncta. Heat maps of the relative DAPI intensity of the nucleus is shown in the rightmost panels. Relative DAPI intensity is shown using the color scale shown at the bottom. (B) DAPI heat maps for nuclei of 8 Arc-positive and 8 Arc-negative neurons. Relative DAPI intensity is shown by the color scale on the left, which was the same for both panels. Whereas chromatin of Arc-negative neurons (right panel) was relative homogenous (turquoise, green yellow), Arc-positive neurons (left panel) was characterized by several areas of high DAPI intensity (red), indication condensed heterochromatin (chromocenters) separating domains with decondensed euchromatin (blue). (C) and (D) Puncta were quantified based on their size and intensity. Arc expression, measured as mean Arc intensity, was used to correlate with the properties of the puncta, generating the boxplots. Boxplots of number of puncta (C) and area of puncta (D) for Arc-negative and Arc-positive neurons. Each • represents the (C) number or (D) mean area of puncta in a nucleus. A total of 167 nuclei were analyzed from three sets of independent experiments. (C) Nuclei of Arc-positive neurons have significantly higher number of puncta with 11.1 ± 0.8 puncta in Arc-negative nuclei and 15.9 ± 0.8 puncta in Arc-positive nuclei. **** indicates P-value < 0.0001, unpaired t test. (D) No significant change in area of puncta was observed: Arc-positive had an area of 488 ± 25 pixels, while the area of Arc-negative neurons was 431 ± 30 pixels (p = 0.15, unpaired t test). N.S. indicates not significant.
Arc associates with dynamic chromatin
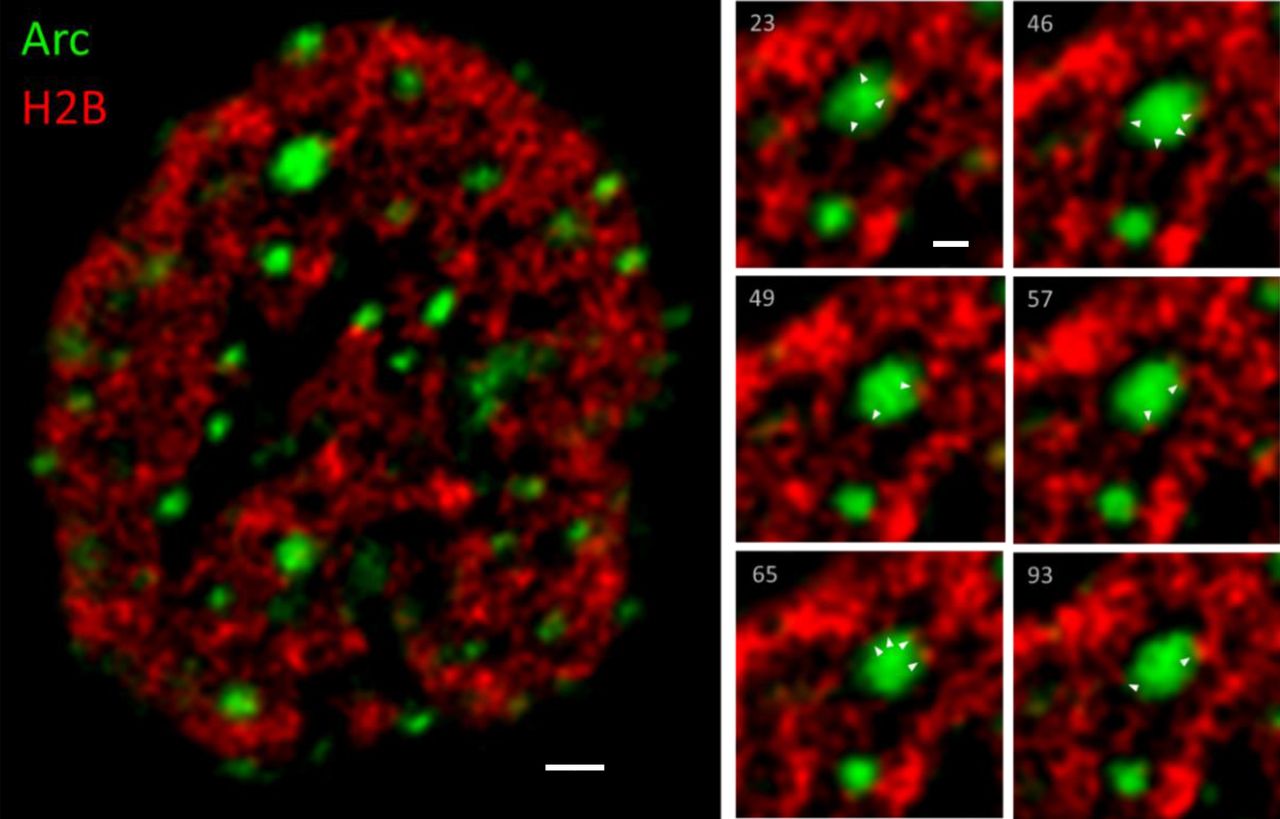
The interaction between Arc and chromatin was studied in more detail using time-lapse fluorescence microscopy of hippocampal neurons expressing Arc and histone 2B (H2B) tagged with YFP and mCherry, respectively (Fig. 3). Arc was induced in 18 days in vitro (DIV) hippocampal neurons by a 4-hour treatment with 4BF. The time-lapse movies of Arc-eYFP and H2B-mCherry revealed a highly dynamic chromatin that constantly reorganizes on a time scale of seconds (Movie 1). Arc is concentrated in small puncta to which the chromatin can be seen to reach out with finger-like structures, which likely represent the dynamic chromatin loops described by others53–55.

Time-lapse movies of Arc-eYFP and H2B-mCherry expressed in hippocampal neurons (18 DIV) were obtained using a spinning disc confocal microscope (100x, 1.49 NA Apo TIRF objective). Z-stacks (5 images) were acquired for both YFP and mCherry channels. 3D blind deconvolution (AutoQuant) was used to remove out-of-focus fluorescence. The movie is 5 minutes long, 3.2 seconds between frames, which was the time required to acquire Z-stacks from both channels. The image on the left shows a single frame of the movie in the centre of the Z-stack of a neuronal nucleus (scale bar= 1 μm). Arc (green) is seen to form puncta, while H2B (red) labels the lattice-like chromatin structure. The panels on the right show six frames of a zoomed-in section illustrating small chromatin structures transiently interact with the two Arc puncta (scale bar = 500 nm). White arrowheads indicate points of contact between Arc and chromatin. The highly dynamic interaction of chromatin with Arc puncta is most clearly seen in the Movie.
Arc associates with a marker of active enhancers
Because Arc was shown to associate with the Tip60 substrate H4K12Ac25, we have examined interactions of Arc with other histone modifications, by comparing Arc-positive and -negative neurons following pharmacological network activation. The ‘histone code’56 is complex and still incompletely understood. We have therefore focused on histone modifications whose function is best studied. In our survey we have found several histone modifications for which there was a difference in nuclear organization between Arc positive and negative neurons, including H3K9Ac, H3K4me3, and H3K14Ac (data not shown). Figure 4 illustrates the close association between Arc and H3K27Ac, which marks active enhancers57,58. Arc and H3K27Ac form two separate lattice-like structures that are closely inter-connected and, in some locations, appear to overlap (yellow areas in Figure 4).

Hippocampal neurons were treated with 4BF for 4 hours, fixed with methanol and stained for Arc and H3K27Ac, which marks sites containing active enhancers. Z-stacks of images were acquired of neuronal nuclei using a spinning disc confocal microscope (60X, 1.49 NA objective). Resolution was increased using 3D blind deconvolution (Autoquant). The enlarged section shows the close interaction between Arc and H3K27Ac.
Arc associates with a marker for active transcription
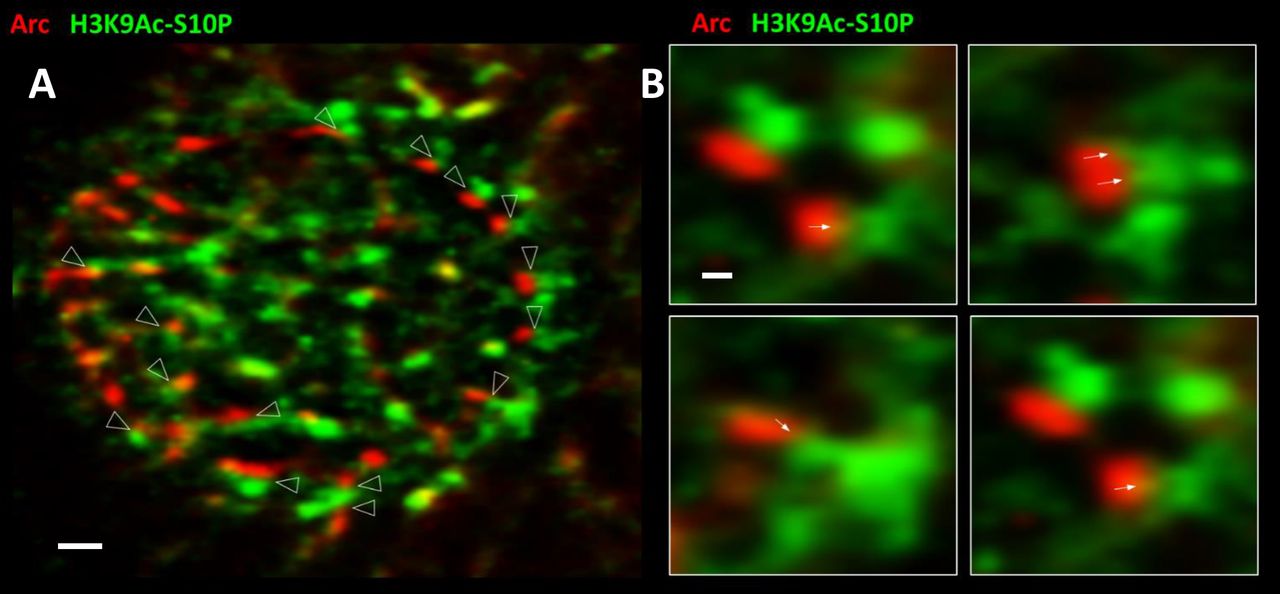
Another histone mark that showed a strong interaction with Arc was H3K9Ac-S10P, which requires the concurrent acetylation of lysine 9 of histone H3 (H3K9Ac) and phosphorylation of the neighboring serine 10 (S10P). This dual marker indicates genomic regions undergoing active transcription22,59,60. Figure 5 illustrates the close interaction between Arc and this histone mark, using Stochastic Optical Reconstruction Microscopy (STORM), a form of super-resolution microscopy with a resolution of ~30 nm61. Both Arc and H3K9Ac-S10P are enriched at the nuclear periphery, where reorganization of chromatin between active and inactive transcriptional states takes place62,63. With the increased resolution of STORM, Arc can be seen to localize to distinct puncta. H3K9Ac-S10P forms an elaborate meshwork, as expected for chromatin, but also is enriched in puncta-like domains. Arrowheads in Figure 5A indicate the close apposition between these two sets of puncta. Close inspection of the interface between the two types of puncta revealed invasions of H3K9Ac-S10P into the Arc puncta (arrows in Fig. 5B), resembling the finger-like chromatin loop structures seen in live cell imaging (Fig. 3,Movie).

Image of a neuronal nucleus obtained using STORM. Cultured hippocampal neurons were treated with 4BF for 4 hours, fixed and stained for Arc and H3K9Ac-S10P, which marks sites undergoing active transcription. (A) Arrowheads point to close appositions between Arc and the histone mark (scale bar = 1μm). (B) Enlarged sections showing the association in greater detail. Arrows point to what appear to be invasions of H3K9Ac-S10P into Arc puncta (scale bar 200 nm).
Arc regulates activity-dependent gene transcription
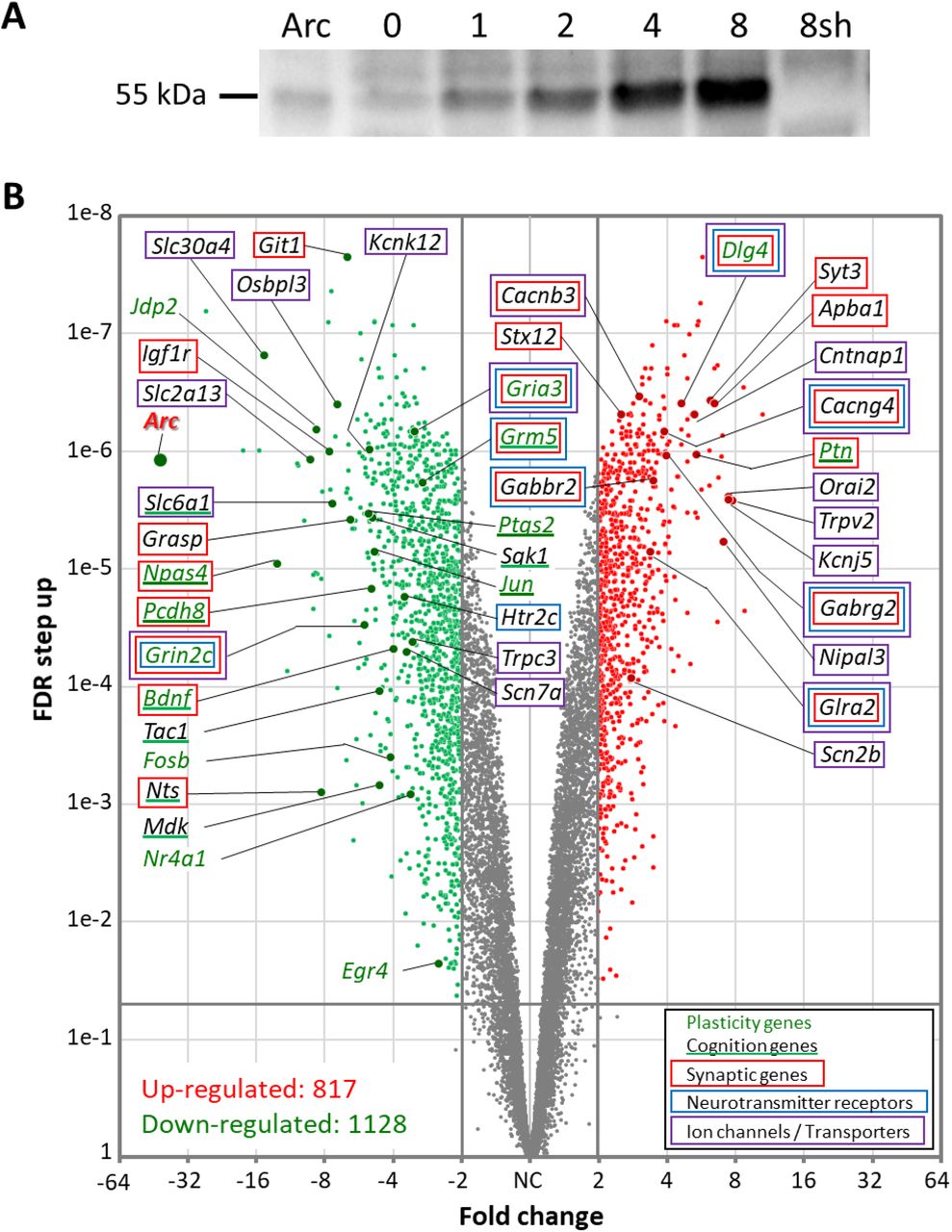
Because Arc was found to associate with histone marks involved in transcription activation, we wanted to investigate whether network activity-induced Arc expression alters the gene expression profile of the neurons. Four short hairpin RNAs (shRNAs) targeting the coding region of Arc were tested for their ability to suppress Arc induction following four hours of 4BF treatment. We selected the most effective shRNA to generate an adeno-associated AAV9 virus. Because AAV9 infection itself may alter the gene expression profile, we also generated a negative control consisting of AAV9 virus encoding a scrambled version of the Arc shRNA. We performed an RNA-Seq analysis of cortical neurons expressing either the Arc shRNA or its scrambled control. When 4BF-mediated Arc expression is prevented using the Arc shRNA (Fig. 6A), mRNA levels for more than 1900 genes were altered (Fig. 6B). Many gene families were affected, including those associated with plasticity genes (Jun, Fosb, Bdnf, Dlg4, Egr4, Npas4 and Nr4a1), synaptic proteins (syntaxin Stx12 and synaptotagmin Syt3), neurotransmitter receptors (NMDA, AMPA, GABA, glycine, serotonin and metabotropic glutamate receptors) (Fig. 6B). Arc also regulated the expression of genes controlling intrinsic excitability: 62 genes encoding ion channels (20 K+, 4 Na+, and 9 Ca2+ channel subunits, 7 transient receptor potential (Trp) channels, 14 ligand-gated ion channels, 7 regulatory subunits and 1 non-selective cation channel), and 139 genes encoding transporters/pumps (for glutamate, GABA, serotonin, ADP, ATP, phosphate, glucose, inositol, alanine, cysteine, glutamine, glycine, proline, Na+, Ca2+, Cl-, H+ and Zn2+). These results suggest that Arc regulates activity-dependent gene expression relevant for synaptic function, neuronal plasticity and intrinsic excitability.

(A) Western Blot showing the time course of Arc protein expression in cultured hippocampal neurons following 4BF treatment (time in hours indicated on top). Lane 7 (8sh) shows that Arc fails to express at 8 hours of 4BF when the cultures are transduced with an AAV9 virus encoding a short-hairpin RNA (shRNA) targeting the coding region of Arc. Lane 1 has purified Arc protein. (B) Volcano plot of RNA-Seq results comparing mRNA isolated from neurons after 8 hours of 4BF that were transduced with AAV9 virus encoding either the Arc shRNA or a scrambled version of this shRNA, done in triplicate. Preventing activity-dependent Arc expression resulted in the upregulation of 817 genes (red), and down-regulation of 1128 genes (green). Genes that are below the cut-off (FDR > 0.05 or absolute fold change < 2) are marked in gray. Some of the highly regulated genes involved in learning and memory are indicated in the plot. Genes are color coded as stated in the legend. Both axes are log scaled.
Table 1 shows the 30 top-ranking genes sorted by absolute fold change (FC) caused by the shRNA-mediated knockdown of Arc expression. Gene names are shown together with a description of their function, their Fold Change, False Discovery Rate (FDR), and references to relevant papers. Many of the top-regulated genes are involved in synapse modulation, neurotransmission, neurogenesis and neurological disorders. Interestingly, 9 out of the top 30 genes have been implicated in the pathophysiology of AD (Fgf1, Slc30a4, Npas4, Cxcl1, Jdp2, Nts, Mmp10, Orai2 and Tomm34) while an additional 5 genes are linked to amyloid beta (Aβ) metabolism (Mmp13, Mmp12, Slc2a13, Igf1r and Apba1).
APOE: apolipoprotein E; p-tau: phosphorylated tau; Aβ: Amyloid beta; PI3K/Akt: phosphatidylinositol 3-kinase/protein kinase B; CSCR2: C-X-C motif chemokine receptor 2; HDAC3: histone deacetylase 3; AP-1: activator protein 1; APP: amyloid precursor protein; CSF: cerebrospinal fluid; STIM: stromal interaction molecule; LTP: long term potentiation; GABA: γ-aminobutyric acid; KO: knockout; 5HT: 5-hydroxytryptamine; ER: endoplasmic reticulum; Cav2: neuronal voltage-gated calcium channels. References: [1-3]232–234, [4]235, [5]236, [6-8]237–239, [9, 10]240,241, [11-13]242–244, [14]245, [15]246, [9, 16]240,247, [17]248, [18-20]249–251, [21, 22]252,253, [23, 24]254,255, [25-27]256–258, [28]259, [29, 30]260,261, [31, 32]262,263, [33-36]264–267, [37, 38]268,269, [39, 40]270,271, [41]272, [42, 43]273,274, [44, 45]275,276, [46, 47]277,278, [48]279, [49-51]280–282, [52, 53]283,284, [54, 55]285,286, [56-58]287–289 and [59-61]290–292.
GO analysis of differentially expressed genes
A Gene Ontology (GO) analysis of the RNA-Seq data identified several biological processes and molecular functions that were affected when Arc expression was prevented during network activation (Fig. 7). Arc knockdown altered many genes involved in the regulation of nervous system development and neuronal differentiation (Fig. 7A). In addition, many of the genes were enriched in biological processes involved in cognition, regulation of cell projection organization and axonogenesis (Fig. 7A), processes which could modulate structural plasticity involved in neural development, learning and memory64,65. While the top ten regulated genes enriched for the regulation of plasma membrane bounded cell projection organization were both up- and down-regulated (Fig. 7Cii), genes enriched for cognition and the regulation of axonogenesis were mostly down-regulated due to the absence of Arc (Fig. 7Ci and iii). Many of the altered genes were also enriched in molecular functions such as ion channel regulator activity, glutamate receptor binding and ligand-gated ion channel activity (Fig. 7B), including Sgk1 (Fig. 7Di), Dlg4, which encodes PSD-95 (Fig. 7Dii), and Grin2c, which encodes the NMDA receptor NR2C subunit(Fig. 7Diii). These molecular functions are well-established to underlie synaptic plasticity processes crucial for formation of memory66,67.

(A) and (B) Gene set enrichment analysis was performed to investigate the Biological Processes (A) and Molecular Functions (B) that the altered genes were involved in. The enrichment score is plotted against the category names. The enrichment score is the negative natural logarithm of the enrichment P-value derived from Fisher’s exact test and reflects the degree to which the gene sets are overrepresented at the top or bottom of the entire ranked list of genes. Bars indicate the enrichment score while the line graph indicates the percentage of genes that are altered under the respective GO term. The top 25 biological processes (B) and molecular functions (C) are shown. Many of the categories are related to synaptic plasticity (underlined blue and orange). (C) and (D) Bar-charts showing genes involved in the stated category from Biological Processes (Ci)-(Ciii) and Molecular Functions (Di)-(Diii) and their respective fold changes. The top 10 regulated genes are shown. Dotted blue line indicates an absolute Fold Change of 2.
Arc regulates expression of synaptic and plasticity genes
From the GO results in Figure 7, we observed that the knockdown of Arc affected many genes involved in synaptic plasticity, as well as genes implicated in processes underlying learning and memory. We have therefore investigated how Arc knockdown affected genes encoding synaptic proteins by manually curating a list of differentially expressed genes whose protein products are located at the presynaptic or postsynaptic compartment. A total of 232 synaptic genes were differentially expressed. These genes are involved in the development and growth of axons and dendrites, including Ephb3, Lrfn2, Lama5, Neurod2, Sema4f, Caprin2, and Unc5c and the modulation of the synapses and dendritic spines, including Npas4, Pcdh8, Ephb3, Lrfn2, Bdnf, Atxn1, Cbln2, Cadps2, Caprin2, C1ql1, C1ql3, and Unc5c (Table 2). Many of these synaptic genes are also involved in neuroplasticity, cognition, learning and memory, including Syt3, Pcdh8, Pdyn, Lrfn2, Dlg4, Kcna4, Bdnf and Mapki8ip2. Figure 8 lists neuroplasticity genes and genes that are involved in cognition, learning and memory whose activity-dependent expression is regulated by Arc. Most of these genes were down regulated when activity-dependent Arc expression was prevented.

Neuronal plasticity genes were manually curated in addition to reference to GO terminology from the Gene Ontology Consortium. Neuronal plasticity genes with absolute FC ? 2.5 are shown. Genes that are involved in cognition or learning and memory are marked by orange boxes.
This table shows the top 40 synaptic genes (out of a total of 323) ranked by absolute fold change. Comments list relevant information about the function and disease association of the genes. An asterisk (*) indicates genes involved in neuroplasticity. A hashtag (#) indicates genes involved in cognition, learning and memory. APP: amyloid precursor protein; LTP: long-term potentiation; CRH: corticotropin-releasing hormone; V-gated: voltage-gated; NMDAR: N-methyl-D-aspartate receptor; TARP-γ4: transmembrane AMPR regulator protein γ4; Aβ: amyloid beta; GPCR: G-protein-coupled receptor. References: [1,2]293,294, [3]295; [4, 5]264,265, [6]292, [7]296, [8]297, [9]298, [10]299, [11, 12]300,301, [13]302, [14]303, [15]304, [16, 17]305,306, [18, 19]307,308, [20]309, [21]310, [22]311, [23]312, [24]313, [25, 26]314,315, [27]316, [28]317, [29]318, [30]319, [31]320, [32, 33]321,322, [34, 35]323,324, [36, 37]325,326, [38]327, [39, 40]328,329, [41, 42]330,331, [43, 44]332,333, [45]334, [46]335, [47]336, [48-50]337’339, [51, 52]340,341, [53]342, [54, 55]343,344 and [56-58]345’347.
Arc knockdown altered synaptogenesis, synaptic plasticity and neuroinflammation pathways
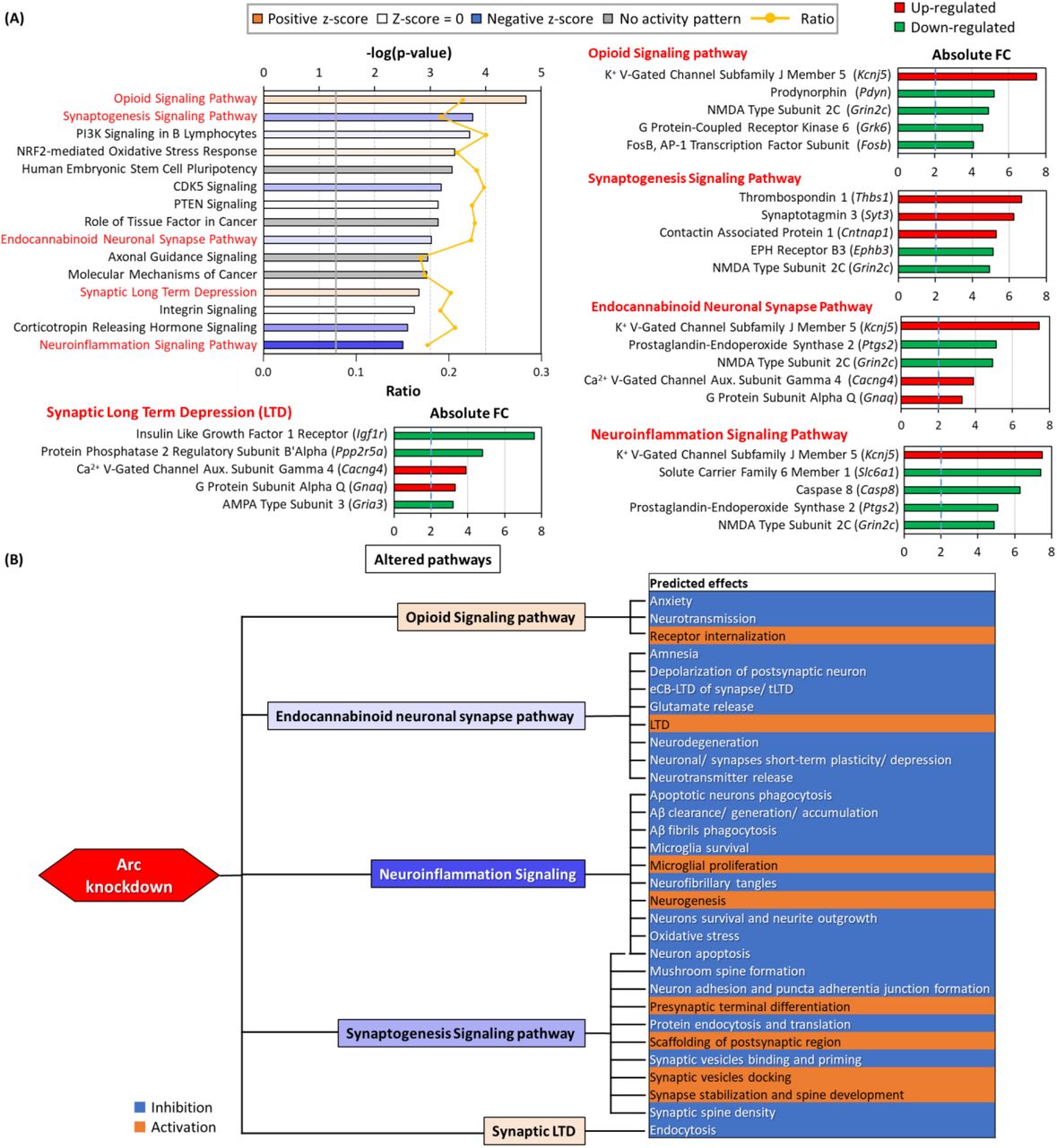
From the GO results and the list of manually curated synaptic genes, we were interested in investigating the signaling pathways and the possible downstream effects resulting from Arc knockdown. We have analyzed the differentially expressed genes and their respective fold changes using IPA. Figure 9A shows the top 15 pathways that were altered due to Arc knockdown. IPA made inferences on the activation or inhibition of the pathways based on the differential expression observed and canonical information stored in the Ingenuity Knowledge Base. The degree of activation or inhibition of each identified pathway is indicated by the z-score. The ratio is calculated as the number of differential expressed genes for each pathway divided by the total number of genes involved in that pathway. Many identified pathways involved cellular signaling cascades, including those mediated by CDK5, PTEN, integrin and corticotropin releasing hormone (Fig. 9A). Pathways predicted to be responsible for the observed differential expression profile include opioid and endocannabinoid signaling, synaptogenesis, synaptic long-term depression (LTD) and neuroinflammation (Fig. 9A and 9B). Kcnj5, Ptgs2, Grin2c, Cacng4 and Gnaq are members of at least two of the pathways shown and are synaptic genes or associated with cognition (Fig. 7, 8, 9A, Table 2). Except for the neuroinflammation signaling pathway, all these pathways are associated with synaptic plasticity. Knockdown of Arc modulated neurotransmission, synaptic plasticity, spine formation/maintenance and neurite outgrowth, processes that are crucial for learning and memory (Fig. 9B)68–70. Interestingly, the two hallmarks of AD, the generation, clearance and accumulation of amyloid beta (Aβ) and the formation of neurofibrillary tangles (NFTs), are both affected by downregulation of the neuroinflammation signaling pathway resulting from Arc knockdown (Fig. 9B). These alterations in the generation and clearance of molecular markers and triggers of AD could indicate a possible role of Arc in the pathophysiology of AD71.

(A) Bar-chart showing altered pathways identified after analysis with IPA. The orange line graph indicates the ratio of genes in our dataset that were involved in the specified pathway. The gray line indicates threshold at P-value 0.05. The orange and blue bars indicate predicted activation and inhibition of pathways respectively (determined by z-score). Top 15 significantly altered pathways are shown. Pathways with predicted activation or inhibition of downstream effects are in red, further elaborated in panel B. The top 5 genes altered in the respective pathways are shown on the right and bottom of the altered pathway bar-chart. (B) Diagram describing the predicted effects of the altered pathways. Five pathways are highlighted, and its downstream effects as predicted by IPA are listed.
Arc knockdown changes the expression of genes involved in the aetiology and pathophysiology of AD
Considering that the generation, clearance and accumulation of amyloid beta and neurofibrillary tangles was predicted to be altered due to the knockdown of Arc, we investigated whether any neurological diseases or psychological disorders were correlated with the profile of differentially expressed genes mediated by Arc knockdown. Table 3 summarized the disease annotation and predicted activation state for two disease/disorder classes whose associated genes were significantly altered by Arc knockdown. Absence of Arc was predicted to increase damage of the cerebral cortex and its neurons and cells. In addition, Arc knockdown was also associated with psychological disorders, including Huntington’s Disease, basal ganglia disorder, central nervous system (CNS) amyloidosis, tauopathy and Alzheimer disease. Of note, CNS amyloidosis and tauopathy are predictors of AD. The activation states of the five psychological disorders were not reported, possibly due to inconsistencies in the literature findings with respect to fold changes of the differentially expressed genes. However, the p-values for all five disorders were highly significant, suggesting that the progression of these disorders may be modulated by the Arc function.
Prevention of activity-dependent expression of Arc resulted in gene expression profile changes that are associated with neurological diseases and psychological disorders, including Alzheimer’s disease.
We next investigated how Arc knockdown could affect genes that were previously identified to increase susceptibility to AD. We have manually curated genes that were found to be genetic risk factors of AD and validated them by referencing the Genome Wide Associations Studies (GWAS) catalogue72. Notably, critical genetic risk factors of AD such as Picalm, Apoe, Slc24a4, and Clu were downregulated upon the knockdown of Arc73–77 (Fig. 10). Out of a total of 39 susceptibility genes identified, 26 were regulated by Arc (Fig. 10).

The expression levels of 26 AD susceptibility genes were affected when activity-dependent Arc expression was prevented by an shRNA. Green bars indicated that the mRNA level was downregulated, while red bars indicate upregulation. The blue line indicates an absolute fold change of 1.5.
Because Arc plays a role in the aetiology of AD by modulating its genetic risk factors, we investigated if Arc regulates genes that are more broadly involved in the pathophysiology of AD. Table 4 lists the results. While some differentially expressed genes control amyloid beta formation/accumulation through the regulation of cleavage and stabilization of amyloid precursor protein (APP) (Mmp13, Slc2a13, Apba1, Casp8, Ptgs2, Gpr3, Pawr, Timp3, Kcnip3, Plk2, Aplp2, Bace2, Apoe and Apba2), others are involved in the hyperphosphorylation of tau and formation of neurofibrillary tangles (Npas4, Cxcl1, Dryrk2, Tril, Pltp, Plk2 and Selenop). Arc knockdown also altered the expression of genes that are associated with the neurodegeneration and neurotoxicity observed in AD (Casp8, Bcl2l11, Alg2, Tac1, Bdnf, Hmox1, Pawr, Ccl2, Selenop and Atf6). Finally, Arc regulated genes associated with altered cognitive function, a characteristic of AD (Mmp13, Pdyn, Tac1, Bdnf, Nr4a2, Penk, Pltp and Ccl2). To date, presenilin 1 (Psen1) and glycogen synthase kinase 3 beta (Gsk3b) are the only AD mediators which have been reported to physically associate and interact with Arc78–80. Arc also interacts with endophilin 2/3 and dynamin and recruits them to early/recycling endosomes to traffic APP and beta secretase 1 (BACE1), crucial determinants of AD progression80. However, the observation that knocking down Arc resulted in more than 100 differentially expressed genes that are either AD susceptibility genes or genes implicated in the pathophysiology of AD (Fig. 10 and Table 4), indicated that Arc could be mediating the expression of these genes via transcriptional regulation and not simply physical interactions. Arc has previously been reported to reside in the nucleus20,24,30,31 and we have shown how Arc physically associates with chromatin and with markers of active transcription and enhancers (Fig. 3, 4 and 5). Therefore, we wanted to investigate how Arc downregulation affects transcription regulation.
Only genes with more than one citation or citations that included mechanisms of regulating AD are presented. BACE1: β-secretase 1; Aβ: amyloid beta; APP: amyloid precursor protein; FoxO3a: forkhead box O3; KO: knock-out; CO: carbon monoxide; ApoER2: apolipoprotein E receptor 2; PSEN2: presenilin 2; Zn2+: zinc; MAPK: mitogen-activated protein kinase; ER: endoplasmic reticulum. References: [1]238, [2]241, [3]248, [4, 5]250,251, [6, 7]292,348, [8]349, [9, 10]350,351, [11]352, [12]353, [13-15]354–356, [1619]85,357–359, [20]304, [21]360, [22-24]361–363, [25, 26]364,365, [1]238, [27-29]366–368, [30]264, [31, 32]321,322, [33, 34]369,370, [35, 36]371,372, [37]373, [38]374, [39, 40]375,376, [41, 42]377,378, [43]379, [44]380, [45-47]381–383, [48-53]384–389, [54, 55]390,391, [56-58]392–394, [59-61]395–397, [62-65]398–401, [66-69]402–405, [70-72]406–408, [73, 74]409,410, [75]411, [76]412, [77-79]413–415, [80, 81]416,417, [82]418, [83, 84]419,420, [85]421, [86]422 and [87]423.
Arc regulates the expression of transcription factors
From our GO analysis and a manual curation based on literature citations, we have identified 369 transcriptional regulators and transcription factors whose expression is controlled by Arc. Table 5 shows the top 40 transcriptional regulators or factors whose mRNA levels were altered when activity-dependent Arc expression was prevented. Some of the transcriptional regulators are involved in neuronal development and differentiation (Fgf1, Tgfb1i1, Fezf2, Jun, Magel2, Neurod2, Atxn1, Gdf15, Prdm1, Mycn, Nr4a2 and Pou2f2), while others are involved in the development of neurological or neurodegenerative diseases (Npas4, Igf1r, Txnip, Lgr4, Cebpd, Pim1, Magel2, Ireb2, Smad7, Sorbs1, Nfil3, Pknox2, Hdac9, Hmox1, Atxn1, Cbfb, Lrp2, Hipk3 and Nr4a2). Many of the transcriptional regulators/factors have been implicated in memory formation and plasticity, such as such as Thbs1, Jun, Tet3, Fosb, Atxn1 and Cbfb. A GO analysis by DAVID81 was carried out to identify the biological processes that these transcription factors could be modulating. Table 6 shows the top 20 biological processes that were regulated by altered transcription factor expression and that have neurological relevance. Corroborating the identified functions of the top 40 transcriptional regulators/factors (Table 5), differentially expressed transcriptional regulators/factors were observed to be highly enriched in biological processes such as differentiation of neurons, nervous system development, learning, long-term memory and aging (Table 6). Some of the transcriptional regulators were involved in multiple processes: Npas4, Jun, Bdnf, Nr4a2 and Elavl4 modulate learning, long-term memory, aging, neuron differentiation and nervous system development (Table 6).
This table shows the top 40 genes with neuronal relevance. * indicates transcription factors. ATF3: activating transcription factor 3; LEF1: lymphoid enhancer binding factor 1; FoxA2: forkhead box A2; APP: amyloid precursor protein; CREB: cAMP response element-binding protein; NMDAR: N-methyl-D-aspartate receptor; SOX2: SRY-box 2; PITX2: paired like homeodomain 2; bZIP: basic leucine zipper domain; C-MYC: MYC proto-oncogene, BHLH transcription factor; NFATc1: nuclear factor of activated T cells 1; FOXP3: forkhead box P3; HIF: hypoxia inducible factor; TGFβ: transforming growth factor beta; PD: Parkinson’s disease; NLS: nuclear localization signal; SMAD: transcription factors forming the core of the TGFβ signaling pathway; AP-1: activator protein 1; ALS: amyotrophic lateral sclerosis; MEF2: monocyte enhancer factor; GABAA: γ-aminobutyric acid type A; JAK/STAT: Janus kinases/ signal transducer and activator of transcription; SCA1: spinocerebellar ataxia type 1; MAPK: mitogen-activated protein kinase; ZNF683: zinc finger protein 683; HTT: huntingtin. References:: [1]424, [2]425, [3]426, [4, 5]264,427, [6]282, [7]428, [8]429, [9]430, [10]431, [11, 12]432,433, [13]434, [14, 15]435,436, [16-19]437–440, [20]441, [21, 22]442,443, [23]444, [24]445, [25, 26]446,447, [27, 28]448,449, [29]450, [30, 31]451,452, [32]453, [33]454, [34, 35]455,456, [36]457, [37]458, [38] 459, [39-43]323,324,460–462, [44-47]463–466, [48, 49]467,468, [50]469, [51]470, [52-54]471–473, [55]474, [56]475, [57, 58]476,477, [59]478, [60, 61]479,480, [62-64]481–483 and [65, 66]484,485.
The top 20 biological processes of neurological relevance are listed. EASE score is a modified fisher exact p-value measuring the gene-enrichment in the annotated terms. Genes are arranged in order of highest to lowest absolute fold change. Genes highlighted in red are up-regulated while those highlighted in green are down-regulated.
Upstream regulators associated with Arc-dependent genes
Because Arc knock down resulted in the differential expression of 1945 genes (Fig. 6), altering downstream pathways (Fig. 9) possibly leading to disease states (Table 3), we identified the upstream modulators that could explain the vast differential expression pattern observed. From the IPA analysis, 11 upstream regulators were predicted to critically contribute to the differential expression profile (Table 7). Except for Sox2, none of these upstream regulators were transcriptionally affected by Arc knockdown, suggesting Arc controls their function through a different mechanism. SOX2 and HDAC4 were both activated by the absence of Arc, while the function of the remaining 9 regulators was inhibited. Of note, the predicted inhibition of CREB1 (z-score = −3.5) and APP (z-score = −2.8) explains the differential expression of 100 and 94 genes, respectively (Table 7). The 11 upstream regulators predicted by IPA control the expression of Nr4a2, Slc6a1 and Igf1r, genes that are also involved in AD progression, neuroinflammation pathways and synaptic LTD (Fig. 9A and Table 4). We have investigated the mechanisms by which Arc could alter the function of the identified upstream regulators resulting in the alteration of downstream pathways and AD progression. The downstream pathways investigated are (i) opioid signaling, (ii) synaptogenesis signaling, (iii) the endocannabinoid neuronal synapse pathway, (iv) synaptic LTD and (v) neuroinflammation signaling (Fig 11). These are also the pathways whose downstream effects we focused on in Figure 9. APP, CREB1 and TNF are three upstream regulators identified by IPA that controlled the highest number of genes involved in the downstream pathways highlighted (Fig. 11). The top five genes regulated by APP were Igf1r (synaptic LTD)82, Ptgs2 (endocannabinoid neuronal synapse pathway; neuroinflammation, AD progression)83–86, Jun (neuroinflammation, AD progression)87–90, Dlg4 (PSD95, synaptogenesis)91,92 and Syn2(synaptogenesis)93 (Fig. 11). In addition to Ptgs2 and Syn2, CREB1 regulated the differential expression of Slc6a1 (neuroinflammation)94, Pdyn (opioid signaling)95 and Fosb (opioid signaling)96 (Fig. 11). Interestingly, TNF, whose transcription was not altered upon knockdown of Arc, regulates 15 genes (Table 7), the top five of which are Casp8 (neuroinflammation)97, Ptgs2 (also regulated by APP and CREB1), Gabrg2 (neuroinflammation)98,99, Bdnf (synaptogenesis; neuroinflammation, AD progression)100–103 and Penk (opioid signaling, AD progression)104. While the top CREB1-regulated genes are mainly associated with the opioid signaling pathway, APP and TNF are implicated in neuroinflammation. Triggering of the neuroinflammation pathway leads to the altered expression of AD-associated genes such as Ptgs2, Jun, Bdnf, Hmox1 and Gabbr2.

The map shows 11 upstream regulators (blue and orange boxes) predicted by IPA to mediate altered gene expression upon Arc knockdown. Genes are positioned in the extracellular space, the plasma membrane, the cytosol or the nucleus, depending on where their associated proteins are located. Arc was positioned at the interface of nucleus and cytoplasm because it can be in either compartment. Only genes that were involved in the following pathways and disease annotations are shown: (i) opioid signaling, (ii) synaptogenesis, (iii) endocannabinoid neuronal synapse pathway, (iv) synaptic LTD, (v) neuroinflammation, (vi) CNS amyloidosis, (vii) tauopathy and (viii) AD. Genes associated with disease annotations are boxed in magenta. The respective fold changes are indicated below each gene.
Arc over-expression alters gene expression in human embryonic kidney cells
The results presented thus far suggest that preventing Arc expression during neuronal network activation results in an altered gene expression profile affecting synaptic plasticity and cellular excitability, as well as neurodegenerative disease state. We therefore tested whether Arc can alter gene transcription, outside the context of neuronal network activation and without viral infection. We induced the expression of the endogenous Arc gene in human embryonic kidney (HEK293T) cells using a CRISPR-Cas9 approach105 (Fig. 12A). Whereas wildtype HEK293T cells expressed Arc at a very low level, targeting a transcription activator complex to its promoter increased Arc mRNA levels nearly 250-fold. This in turn altered the expression of 57 genes (absolute FC > 2, p < 0.05), with 54 genes up-regulated and 3 genes down-regulated. Many of the genes have neuronal functions (Fig. 12B). We have performed a GO analysis to understand the cellular components (Fig. 12C) and biological processes (Fig. 12D) these differentially expressed genes were involved in. We observed many genes that are typically expressed in neurons or are synaptic components, as indicated by the following GO terms: i) synapse part (p = 1.1E-04), ii) presynapse (p = 1.0E-03), iii) neuron part (p = 1.5E-03) and iv) postsynaptic membrane (p = 2.3E-03) (Fig. 12C). Differentially expressed genes upon the induction of Arc in HEK293T cells are involved in synaptic transmission processes or neuronal development, including i) chemical synaptic transmission (p = 2.5E-04), ii) signal release from synapse (p = 1.9E-03), iii) interneuron precursor migration (p = 3.2E-03) and iv) axon guidance (p = 3.2E-03) (Fig. 12D). Genes that are associated with these cellular components and processes were also highly altered, including i) Chat (p=4.7E-85, choline acetyltransferase) located at presynaptic terminals, synthesizes acetylcholine, ii) Oprd1 (p=2.6E-62, δ-opioid receptor), activation reduces pain and improves negative emotional states, iii) Arx (p=1.1E-70, Aristaless Related Homeobox), a transcription factor involved in neuronal migration and development, iv) Scn1b (p=6.6E-22, Na channel β1 subunit), involved in axonal guidance, v) Foxa3 (p=3.3E-24, Forkhead Box A3), a transcription factor involved in the determination of neuronal fate106,107, vi) Pllp (p=1.5e-25, Plasmolipin), involved in membrane organization and ion transport, vii) Slc18a3 (p=1.6E-16), a vesicular acetylcholine transporter at the presynapse, viii) Fndc11 (p=4.4E-14, Fibronectin Type III Domain Containing 11), a vesicular gene, and ix) Adgrb1 (p=3.7E-12, Adhesion G Protein-Coupled Receptor B1), localized at the postsynapse, involved in synapse organization and cell projection morphogenesis (Fig. 12B).

(A) Endogenous Arc expression was enhanced in HEK293T cells by targeting two single guide RNAs (sgRNAs) containing MS2 aptamers to the Arc promoter with the CRISPR/Cas9 Synergistic Activation Mediator system (see Methods for details). As a negative control, we used two sgRNAs targeting the promoter of the lac operon. Control cells (top) and Arc-induced cells (bottom) stained for Arc (green) and DNA was labeled with DAPI (blue). About 90% of the cells expressed Arc. (B) Graph showing the top 20 differentially expressed genes upon the induction of endogenous Arc in HEK293T cells. RNA-Seq was used to compare the mRNA levels between the Arc-induced and control HEK293T cells. Neuronal genes are bolded and highlighted in red. (C) and (D) GO analysis of the differential expressed genes upon overexpression of Arc. The top 20 cellular components (C) and biological processes (D) are presented. Neuronal features were bolded and highlighted in red. Na+: sodium; V-gated: voltage-gated; Postsynapt: postsynaptic; Memb: membrane; Synapt: synaptic; Clath: clathrin; Ach: acetylcholine; Presynapt: presynaptic; Musc: muscle; Develop: development; Neg: negative; Reg: regulation; Prolif: proliferation; Skelet: skeletal; Contract: contraction and Mech: mechanical; Conv: conversion.
Together with the results obtained with Arc knockdown in neurons, this finding strongly implicates Arc as a transcriptional regulator of neuronal development, synaptic function, plasticity and intrinsic excitability.
DISCUSSION
Activity-regulated cytoskeleton-associated protein (Arc) was discovered in 1995 as a neuronal activity-dependent immediate-early gene1,2, which is rapidly transcribed in response to network activation associated with novel experiences7–11. Knockdown of Arc expression interferes with stabilization of short-term memory, indicating that Arc plays a critical role in memory consolidation3,4. Arc’s function has been most widely studied in excitatory synapses, where it regulates endocytosis of AMPA receptors14,17. Interestingly, AMPA receptor removal also underlies Aβ-induced synaptic depression and dendritic spine loss108, processes thought to be associated with cognitive dysfunction in Alzheimer’s disease (AD)109. In Arc knockout mice, long-term potentiation (LTP) is not stable, and dissipates within a few hours, consistent with the impaired memory consolidation observed in these mice3–6. However, the absence of the late form of LTP in Arc knockout mice cannot be explained by an AMPA receptor endocytosis deficit4, indicating that Arc must have additional functions. The data presented here identify a second function for Arc: regulation of neuronal activity-dependent transcription for genes associated with synaptic plasticity, intrinsic excitability and cellular signaling. Analysis of the differentially expressed genes points to Arc’s involvement of several neurological disorders, including Autism, Huntington’s and Alzheimer’s disease. This newly proposed role for Arc is supported by its interaction with chromatin and histone markers reported here (Fig. 2-5, Movie).
Arc and chromatin
Pharmacological network stimulation induces Arc in a subset of cultured neurons (Fig. 1). Whereas chromatin in cultured hippocampal neurons is relatively uniform, Arc positive neurons are characterized by a larger number of densely packed heterochromatin puncta (chromocenters), likely harbouring silent genes, interspersed with highly open euchromatin domains, which are capable of active transcription (Fig. 2). This result is consistent with what has been observed in vivo, where Arc-deficient mice were found to have decreased heterochromatin domains31. These significant changes in chromatin structure observed in Arc-positive neurons are likely associated with equally substantial alterations in gene expression profiles. The correlation between Arc expression and chromatin remodeling that we observed does not establish a causative relationship. It is possible that Arc expression requires an alteration in chromatin structure, or alternatively, Arc expression may cause chromatin remodeling. Additional experiments are needed to decide on the underlying mechanism. It is also not clear at this time what determines which neurons will express Arc following network activation, although it likely has to do with the degree of participation of individual neurons in the enhanced network activity, which in turn depends on their synaptic connectivity.
Arc appears to physically interact with DNA: timelapse movies show dynamic chromatin loops that appear to invade Arc puncta (Fig. 3, Movie). The interaction is transient, lasting only a few seconds. Because these Arc puncta likely contain the histone acetylase Tip6025, it is conceivable that this interaction alters chromatin accessibility, thereby facilitating transcription. This idea is further strengthened by the association of Arc puncta with a histone marker for active enhancers (Fig. 4), as well as the close apposition between Arc puncta and puncta for a dual histone marker (H3K9Ac-S10P) that labels sites of active transcription (Fig. 5). A similar result has been obtained in vivo, where cocaine administration in rats results in an increase in nuclear Arc, which then associates with H3S10P31. Taken together, the data presented here on the interaction of Arc and chromatin may provide a mechanism for epigenetic regulation of gene transcription as the basis for memory consolidation.
How does Arc regulate transcription?
Preventing Arc induction during neuronal network activation affects the transcription of a very large number of genes (Fig. 6). The domain structure of Arc protein appears to rule out that it can function as a transcription factor78. This raises the question: how does Arc regulate transcription? One possible mechanism, discussed above, is that Arc epigenetically controls gene transcription by regulating chromatin structure (through Tip60 or other chromatin remodelers) and modification of histones (e.g. H4K12Ac25). However, the differential gene expression associated with Arc knockdown is mediated through eleven upstream regulators identified by IPA (Table 7, Fig. 11). This suggests that Arc has additional, less direct ways of regulating transcription. Interestingly, to date, none of the eleven upstream regulator proteins have been shown to either directly interact with or be modulated by Arc. They are also not transcriptionally controlled by Arc (except for Sox2) (Table 7). How then does Arc regulate transcription by activating or inhibiting these upstream regulators? Using IPA and its Ingenuity Knowledge Base, we were able to identify several known interactors of Arc that can modulate the action of the upstream regulators, which could then subsequentially alter gene transcription (Fig. 13A). Next, we will discuss the mechanisms by which four identified Arc interactors, NOTCH1, TIP60/Kat5, APP and GSK3B, could modulate the upstream regulators.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A) Schematic diagram illustrating how interactors of Arc could bring about changes in activity of upstream regulators identified by IPA, which in turn results in alteration of gene transcription. The dotted line indicates an indirect effect on transcription through the regulation of a transduction cascade. (B) Diagram showing the functional connectivity between Arc interactors (KAT5, NOTCH1, GSK3B and APP), upstream regulators highlighted in orange (activated) or blue (inhibited) and genes that mediate their interaction. Connections of Arc interactors with other genes are highlighted are highlighted in cyan. Connections between KAT5, NOTCH1, GSK3B and APP are highlighted in blue.
Activation z-score indicates the predicted activity of upstream regulators by IPA analysis. Upstream regulators that were predicted to be inhibited are highlighted in blue while those activated are highlighted in orange. Regulated genes were highlighted in green (down-regulated) and red (up-regulated).
NOTCH1
NOTCH1 is a transmembrane receptor capable of signaling to the nucleus. Arc is required for the proteolytic cleavage of NOTCH1 to release its intracellular domain (NICD), which can translocate to the nucleus and alter transcription110. NICD regulates the expression of the transcriptional repressor BCL6111 and the activity of the calcium-dependent kinase CAMK4112, which in turn alter the localization and the nuclear-cytoplasmic shuttling of the histone deacetylase HDAC4, thereby affecting its downstream interactions/modulation113,114 (Fig. 13B). NOTCH1 could regulate the stability, nuclear localization and signaling of the transcription factor SOX2 through regulation of the protein kinase AKT1 and cell-surface glycoprotein CD44115–118 (Fig. 13B).
NOTCH1, through NICD, controls the expression of plasminogen activator inhibitor-1 (SERPINE1)119, an inhibitor of thrombin (F2)120 (Fig. 13B). NOTCH1 regulates the transcriptional activity of T-cell factor 4 (TCF7L2)121, through its interaction with the DNA-repair protein Ku70 (XRCC6)122 (Fig. 13B). NOTCH1 interacts with the nerve growth factor NR4A1/Nur77123, thereby modulating expression levels of the cytokine tumor necrosis factor alpha (TNF)124. Finally, NOTCH1 regulates the expression level of the inhibitor of apoptosis protein cIAP1/Birc2125, which also affects TNF expression126 (Fig. 13B).
TIP60/Kat5
The Kat5 gene encodes TIP60, a member of the MYST family of histone acetyl transferases, which plays important roles in chromatin remodeling and transcription regulation127. In the fruit fly Drosophila, TIP60 has been implicated in epigenetic control of learning and memory128, while it mediates APP-induced apoptosis and lethality in a fly AD model129. Nuclear Arc interacts with TIP60 at perichromatin regions and recruits TIP60 to PML bodies, sites of epigenetic transcription regulation25. Arc levels correlate with acetylation status of H4K12, a substrate of TIP60 and a memory mark that declines with aging29, suggesting that Arc mediates activation of TIP60. TIP60/Kat5 facilitates the repressive action of HDAC4 through the formation of complexes with the zinc-finger transcription factor KLF4121,130, the cAMP-dependent transcription factor ATF3131–133 and the neurodegenerative disease protein ataxin-1 (ATXN1)134,135 (Fig. 13B). Arc’s interaction with TIP60/Kat5 may result in a complex being formed at the cIAP1/Birc2 promoter region136 to mediate downstream signaling of TNF126 (Fig. 13B). TIP60/Kat5 forms a complex with the Kaiso transcription factor ZBTB33137 resulting in the inhibition of the TCF7L2 transcriptional complex138 (Fig. 13B). Complexing of TIP60 with ARID1B could affect SOX2 signaling139–141. The regulation of SOX2 by TIP60/Kat5 could also have an implication on the transcriptional activity of Achaete-Scute homolog 1 (ASCL1), as SOX2 and ASCL1 regulate each other, possibly as a feedback loop142,143.
APP
The functional interaction between APP and Arc is crucial for Arc’s modulation of upstream regulators (Fig. 13A). Arc interacts with endophilin 2/3 (SH3GL3) and dynamin on early/recycling endosomes to alter the trafficking and localization of APP. The association of Arc with presenilin 1 (PSEN1) promotes the trafficking of γ-secretase to endosomes and enzymatic cleavage of APP80 (Fig. 13B). The generation of amyloid beta through APP cleavage leads to an altered downstream signaling, activity and production of HDAC4, SOX2 and F2 through changes of caspase-3 (CASP3)144,145, JUN146,147 and thrombospondin-1 (THBS1)148,149, respectively (Fig. 13B). Cleavage of APP generates a cytosolic fragment, AICD, which forms a transcriptionally active complex with TIP60 and the transcription factor FE65150. AICD also modulates the ubiquitin-proteasome system (UPS) via UBE2N151, to change downstream signaling induced by TNF152 (Fig. 13B). The modulation of the UPS via UBE2N, UBC and UBE3A153 could implicate the ubiquitination of serum- and glucocorticoid-regulated kinase-1 (SGK-1)154–156 and polyglutamine-expanded ataxin 3 (ATXN3)157 and their ability to regulate the transcription factor cAMP responsive element binding protein 1 (CREB1)158,159 (Fig. 13B). The modulation of CREB1 would further implicate changes in expression levels of cAMP responsive element modulator CREM160–162 (Fig. 13B). Finally, APP has a role in the regulation of TNF through indirect modulation of CREM163 and direct interactions with laminin could regulate the production of TNF164,165 (Fig. 13B).
GSK3B
Although glycogen synthase kinase 3 beta (GSK3B) is not regulated by Arc, promotion of cleavage of APP to amyloid beta enhances the induction and activation of GSK3B166–168. This could lead to modified downstream signaling of CREB1169 (Fig. 13B). GSK3B is also a downstream mediator of NOTCH1170, PSEN1171, and CAMK2B172, all of which are Arc interactors80,110,173. This creates an interesting situation as APP/amyloid beta is positively regulated by GSK3B174,175, creating a positive feedback loop for amyloid beta production and its downstream signaling166–168 (Fig. 13B).
Interactions among TIP60, NOTCH1 and APP
A delicate regulatory network exists among Arc’s interactors TIP60/Kat5, NOTCH1 and APP (Fig. 13B). Arc’s activation of the γ-secretase PSEN1 to promote cleavage of APP not only increases amyloid beta load, but also results in an increased level of the APP intracellular domain (AICD)80,176. AICD forms a complex with TIP60/Kat5 to alter transcriptional activity crucial for AD progression150,177–182 (Fig. 13B). This AICD-TIP60 interaction is disrupted by NICD, formed when Arc activates NOTCH1110, thereby downregulating AICD signaling while promoting NICD signaling183,184 (Fig. 13B). The formation of NICD and AICD is competitive, as NOTCH1 and APP are both substrates of γ-secretase185, whose activity is regulated by Arc80. In addition, the induction of TIP60 histone acetylation activity by Arc25 could also increase the negative regulation of NOTCH1183 (Fig. 13B). This highlights Arc as an important modulator of the relationship and downstream signaling mediated by NOTCH1, TIP60/Kat5 and APP. Of note, the mRNA levels of Notch1, Kat5 and App were not significantly altered upon knockdown of Arc, indicating that the transcriptional changes brought about were due to protein interaction and activation (Fig. 13B), which is upstream of transcription (Fig. 13A). However, the modulation of upstream regulators by Arc is also dependent on the its subcellular localization.
Arc’s subcellular localization determines its function
When Arc is localized outside the nucleus it tends to accumulate in dendrites and spines, small membrane protrusions that harbour excitatory synapses. Here, Arc controls the removal of AMPA receptors by endocytosis, allowing it to regulate synaptic efficacy14,17. Synaptic Arc also associates with the synaptic scaffolding protein PSD-95/Dlg4, which complexes with the tyrosine kinase FYN186–188, allowing it to regulate brain-derived neurotrophic factor (BDNF) signalling through tyrosine receptor kinase B (TrkB), a major pathway for synapse maturation, plasticity and neurodevelopmental disorders189. Activation of FYN could also mediate the secretion of TNF190(Fig. 13B). A high-affinity interaction with calcium-calmodulin kinase 2 beta (CAMK2B) targets Arc to inactive synapses, where it removes GluA1 AMPA receptors from the postsynaptic membrane surface173.
Arc has been shown to possess both a nuclear localization signal (NLS) and a nuclear retention domain24, allowing it to translocate to the nucleus autonomously. Once in the nucleus, Arc has access to several other potential binding partners, including a nuclear spectrin isoform (βSpectrinIV∑5)20 and TIP60, a subunit of a chromosome remodeling complex25. Association with Amida, encoded by the Tfpt gene (Fig. 13B) facilitates Arc’s entry into the nucleus191. Amida is a subunit of the INO80 chromatin remodeling complex, which contains the transcriptional regulator MCRS1192,193. MCRS2, an isoform of MCRS1, is associated with the MLL chromatin remodeling complex, which also contains KMT2A (MLL1) (Fig. 13B). Arc’s association with Amida and possibly the INO80 and MLL complexes may provide Arc with yet another opportunity to control gene expression by altering chromatin structure.
The ability of Arc to translocate between the synapse and the nucleus, with unique functions in each subcellular compartment, further strengthens its role in memory consolidation, which requires both alterations of synaptic function and de novo gene transcription194.
Arc controls synaptic plasticity and intrinsic excitability
Arc’s well-studied ability to alter synaptic efficacy by endocytosis of AMPA receptors established it as a critical regulator of synaptic plasticity14,17,188,195. Whereas this mechanism of activity-dependent removal of glutamate receptors supports Arc’s role in mediating long-term depression (LTD)196–199, it does not explain the absence of stable long-term potentiation (LTP) observed in Arc knock-out mice4. Because late-LTP is considered a critical cellular mechanism underlying memory consolidation, the molecular and cellular mechanism by which Arc supports memory stabilization has remained elusive. The data presented here showing that Arc transcriptionally regulates the expression of a large number of synaptic proteins, with functions in both the pre- and post-synaptic compartment (Table 2), provides a new mechanism by which Arc can control long-lasting changes in synaptic structure and function required for memory consolidation.
Formation of a memory trace not only requires longterm changes in the strength of the synapses connecting the neurons that constitute the engram, but also stable changes in their intrinsic excitability200–202. Because Arc controls the expression of a large number of ion channels and pumps/transporters, it appears that Arc is capable of supporting this functional aspect of memory consolidation as well.
Arc and Alzheimer’s disease
Alzheimer’s Disease (AD) is a devastating neurodegenerative disorder203,204 characterized by the progressive loss of both synaptic function205 and long-term memory formation206. There is currently no therapy that prevents, stabilizes or reverses the progression of this disease, which is projected to take on epidemic proportions as the world population ages207,208. Several previous studies have revealed an association between Arc and AD. A landmark study published in 2011 showed that Arc protein is required for the formation of amyloid (Aβ) plaques80. Moreover, Arc protein levels are aberrantly regulated in the hippocampus of AD patients209, and are locally upregulated around amyloid plaques210, whereas a polymorphism in the Arc gene confers a decreased likelihood of developing AD211. It has been shown that spatial memory impairment is associated with dysfunctional Arc expression in the hippocampus of an AD mouse model212. These published results together with the data presented here, suggests that aberrant expression or dysfunction of Arc contributes to the pathophysiology of AD205,213.
Arc and AD therapy
Arc’s ability to transcriptionally regulate AD susceptibility and AD pathophysiology related genes indicates a possibility for modifying expression and activity of Arc as a therapy for AD. Current treatments are symptomatic, not effective disease-modifying cures214. Many hypotheses have been proposed to underlie the development of AD, including i) amyloid beta aggregation, ii) tau hyperphosphorylation, iii) neuroinflammation, iv) neurotransmitter dysfunction, v) mitochondria dysfunction, vi) glucose metabolism, vii) vascular dysfunction and viii) viral infection214–217. These hypotheses have generated many new compounds, none of which showed efficacy in slowing cognitive decline or improving global functioning214,216. Arc appears a good therapeutic candidate for AD, because of its involvement in amyloid beta production, tau phosphorylation, neuroinflammation and neurotransmission. Moreover, we have shown that Arc can modulate the expression of many AD genetic risk factors and genes associated with the pathophysiology of AD (Fig. 9, Fig. 10, Fig. 12 and Table 4). Currently, known drugs that could increase mRNA or protein expression of Arc include antidepressant drugs218, phencyclidine219 and corticosterone, a memory enhancing drug220. Arc expression could be altered by targeting TIP60 and PHF8, two histone modifiers that together control Arc transcription22. Drugs could also modulate Arc’s effect via its interactors such as TIP60 and NOTCH1. Natural and synthetic drug molecules targeting TIP60 exist, but they are currently used for cancer treatment221. Modulation of NOTCH1 function often involves inhibitors of γ-secretase, which would also affect APP cleavage185,222. These pharmaceutical modifications of Arc expression and activity could present a promising starting point for development of a more effective AD therapy.
METHODS
Animals and chemicals
All work involving the use of animals were performed according to the guidelines of the Institutional Animal Care and Use Committee (IACUC) and were approved by the IACUC at the SingHealth in Singapore. Time-mated E18 Sprague Dawley rats were purchased through the SingHealth Experimental Medicine Centre (SEMC). They were sacrificed immediately after delivery to the vivarium. All chemicals were purchased from Sigma-Aldrich unless otherwise stated.
Culturing hippocampal and cortical neurons
Hippocampal and cortices were dissected from the E18 embryos of Sprague Dawley rats. Hippocampi or cortices underwent papain dissociation based on the protocol from the Papain Dissociation System (Worthington Biochemical Corporation). Gentle mechanical trituration was performed to ensure complete dissociation of tissues. Dissociated cells were plated on poly-D-lysine (Sigma) coated dishes at a plating density of 1.5 x 105/cm2 in Neurobasal medium (Gibco) supplemented with 10% (v/v) foetal-bovine serum (FBS, Sigma), 1% (v/v), penicillin-streptomycin (P/S, Gibco) and 2% (v/v) B27 supplement (Gibco) for 2 hours. FBS-containing medium was then removed and replaced with FBS-free medium and cells were cultured FBS-free subsequently to prevent astrocytic over-growth. Medium was changed on Days In Vitro (DIV) 5. Subsequently, medium was changed every three to four days. Experiments were carried out on DIV 18-22.
Inhibition of Arc expression by an shRNA
Four Arc shRNA plasmids (SureSilencing, Qiagen) were transfected into neuronal cultures using Lipofectamine 2000 (Qiagen). Pharmacological LTP was induced in neuronal cell cultures using a 4-hr treatment with 4BF. Cells were fixed and stained for Arc protein. Immunofluorescence images were obtained using wide-field microscopy. Effectiveness of inhibition of Arc expression was based on co-occurrence of expression of the plasmids and the absence of Arc immunofluorescence. The most effective shRNA plasmid was chosen and adeno-associated virus AAV9 constructs harbouring an Arc shRNA and a scrambled version of this shRNA were synthesized using the annealed oligo cloning method. The oligos for the Arc shRNA were: i) 5′-GAT CCG GAG GAG ATC ATT CAG T-3′, ii) 5′-ATG TCT TCC TGT CAA CAT ACT GAA TGA TCT CCT CCT TTT TG-3′, iii) 5′-AAT TCA AAA AGG AGG AGA TCA TTC AGT-3′ and iv) 5′-ATG TTG ACA GGA AGA CAT ACT GAA TGA TCT CCT ccG-3′. The oligos for Arc scrambled shRNA were i) 5′-GAT CCG GTA ATT TCG GAG GAT C-3′, ii) 5′-AAG TCT TCC TGT CAA CTT GAT CCT CCG AAA TTA CCT TTT TG-3′, iii) 5′-AAT TCA AAA AGG TAA TTT CGG AGG ATC-3′ and iv) 5′-AAG TTG ACA GGA AGA CTT GAT CCT CCG AAA TTA CCG-3′. The ends of the annealed oligos harbor overhangs of the restriction sites for BamH1 and EcoR1. Oligos for the Arc shRNA were annealed in buffer A (mM) 100 NaCl and 50 HEPES, pH 7.4 while oligos for Arc scrambled shRNA were annealed in buffer B (mM) 10 Tris, pH 7.5-8.0, 50 NaCl and 1 EDTA at equimolar concentration by heating to a temperature of 95°C for 5 min then cooling it down to room temperature (rtp). The annealed oligos were ligated using T4 ligase (New England Biolabs) into the BamH1/EcoR1-cut vector pENN.AAV.U6.shRLuc.CMV.eGFP.SV40, generously provided by the University of Pennsylvania, Vector Core. Ligated products were transformed into Stbl3 competent cells (Thermo Fisher Scientific). Successful constructs were identified by restriction enzymes digestion and verified by sequencing. AAV9 virus harbouring the transgenes (concentrations at 1 x 1013– 1 x 1014 GC/ml range) were synthesized by University of Pennsylvania, Vector Core. Arc expression was prevented by treating neuronal cultures with 3 x 106 multiplicity of infection (MOI) AAV9 Arc shRNA virus on DIV14. Induction of Arc expression by pharmacological LTP (see below) was performed between DIV19-22.
Pharmacological LTP and immunofluorescence
Hippocampal or cortical neuronal cultures were treated with a combination of 100 μM 4-aminopyridine (4AP), 50 μM bicuculline (Bic) and 50 μM forskolin for the respective time stated to induce pharmacological LTP21,32,33. This drug combination will be referred to as 4BF henceforth. At the end of the treatment, cells were fixed with 100 % ice-cold methanol at −20°C for 10 min. Cells were washed three times with 1x Phosphate Buffered Saline (PBS, in mM: 137 NaCl, 2.7 KCl and 12 phosphate buffer) containing 0.1% (v/v) Triton X-100 (PBS-Tx). Depending on the antibodies used, some cells were fixed again with 4% (w/v) paraformaldehyde (PFA) in 1x PBS containing 4% (w/v) sucrose. Cells were washed three times in 1x PBS-Tx and blocked in 2% (w/v) Bovine Serum Albumin (BSA) in 1x PBS for 1 h at rtp. Depending on the species the secondary antibodies were raised in, 10% (v/v) serum of the corresponding species was added to the blocking buffer. Cells were probed with primary antibodies as indicated for the experiments: i) anti-Arc (1:300, Santa Cruz, sc-17839), ii) anti-Arc (1:300, Synaptic Systems, 156 003), iii) anti-MAP2 (1:300, Millipore, AB5622), iv) anti-H3K27Ac (1:300, Wako, 306-34849) and v) anti-H3K9Ac-S10P (1:300, Abcam, ab12181) in antibody dilution buffer (1x PBS containing 1% (w/v) BSA, 5% (v/v) serum and 0.05% (v/v) Triton X-100) for 1 h at rtp. Cells were washed three times in 1x PBS-Tx. Cells were then probed with 1:1000 Alexa-Fluor 647, Alexa Fluor 568 or Alex-Fluor 488 (Molecular Probes) for 1 h at rtp. Cells were washed three times, followed by staining of DNA with 50 μM DAPI for 20 min at rtp. Cells were mounted in FluorSave (Calbio-chem). For immunofluorescence staining for STORM imaging, cells were fixed with 3% paraformaldehyde and quenched with 0.1% NaBH4 as described in Oey et al. (2015)22. Blocking, primary and secondary antibody staining were carried out as above. A post-fixation, as described in Oey et al. (2015)22, was also carried out after secondary antibody binding.
Transfection of neuronal cultures
The Arc-eYFP construct was generated as described in Bloomer et al. (2007)20. Neuronal cultures (DIV16) were transfected with Arc-eYFP and H2B-mCherry (Addgene, 20972) with Lipofectamine 2000 (Invitrogen) according to manufacturer’s protocol with some adjustment. Arc-eYFP:H2B-mCherry DNA was added to Lipofectamine at a ratio of 1:1. The Lipofectamine:DNA complex was incubated at rtp for 20 min before been added to the cells. The complex was added dropwise such that it was evenly distributed on the cell culture. Culture medium was added after 20 min and experiments were performed on DIV19.
Widefield microscopy
Fluorescence images were obtained using widefield microscopy as detailed in Oey et al. (2015)22. Images obtained were analyzed using NIS Elements AR version 4.1 (Nikon) to perform background subtraction. Out-offocus fluorescence was removed using 3D deconvolution (AutoQuant, Media Cybernetics). The Region-Of-Interest (ROI) analysis tool was used to mark nuclei based on DAPI intensity. Corresponding mean Arc intensity of each nucleus was also measured using the automated measurement module. The average of mean Arc intensity for all neurons from non-4BF stimulated controls were obtained for each set of experiments. This would be used as a cut-off threshold between Arc-positive and Arc-negative neurons for each set of experiments since Arc expression was only observed upon stimulation223,224. Nuclei images were cropped individually and analyzed using a custom MATLAB (Mathworks) program. Size and intensity thresholds were applied to identify and quantitate puncta in each nucleus. Batch processing using the same size and intensity threshold was performed. Mean size of the puncta and number of puncta were recorded. ROI ID for each nucleus was used to correlate the mean Arc intensity with the mean area or number of puncta. Statistical analysis was performed using GraphPad Prism Version 6.01. Statistical data shown are mean ± S.E.M. (standard error of the mean) across experiments.
Spinning Disc confocal microscopy
Fluorescence images and time-lapse movies were obtained using a motorized Ti-E inverted microscope (Nikon) with a 60X oil Plan-Apo objective (1.49 NA) and a 100X Apo-TIRF objective (1.49 NA). Spinning disk confocal microscopy was achieved using the CSU-W1 Nipkow spinning disk confocal unit (Yokogawa Electric). A sCMOS camera (Zyla, Andor) was used to capture the confocal images. Laser lines used were 488 nm (100 mW) for GFP, 515 nm (100 mW) for eYFP and 561 nm (150 mW) for mCherry (Cube lasers, Coherent). Fast excitation/emission switching was obtained using a dichroic beam splitter (Di01-T405/488/568/647-13 x 15 x 0.5, Semrock) and filter wheels controlled by a MAC6000DC (Ludl). The Perfect Focus System (Nikon) was applied to ensure minimal focus drift during image acquisition. Z stacks were obtained using step sizes recommended for objectives used, which were processed using 3D blind deconvolution (AutoQuant) to remove out-of-focus fluorescence.
Stochastic Optical Reconstruction Microscopy (STORM)
Dual color STORM image sequences were obtained using a Zeiss ELYRA PS.1 platform. Endogenous Arc and the dual histone marker H3K9Ac-S10P were labeled with primary antibodies and visualized using Alexa 488 and Alexa 647 secondary antibodies. Time-lapse movies of 10,000 frames were obtained of neuronal nuclei expressing Arc capturing the blinking of individual Alexa 488 and 647 molecules brought into the dark state by intense laser illumination. Fitting of a 2D Gaussian to each blinking dot allowed their XY localization to be determined with high precision (typically 30 nm). Super resolution images are generated from the localizations by superimposing a 2D Gaussian (green for 488 nm, red for 647 nm) for each localized position. Molecule localization and image rendering were performed by Zen software (Zeiss).
Cell lysate preparation and Western blotting
Following 4BF stimulation, neuronal cultures were washed gently with 1x PBS. Cells were gently scraped off and harvested in an Eppendorf tube. Cells were spun down at 10000 g for 5 min at 4°C to obtain the cell pellet. Total protein was isolated using an RNA-protein extraction kit (Macherey-Nagel), as specified by the manufacturer. A BCA kit (Pierce) was used to measure the concentration of proteins. 30 μg of each protein sample was denatured and reduced by boiling at 95°C for 5 min in 10% (v/v) 2-mercaptoethanol-containing Laemmli sample buffer (Bio-Rad). Samples were resolved by SDS-PAGE with a precast Tris-glycine gel (Bio-Rad) and transferred onto PVDF membranes using the Trans-Blot Turbo transfer System (BioRad) as indicated by manufacturer. Membranes were blocked for 1 h at rtp with 5% (w/v) non-fat milk block (Bio-Rad) in 1x TBS (in mM) (140 NaCl, 3 KCl, 25 Tris base) (First Base) containing 0.1% (v/v) Tween-20 (TBST), followed by primary antibodies incubation for 1 h (anti-Arc, 1:1000, Santa Cruz, sc-17839) in 1x TBST at rtp. Membranes were washed three times, each for 5 min in 1x TBST at rtp. Secondary antibodies binding was performed using the corresponding HRP-conjugated secondary (1:10000, Invitrogen) for 1 h in 1x TBST at rtp. Protein bands were detected with chemiluminescence substrate (Pierce) visualized with a Gel Doc XRS imaging system (Bio-RAD) or developed on scientific imaging film (Kodak).
RNA sample preparation, library construction, RNA-Seq and analysis
4BF treated neuronal cells were washed, scraped and spun down as above. RNA samples were obtained from the cell pellet using the RNA-protein extraction kit as specified by manufacturer (Macherey-Nagel). Library construction and RNA sequencing were performed by the Duke-NUS Genome Biology Facility. 2.2 μg of RNA was used for library construction. Prior to library construction, quality of the RNA was analyzed with an Agilent Bioanalyzer. Following poly-A enrichment, recovered RNA was processed using the Illumina TruSeq stranded mRNA kit to generate the adaptor-ligated libraries. A total of 9 samples were analyzed. These samples came from 3 different sets of experiments (n = 3). Each set contained samples treated with i) 8 h 4BF, ii) Arc shRNA + 8 h 4BF and iii) Arc scrambled shRNA + 8 h 4BF. Six samples were sequenced per lane on the HiSeq 3000 sequencer using 150 pair-end reads. For the HEK293T cells, RNA was obtained similarly. Three samples were analyzed, with two Arc induced samples and one control sample. The samples were processed as described above and sequenced on 1 lane on the HiSeq 3000 sequencer.
Computational analyses of RNA-Seq data
FASTQ files obtained from the RNA-sequencing were mapped to the rat genome using Partek Flow (version 7.0.18.1210). Adapter sequences were trimmed. Contaminant reads contributed from rDNA, tRNA and mtDNA were filtered out using Bowtie2 (version 2.2.5). Filtered, trimmed reads of high quality (Phred score > 30) were then mapped onto the Rattus norvegicus genome (rn6) for the rat samples or Homo sapiens genome (hg38) for the HEK293T samples with Star (version 2.5.3a)225. Post alignment QA/QC were performed after the alignment step to determine if alignment had a good average coverage and were uniquely aligned. The unique, paired reads were used for gene expression quantification. Reads were assigned to genes using the Expectation/ Maximization (E/M) algorithm in Partek Flow226 based on the annotation model rn6 (Ensembl transcripts release 93) for the rat samples and the annotation model hg38 (Ensembl transcripts release 94) for the HEK293 samples. To ensure only informative genes were included in the downstream analysis, noise (maximum feature counts ≤ 30) was filtered out. Read counts between samples were normalized with the Upper quantile method227. As genes with very low expression might be inadequately represented and incorrectly identified as differentially expressed, a constant of 1 was added to normalized counts for rectification. Statistical analysis was performed using the Gene Specific Analysis (GSA) module in Partek Flow to identify differential gene expression. P-value and fold changes of differentially expressed genes were calculated based on the lognormal with shrinkage distribution. Average coverage of less than 3 were also filtered out prior to statistical analysis. Differential gene expression with a cut-off value of false discovery rate (FDR) step-up < 0.05228 and an absolute fold change ≥ 2 were considered for further gene ontology analysis in Partek Flow that is based on the GO Consortium (version 2018_08_01)229,230. GO analysis was also performed on the transcriptional regulators/factors that were observed to be altered upon Arc knockdown using DAVID (version 6.8)81,231. The EASE score obtained from the DAVID analysis is a modified Fisher Exact P-value to indicate gene enrichment in the annotation terms. Functional analysis on the statistically significant differential gene expression (FDR step-up < 0.05; absolute fold change ≥ 2) was performed by Ingenuity Pathway Analysis (IPA) (https://tinyurl.com/y6672c22, Qiagen). Pathways and their associated downstream effects, diseases, regulator networks and upstream regulators were identified by IPA. Predictions on the possible activation and inhibition of pathways, downstream effects and upstream regulators were inferred from the degree of consistency in the expression of the target genes compared to the fold changes in the differentially expressed gene list. This activation or inhibition status was expressed as a z-score, with z ≥ 2 indicating activation and z ≤ 2 indicating inhibition. Inferences made were based on at least one publication or from canonical information stored in the Ingenuity Knowledge Base. Fisher’s exact test was used to calculate the P-value for all analyses in IPA.
Plasmid construction for Arc expression in HEK293T cells
The following plasmids were used: pSBbi-Hyg (Addgene #60524) and pSBbi-Pur (Addgene #60523) were a gift from Eric Kowarz. pCMV(CAT)T7-SB100 (Addgene #34879) was a gift from Zsuzsanna Izsvak. sgRNA(MS2) cloning backbone plasmid, (Addgene #61424), MS2-P65-HSF1_GFP, (Addgene #61423), and dCAS9-VP64_GFP (Addgene #61422) were a gift from Feng Zhang. The psBbi-Hyg-dCAS9-VP64 and the pSBbi-MS2-P65-HSF1-Pur plasmids were constructed by isolating the dCAS9-VP64 and MS2-P65-HSF1 sequences via PCR from the MS2-P65-HSF1_GFP and dCAS9-VP64_GFP plasmids and annealed into the SfiI-linearised pSBbi-Hyg and pSBbi-Pur plasmids. psBbi-Hyg-dCAS9-VP64 and pSBbi-MS2-P65-HSF1-Pur were then co-transfected with pCMV(CAT)T7-SB100 into HEK293T using JetPrime (Polyplus-Transfection) according to manufacturer’s instructions. HEK293T cells with successful transposition of both genes were selected with a combination of 0.75ug/ml puromycin (Gibco) and 200ug/ml hygromycin B (Nacalai) in DMEM + 10% FBS (Gibco) over several passages for a month.
Transfection for endogenous Arc overexpression and purification of mRNA
sgRNA(MS2) backbone plasmids containing guide RNAs complementary to human Arc promoters were transfected into the mutated HEK293T cells. A separate control well was transfected with sgRNA(MS2) backbone containing LacZ promoter sgRNAs. After 48 hours, mRNA was purified using the NucleoSpin RNA kit (Macherey-Nagel) and submitted for RNA-sequencing by the Duke-NUS Genomics Core Facility. The table below lists the guide RNAs (sgRNAs) used:
ACKNOWLEDGEMENTS
We would like to thank the SingHealth Advanced BioImaging Core for excellent support with our microscopy experiments. The experiments were supported by grant OFIRG/0019/2016 from the National Medical Research Council (NMRC) to AMJ VanDongen.
Footnotes
REFERENCES
- 1.↵
- 2.↵
- 3.↵
- 4.↵
- 5.
- 6.↵
- 7.↵
- 8.↵
- 9.
- 10.
- 11.↵
- 12.↵
- 13.↵
- 14.↵
- 15.↵
- 16.↵
- 17.↵
- 18.
- 19.↵
- 20.↵
- 21.↵
- 22.↵
- 23.↵
- 24.↵
- 25.↵
- 26.↵
- 27.
- 28.↵
- 29.↵
- 30.↵
- 31.↵
- 32.↵
- 33.↵
- 34.↵
- 35.↵
- 36.↵
- 37.
- 38.
- 39.
- 40.↵
- 41.↵
- 42.↵
- 43.↵
- 44.↵
- 45.↵
- 46.
- 47.
- 48.
- 49.
- 50.
- 51.
- 52.↵
- 53.↵
- 54.
- 55.↵
- 56.↵
- 57.↵
- 58.↵
- 59.↵
- 60.↵
- 61.↵
- 62.↵
- 63.↵
- 64.↵
- 65.↵
- 66.↵
- 67.↵
- 68.↵
- 69.
- 70.↵
- 71.↵
- 72.↵
- 73.↵
- 74.
- 75.
- 76.
- 77.↵
- 78.↵
- 79.
- 80.↵
- 81.↵
- 82.↵
- 83.↵
- 84.
- 85.↵
- 86.↵
- 87.↵
- 88.
- 89.
- 90.↵
- 91.↵
- 92.↵
- 93.↵
- 94.↵
- 95.↵
- 96.↵
- 97.↵
- 98.↵
- 99.↵
- 100.↵
- 101.
- 102.
- 103.↵
- 104.↵
- 105.↵
- 106.↵
- 107.↵
- 108.↵
- 109.↵
- 110.↵
- 111.↵
- 112.↵
- 113.↵
- 114.↵
- 115.↵
- 116.
- 117.
- 118.↵
- 119.↵
- 120.↵
- 121.↵
- 122.↵
- 123.↵
- 124.↵
- 125.↵
- 126.↵
- 127.↵
- 128.↵
- 129.↵
- 130.↵
- 131.↵
- 132.
- 133.↵
- 134.↵
- 135.↵
- 136.↵
- 137.↵
- 138.↵
- 139.↵
- 140.
- 141.↵
- 142.↵
- 143.↵
- 144.↵
- 145.↵
- 146.↵
- 147.↵
- 148.↵
- 149.↵
- 150.↵
- 151.↵
- 152.↵
- 153.↵
- 154.↵
- 155.
- 156.↵
- 157.↵
- 158.↵
- 159.↵
- 160.↵
- 161.
- 162.↵
- 163.↵
- 164.↵
- 165.↵
- 166.↵
- 167.
- 168.↵
- 169.↵
- 170.↵
- 171.↵
- 172.↵
- 173.↵
- 174.↵
- 175.↵
- 176.↵
- 177.↵
- 178.
- 179.
- 180.
- 181.
- 182.↵
- 183.↵
- 184.↵
- 185.↵
- 186.↵
- 187.
- 188.↵
- 189.↵
- 190.↵
- 191.↵
- 192.↵
- 193.↵
- 194.↵
- 195.↵
- 196.↵
- 197.
- 198.
- 199.↵
- 200.↵
- 201.
- 202.↵
- 203.↵
- 204.↵
- 205.↵
- 206.↵
- 207.↵
- 208.↵
- 209.↵
- 210.↵
- 211.↵
- 212.↵
- 213.↵
- 214.↵
- 215.
- 216.↵
- 217.↵
- 218.↵
- 219.↵
- 220.↵
- 221.↵
- 222.↵
- 223.↵
- 224.↵
- 225.↵
- 226.↵
- 227.↵
- 228.↵
- 229.↵
- 230.↵
- 231.↵
- 232.↵
- 233.
- 234.↵
- 235.↵
- 236.↵
- 237.↵
- 238.↵
- 239.↵
- 240.↵
- 241.↵
- 242.↵
- 243.
- 244.↵
- 245.↵
- 246.↵
- 247.↵
- 248.↵
- 249.↵
- 250.↵
- 251.↵
- 252.↵
- 253.↵
- 254.↵
- 255.↵
- 256.↵
- 257.
- 258.↵
- 259.↵
- 260.↵
- 261.↵
- 262.↵
- 263.↵
- 264.↵
- 265.↵
- 266.
- 267.↵
- 268.↵
- 269.↵
- 270.↵
- 271.↵
- 272.↵
- 273.↵
- 274.↵
- 275.↵
- 276.↵
- 277.↵
- 278.↵
- 279.↵
- 280.↵
- 281.
- 282.↵
- 283.↵
- 284.↵
- 285.↵
- 286.↵
- 287.↵
- 288.
- 289.↵
- 290.↵
- 291.
- 292.↵
- 293.↵
- 294.↵
- 295.↵
- 296.↵
- 297.↵
- 298.↵
- 299.↵
- 300.↵
- 301.↵
- 302.↵
- 303.↵
- 304.↵
- 305.↵
- 306.↵
- 307.↵
- 308.↵
- 309.↵
- 310.↵
- 311.↵
- 312.↵
- 313.↵
- 314.↵
- 315.↵
- 316.↵
- 317.↵
- 318.↵
- 319.↵
- 320.↵
- 321.↵
- 322.↵
- 323.↵
- 324.↵
- 325.↵
- 326.↵
- 327.↵
- 328.↵
- 329.↵
- 330.↵
- 331.↵
- 332.↵
- 333.↵
- 334.↵
- 335.↵
- 336.↵
- 337.↵
- 338.
- 339.↵
- 340.↵
- 341.↵
- 342.↵
- 343.↵
- 344.↵
- 345.↵
- 346.
- 347.↵
- 348.↵
- 349.↵
- 350.↵
- 351.↵
- 352.↵
- 353.↵
- 354.↵
- 355.
- 356.↵
- 357.↵
- 358.
- 359.↵
- 360.↵
- 361.↵
- 362.
- 363.↵
- 364.↵
- 365.↵
- 366.↵
- 367.
- 368.↵
- 369.↵
- 370.↵
- 371.↵
- 372.↵
- 373.↵
- 374.↵
- 375.↵
- 376.↵
- 377.↵
- 378.↵
- 379.↵
- 380.↵
- 381.↵
- 382.
- 383.↵
- 384.↵
- 385.
- 386.
- 387.
- 388.
- 389.↵
- 390.↵
- 391.↵
- 392.↵
- 393.
- 394.↵
- 395.↵
- 396.
- 397.↵
- 398.↵
- 399.
- 400.
- 401.↵
- 402.↵
- 403.
- 404.
- 405.↵
- 406.↵
- 407.
- 408.↵
- 409.↵
- 410.↵
- 411.↵
- 412.↵
- 413.↵
- 414.
- 415.↵
- 416.↵
- 417.↵
- 418.↵
- 419.↵
- 420.↵
- 421.↵
- 422.↵
- 423.↵
- 424.↵
- 425.↵
- 426.↵
- 427.↵
- 428.↵
- 429.↵
- 430.↵
- 431.↵
- 432.↵
- 433.↵
- 434.↵
- 435.↵
- 436.↵
- 437.↵
- 438.
- 439.
- 440.↵
- 441.↵
- 442.↵
- 443.↵
- 444.↵
- 445.↵
- 446.↵
- 447.↵
- 448.↵
- 449.↵
- 450.↵
- 451.↵
- 452.↵
- 453.↵
- 454.↵
- 455.↵
- 456.↵
- 457.↵
- 458.↵
- 459.↵
- 460.↵
- 461.
- 462.↵
- 463.↵
- 464.
- 465.
- 466.↵
- 467.↵
- 468.↵
- 469.↵
- 470.↵
- 471.↵
- 472.
- 473.↵
- 474.↵
- 475.↵
- 476.↵
- 477.↵
- 478.↵
- 479.↵
- 480.↵
- 481.↵
- 482.
- 483.↵
- 484.↵
- 485.↵




