

SUMMARY
Several lipids of the pathogen Mycobacterium tuberculosis are known to promote virulence at various stages of disease. However, the inability to probe these lipids during in vivo infection makes elucidation of their pathogenic mechanisms difficult. Using chemical extraction and reconstitution methods, we were able to define the lipid composition of the outer mycomembrane of Mycobacterium marinum prior to infection. Combining this approach with the synthesis of clickable, semi-synthetic lipids, we introduced a chemically tractable, biologically active variant of the virulence lipid phthiocerol dimycocerosate (PDIM) into the mycomembrane. We find that following infection of zebrafish larvae, PDIM spreads away from bacterial surfaces into the membranes of both macrophage and epithelial cells that it contacts. This spreading facilitates PDIM’s role in preventing bacterium-detrimental immune activation at the site of infection.

INTRODUCTION
Mycobacterium tuberculosis, the causative pathogen of the pulmonary disease Tuberculosis (TB), is estimated to have evolved within the confines of the human lung for millenia1. A result of this host-pathogen co-evolution is a choreographed response of innate and adaptive immune cells that ultimately results in the formation of granulomas, the hallmark pathological structures of TB, that permit bacterial replication and ultimately promote transmission2. A key strategy used by mycobacterium throughout infection is to avoid and manipulate host immune pathways so as to get the pathogen safe harbor in the typically bactericidal myeloid cell compartment3,4. Work in the zebrafish-Mycobacterium marinum model of TB showed that at the earliest stage of infection, mycobacteria use their outer membrane lipid phthiocerol dimycocerosate (PDIM) to avoid bactericidal monocytes that are recruited via toll-like receptor (TLR) signaling in response to bacterial pathogen associated molecular patterns 5. Instead the bacteria are phagocytosed by tissue resident macrophages from which they then transfer into permissive monocytes, mycobacteria’s preferred intracellular niche6. In the absence of PDIM, bactericidal monocytes were recruited rapidly, which phagocytosed and killed the mycobacteria 6. To address how PDIM mediates avoidance of TLR-signaled microbicidal monocytes, we developed an in vivo-compatible chemical approach to directly manipulate the non-covalently attached outer mycomembrane lipids of M. marinum. This approach enables loss of function studies on these lipids as well as the introduction of chemically modified lipids into the mycomembrane. Using bioorthogonal chemistry, we introduced a modified PDIM lipid that allowed us to visualize PDIM’s distribution during infection in zebrafish. We found that upon contact with host cells, PDIM spreads away from the bacterial surface into host membranes. This spreading was found to occur at macrophage and epithelial surfaces and was required for PDIM’s ability to prevent TLR-dependent microbicidal monocyte recruitment following infection.
RESULTS
While the inner envelope of the mycobacterial cell wall comprises the prototypical inner membrane and peptidoglycan layers found is most bacteria, the molecular composition of the outer envelope is unique to mycobacteria7. Arabinogalactan chains, covalently attached to peptidoglycan, are esterified with mycolic acids up to 90 carbons in length. This dense hydrophobic barrier constitutes the inner mycomembrane, while the outer mycomembrane consists of several lipids and glycolipids non-covalently associated with the inner mycomembrane8. Almost all of these outer mycomembrane lipids have known roles in virulence and the proteins responsible for their biosynthesis and localization are well known8. However, in addition to biosynthetic proteins, the stoichiometry of precursor metabolites also influences the mycomembrane composition. Therefore, genetic mutants in one biosynthesis pathway will often lead to alterations in non-target lipid levels due to changes in precursor metabolite abundance9. Furthermore, genetic approaches cannot be used to label non-gene encoded macromolecules in their native environments, hindering studies on their mechanism(s) of action. In order to address these shortcomings, our group developed metabolic labeling strategies where unnatural metabolic precursors to the macromolecule of interest are fed to growing bacteria10. Essential to this approach, is that the unnatural metabolite contains a bioorthogonal functional group. This bioorthogonal functional group will form covalent bonds with reaction partners under physiological conditions, allowing for the direct visualization of the macromolecule of interest in living bacteria11. An example of this approach is the labeling of trehalose containing lipids with azide functionalized trehalose12. This approach led to the labeling of trehalose monomycolate (TMM) and has since been elaborated on with various functional probes13,14. While functionalized trehalose incorporation is highly effective, there are several obstacles to overcome for successful metabolic labeling10. The metabolite needs to be actively taken up by the bacteria and the metabolic machinery needs to be promiscuous enough to incorporate the altered metabolite. Furthermore, labeling efficiency depends on the ability of the altered metabolite to out compete the native metabolite for incorporation into the target of interest. While metabolic labeling has been successful for trehalose containing lipids, its use for labeling other outer mycomembrane lipids has been challenging.
To target virulence lipids that are not amenable to metabolic labeling strategies, we wondered if we could bypass biosynthetic machinery and instead directly incorporate labelled macromolecules into the outer mycomembrane. We reasoned that such an approach would give us complete control over the chemical composition of the outer mycomembrane, allow us to probe specific lipids at near perfect efficiency, and would maintain the native composition of the remaining mycomembrane lipids. To this end, we noted that previous studies evaluating mycomembrane lipid extractions, found that mycobacteria can survive extraction with non-polar solvents such as petroleum ether15. Prior to elucidation of the biosynthetic pathway of trehalose mycolates, petroleum ether extraction was used as a loss of function approach to study the role of these lipids during infection in macrophages16. While these original studies reported that only trehalose mycolates were extracted with petroleum ether, more recent studies found evidence of other mycomembrane lipids being extracted, including PDIM, phenolic glycolipid, mycoside B, and triacylglycerols17. We decided to adapt this approach for studying mycomembrane virulence lipids in the zebrafish/M. marinum model system.
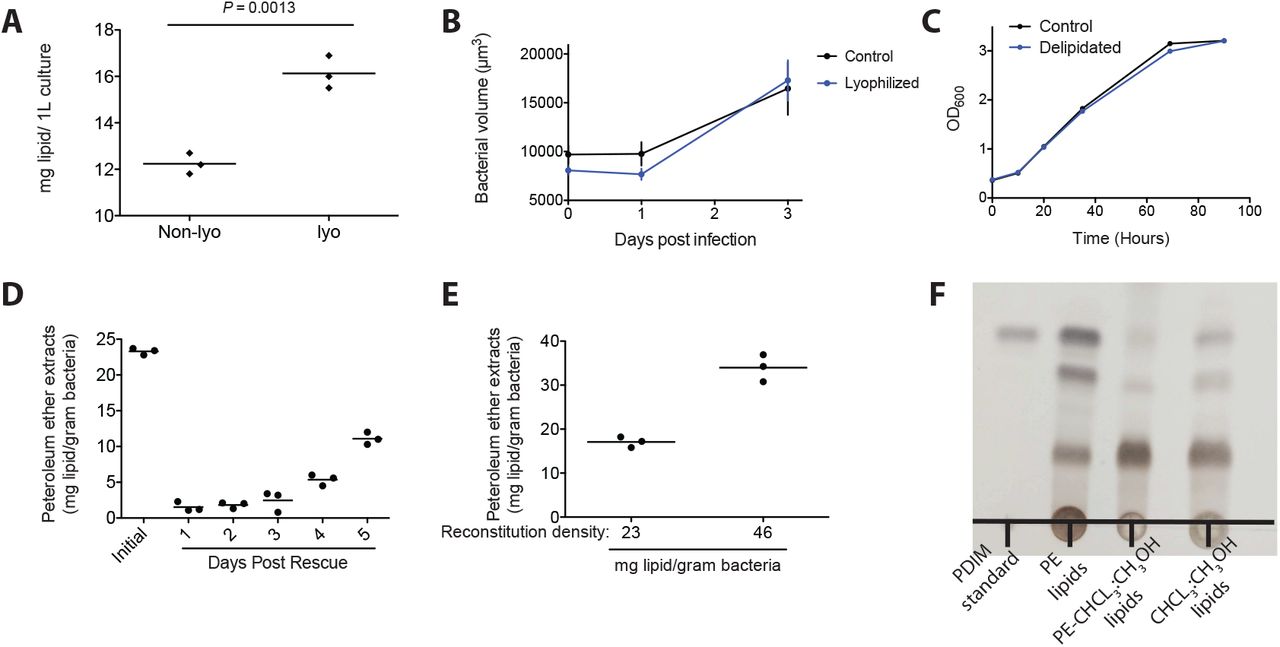
The original approaches extracted wet bacterial pellets and we wondered if complete removal of aqueous medium would increase the total lipid extracted. We found that lyophilization resulted in a 30% increase in the total amount of lipid removed (Figure S1A) and therefore increased the total lipid space that we could potentially interrogate. Importantly, following hindbrain ventricle infection, lyophilization did not alter mycobacteria’s ability to grow (Figure S1B). Similar to previous reports using M. tuberculosis and M. bovis, petroleum ether extraction did not affect M. marinum’s ability to grow in culture (Figure S1C). Another surprising finding from the petroleum ether extraction studies is that, even in the face of bacterial replication, the extracted non-covalently attached lipids do not repopulate the mycomembrane following the first few days in culture16. We found this to also be the case with M. marinum. On average we found that the first extraction yields around 23mg of lipid per gram of dry bacteria (Figure S1D). However, over the first three days following the initial extraction, lipid levels did not recover, and by five days post extraction they were only at 50% of the initial extractant (Figure S1D). Given the rate of pathogenesis in the zebrafish model, where early granulomas form around 3-5 days post infection, this approach is well suited to study potential loss of function of these mycomembrane lipids. Finally, previous studies also found that extracted lipids could be mixed with the bacteria in petroleum ether followed by drying to reconstitute these lipids into the mycomembrane16. To test the feasibility and efficiency of reconstitution, we immediately treated delipidated bacteria with lipids in petroleum ether and dried them down prior to recovering in axenic culture. Following recovery bacteria were again lyophilized and extracted. We found that we could add back lipids and that the efficiency of reconstitution was ~75% (Figure S1E). Knowing the amount of lipid extracted and the reconstitution efficiency we were ready to evaluate this approach in vivo.
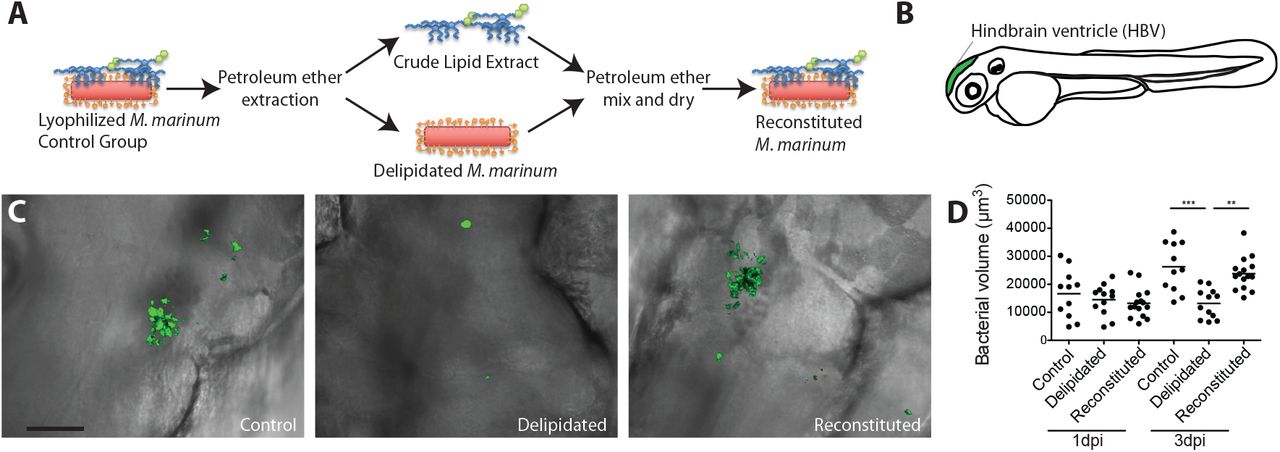
Following lyophilization bacteria were split into three groups. A control group that was not subjected to extraction prior to aqueous recovery, a delipidated group that was extracted with petroleum ether prior to recovery, and a reconstituted group that was extracted with petroleum ether and then mixed with the appropriate amount of extracted lipids (~133% of normal) prior to recovery (Figure 1A). Again, similar to previous reports showing an attenuation of delipidated bacterial growth in macrophages16, we saw that following hindbrain ventricle infection in zebrafish (Figure 1B) that delipidated bacteria were severely attenuated for growth and that this growth attenuation was rescued upon reconstitution (Figure 1C and D).

(A) Model of petroleum ether extraction and reconsitution. (B) Model of zebrafish larva showing the hindbrain ventricle (HBV) injection site in green. (C) Representative images of HBV infection with control, delipidated, or reconstituted bacteria at 3 days post infection, scale bar = 50μm. (D) Mean bacterial volume after HBV infection of wildtype fish with 100 control, delipidated, or reconstituted M. marinum. Representative of three separate experiments. Significance testing done using one-way analysis of variance (ANOVA), with Bonferroni’s post test for comparisons shown. **P < 0.01, ***P < 0.001.
We next asked if this approach could be used to study PDIM’s role in virulence. We first needed to determine if our extraction protocol was removing PDIM. To test this, we extracted bacteria with petroleum ether followed by a total lipid extraction in choloroform:methanol (1:1). Following petroleum ether extraction, we confirmed by thin-layer chromatography (TLC) that PDIM was present in the extract (Figure S1F), and very little, if any, PDIM was further extracted with choloroform:methanol but was present in extracts of bacteria only treated with choloroform:methanol (Figure S1F). These data suggest that PDIM is absent in our delipidated bacteria.
To date both a mutant in PDIM synthesis (Δmas) and a mutant in PDIM localization to the mycomembrane (ΔmmpL7) have been shown to be attenuated for growth in zebrafish5. PDIM mutants were shown to lead to a TLR-dependent recruitment of bactericidal monocytes. Furthermore, wildtype bacteria were attenuated for growth when co-infected with PDIM mutants, suggesting that the immune activation in the absence of PDIM was responsible for limiting bacterial growth5. These studies led us to hypothesize that the lipid profile present on the bacteria at the onset of infection was required for virulence. To test this, we performed a lipid swap experiment. Wildtype and ΔmmpL7 M. marinum were either untouched (control) or extracted and reconstituted with their native lipids or the lipids from the other strain (Figure 2A). TLC analysis showed that wildtype bacteria contained both dimycocerosic acid (DIM) containing lipids, PDIM and the closely related phthiodiolone dimycocerosate (PNDIM), and that ΔmmpL7 bacteria lacked both (Figure 2B). Following infection, we found that wildtype control and wildtype bacteria reconstituted with wildtype lipids grew normally, however wildtype bacteria reconstituted with ΔmmpL7 lipids were attenuated for growth (Figure 2C). And, as expected, ΔmmpL7 control bacteria and those reconstituted with ΔmmpL7 lipids were also attenuated for growth. However, ΔmmpL7 bacteria reconstituted with wildtype lipids grew at wildtype bacterial rates (Figure 2C), suggesting that the presence of DIMs at the onset of infection is required for virulence. Using an antisense morpholino to knockdown the TLR-adaptor MyD88, we also found that the dependence on TLRs to recruit monocytes to ΔmmpL7 bacteria was abolished with wildtype lipids (Figure 2D). We also found that the ability of these strains to transfer attenuation to wildtype bacteria was dependent on having a DIM-negative lipid profile (Figure 2E). Taken together these experiments highlight the strengths of the extraction and reconstitution approach. Not only does this approach recapitulate known phenotypes of PDIM genetic mutants but also shows that the PDIM present in the mycomembrane from the onset of infection is required for virulence.

(A) Model of lipid swap experiment. (B) TLC of PDIM and PNDIM standards and petroleum ether extracts from wildtype (WT) and ΔmmpL7 M. marinum. TLC was ran twice in 98:2 petroleum ether:ethyl acetate, and sprayed with an aquous 1.3M phosphoric acid 10% cupric sulfate solution followed by heating to develop. (C) Mean bacterial volume after HBV infection of wildtype fish with ~100 WT or ΔmmpL7 M. mairnum treated as follows: non-extracted control (black), extracted and reonstituted with WT lipids (green), or extracted and recontituted with Δmmp17 lipids (red). (D) Mean macrophage recruitment at 3 hours post infection in the HBV of wildtype or MyD88-deficient fish with ~100 ΔmmpL7 M. marinum treated as in (C). (E) Mean bacterial volume of red fluorescent WT M. marinum (infection inoculum 50-70) when co-infected with 50-70 green fluorescent WT or ΔmmpL7 M. marinum treated as in (C) at 3 days post infection in the HBV of wildtype fish. (C)-(E) representative of three separate experiments.
Given that our extraction methods resulted in removing most of the PDIM from the mycomembrane, we asked if we could replace endogenous PDIM with a bioorthogonal functionalized PDIM to further probe its mechanism of action. Given our findings that PDIM must be present in the mycomembrane from on the onset of infection to mediate virulence, we reasoned that labeling this pool of PDIM would allow us to observe virulence relevant behaviors. Additionally, our labelled PDIM needs to retain virulence in order to use it as a surrogate for PDIM localization studies. As mentioned above, two DIM variants are present in the outer mycomembrane. The more abundant PDIM, the diol backbone of which is phthiocerol and has a methyl ether in the 3-carbon position, and PNDIM where the diol backbone is phthiodiolone and has a ketone in the 3-carbon position. Previous studies on the role of these variants found that either PDIM or PNDIM was sufficient for virulence in mice18. Therefore, we hypothesized that replacing the methyl ether in PDIM would have little effect on its ability to promote virulence. Given the relatively unreactive nature of the rest of PDIM, we hypothesized that the established dealkylation of alkyl ethers with trimethylsilyl iodide (TMSI) would result in a more reactive hydroxyl or alkyl halide functional group in that position19. Monitoring this reaction by TLC, we found that eventually the vast majority of the product produced was the alkyl halide (Figure 3A). This was then easily replaced to form the alkyl azide by the addition of sodium azide, giving the product azido-DIM (Figure 3A and Supplementary Methods). To test the reactive potential of this bioorthogonal handle we reconstituted delipidated bacteria with native lipids with or without the addition of azido-DIM. Bacteria were then recovered and treated for single cell suspensions20, to prevent clumping and to maximize mycomembrane access to bioorthogonal probes. Bacteria with or without azido-DIM were then treated with the azide reactive cyclooctyne fluorophore conjugate DIBO-488 (Figure 3B). Only bacteria that had azido-DIM showed a roughly 100-fold increase in fluorescence (Figure 3C). Furthermore, only azido-DIM bacteria displayed a membrane associated fluorescent signal (Figure 3D and E).

(A) Synthesis of azido-DIM. (B) Model of delipidation and reconstitution of bacteria with or without azido-DIM followed by treatment with an azide-reactive cyclooctyne DIBO-488. (C) Flow cytometry analysis of control or azido-DIM reconstituted bacteria treated or untreated with DIBO-488. Image of (D) control or (E) azido-DIM recontituted bacteria treated with DIBO-488. (F) TLC of native, DIM depleted (DIM−), DIM depleted plus native DIMs (+DIMs), or DIM depleted plus azido-DIM (+Azido-DIM) lipids prior to reconstitution onto delipidated bacteria. TLC was ran once in 98:2 petroleum ether:ethyl acetate. (G) Mean bacterial volume after HBV infection of wildtype fish with ~100 M. marinum reconstituted with the lipids described in (F), representative of three separate experiments.
While the labeling approach was successful, in order to use azido-DIM as a model to probe PDIM’s role in virulence we first needed to show that it was biologically active. Given that we could completely remove DIMs, along with other outer mycomembrane lipids by extraction, we could also control the chemical composition of these extracts. Using preparative TLC, we were able to deplete DIMs from the native extracted lipids and found that they represented around 30% of the total lipids removed. With the DIM-negative extracts in hand we could then add back physiologically relevant amounts of native DIMs or azido-DIM prior to reconstitution (Figure 3F). Following infection in zebrafish we found that bacteria reconstituted with DIM-negative lipids were attenuated for growth compared to those reconstituted with native lipids, and by adding back native DIMs or azido-DIM to the DIM-negative extracts prior to reconstitution we could rescue this attenuation (Figure 3G). With this chemical approach in hand we now had fully pathogenic bacteria with nearly all of their mycomembrane DIMs containing a bioorthogonal functional handle to further probe PDIM’s virulence mechanisms.
To perform in vivo studies of PDIM distribution we used M. marinum expressing a blue fluorescent protein that were reconstituted with azido-DIM followed by labelling with DIBO-488. Following hindbrain ventricle infection of zebrafish, we found that immediately upon phagocytosis by macrophages azido-DIM appeared to spread away from bacterial membranes onto host membranes (Movie S1). To better visualize spreading of azido-DIM onto macrophage membranes, we used the transgenic zebrafish line Tg(mfap4:tdTomato) where macrophages express the red fluorescent protein tdTomato21. Spreading of azido-DIM was apparent in phagosome membranes immediately surrounding bacteria (Figure 4A, arrows) and was also dispersed throughout infected macrophages as early as 3 hours post infection (Figure 4A, arrow heads). The spreading appeared more ubiquitous as infection progressed, as azido-DIM appeared to incorporate across entire macrophage membranes by 3 days post infection (Figure 4B). Similar spreading dynamics were seen when azido-DIM was conjugated to DIBO-647 (Figure S2), suggesting that the DIM lipid was responsible for the localization and not the fluorescent probe. To better quantify this spread we imaged the entire infected hindbrain and calculated the percent of azido-DIM no longer colocalizing with bacteria (Figure S3). As infection progressed, the more azido-DIM spread onto host membranes (Figure 4C). Spreading was not the result of an active bacterial process as heat-killed bacteria also dispersed azido-DIM (Figure 4D). We next wanted to determine whether this spreading represented a true biological phenomenon or if it was an artifact of our reconstitution approach. To address this we used an established nonspecific glycolipid labeling approach where bacteria are treated with periodate and then reacted with a fluorescent hydroxylamine22. Both control and reconstituted bacteria were labelled in this manner prior to infection. We found that spreading of this non-specific label was equal between control and reconstituted bacteria (Figure 4E), suggesting that the reconstitution approach did not influence global spreading dynamics. We next wondered if PDIM spreading was restricted to macrophage cells. To test this we depleted macrophages from zebrafish larva using clodronate loaded liposomes23. To our surprise we found that azido-DIM spreading still occurred in the absence of macrophages (Figure 4F). To better visualize spreading onto non-macrophage host cells, we used the transgenic zebrafish line Tg(flk1:mcherry), whose endothelium express the red fluorescent protein mCherry24. Following intravenous infection, we found bacteria in contact with endothelium tissue with azido-DIM having spread away from the bacteria onto the endothelium (Figure 4G, arrows). Similar azido-DIM spreading dynamics were observed when we infected THP-1 macrophages and A549 epithelial cells in vitro (Figure S4). Taken together these data suggest that PDIM has a high affinity for host membranes and that spreading into host membranes may underscore its role in virulence.

Images of M. marinum expressing a cytosolic cyan flourescent protein reconstituted with DIBO-488 labelled azido-DIM at (A) 3 hours post infection and (B) 3 days post infection of ~100 M. marinum in the HBV of transgenic fish whose macrophages express the red fluorescent protein tdTomato. Scale bar = 10μm. Arrows, azido-DIM spreading in vicinity of phagosome, arrowheads, azido-DIM spread throughout macrophage. (C) Percent azido-DIM signal no longer colocalized with bacteria following HBV infection of wildtype fish with ~100 M. marinum. (D) Percent azido-DIM signal no longer colocalized with bacteria following HBV infection of wildtype fish with ~100 live or heat-killed M. marinum. (E) Percent fluorescent signal no longer colocalized with bacteria following HBV infection of wildtype fish with ~100 control or reconstituted M. marinum labelled with peri-odate-hydrazide chemistry. (F) Percent azido-DIM signal no longer colocalized with bacteria following HBV infection of lipo-PBS or lipo-clodronate treated fish with ~100 M. marinum. (G) Image of M. marinum expresisng a cytosolic cyan fluorescent protein recontituted with DIBO-488 labelled azido-DIM at 1 day post intravenous infection of transgenic fish whose endothelium express the red fluorescent protein mCherry. Scale bar = 5μm. Arrows, azido-DIM spread onto endothelium. (C) - (F) representative of three separate experiments.
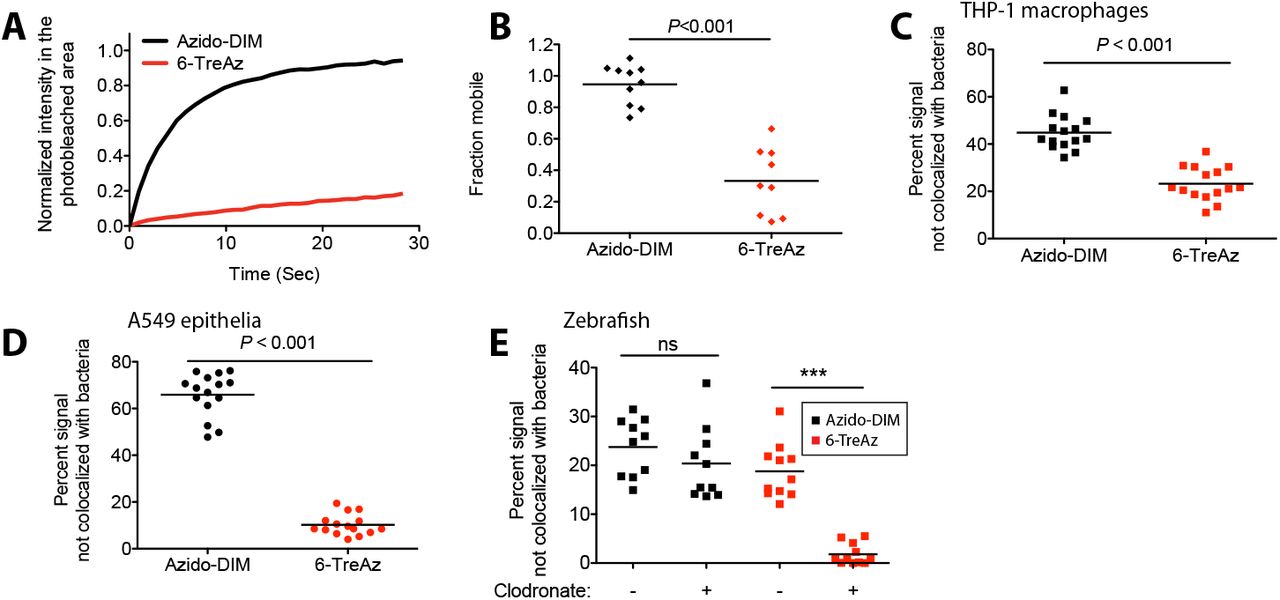
Next we wondered if PDIM’s biophysical properties on the bacterial surface were at all related to its propensity to spread into host membranes. Previous work in our lab has used metabolic labeling of trehalose monomycolate (TMM) to monitor TMM’s fluidity as a surrogate for mycomembrane dynamics. Using fluorescence recovery after photobleaching (FRAP), it was found that the fluidity of TMM correlated to its mycolic acid chain length, with short chain mycolic acids lending to a more fluid TMM compared to long chain containing variants13. Using a similar approach, we metabolically labelled M. marinum with 6-azido-trehalose (6-TreAz) 12 and used DIBO-488 to visualize TMM. Similar to previous findings in Mycobacterium smegmatis, we found very little recovery following photobleaching of 6-TreAz labeled M. marinum, with only around 40% of the signal being mobile (Figure 5A and B). In contrast, azido-DIM recovery was very efficient, with a half-life of 3 seconds and around 95% of the signal being mobile (Figure 5A and B). These results demonstrate that different lipid species of the outer mycomembrane have different biophysical traits. To determine if TMM also spreads onto host membranes we first compared spreading between PDIM and TMM in cultured cells. We found that both azido-DIM and 6-TreAz labels appeared to spread away from bacteria during macrophage infection, with the 6-TreAz label spreading to a lesser extent (Figure 5C). In sharp contrast to azido-DIM, 6-TreAz failed to spread onto A549 epithelial cells (Figure 5D). Finally, following zebrafish infection we found that 6-TreAz spread to a similar extent as azido-DIM in the presence of macrophages, however when macrophages were depleted with clodronate, 6-TreAz no longer spread (Figure 5E). These data demonstrate lipid specific differences in mycomembrane fluidity and that these differences in fluidity correlate with the propensity of these lipids to spread into host tissues. These data also highlight the fact that different mycomembrane lipids occupy unique host cell niches during infection.

(A) Fluorescent recovery curves after photo-bleaching of 6-TreAz and azido-DIM labelled M. marinum, lines represent the average signal from n = 10 cells. (B) Fraction mobile following fitting of FRAP curves to data generated in (A). Percent fluorescent signal no longer colo-calized with bacteria following infection of (C) THP-1 macrophages (D) A549 epithelial cells or (E) lipo-PBS or lipo-clodronate treated fish with 6-TreAz or azido-DIM labelled M. marinum. Significance testing done using (C)-(D) Student’s t-test and (E) one-way ANOVA with Bonferonni’s post test for the comparisons shown. ns = not significant, ***P < 0.001. (A)-(E) representative of three separate experiments.
Our observation that PDIM is more fluid than TMM and only PDIM can spread to epithelial cell membranes led us to hypothesize that PDIM’s increased fluidity was responsible for its ability to spread into epithelial cell membranes. To test this, we used FRAP to identify conditions that decreased PDIM fluidity. Consistent with our in vivo data showing that azido-DIM still spread when bacteria were heat-killed (Figure 4D), heat-killed bacteria had only a partial reduction in azido-DIM fluidity compared to live bacteria (Figure 6A and B). Turning to chemical fixation methods, we found that 4% paraformaldehyde (PFA) fixation also did not reduce azido-DIM fluidity (Figure 6A and B). However, following fixation with a combination of 4% PFA and 1% glutaraldehyde (GA), a condition that has recently been shown to more efficiently fix membrane associated proteins25, resulted in an almost complete loss of recovery and an 80% reduction in the mobile phase (Figure 6A and B). We could now ask if loss of azido-DIM fluidity would prevent it from spreading on epithelial cells. We found that PFA+GA treatment of bacteria dramatically decreased azido-DIM spread following infection of A549 epithelial cells (Figure 6C). Following infection of macrophage-depleted zebrafish, we found that fixed azido-DIM was no longer able to spread (Figure 6D). These results linked azido-DIM’s fluidity to its ability to spread on epithelial cells.

(A) Fluorescent recovery curves after photobleaching of live, heat-killed, 4% paraformaldehyde (PFA) fixed, or 4% paraformaldehyde plus 1% glutaraldehyde (PFA+GA) fixed azido-DIM labelled M. marinum, lines represent the average signal from n = 14-16 cells. (B) Fraction mobile following fitting of FRAP curves to data generated in (A). Percent azido-DIM signal no longer colocalized with bacteria following infection of (C) A549 epithelial cells or (D) lipo-clodronate treated fish with live or PFA+GA fixed azido-DIM labelled M. marinum. Significance testing done using Student’s t-test. (E) Mean macrophage recruitment at 3 hours post infection in the HBV of wildtype or MyD88-deficient fish with ~100 live, heat-killed (HK), PFA fixed, or PFA+GA fixed M. marinum. (F) Mean bacterial volume of red fluorescent WT M. marinum (infection inoculum 50-70) when co-infected with 50-70 green fluorescent live, heat-killed, PFA fixed, or PFA+GA fixed M. marinum. (A)-(F) representative of three separate experiments.
We next wanted to test whether PDIM spreading on epithelial cells was required to avoid TLR-dependent immune activation. Our prior work had suggested the following sequence of infection events for wildtype and PDIM-deficient bacteria: all mycobacteria recruit tissue resident macrophages which phagocytose them, a recruitment response that is induced in response to all bacteria through a ubiquitous heat-stable determinant6. Wildtype bacteria reprogram these microbicidal cells so as to recruit permissive monocytes and escape into them. Importantly, mycobacteria needed to be alive to promote this response, as heat-killed wildtype mycobacteria only recruit tissue resident macrophages6. In the context of PDIM-deficient bacteria, TLR-dependent recruitment of monocytes occurs independent of bacterial viability. Furthermore, unlike the permissive monocyte response to wildtype bacteria, TLR-dependent monocytes responded in the absence of tissue resident macrophages6. In light of our new data on PDIM spreading, these observations led us to wonder if PDIM spreading onto epithelial cells immediately following infection was required to prevent the early recruitment of bactericidal monocytes. To test this hypothesis, we evaluated macrophage recruitment towards bacteria with or without the ability to spread their PDIM.
Following infection, we found that bacteria killed by heat or by PFA, conditions that retained PDIMs fluidity and membrane spreading, both resulted in a decrease in macrophage recruitment compared to live bacteria (Figure 6E), consistent with their recruiting only tissue resident macrophages6. However, PFA+GA-treated bacteria, where PDIM is no longer able to spread, behaved like PDIM-deficient bacteria - they recruited similar numbers of macrophages as live bacteria even though they were also not viable (Figure 6E). This led us to hypothesize that now because PDIM is no longer spreading onto epithelial cells, that TLR-dependent responses were driving macrophage recruitment. We found this to be the case. In the absence of the TLR-adaptor MyD88, PFA+GA treated bacteria recruited lower numbers of macrophages comparable to the other non-viable bacteria (Figure 6E). These results suggested that PDIM spreading onto host cell membranes was preventing the recruitment of monocytes through TLR signaling. To further test this model, we used our prior knowledge that TLR-dependent immune responses to PDIM-deficient bacteria are able to attenuate wildtype M. marinum growth5. If PDIM-spreading is required to avoid TLR-dependent immune responses, we hypothesized that PDIM-fluid conditions should not attenuate wildtype M. marinum while PDIM-fixed conditions should attenuate wildtype M. marinum. Following co-infection, we found that only PFA+GA treated bacteria decreased wildtype M. marinum growth (Figure 6F). Taken together these results suggest that PDIM’s ability to spread into host cells, in particular epithelial cells, is required to avoid TLR-dependent immune activation and subsequent growth attenuation.
DISCUSSION
By combining bioorthogonal chemistry with a modified chemical extraction and reconstitution method we have been able to visualize PDIM’s distribution during infection in zebrafish larvae. While PDIM is a predominant lipid of the outer mycomembrane, the idea that PDIM is restricted to this niche during infection is inaccurate. In fact, PDIM’s ability to leave the outer mycomembrane and incorporate into host membranes is required for its role in promoting infection. Unlabeled PDIM was recently shown to incorporate into host membranes following in vitro infection of macrophages26. Here PDIM was found to adopt a conical shape that altered the lipid phase of model membranes and was found to affect the phagocytosis of foreign objects by macrophages. Our studies confirm these findings that PDIM does in fact incorporate into host cell membranes during in vivo infection. Furthermore, this incorporation into epithelial cells prevents canonical bactericidal immune responses downstream of TLR signaling. Exactly how PDIM’s incorporation into host membranes disrupts TLR signaling is still not clear. TLRs have been shown to aggregate into lipid rafts to facilitate downstream signaling events27. Whether or not PDIM disrupts these lipid rafts to prevent TLR signaling is yet to be determined.
Additional studies have found that PDIM promotes phagosome rupture and cytosolic escape28. In addition to spreading of PDIM on epithelial surfaces, we found robust spreading of PDIM on macrophage membranes. Therefore, PDIM’s proclivity for host membranes may underscore its multiple roles in pathogenesis.
These studies on PDIM are not the first reports of mycobacterial lipids having a propensity to interact with host membranes. A non-specific glycolipid labeling approach first documented the spread of mycobacterial lipids in macrophages22, however the localization of specific lipid species and the impact they might have on pathogenesis was unclear. Our approach, that allows us to evaluate specific lipids during infection in vivo, has already demonstrated unique distributions of two mycobacterial virulence lipids across host membranes. Future detailed studies of these and other mycomembrane lipids in the genetically tractable and optically transparent zebrafish/M. marinum model of TB will allow us to better understand the role these lipids play during mycobacterial pathogenesis.
Author contributions
C.J.C., and C.R.B. conceived the project. C.J.C., S.M.B. and J.A.B. carried out experiments and interpreted data. C.J.C. and C.R.B. wrote the manuscript with input from all authors. C.R.B. provided supervision.
List of Supplementary Materials
Materials and Methods
Figure S1-S4
Supplementary Methods
Materials and Methods
Procedures and materials for synthetic chemistry
All reactions were performed in dry standard glassware fitted with rubber septa under an inert atmosphere of nitrogen. Preparative thin-layer chromatography (TLC) was performed with Millipore’s 1mm and 0.2mm silica gel 60 pre-coated glass plates. Analytical TLC was used for reaction monitoring and product detection using pre-coated glass plates covered with 0.20 mm silica gel with fluorescent indicator; visualized by UV light and 10% CuSO4 in 1.3M phosphoric acid in water. Reagents were purchased in reagent grade from commercial suppliers and used as received, unless otherwise described. Anhydrous dichloromethane (DCM) was prepared by passing the solvent through an activated alumina column.
Chemical Analysis Instrumentation
Proton (1H NMR) and proton-decoupled carbon-13 (13C {1H} NMR) nuclear magnetic resonance spectra were recorded on an Inova-500 spectrometer at 25 °C, are reported in parts per million downfield from tetramethylsilane, and are referenced to the residual protium (CDCl3: 7.26 [CHCl3]) and carbon (CDCl3: 77.16) resonances of the NMR solvent. Data are represented as follows: chemical shift, multiplicity (br = broad, s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, sept = septet, m = multiplet), coupling constants in Hertz (Hz), integration. Mass spectra were obtained on a Bruker Microflex MALDI-TOF by mixing 0.5μl of 1mg/ml sample in chloroform with 0.5μl of 10mg/ml 2,5-dihydroxybenzoic acid before spotting onto a 96-well MALDI plate.
Isolation of PDIM
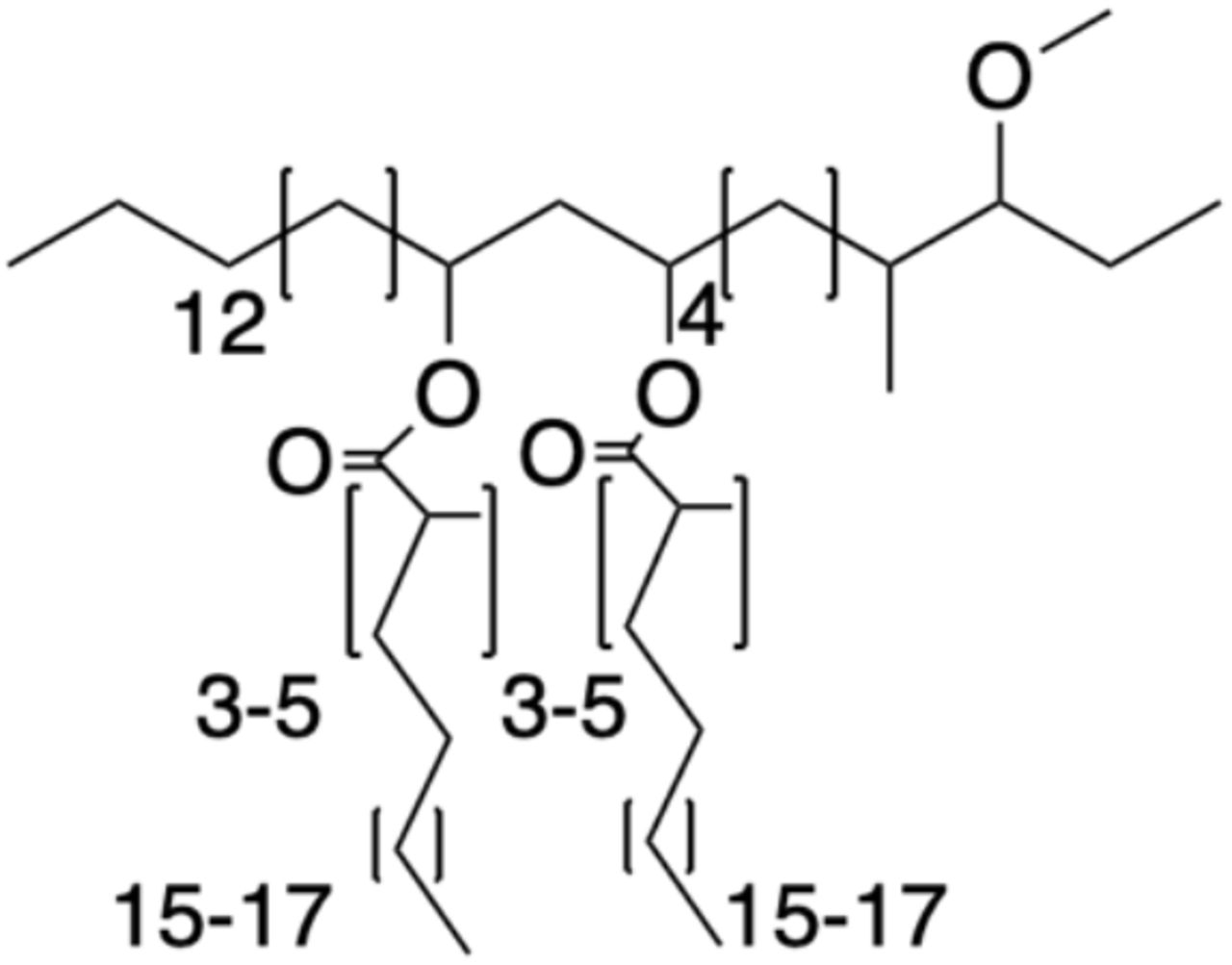
Wildtype M. marinum was grown in GAS medium without Tween-80 to an OD600 of 1.2. Bacteria were pelleted, frozen and lyophilized on a Labconoco FreezeZone 4.5 plus. Bacterial lipids were then extracted by stirring lyophilized pellet in petroleum ether for 1hr at room temp. Bacteria were allowed to settle, and solvent was collected. Remaining bacteria were re-extracted 3-5 times. The solvent extract was then passed through a 0.2μm PTFE filter, and crude lipids were concentrated under reduced pressure. Crude lipid extracts were separated by preparative TLC in the solvent system 98:2 petroleum ether:ethyl acetate. The band correlating to PDIM at an Rf = 0.4 was isolated. Preparative TLC was repeated twice more to further purify PDIM. Average PDIM isolated from M. marinum was 60mg per 10 liters of culture. 1H NMR (500 MHz, CDCl3) δ 4.93-4.87 (m, 2H), 3.32 (s, 3H), 2.87-2.83 (m, 1H), 2.57-2.51 (m, 2H), 1.90-0.81 (m, 166H). 13C NMR (126 MHz, CDCl3) δ 176.69, 176.63, 86.78, 86.73, 77.41, 77.37, 77.16, 76.90, 70.72, 70.67, 57.51, 45.56, 45.51, 41.41, 41.33, 38.56, 37.85, 37.73, 37.09, 36.77, 34.91, 34.18, 34.07, 32.83, 32.08, 30.24, 29.96, 29.87, 29.82, 29.75, 29.62, 29.53, 28.34, 27.64, 27.36, 27.25, 27.16, 27.12, 25.74, 25.35, 22.85, 22.74, 22.43, 20.81, 20.57, 20.52, 20.37, 20.27, 18.67, 18.58, 18.53, 14.80, 14.75, 14.28, 14.22, 10.26, 1.16. MALDI-TOF for: C82H162O5 Calc’d [M+Na+]=1250.23; found 1250.41. C84H166O5 Calc’d [M+Na+]=1278.26, found 1278.40. C86H170O5 Calc’d [M+Na+]=1306.29, found 1306.55.
Synthesis of azido-DIM
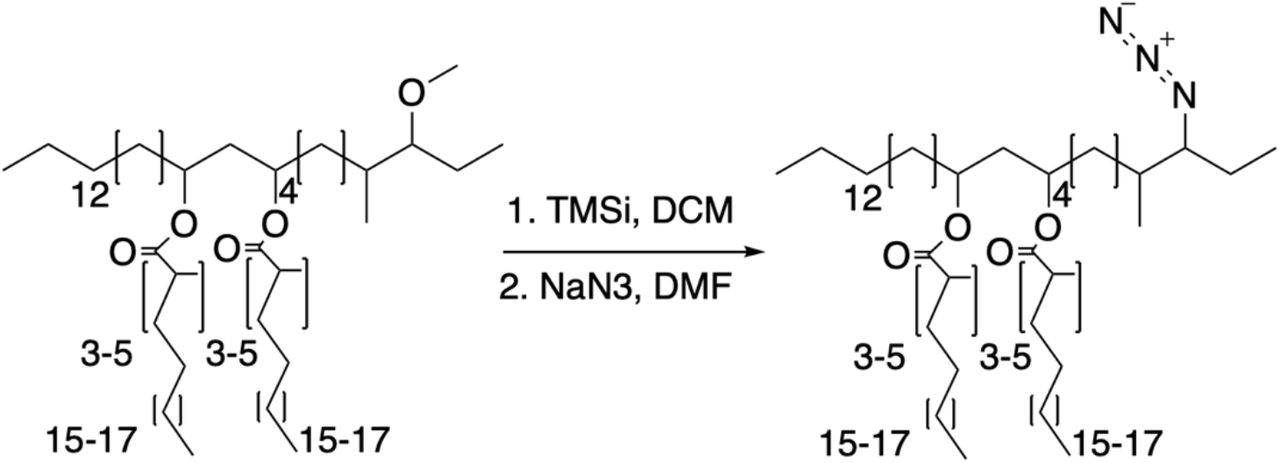
To PDIM (75.6mg, 60.2μmol, 1.0 equiv.) was added 0.3ml of DCM, the mixture was stirred and iodotrimethylsilane (TMSi, 258μl, 1.8mmol, 30.0 equiv.) was added and allowed to react for 12hrs at room temperature. The reaction was concentrated under reduced pressure. NaN3 (39.1mg, 602μmol, 10.0 equiv.) was added followed by 0.3ml of anhydrous dimethylformamide (DMF) and stirred for 12hrs at room temperature. The solvent was removed under reduced pressure and the product was purified using preparative TLC in 98:2 petroleum ether:ethyl acetate Rf=0.5 as a white wax (37.7mg, 29.8μmol, 50%). 1H NMR (500 MHz, CDCl3) δ 5.06-4.86 (m, 2H), 3.30-3.21 (m, 1H), 2.58-2.50 (m, 2H) 2.1-0.81 (m, 163H). 13C NMR (126 MHz, CDCl3) δ 176.73, 176.63, 77.41, 77.36, 77.16, 76.91, 71.08, 70.74, 59.86, 45.56, 41.96, 41.45, 41.35, 39.27, 37.84, 37.73, 37.24, 37.08, 37.01, 35.28, 34.82, 34.18, 32.08, 30.32, 30.24, 30.22, 30.04, 29.96, 29.87, 29.82, 29.75, 29.72, 29.67, 29.62, 29.52, 29.39, 28.40, 28.34, 27.72, 27.36, 27.25, 27.18, 27.16, 26.20, 25.35, 25.27, 25.23, 22.85, 21.28, 21.13, 20.35, 20.32, 20.27, 18.73, 18.66, 18.58, 18.53, 14.56, 14.29, 13.87. MALDI-TOF for: C81H159N3O4 Calc’d [M+Na+]=1261.22; found 1260.92. C83H163N3O4 Calc’d [M+Na+]=1289.25, found 1289.02. C85H167N3O4 Calc’d [M+NH4+]=1312.33, found 1312.37.
Zebrafish Husbandry and Infections
Wild-type AB (Zebrafish International Resource Center), Tg(mfap4:tdTomato), and Tg(flk1:mCherry) lines were maintained in buffered reverse osmotic water systems. Fish were fed twice daily a combination of dry feed and brine shrimp and were exposed to a 14 hr light, 10 hr dark cycle to maintain proper circadian conditions. Larvae (of undetermined sex given the early developmental stages used) were infected at 48 hr post-fertilization (hpf) via caudal vein (CV) or hindbrain ventricle (HBV) injection using single-cell suspensions of known titer. Number of animals to be used for each experiment was guided by pilot experiments or by past results with other bacterial mutants and/or zebrafish. On average 15 to 40 larvae per experimental condition were required to reach statistical significance and each experiment was repeated at least twice. Larvae were randomly allotted to the different experimental conditions. The zebrafish husbandry briefly described above and all experiments performed on them were in compliance with the U.S. National Institutes of Health guidelines and approved by the Stanford Institutional Animal Care and Use Committee.
Bacterial Strains and Methods
M. marinum strain M (ATCC BAA-535) and ΔmmpL7 mutants expressing either TdTomato, Wasabi, or EBFP2 under the control of the msp12 promoter were grown under hygromycin (Mediatech) selection in 7H9 Middlebrook’s medium (Difco) supplemented with OADC (Fisher), glycerol, and 0.05% Tween-80 (Sigma). Where noted bacteria were also grown in glycerol-alanine-salts (GAS) medium, recipe (%weight/volume) in 18mM sodium hydroxide in Milli-Q water pH 6.6 +/− 0.05% Tween-80: 0.03% BactoCasitone (BD Science), 0.005% ferric ammonium citrate (Sigma), 0.4% potassium phosphate, dibasic anhydrous (VWR), 0.2% citric acid, anhydrous (VWR), 0.1% L-alanine (Sigma), 0.12% magnesium chloride, heptahydride (VWR), 0.06% potassium sulfate (VWR), 0.2% ammonium chloride (VWR), and 1% glycerol. To prepare heat-killed M. marinum, bacteria were incubated at 80°C for 20 min. To prepare fixed bacteria, bacteria were incubated in described concentrations of glutaraldehyde (Sigma) and/or paraformaldehyde (Sigma) for 1hr at 23°C, followed by 3 washes with PBS prior to experimental use.
Cell Lines and infections
Cells were grown in T75 flasks (Thermo Fisher) and maintained at 37 °C and 5% CO2. THP-1 were grown in RPMI supplemented with 10% fetal bovine serum (FBS). A549 were grown in DMEM supplemented with 10% FBS. Two days prior to infection, THP-1s were plated on 8-well Nunc Lab-Tek II chamber slides at density of 150,000 cells per well with 100nM phorbol 12-myristate 13-acetate in growth media and incubated at 37°C and 5% CO2 for 24hrs. One day prior to infection, the media on THP-1s was replaced with fresh growth media and the cells were incubated at 33°C and 5% CO2 for 24hrs, cells were then infected with M. marinum at a multiplicity of infection (MOI) of 2 in growth media. Infection was allowed to progress for 6hrs prior to washing twice with PBS and replacing with growth media. THP-1s were then incubated for 24hrs at 33°C and 5% CO2 prior to experimental end point. One day prior to infection, A549s were plated on 8-well chamber Nunc Lab-Tek II chamber slides at a density of 50,000 cells per well in growth media and incubated at 37°C and 5% CO2. Day of infection cells were moved to 33°C and 5% CO2 for 3 hours, and then were infected with M. marinum at a MOI of 5 and incubated at 33°C and 5% CO2 for 24hrs. Infected THP-1s and A549s were then imaged as described below.
Extraction and reconstitution of M. marinum
Extraction
1 liter of M. marinum were grown in GAS medium plus Tween-80 to an OD600 of 1.2. Bacteria were pelleted in a glass 50mL conical tubes (Fisher) of known weight, frozen and lyophilized. The dry bacteria in 50mL conical tubes were then weighed and the dry bacterial weight was calculated. 25mL of petroleum ether were then added to the bacteria, and the conical tube was capped with a PTFE lined lid (Sigma) and the suspension was vortexed for 3min. An additional 25mL of petroleum ether was added and the sample was centrifuged for 3min at 1000xg at 4°C. The extract was then saved or discarded depending on the downstream experimental applications and the bacteria were extracted once more as above. For total lipid extractions, bacteria were treated with 1:1 chloroform:methanol for 12hrs at 60°C. Extracts were filtered, back extracted with water to remove water soluble contaminants, dried under reduced pressure and used for downstream experiments.
Reconstitution
Prior to extractions the following calculation was used to determine the amount of lipids to add back to each pellet.

Where 23mg of lipid per gram of dry bacteria is the experimentally determined (Figure S1D) average amount of lipid removed during petroleum ether extraction of bacteria grown in these conditions and the 0.75 reconstitution efficiency was also experimentally determined (Figure S1E). If reconstituting with DIM variants, 30% of the above weight consists of DIMs and the remainder will consist of DIM-depleted petroleum ether extract. Following two rounds of petroleum ether extraction as detailed above, bacteria were immediately mixed with pre-determined lipid mixtures (or no lipid at all for delipidated bacteria) in petroleum ether followed by drying under reduced pressure. Dried bacteria were then rescued into 7H9 complete media without Tween-80 (prep media) and subjected to single cell preparation protocol.
M. marinum single cell preparation
Bacteria were washed once with 15mL of prep media followed by resuspension in 500μl of prep media. Bacteria were then passed through a 27-gauge needle 10 times, followed by the addition of 1ml of prep media and centrifugation at 100xg for 3min. 1ml of supernatants were saved. This process was repeated 3-5 times. Collected supernatants were then passed through a 5.0μm Acrodisc Versapor membrane syringe filter (VWR). The filtrate was then pelleted at 16,000xg for 2min, pellets were resuspended in prep media to a concentration of around 1×10^8-9 bacteria per ml, aliquoted and stored at −20C for future use, or immediately subjected to copper-free click chemistry reactions.
Periodate-hydroxylamine staining of mycobacterial surfaces
Surface-exposed terminal oxidizable carbohydrates were labeled with hydroxylamine following periodate oxidation. Lyophilized control or reconstituted M. marinum were washed twice with PBS and resuspended in 0.1 M sodium acetate, pH 5.5 containing 1 mM sodium periodate (Sigma). Following a 20-min incubation at 4°C with gentle rotation, 0.1 mM glycerol was added to stop the reaction. Cultures were washed three times with PBS and then subjected to single cell preparation. Following single cell preparation, pellets were transferred to a 96-well plate and then incubated with PBS containing 1 mM Alexa-647 hydroxylamine (ThermoFisher). Following a 2-h incubation at room temperature, the cultures were washed 5 times with PBS and twice with prep media.
Copper-free click chemistry of M. marinum
Following reconstitution or metabolic labeling, bacteria were treated for single cell preparation. Bacteria were then transferred to 96-well v-bottom dishes and were washed twice with PBS using centrifugation at 3000xg for 3min between washing. Bacteria were then stained with either 5μM DIBO-488 (ThermoFisher), or 30μM DIBO-647 (ThermoFisher) in 200μl PBS for 90min at room temperature. Bacteria were then washed 5 times in PBS followed by 2 washes in prep media. Bacteria were then aliquoted and stored at −20C for future use. Staining efficiency was evaluated by flow cytometry on a BD-Accuri C6 Plus and analysis was performed using the FlowJo software package. Staining was also evaluated by microscopy with a 60x oil-immersion Plan Apo 1.4 NA objective on the Nikon A1R confocal microscope.
MyD88 morpholino and liposome injections
To generate MyD88 knockdown zebrafish larvae, the MyD88 morpholino 5’GTTAAACACTGACCCTGTGGATCAT3’ previously described was injected into the 1-4 cell stage of the developing embryo. Lipo-PBS and lipo-clodronate (http://clodronateliposomes.org) were diluted 1:10 in PBS and injected into 2-dpf-old larvae in ~10 nl via the caudal vein.
Confocal microscopy and image-based quantification of infection
Larvae were embedded in 1.5% low melting point agarose (ThermoFisher) and a series of z stack images with a 2μm step size was generated through the infected HBV. For infected THP-1 and A549 cells, a series of z stack images with a 1μm step size was generated. Images were captured using the galvo scanner (laser scanner) of the Nikon A1R confocal microscope with a 20x Plan Apo 0.75 NA objective. Higher resolution images were generated using a 40x water-immersion Apo 1.15 NA objective. Bacterial burdens were determined by using the 3D surface-rendering feature of Imaris (Bitplane Scientific Software). Spread lipid images were generated by subtracting the bacterial surface from the lipid channel. 3D surface-rendering was then done on both the total lipid and spread lipid images to generate a percent spread value.
Macrophage hindbrain recruitment assay
2 dpf zebrafish were infected in the HBV with M. marinum at the dose reported in the figure legends. At 3 hours post infection, the number of myeloid cells in the HBV was quantified using differential interference contrast microscopy using a 20x Plan Fluor 0.75 NA objective on a Nikon’s Ti eclipse inverted microscope.
Fluorescence recovery after photobleaching (FRAP) experiments
FRAP experiments were performed using the galvo scanner (laser scanner) of the Nikon A1R confocal microscope with a 60x oil-immersion Plan Apo 1.4 NA objective. Photobleaching was performed with the 405 nm laser for 200ms on a region of interest (ROI) encompassing ~1μm from one pole of a single bacteria. A series of images was taken every second over the course of 31 seconds, one prior to bleaching. Labeled cells were mounted on a slide and coverslip in 0.75% low melting agarose. Nikon Elements software was used to analyze the FRAP data to extract the fluorescence recovery kinetics. Briefly, the first image before photobleaching was used to generate an ROI for the entire cell and a second ROI was generated in the photobleached area. Total fluorescence intensities in both the whole cell area and the bleached area were extracted and normalized to correct for photobleaching of the dyes due to acquisition. The normalized fluorescence intensities of the bleached area were then fitted to a non-linear regression with a one-phase association, with the plateau values from each sample plotted to represent the mobile fraction.


Supplementary Figures

(A) Mean mg of lipid extracted from a wet or lyophilized bacterial pellet isolated from a 1L culture of M. marinum grown to an OD600 of ~1-1.2. Significance testing done with Student’s t-test. (B) Mean bacterial volume after HBV infection of wildtype fish with 100 control or lyophilized M. marinum. (C) Mean OD600 values of control or lyophilized M. marinum recovered in complete growth medium. (D) Mean mg of lipid extracted per gram of dry bacteria, showing the initial extracted lipid and the lipid extracted following recovery of the extracted bacteria into complete growth medium over time. (E) Mean mg of lipid extracted following reconstitution with designated lipid densities. (F) Thin-layer chromatography (TLC) of lipid extracts. PDIM standard, petroleum ether extract (PE lipids), chloroform:methanol extract of the bacterial pellet previously extracted with petroleum ether (PE-CHCL3:CH3OH lipids), and chloroform:methanol extract of a fresh bacterial pellet (CHCL3:CH3OH lipids). TLC was ran twice in 98:2 petroleum ether:ethyl acetate, and sprayed with an aquous 1.3M phosphoric acid 10% cupric sulfate solution followed by heating to develop.

Images of M. marinum expresisng a cytosolic wasabi flourescent protein reconstituted with DIBO-647 labelled azido-DIM at (A) 3 hours post infection and (B) 3 days post infection of ~100 M. marinum in the HBV of transgenic fish whose macrophages express the red flourescent protein tdTomato. Scale bar = 10μm

A surface rendering of the cytosolic expressing protein of the flourescent bacteria is subtracted from the total azido-DIM signal to give the spread azido-DIM signal. The total volume of the spread azido-DIM signal is then divided by the volume of the total azido-DIM signal to calculate percent spread.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Images of M. marinum expresisng a blue flourescent protein reconstituted with DIBO-488 labelled azido-DIM 24 hours post infection of (A) A549 epithelial and (B) THP-1 macrophages at an MOI of 2. Scale bar = 10μm.
Movie S1: Azido-DIM dynamics. Real-time video of M. marinum expressing blue fluorescent protein reconstituted with DIBO-488 labelled azido-DIM.
Acknowledgements
We thank Karen Dobos (Colorado State University) for help in designing the PDIM purification protocol. We thank David Tobin (Duke University) for sending us zebrafish lines. This work was supported in part by National Institutes of Health grant (AI51622) to C.R.B. C.J.C. was supported by a Damon Runyon Postdoctoral Fellowship. S.M.B was supported by a National Institute of General Medical Sciences F32 Postdoctoral Fellowship. J.A.B. was supported by a National Institute of General Medical Sciences F32 Postdoctoral Fellowship
REFERENCES




