
Abstract
This article outlines the need for a homeostatic response to alterations in cellular oxygenation. It describes work on erythropoietin control that led to the discovery of the hypoxia-inducible transcription factor (HIF-1) and the parallel recognition that this system was responsive to a widespread oxygen-sensing mechanism. Subsequently, multiple HIF isoforms have been shown to have overlapping but non-redundant functions, controlling expression of genes involved in diverse processes such as angiogenesis, vascular tone, metal transport, glycolysis, mitochondrial function, cell growth and survival. The major role of prolyl and asparaginyl hydroxylation in regulating HIFs is described, as well as the identification of PHD1-3 and FIH as the oxygen-sensing enzymes responsible for these hydroxylations. Current understanding of other processes that modulate overall HIF activity, including influences from other signalling mechanisms such as kinases and nitric oxide levels, and the existence of a variety of feedback loops are outlined. The effects of some mutations in this pathway are documented as is knowledge of other substrates for these enzymes. The importance of PHD1-3 and FIH, and the large family of 2-oxoglutarate and iron(II)-dependent dioxygenases of which they are a part, in biology and medicine are discussed (part of a multi-author review).
Similar content being viewed by others

Introduction
Oxygen is the most abundant element in our planet as a whole. The majority of life has evolved over a period during which atmospheric oxygen has risen from very low levels to current levels of just under 21%, with perhaps a higher peak in the dinosaur era. For the majority of eukaryotic life forms oxygen has become the preferred terminal electron acceptor in metabolism, whereas many prokaryotes can utilise alternative electron acceptors. However, oxygen is a highly reactive element, and activated oxygen species are extremely toxic, used to benefit in bacterial killing by professional phagocytes, but with the potential for detrimental effects, including DNA, RNA and protein damage resulting in cell death. Oxygen supply and consumption, therefore, need to be tightly balanced to ensure optimal usage of this critically important element.
Mechanisms for regulating behaviour in response to changes in oxygen availability have been identified in both unicellular and multicellular organisms. Unicellular organisms are exposed to ambient levels of oxygen and have to adjust their metabolism accordingly. Environmental oxygen levels may vary in a diurnal pattern due to light-driven photosynthesis; the ambient level may be affected by the rate at which oxygen is consumed by the organism when the rate of supply is limited. Those organisms in which oxygen is the ultimate electron acceptor in energy metabolism can only tolerate anoxia, the total lack of oxygen, for very brief periods. In contrast, hypoxia, a reduction in oxygen availability, is an important stimulus for a variety of adaptive responses. Several regulatory mechanisms have been dissected, examples including ArcA, Fnr, OxyR, SoxR and FixL in various bacteria and Hap1 (Cyp1) in Saccharomyces cerevisiae [1–6].
A challenge for complex multicellular organisms, such as man, is to ensure optimal oxygen availability (and the disposal of waste products from oxygen consumption) is maintained to every cell in the organism throughout development and post-natal life. Tight linkage must therefore exist between oxygen availability and growth during development, and oxygen availability and metabolism in adulthood. In man, the placenta and the entire cardiovascular and respiratory systems are devoted to this purpose. These systems are controlled by neuronal, hormonal and autocrine mechanisms, allowing the organism to defend against both too little and too much oxygen being available. Responses to hypoxia can be triggered by very modest variations in blood oxygen supply, such as the effects of donating a unit of blood or moderate changes in altitude, and deviations in oxygen supply and demand are either causes or consequences of the vast majority of diseases. Adaptive responses can occur at the level of individual cells, individual tissues and organs, the organism as a whole, and ultimately over prolonged periods of time within and between species (see articles by Joseph, Calbet and Lundby in this issue). Many of the adaptive responses require changes in expression of multiple genes, sometimes across several cell types, implying the need for common master regulators and hierarchical control systems. Sensing mechanisms are required that can control oxygen delivery and consumption mechanisms, but the time frame required for different adaptive responses to hypoxia will vary enormously, needing to be very short when controlling the acute ventilatory response, but much longer-term in determining the morphogenesis of entire tissues, organs or organisms. Since oxygen can only reach the most distal parts of an organism by transfer down a concentration gradient, the oxygen level experienced by different cells in the body varies substantially, although that experienced by a particular cell type in a particular tissue tends to remain relatively constant. Since individual cells must retain the ability to sense and respond to changes in oxygen availability, an implication of this observation is that at a cellular level the particular oxygen level required to trigger an active response would be expected to vary.
Hypoxia-inducible factors (HIF)
One paradigm for the investigation of the oxygen-sensing mechanisms has been control of erythropoietin gene expression. Erythropoietin is a glycoprotein hormone which, by acting as a survival factor for red cell precursors, is dominantly important in controlling erythropoiesis and thus the ability of higher organisms to transport oxygen effectively in the blood [7]. A substantial body of evidence accumulated to indicate that erythropoietin levels were determined by the rate of erythropoietin gene transcription and responsive directly to changes in oxygen level, rather than indirectly via compromise of metabolism [8, 9]. In addition to up-regulation by hypoxia, erythropoietin production can also be enhanced by iron chelation and treatment with some, but not all, transition metals [10–12]. Erythropoietin is produced predominantly by specialised cells within the kidney, although liver, brain and testis cells may also contribute [13–19]. The discovery that oxygen-sensitive changes in erythropoietin gene transcription could be encapsulated in particular human hepatoma-derived tissue culture lines [20] led ultimately to the discovery of the hypoxia-inducible factor (HIF) family of transcriptional activators [21, 22].
The route that led to this discovery was initially the identification of an oxygen-responsive transcriptional enhancer lying 3′ to the gene [hypoxia response element (HRE)] [23–25], followed by affinity purification of the associated DNA binding proteins, protein sequencing and subsequent PCR with degenerate oligonucleotides to identify cDNAs and thus the gene [21]. The importance of this system was emphasised by the early discovery that HREs were widely operative, even in cells that did not make erythropoietin, implying that the sensing mechanisms were widely expressed and would be controlling processes other than erythropoiesis [26]. These predictions have subsequently been vindicated.
Hypoxia-inducible factor-1 (HIF) is an alpha/beta heterodimeric protein complex that binds to the HRE consensus, RCGTG, and regulates the expression of many genes in response to changes in oxygen availability. In addition to controlling erythropoiesis HIF-responsive genes influence processes including angiogenesis, vascular tone, metal transport, glycolysis, mitochondrial function, cell growth and survival, emphasising HIF’s central involvement in oxygen homeostasis. HIF has earned the epithet of being the ‘master regulator’ of hypoxically regulated transcriptional systems [27–32]. However, it is clear that a variety of other transcriptional systems, including NFκB, ATFs and p53, are responsive to ambient oxygen levels [33–37]. This raises the question of whether these other effects are indirectly driven by HIF or arise directly, perhaps via shared, overlapping or independent oxygen-sensing mechanisms [38]. Array analysis in the presence and absence of siRNA against HIF shows that in MCF7 breast cancer cells the majority of transcripts that were significantly up-regulated by hypoxia were HIF-dependent, but a significant minority (105/246) were not [39]. This analysis probably flatters the importance of HIF since the conditions used (16-h exposure to 1% oxygen above the culture medium) are known to be particularly good at activating HREs. Under different stimuli other systems will predominate. Examples include signalling via the NFkappaB pathway in preference to HIF activation during intermittent hypoxia and re-oxygenation [37] and ATF signalling triggered by absolute anoxia [40, 41], although the independence of this effect from the unfolded protein stress response is debated.
The HIF alpha and beta proteins contain basic helix–loop–helix (bHLH) and PAS (Per; AHR; ARNT; SIM) domains and are part of a large family of proteins with these features [42]. Intriguingly, the PAS domain has primitive origins and is a component of several proteins involved in circadian rhythms, perhaps indicating links between the circadian light–dark cycle via variations in photosynthetic rate to oxygen availability and thus the response to oxygen levels [42, 43]. The HIF alpha chains confer oxygen regulation on the complex as a whole. The HIF beta chains are constitutively expressed and had previously been identified as the aryl hydrocarbon receptor nuclear translocator (ARNT), contributing in combination with the aryl hydrocarbon receptor (AHR) to the transcriptional response to xenobiotic chemicals, such as dioxin [44–47]. Under stimulatory conditions HIF-alpha chains enter the nucleus and heterodimerise with HIF-beta. Active complexes bind DNA and where appropriate recruit CBP/p300 transcriptional co-activators to the transcriptional complex, thereby augmenting their inherent trans-activating abilities and influencing transcription of downstream genes [48]. The details of these processes and their regulation are discussed below.
Multiple HIF-alpha chains
Evolutionary precursors of the HIF system are found in Caenorhabditis elegans and Drosophila melanogaster, where the HIF-alpha chain is termed hif-1 [49] and Similar [50, 51] and the HIF-beta chain aha-1 [49] and Tango [52], respectively. Following the initial identification of HIF-1 several different genes encoding HIF-alpha and -beta subunits, and differentially spliced versions of these transcripts, have been found in mammalian systems. Three alpha chains, now commonly referred to as HIF-1 alpha [21], HIF-2 alpha [53–55] and HIF-3 alpha [56–60] and three beta chains, ARNT1, 2 and 3 [44, 61–64] are encoded at distinct genetic loci.
These components have different gene expression patterns. HIF-1alpha is widely expressed in normal tissues. HIF-2alpha was initially defined as an endothelial cell-specific isoform [53], though it is now clear that its expression is more widespread [65]. ARNT1 is widely expressed [66], whereas ARNT2 is highly expressed in the central nervous system and kidney [67], and ARNT3 in brain and muscle [68]. The expression pattern of HIF-3 alpha has been less fully studied [56].
Knock-out studies have thrown light on the function of these components, and this work is now being complemented by gene array studies and molecular dissection of the differential responses. HIF-1 alpha, HIF-2 alpha and ARNT have non-redundant functions and are all essential for normal embryogenesis. Targeted inactivation of HIF-1 alpha and HIF-2 alpha genes produces significantly different phenotypes. HIF-1 alpha null mice die around day 10.5 of embryogenesis with defective cardiac morphogenesis, vascularisation and neural tube closure [69, 70]. HIF-2 alpha inactivation has been reported to cause diverse phenotypes in the context of different genetic backgrounds, with death occurring in utero with defective catecholamine production [71] or vascularisation [72] or a few days or weeks after birth due to respiratory distress [73] or mitochondrial dysfunction [74]. ARNT1 negative mice also suffer embryonic death by day 10.5 associated with placental, vascular and haematopoietic defects [75]. ARNT2 null embryos die perinatally and exhibit impaired hypothalamic development [76]. Whilst differences in phenotype between HIF-1 and -2 alpha null animals may arise because of differences in their temporal and spatial expression, it is now evident that they also activate overlapping but distinct sets of genes, even in a single cell type.
Despite similar activity in promoting transcription of reporter genes linked to minimal multimerized HREs, endogenous target genes are often differentially activated by either HIF-1 alpha or HIF-2 alpha in individual cell types. In MCF7, a breast cancer cell line, these effects have been systematically studied using RNA arrays to assess transcription in a single cell type after RNA interference-mediated knock-down of individual HIF-alpha isoforms [39]. In other contexts, examples of specific genes with clear-cut preferential activation by one or other HIF isoform include activation of the Tie-2 promoter and the erythropoietin gene by HIF-2 alpha, but not HIF-1 alpha [53] and activation of glycolytic genes and carbonic anhydrase IX by HIF-1 alpha, but not HIF-2 alpha, in cell lines of kidney origin [77–79].
Differential gene activation inevitably leads to altered cellular phenotypes, although precise effects are likely to depend on the cellular background. Forced expression studies have revealed that HIF-2 alpha, but not HIF-1 alpha, is sufficient to promote renal clear cell tumourigenesis [79–82]. Analysis of different types of lesions in the kidneys of von Hippel-Lindau (VHL) patients has revealed that more advanced neoplastic lesions show proportionally greater expression of the HIF-2 alpha isoform, again suggesting that the HIF-2 alpha-induced phenotype has a selective advantage in this context [83]. Similarly, a recent study of mouse ES cell teratoma xenografts demonstrated enhanced tumour growth of cells bearing a HIF-2 alpha ‘knock-in’ allele at the HIF-1 alpha locus [84]. In contrast, it has been reported that genetic ablation of HIF-1 alpha, but not HIF-2 alpha, in murine embryonic stem cells produces insensitivity to hypoxia-induced apoptosis [85].
Taken together these findings indicate that, despite many similarities, HIF-1 alpha and HIF-2 alpha isoforms have contrasting roles. Interest has centred around the mechanisms responsible for these effects. It has been shown that a post-DNA binding mechanism is responsible [86]. It has been suggested that differential gene regulation may result from the relative activities of the N-terminal and C-terminal transactivation domains of the HIF-1 alpha and HIF-2 alpha [87]. Titratable binding partners that suppress the activity of one or the other HIF isoform have been reported in different contexts [77], and more recently differences between HIF-1 and HIF-2 alpha in interactions with c-myc signalling have been shown [88].
Further complexity may arise from effects of HIF-3 alpha, a protein that exhibits a high degree of sequence similarity with HIF-1 and HIF-2 alpha over the bHLH and PAS regions but a low degree of sequence similarity across the C-terminus [89]. Transfection assays revealed that HIF-3 alpha could suppress basal and hypoxia-induced transactivation of a reporter gene linked to a promoter containing previously defined HIF-1 binding sites and antagonised hypoxia-inducible gene expression mediated by HIF-1 or HIF-2 alpha [90]. HIF-3 alpha also dimerises with ARNT1 and recognises the same DNA sequences as, and may compete for binding with, heterodimers containing HIF-1 and HIF-2 alpha [89]. However, alternative splicing of the HIF-3 alpha transcript produces a protein, termed inhibitory PAS domain protein (IPAS), that forms transcriptionally inactive heterodimers with HIF-1 alpha [59, 60] that could potentially bias the transcriptional response towards HIF-2 alpha-dependent genes.
Regulation of HIF-alpha chain activity—amino acid hydroxylation
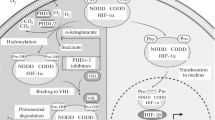
HIF activation in hypoxia requires many processes including HIF-alpha chain synthesis, stabilisation, nuclear import, dimerisation, DNA binding and co-activator recruitment. Whilst HIF-alpha chain synthesis may be maintained under the cellular stress of hypoxia by the presence of an IRES sequence in the 5′-UTR, obviating the need for cap-directed translational initiation [91], and nuclear accumulation may be governed by hypoxia-sensitive mechanisms [92], it is widely agreed that the dominant regulation of HIF-alpha chain activity arises from the combination of protein stabilisation and enhanced co-activator recruitment in hypoxia (Fig. 1).

Scheme showing the regulation of HIF alpha chain activity by HIF hydroxylases. When oxygen levels are high the HIF hydroxylases are active. Prolyl hydroxylases modify specific prolyl residues within the oxygen-dependent degradation domain of HIF alpha chains, allowing enhanced recognition by the pVHL ubiquitin ligase, ubiquitylation of the HIF alpha chain and subsequent proteasomal destruction. Factor-inhibiting HIF (FIH), the HIF asparaginyl hydroxylase, hydroxylates a C-terminal asparaginyl residue. This blocks recruitment of the transcriptional co-activators p300/CBP. As oxygen levels fall the HIF hydroxylases become progressively less active. In the absence of prolyl hydroxylation HIF alpha chains are stable and can heterodimerise with HIF beta chains to form a transcriptional complex that binds to hypoxia response elements (HREs). In the absence of asparaginyl hydroxylation p300/CBP can be recruited to this transcriptional complex, enhancing its activity and leading to increased transcription of downstream genes
Under normoxic conditions HIF-alpha chains are rapidly degraded by the proteasome [93] following promiscuous ubiquitylation at several sites [94, 95] by an E3 ligase in which the von Hippel-Lindau tumour suppressor protein (pVHL) acts as the substrate recognition component [96]. Mutations of pVHL found in renal clear cell cancer disable this process, leading to accumulation of high levels of HIF alpha proteins and contributing to the phenotype of these tumours [96–99]. Other mutations, associated with phaeochromocytoma (type 2C) [97, 100] and the rare recessive mutation found in Chuvash polycythaemia (R200W) [101], appear to distort HIF signalling without completely ablating HIF ubiquitylation and consequent degradation. Recognition of normoxic HIF-alpha proteins by pVHL arises from the enzymatic hydroxylation of either of two critical prolyl residues, residing within the N-terminal oxygen-dependent degradation domain (NODD) and the C-terminal oxygen-dependent degradation domain (CODD) of HIF-1 and HIF-2 alpha chains (HIF-3 alpha only contains a single prolyl residue) [102–106]. The presence of a hydroxyl group excludes a water molecule from the pVHL:HIF-alpha interface and allows the formation of two additional hydrogen bonds between HIF-alpha and pVHL, thereby stabilising the interaction and enhancing ubiquitylation [107]. Co-activator recruitment is also blocked by oxygen-dependent enzymatic hydroxylation of an asparaginyl residue in the carboxy-terminal activation domain (CAD) of HIF-1 and HIF-2 alpha chains [108–110].
This amino acid hydroxylation is a function of a family of 2-oxoglutarate and iron(II)-dependent dioxygenases, the HIF hydroxylases. In each case part of the HIF-alpha chain enters the active site of the relevant hydroxylase in an extended conformation, bringing the residue to be hydroxylated into close proximity with the iron atom, which is essential for catalysis [111, 112]. During hypoxia, iron chelation or treatment with transition metal ions such as cobalt(II), HIF hydroxylase activity is reduced, allowing HIF to escape from both destruction and blockade of co-activator recruitment, thereby up-regulating genes involved in cellular and systemic hypoxia responses such as angiogenesis, apoptosis, erythropoiesis, glycolysis, inflammation, matrix metabolism, metal transport, mitochondrial function, proliferation, control of vascular tone and ventilation [113] and micro RNAs [114].
The mammalian genome encodes three HIF prolyl hydroxylases, named PHDs 1-3 [103], whereas in Drosophila melanogaster and Caenorhabditis elegans there are single proteins with this function named Fatiga and EGL9, respectively [103, 115]. A mammalian endoplasmic reticulum prolyl hydroxylase has also been reported to influence HIF activity [116], and mammals have a single HIF asparaginyl hydroxylase, factor-inhibiting HIF (FIH) [108–110]. Neither the endoplasmic reticulum HIF prolyl hydroxylase activity nor the asparaginyl hydroxylase activity is present in Drosophila melanogaster or Caenorhabditis elegans.
Function, regulation and mutations of the HIF hydroxylases
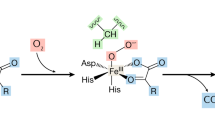
The HIF hydroxylases are part of a family of enzymes that all use dioxygen to hydroxylate their prime substrate and simultaneously convert 2-oxoglutarate into succinate with the release of carbon dioxide. Structurally their active site contains eight beta strands folded into a beta barrel jelly roll motif that positions oxoglutarate and iron, generally liganded to two histidine residues and a carboxylate group (HXD….H) [111, 112]. Somewhat different estimates of the K m of these enzymes have been published using a variety of partially physiological assays. However, in all cases the K m is thought to be significantly higher than the ambient oxygen level in cells, making enzyme activity exquisitely dependent on oxygen concentration, and allowing these enzymes to act as true oxygen sensors. The K m of FIH, which functions as a dimer [117], has been measured to be less than that of the prolyl-hydroxylases, theoretically allowing FIH to regulate the transcriptional activity of any HIF-alpha chains that survive the degradative pathway [118–120]. However, practically it has been shown that HIF transcriptional targets can be up-regulated by inhibition of either PHD2 or FIH even when the other enzyme is fully active [121].
These enzymes catalyse the forward reaction but not the reverse reaction, and thus the net reaction rate is increased with the enzyme level (rather than this simply determining the speed with which equilibrium is achieved). In general in normoxic conditions PHD2 is the most abundant HIF prolyl-hydroxylase, and in keeping with this it has been reported to be the dominantly active enzyme [122]. However, it has been clearly established that when their abundance is increased PHD1 and PHD3 both also contribute to the control of HIF level and thus activity [123]. PHD1 production can be induced by oestrogens [124], and in some species at least, two variants with different translational initiations are produced, although the functional significance of this is not known [124]. PHD2 and PHD3 levels are both themselves up-regulated in response to hypoxia [103]. PHD2 and PHD3 regulation is achieved both by transcriptional and protein stability effects. The rate of degradation of PHD2 and PHD3 is mediated at least in part by ubiquitin ligases of the SIAH family [125, 126], which have more recently also been implicated in the regulation of FIH [127]. Additionally, proteolytic regulation of PHD2 through interaction with FKBP38, a peptidyl prolyl cis/trans isomerase, has been reported [128].
Enzyme activity can be regulated by a variety of factors including the availability of the prime substrate, the critical co-factor, iron, co-substrate, oxoglutarate, and the removal of the products, succinate (and possibly carbon dioxide). Modulation of iron(II) levels, either directly by supplementation or chelation, or indirectly using ascorbate or other redox agents, clearly affects enzyme activity [129]. Similarly, succinate has been shown to inhibit enzyme activity, and other Krebs’ cycle intermediates also affect the system [130]. To date no convincing post-translational modifications have been shown to modify the specific activity of these enzymes, although evidence for changes in specific activity has been put forward [131].
The relative amounts of the different PHD enzymes present in a particular cell type at a particular time may also contribute to the transcriptional bias between HIF-1 alpha and HIF-2 alpha responsive genes. Despite highly conserved sites of prolyl hydroxylation between HIF-1 and HIF-2 alpha proteins, the effects of PHD enzymes on the levels of these proteins are not equivalent, with PHD2 contributing more to the regulation of HIF-1 alpha than HIF-2 alpha, while PHD1 and PHD3 contribute somewhat more to the regulation of HIF-2 alpha than HIF-1 alpha. Differential action of the PHD proteins on the NODD and CODD of HIF-1 alpha and HIF-2 alpha may contribute to this bias [123].
The tissue distribution of the different HIF hydroxylase isoforms is gradually becoming clear. At the RNA level under basal conditions PHD1 is peculiarly abundant in the testis, PHD2 and FIH are ubiquitous, and PHD3 is highly expressed in heart and smooth muscle [132, 133]. Analysis of protein levels has lagged behind due to the relatively slow development of antibodies capable of specifically recognising endogenous levels of these proteins by immunoblotting and immunohistochemistry, and levels of protein responsible for functional effects are still close to the limits of detection, particularly for rodent PHD1. Protein levels generally match those of mRNAs in those tissue studied to date. Over-expression and physiological studies have shown that PHD1 is dominantly nuclear, PHD2 and FIH are mainly present in the cytoplasm, and PHD3 is present in both nucleus and cytoplasm [133, 134]. The significance of these different intracellular localisation patterns is unproven, although it is tempting to speculate that whilst cytoplasmic enzymes keep the system under check in normoxic conditions, the hypoxically induced intra-nuclear PHD3 enzyme may be particularly important for inactivating HIF, and perhaps particularly HIF-2 alpha, following re-oxygenation, although this would logically also require nuclear ubiquitylation and proteasomal destruction.
Functionally relevant mutations of PHD2 have now been reported in patients with primary polycythaemia [112, 135, 136], and it is likely that further mutations will also be found as other populations of people are surveyed. Additionally, given the genetic instability of cancer cell lines and the importance of the HIF system for tumour growth it seems inevitable that cancer cells will select HIF hydroxylase variants.
Knock-out mouse models provide clues about unique functions of individual HIF hydroxylases and may thus provide pointers to the existence, and possible nature, of alternative substrates. Studies of constitutive gene inactivation have shown that PHD2 is required for embryonic development. PHD2−/− embryos suffer defects in placentation and die in utero at dpc 12.5–14.5, but PHD2+/− animals survive to term and appear to develop relatively normally [137]. Somatic inactivation of PHD2 results in erythrocytosis and congestive cardiac failure [138]. PHD1−/− and PHD3−/− animals survive to term. PHD1−/− animals appear relatively normal in unstressed conditions, but their tissues have an enhanced ability to survive ischaemic insults [139]. PHD3−/− animals have a hyperplastic, but hypofunctional sympatho-adrenal axis, leading to relative hypotension in mid-life, but they develop cardiac failure in later life [140]. Further studies using tissue-specific conditional gene inactivation are required to assess the interplay between these phenotypes. FIH−/− mice have only recently been produced; preliminary reports suggest they are viable but of reduced size (R. Johnson; personal communication).
Other substrates for the HIF hydroxylases
Several laboratories are searching for other HIF hydroxylase substrates to discover novel oxygen-sensitive pathways or equally pathways regulated by iron or oxoglutarate availability. Other substrates for PHDs have been suggested to include the large sub-unit of RNA polymerase II [141, 142], IKK beta [143], ATF4 [38] and beta(2)-adrenergic receptors [144]. FIH hydroxylates many ankyrin repeat domains (ARD), and evidence exists to show this happens under endogenous circumstances [145–148]. FIH substrates appear to conform to a consensus motif that is present in most of the 300 human ARD-containing proteins, implying that FIH-mediated ARD hydroxylation is common. It is currently unclear what the functional consequences of FIH are on ARD biology since reported effects of FIH on the activity of ARD containing proteins are either subtle [145, 146] or may not be attributable to hydroxylation [147, 148]. However, it has been suggested that ARD proteins act as competitive inhibitors of FIH-dependent HIF alpha CAD hydroxylation: the ARD of Notch1 binds to FIH more tightly than HIF alpha CAD, and is a better substrate [146]. Overexpression of Notch1 ARD inhibits HIF1 alpha CAD hydroxylation, resulting in enhanced HIF1 alpha transcriptional activity (see “Attenuation of HIF activity in sustained hypoxia” below). The role of FIH in regulating other ARD pathways is unknown, but the notion that FIH-mediated ARD hydroxylation has a HIF-independent function is supported by bioinformatic analysis, which suggests that FIH and ARDs predate HIF-alphas (C. Loenarz; personal communication).
Other influences on HIF activity
The HIF system has pleiotropic actions and must be under subtle control. Whilst the actions of the HIF hydroxylases are vital, particularly for the hypoxic regulation of these transcriptional cascades, other mechanisms contribute to basal and induced activity.
HIF-alpha proteins are heavily phosphorylated [21], particularly in their oxygen-dependent degradation domains. In lower organisms such as nitrogen-fixing bacteria hypoxia-dependent phosphorylation activates transcription factors. A variety of kinase/phosphatase systems, including casein kinase II, AMP-activated protein kinase (AMPK), p42/44 mitogen-activated kinase (MAPK) and PI3K, have been reported to affect HIF transcriptional activity. Some sites of phosphorylation exert relatively direct influences on the transcriptional activity of HIF-alpha proteins. For example, phosphorylation of HIF-1 alpha at Thr-796 by casein kinase II appears to enhance HIF transcriptional activity [149], and consistent with this observation a synthetic peptide phosphorylated at this position is a poor substrate for FIH in vitro [117]. Similarly, MAPK also affects activity of the C-terminal activation domain [150]. However, more indirect effects also occur. Examples include MAPK also suppressing CRMI-mediated nuclear export of HIF [151, 152] and AMPK phosphorylating tuberous sclerosis complex-2 (TSC2), thereby inhibiting the mammalian target of rapamycin (mTOR), which in turn, independently of hypoxia, affects HIF-alpha protein levels [153–155].
In addition to phosphorylation, hydroxylation and ubiquitylation HIF has been reported to be subject to acetylation [156] and SUMOylation [157], although more recent data refuted the presence of ARD1-driven acetylation [158–160] or the functional significance of SUMOylation [161].
Redox chemistry also plays a critical role in the trans-activation of oxygen-responsive genes in unicellular organisms and modulates the activation of HIF-1, although it remains controversial as to whether these effects are direct, mediated via HIF hydroxylases or by entirely separate routes [162].
Nitric oxide, nitric oxide donors, scavengers and iNOS levels also influence the HIF system. The effects are complex, and some observations appear paradoxical [163, 164]. Proposed mechanisms include inhibition of HIF hydroxylases in normoxic conditions [165, 166], inducing HIF hydroxylase levels [167], possibly acting as partial HIF hydroxylase substrates in hypoxia and influencing intracellular oxygen levels available to the HIF hydroxylases by affecting mitochondrial function [168].
A wide variety of tumour suppressor and oncogenic proteins, including PTEN, p53, MDM2, Ha-Ras, v-Src and c-Myc, influence HIF activity, as do a variety of growth factors, including epidermal growth factor, insulin, insulin-like growth factors 1 and 2, angiotensin II, thrombin and PDGF.
Recent work has expanded the number of components of the HIF pathway. OS-9 (amplified in osteosarcoma-9) [169], inhibitor of growth family member 4 (ING4) [170], MAPK organizer 1 (Morg1) [171], TCP-1 ring complex (TRiC) [172] and NEMO (NF-KB essential modulator) [173] have been reported to interact with HIF and/or HIF hydroxylases. It is claimed that these interactions contribute to regulation of the system, although further work is required to clarify the general scope of these actions.
Attenuation of HIF activity in sustained hypoxia
Like other biological systems the HIF system is subject to a variety of negative feedback mechanisms. These both attenuate HIF activity in sustained hypoxia and contribute to adjusting the system to sense relative hypoxia in tissues with different ambient oxygen levels.
HIF transcriptional targets include a variety of genes, the products of which, directly or indirectly, attenuate the HIF response. The first example identified was CITED2, previously known as p35-srj, which blocks co-activator recruitment [174]. A second mechanism is the induction of an antisense RNA to HIF-1 alpha (aHIF) driven by an HRE 3′ to the HIF-1 alpha gene. A third mechanism involves the hypoxic induction of HIF hydroxylase levels, modulated at the transcriptional level by HIF, but also affected by hypoxic influences on protein stability (Fig. 2). More indirect effects occur because HIF exerts major influences on glycolysis, mitochondrial metabolism and iron transport, which in turn affect HIF hydroxylase activity by determining the availability of oxoglutarate and iron and the levels of downstream products (succinate, fumarate) [130, 175–177]. HIF-driven induction of nitric oxide synthase may also contribute to these effects. The involvement of HIF-induced microRNAs in further modulating the transcriptional outflow from this system is just being recognised [114]. Differential effects of these feedback mechanisms on the various components of the HIF system clearly have the potential to adjust the balance of HIF-1 versus HIF-2 activity. This is most clearly illustrated by effects of aHIF [178]; a more complex example is the preferential hydroxylation of HIF-2 alpha by PHD3, itself a preferential transcriptional target of HIF-2 alpha [86].

Negative feedback systems affect HIF activity at multiple levels. Under hypoxic conditions HIF is active and leads to the production of a variety of transcripts. HIF-induced antisense HIF (aHIF) can specifically reduce HIF-1 alpha mRNA levels. HIF-induced up regulation of PHD-2 and PHD-3 production can increase the total PHD pool in the cell. The increased amount of prolyl hydroxylase protein does increase the amount of HIF alpha hydroxylation, and thus destruction, even though oxygen levels are low. Active HIF leads to production of CITED2, which can bind to p300 in its CH-1 domain, thereby blocking the ability of this co-activator to bind to the HIF transcriptional complex
HIF-3 alpha induction also has the ability to negatively regulate HIF activity by heterodimerising with HIF alpha or HIF beta chains [59, 60, 89], but the generality of these effects has not been fully elucidated.
Clearly further adjustment of the system to sense relative hypoxia in tissues with different ambient oxygen levels can be mediated by the effects of phosphorylation, redox, nitric oxide, tumour suppressors and oncogenes discussed above. A new insight into tuning of this system has come from the identification of alternative substrates for the HIF hydroxylases. It has been shown that ARDs competitively inhibit FIH-mediated HIF-alpha CAD hydroxylation [146]. As these alternative HIF hydroxylase substrates have relatively long half-lives compared with HIF-alpha proteins, their hydroxylation status, and thus their ability to compete with HIF for hydroxylase activity, will depend on the longer term oxygenation of the cell (Fig. 3).

Effects of ankyrin repeat domain hydroxylation on FIH and HIF activity. FIH will preferentially hydroxylate asparaginyl residues in ankyrin repeat domains (ARD) over those in HIF alpha chains. Under circumstances where ankyrin repeat domains have not been hydroxylated, the majority of FIH activity will be directed towards ankyrin hydroxylation, leaving the asparaginyl residue of HIF alpha chains unhydroxylated, allowing p300 recruitment and thus HIF transcriptional activity. Conversely, once ankyrin repeat domains have become fully hydroxylated FIH activity will be directed towards HIF alpha chains, blocking p300 recruitment and reducing the activity of the transcriptional complex
HIF hydroxylases and medicine
HIF has been implicated in disease processes, including cancer, ischaemic heart disease, pulmonary hypertension, mountain sickness, diabetic eye disease and arthritis [179, 180].
In human diseases some HIF effects are beneficial to patients, and medical benefit would be gained by augmenting these responses. For example, occlusive vascular disease is a major health problem in the western world, presenting as stroke, heart attack or peripheral vascular disease. Current therapies, including thrombolysis, percutaneous angioplasty or surgical revascularisation all aim to restore the blood supply to the tissue at risk. Promotion of angiogenesis in ischaemic tissues has been attempted using exogenous growth factor therapies. This has been of limited use, since it only drives certain aspects of the angiogenic response and has no direct cytoprotective effects. An alternative approach is to enhance the normal physiological response to ischaemia, driven at least in part by the HIF pathway. Several gene therapy approaches have adopted this strategy [181, 182]. Additionally, both desferrioxamine [183] and cobalt(II) chloride [184], known to activate the HIF system, have been shown to protect the rodent myocardium in ischaemia models. Therapeutic manipulation of the HIF pathway by inhibition of HIF hydroxylase activity is under active investigation. Many experiments reported to date have all used generic 2-oxoglutarate analogue based hydroxylase inhibitors, such as FG-0041 [185, 186], the plant extract l-mimosine [187], dimethyloxalylglycine [188], FG-2216 [189] or FG-4487 [190] to produce benefits in various ischaemic models. Other HIF responses are detrimental to the patient, for example, the growth of new vessels to supply an expanding cancer, and medical benefit would be gained by their inhibition. Benefits have been reported from both peptide-based and small molecule-induced inhibition of co-activator recruitment in experimental models of cancer [191, 192].
Whether up-regulating or down-regulating the HIF pathway for therapeutic benefit, the presence of negative feedback loops and antagonists in the HIF pathway (discussed above) will all need to be considered in evaluating therapeutic approaches and optimising dosing schedules. Differential gene activation by different HIF alpha isoforms in a particular tissue also needs to be taken into account.
For HIF up-regulation differential effects of individual HIF hydroxylases on particular HIF alpha isoforms raise the possibility that therapeutic use of inhibitors of individual HIF hydroxylases could be tailored to bias the expression of hypoxia responsive genes. Indeed, analysis of the protective response of PHD-1 knockout mice to hind-limb [139] and hepatic ischaemia (M. Schneider; personal communication) not only provides further evidence in favour of HIF hydroxylase inhibition as a treatment for ischaemia diseases but also suggests that this hydroxylase isoform should be specifically inhibited for this purpose. Studies in renal cancers discussed above suggest that benefit may be derived from upsetting the balance of HIF-1 versus HIF-2 and that it is naïve to think simply in terms of overall up-regulation or down-regulation of the pathway [79, 88, 193, 194].
The wider 2-oxoglutarate and iron(II)-dependent dioxygenase (2OG oxygenase) family
Since most enzyme inhibitors will not be totally specific, knowledge of the consequences of down-regulating the activity of closely related enzymes is likely to be of benefit in anticipating potential toxicity and interpreting off target effects. The 2-oxoglutarate (2OG) and iron(II)-dependent dioxygenase (2OG oxygenase) families are widespread in biology [195], with approximately 60 members in humans. They catalyse a wide range of oxidative reactions, including hydroxylations, desaturations and ring closures [196]. They are involved in a variety of medicinally critical processes beyond oxygen-sensing, including DNA repair, chromatin remodelling, biosynthesis of antibiotics, the assembly of collagen and effects on lipid metabolism, diabetes and obesity [197–201]. Since enzyme activity can be regulated by a variety of factors, including enzyme level, the availability of the prime substrate and the critical co-factors and co-substrates, oxygen, iron and oxoglutarate and probably the removal of the products, succinate and carbon dioxide it is highly likely that these enzymes are involved in sensing, regulating and integrating many vital functions including oxygen supply and demand, iron balance, energy metabolism, ventilation and perhaps pH regulation.
The best characterised target of posttranslational hydroxylation by a 2OG oxygenase is the extracellular protein collagen [200]. The critical need for adequate co-factor availability for the proper function of this enzyme is illustrated by the effects of ascorbate deficiency, which lead to the disease scurvy. A second well-defined example of a disease associated with a 2OG oxygenase is Refsum’s disease, a neurological syndrome characterised by adult-onset retinitis pigmentosa, anosmia, sensory neuropathy and phytanic acidaemia. Many cases are caused by mutations in peroxisomal oxygenase phytanoyl-CoA 2-hydroxylase (PAHX), a 2-oxoglutarate dependent dioxygenase that catalyses the initial β-oxidation step in the degradation of phytanic acid [202, 203].
There is now great interest in the roles of 2OG oxygenase enzymes in DNA repair and epigenetic control of gene expression. DNA is damaged by a variety of mutagenic alkylating agents. The ABH enzymes are human homologues of E. coli AlkB that repair 1-methyladenine and 3-methylcytosine [197]. Hydroxylation of the methyl group produces an unstable intermediate that decomposes to the repaired base plus formaldehyde. Inactivity of these enzymes in the hypoxic microenvironment of cancers may contribute to the general phenomenon of genetic instability of cancers, which makes them so hard to treat.
Histone methylation regulates gene expression by modulating transcription factor recruitment. Methyltransferases catalyse the methylation of H3 and H4 at specific residues, with the site of methylation dictating whether modification results in transcriptional activation or silencing. Recent studies suggest that 2OG oxygenases can reverse this process [204, 205]. Histone demethylases of the 2OG oxygenase family have been shown to regulate gene expression and the proliferation of tumour cells [206].
Other 2OG oxygenases also have important roles in metabolism. Gamma butyrobetain hydroxylase and trimethyl lysine hydroxylase are both involved in carnitine biosynthesis of particular relevance in muscles and cardiac disease [207–210]. The effects of oxoglutarate analogues on iron-regulatory protein 2 signalling also suggest an as yet unidentified member of this enzyme class is involved in regulating iron levels [211]. Excitingly, links between 2OG oxygenases and diabetes and obesity have recently been suggested. Genome-wide searches for type 2 diabetes-susceptibility genes have identified a common variant in the FTO (fat mass and obesity associated) gene that predisposes to diabetes through an effect on body mass index. Sequence analysis suggested that FTO is a Fe(II)- and 2-oxoglutarate-dependent oxygenase, and recombinant murine Fto catalyses the Fe(II)- and 2OG-dependent demethylation of 3-methylthymine in single-stranded DNA. In mice Fto messenger RNA is most abundant in the brain, particularly in hypothalamic nuclei governing energy balance, and Fto mRNA levels in the arcuate nucleus are regulated by feeding and fasting [201]. Further studies are now required to determine the physiologically relevant FTO substrate and how nucleic acid methylation status is linked to increased fat mass.
Clearly this important and fascinating family of enzymes warrants much further investigation in its own right as well as because of its interplay with the HIF signalling system.
References
Gilles-Gonzalez MA, Gonzalez G, Perutz MF (1994) Heme-based sensors, exemplified by the kinas FixL, are a new class of heme protein with distinctive ligand binding and autoxidation. Biochemistry 33:8067–8073
Bunn HF, Poyton RO (1996) Oxygen sensing and molecular adaptation to hypoxia. Physiol Rev 76:839–885
Zitomer RS, Lowry CV (1992) Regulation of gene expression by oxygen in Saccharomyces cerevisiae. Microbiol Rev 56:1–11
Hidalgo E, Ding H, Demple B (1997) Redox signal transduction: mutations shifting [2Fe-2S] centers of the SoxR sensor-regulator to the oxidized form. Cell 88:121–129
Spiro S, Roberts RE, Guest JR (1989) FNR-dependent repression of the ndh gene of Escherichia coli and metal ion requirement for FNR-regulated gene expression. Mol Microbiol 3:601–608
Spiro S, Guest JR (1990) FNR and its role in oxygen-regulated gene expression in Escherichia coli. FEMS Microbiol Rev 6:399–428
Jelkmann W (1992) Erythropoietin: structure, control of production, and function. Physiol Rev 72:449–489
Necas E, Neuwirt J (1972) The effect of inhibitors of energy metabolism on erythropoietin production. J Lab Clin Med 79:388–396
Necas E, Thorling EB (1972) Unresponsiveness of erythropoietin-producing cells to cyanide. Am J Physiol 222:1187–1190
Wang GL, Semenza GL (1993) Desferrioxamine induces erythropoietin gene expression and hypoxia-inducible factor 1 DNA-binding activity: implications for models of hypoxia signal transduction. Blood 82:3610–3615
Goldwasser E, Jacobson LO, Fried W, Plazk LF (1958) Studies on erythropoiesis V: the effect of cobalt on the production of erythropoietin. Blood 13:55–60
Ho VT, Bunn HF (1996) Effects of transition metals on the expression of the erythropoietin gene: further evidence that the oxygen sensor is a heme protein. Biochem Biophys Res Commun 223:175–180
Tan CC, Eckardt K-U, Ratcliffe PJ (1991) Organ distribution of erythropoietin messenger RNA in normal and uremic rats. Kidney Int 40:69–76
Maxwell PH, Osmond MK, Pugh CW, Heryet A, Nicholls LG, Tan CC, Doe BG, Ferguson DJP, Johnson MH, Ratcliffe PJ (1993) Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int 44:1149–1162
Maxwell PH, Ferguson DJP, Osmond MK, Pugh CW, Heryet A, Doe BG, Johnson MH, Ratcliffe PJ (1994) Expression of a homologously recombined erythropoietin-SV40 T antigen fusion gene in mouse liver: evidence for erythropoietin production by Ito cells. Blood 84:1823–1830
Koury ST, Bondurant MC, Koury MJ (1988) Localization of erythropoietin synthesizing cells in murine kidneys by in situ hybridization. Blood 71:524–527
Koury ST, Bondurant MC, Koury MJ, Semenza GL (1991) Localization of cells producing erythropoietin in murine liver by in situ hybridization. Blood 77:2497–2503
Eckardt K-U, Koury ST, Tan CC, Schuster SJ, Kaissling B, Ratcliffe PJ, Kurtz A (1993) Distribution of erythropoietin producing cells in rat kidneys during hypoxic hypoxia. Kidney Int 43:815–823
Bachmann S, Le Hir M, Eckardt K-U (1993) Co-localization of erythropoietin messenger RNA and ecto-5′-nucleotidase immunoreactivity in peritubular cells of rat renal cortex indicates that fibroblasts produce erythropoietin. J Histochem Cytochem 41:335–341
Goldberg MA, Glass GA, Cunningham JM, Bunn HF (1987) The regulated expression of erythropoietin by two human hepatoma cell lines. Proc Natl Acad Sci USA 84:7972–7976
Wang GL, Jiang B-H, Rue EA, Semenza GL (1995) Hypoxia-inducible factor 1 is a basic-helix–loop–helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 92:5510–5514
Wang GL, Semenza GL (1995) Purification and characterisation of hypoxia-inducible factor 1. J Biol Chem 270:1230–1237
Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE (1991) Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci USA 88:5680–5684
Beck I, Ramirez S, Weinmann R, Caro J (1991) Enhancer element at the 3′-flanking region controls transcriptional response to hypoxia in the human erythropoietin gene. J Biol Chem 266:15563–15566
Pugh CW, Tan CC, Jones RW, Ratcliffe PJ (1991) Functional analysis of an oxygen-regulated transcriptional enhancer lying 3′ to the mouse erythropoietin gene. Proc Natl Acad Sci USA 88:10553–10557
Maxwell PH, Pugh CW, Ratcliffe PJ (1993) Inducible operation of the erythropoietin 3′ enhancer in multiple cell lines: evidence for a widespread oxygen sensing mechanism. Proc Natl Acad Sci USA 90:2423–2427
Firth JD, Ebert BL, Ratcliffe PJ (1995) Hypoxic regulation of lactate dehydrogenase A: interaction between hypoxia inducible factor 1 and cAMP response elements. J Biol Chem 270:21021–21027
Ebert BL, Firth JD, Ratcliffe PJ (1995) Hypoxia and mitochondrial inhibitors regulate expression of glucose transporter-1 via distinct cis-acting sequences. J Biol Chem 270:29083–29089
Gleadle JM, Ebert BL, Firth JD, Ratcliffe PJ (1995) Regulation of angiogenic growth factor expression by hypoxia, transition metals, and chelating agents. Am J Physiol 268:C1362–C1368
Firth JD, Ebert BL, Pugh CW, Ratcliffe PJ (1994) Oxygen-regulated control elements in the phosphoglycerate kinase 1 and lactate dehydrogenase A genes: similarities with the erythropoeitin 3′ enhancer. Proc Natl Acad Sci USA 91:6496–6500
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3:721–732
Liu Y, Cox SR, Morita T, Kourembanas S (1995) Hypoxia regulates vascular endothelial growth factor gene expression in endothelial cells. Circ Res 77:638–643
Blais JD, Filipenko V, Bi M, Harding HP, Ron D, Koumenis C, Wouters BG, Bell JC (2004) Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol Cell Biol 24:7469–7482
Graeber TG, Peterson JF, Tsai M, Monica K, Fornace AJ Jr, Giaccia AJ (1994) Hypoxia induces accumulation of p53 protein, but activation of a G1-phase checkpoint by low-oxygen conditions is independent of p53 status. Mol Cell Biol 14:6264–6277
Schmedtje JF Jr, Ji Y-S (1998) Hypoxia and molecular cardiovascular medicine. Trends Cardiovasc Med 8:24–33
Zampetaki A, Mitsialis SA, Pfeilschifter J, Kourembanas S (2004) Hypoxia induces macrophage inflammatory protein-2 (MIP-2) gene expression in murine macrophages via NF-κB: the prominent of p42/p44 and PI3 kinase pathways. FASEB J 18:1090–1092
Cummins EP, Comerford KM, Scholz C, Bruning U, Taylor CT (2007) Hypoxic regulation of NF-kappaB signaling. Methods Enzymol 435:479–492
Koditz J, Nesper J, Wottawa M, Stiehl DP, Camenisch G, Franke C, Myllyharju J, Wenger RH, Katschinski DM (2007) Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood 110:3610–3617
Elvidge GP, Glenny L, Appelhoff RJ, Ratcliffe PJ, Ragoussis J, Gleadle JM (2006) Concordant regulation of gene expression by hypoxia and 2-oxoglutarate dependent dioxygenase inhibition; the role of HIF-1alpha, HIF-2alpha and other pathways. J Biol Chem 281:15215–15226
Ameri K, Hammond EM, Culmsee C, Raida M, Katschinski DM, Wenger RH, Wagner E, Davis RJ, Hai T, Denko N, Harris AL (2007) Induction of activating transcription factor 3 by anoxia is independent of p53 and the hypoxic HIF signalling pathway. Oncogene 26:284–289
Ameri K, Lewis CE, Raida M, Sowter H, Hai T, Harris AL (2004) Anoxic induction of ATF-4 through HIF-1-independent pathways of protein stabilization in human cancer cells. Blood 103:1876–1882
Zhulin IB, Taylor BL, Dixon R (1997) PAS domain S-boxes in Archaea, bacteria and sensors for oxygen and redox. Trends Biol Sci 22:331–333
Taylor BL, Zhulin IB (1999) PAS domains: internal sensors of oxygen, redox potential, and light. Microbiol Mol Biol Rev 63:479–506
Hoffman EC, Reyes H, Chu F-F, Sander F, Conley LH, Brooks BA, Hankinson O (1991) Cloning of a factor required for activity of the Ah (Dioxin) receptor. Science 252:954–958
Reyes H, Reisz-Porszasz S, Hankinson O (1992) Identification of the Ah receptor nuclear translocator protein (Arnt) as a component of the DNA binding form of the Ah receptor. Science 256:1193–1195
Reisz-Porszasz S, Probst MR, Fukunaga BN, Hankinson O (1994) Identification of functional domains of the aryl hydrocarbon receptor nuclear translocator protein (ARNT). Mol Cell Biol 14:6075–6086
Hankinson O (1995) The aryl hydrocarbon complex. Annu Rev Pharmacol Toxicol 35:307–340
Arany Z, Huang LE, Eckner R, Bhattacharya S, Jiang C, Goldberg MA, Bunn HF, Livingston DM (1996) An essential role for p300/CBP in the cellular response to hypoxia. Proc Natl Acad Sci USA 93:12969–12973
Jiang H, Guo R, Powell-Coffman JA (2001) The Caenorhabditis elegans hif-1 gene encodes a bHLH-PAS protein that is required for adaptation to hypoxia. Proc Natl Acad Sci USA 98:7916–7921
Bacon NC, Wappner P, O’Rourke JF, Bartlett SM, Shilo B, Pugh CW, Ratcliffe PJ (1998) Regulation of the Drosophila basic helix–loop–helix PAS protein Sima by hypoxia: functional evidence for homology with mammalian HIF-1 alpha. Biochem Biophys Res Commun 249:811–816
Lavista-Llanos S, Centanin L, Irisarri M, Russo DM, Gleadle JM, Bocca SN, Muzzopappa M, Ratcliffe PJ, Wappner P (2002) Control of the hypoxic reponse in Drosophila melanogaster by the basic helix–loop–helix PAS protein similar. Mol Cell Biol 22:6842–6853
Sonnenfeld M, Ward M, Nystrom G, Mosher J, Stahl S, Crews S (1997) The Drosophila tango gene ecodes a bHLH-PAS protein that is orthologous to mammalian Arnt and controls CNS midline and tracheal development. Development 124:4571–4582
Tian H, McKnight SL, Russell DW (1997) Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev 11:72–82
Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y (1997) A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA 94:4273–4278
Flamme I, Fröhlich T, von Reutern M, Kappel A, Damert A, Risau W (1997) HRF, a putative basic helix–loop–helix-PAS-domain transcription factor is closely related to hypoxia-inducible factor-1α and developmentally expressed in blood vessels. Mech Dev 63:51–60
Hara S, Hamada J, Kobayashi C, Kondo Y, Imura N (2001) Expression and characterization of hypoxia-inducible factor (HIF)-3α in human kidney: suppression of HIF-mediated gene expression by HIF-3α. Biochem Biophys Res Commun 287:808–813
Maynard MA, Qi H, Chung J, Lee EHL, Kondo Y, Hara S, Conaway RC, Conaway JW, Ohh M (2003) Multiple splice variants of the human HIF-3α locus are targets of the von Hippel-Lindau E3 ubiquitin ligase complex. J Biol Chem 278:11032–11040
Heidbreder M, Frohlich F, Johren O, Dendorfer A, Qadri F, Dominiak P (2003) Hypoxia rapidly activates HIF-3α mRNA expression. FASEB J 17:1541–1543
Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, Cao Y, Berkenstam A, Poellinger L (2001) Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 414:550–554
Makino Y, Kanopka A, Wilson WJ, Tanaka H, Poellinger L (2002) Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3α locus. J Biol Chem 277:32405–32408
Hirose K, Morita M, Ema M, Mimura J, Hamada H, Fujii H, Saijo Y, Gotoh O, Sogawa K, Fujii-Kuriyama Y (1996) cDNA cloning and tissue-specific expression of a novel basic helix–loop–helix/PAS factor (Arnt2) with close sequence similarity to the aryl hydrocarbon receptor nuclear translocator (Arnt). Mol Cell Biol 16:1706–1713
Ikeda M, Nomura M (1997) cDNA cloning and tissue-specific expression of a novel basic helix–loop–helix/PAS protein (BMAL1) and identification of alternatively spliced variants with alternative translation initiation site usage. Biochem Biophys Res Commun 233:258–264
Hogenesch JB, Guy Y–Z, Jain S, Bradfield CA (1998) The basic-helix–loop–helix-PAS orphan MOP3 forms transcriptionally active complexes with circadian and hypoxia factors. Proc Natl Acad Sci USA 95:5474–5479
Hogenesch JB, Gu Y–Z, Moran SM, Shimomura K, Radcliffe LA, Takahashi JS, Bradfield CA (2000) The basic helix–loop–helix-PAS protein MOP9 is a brain-specific heterodimeric partner of circadian and hypoxia factors. J Neurosci 20:1–5
Wiesener MS, Jurgensen JS, Rosenberger C, Scholze C, Horstrup JH, Wamecke C, Mandriota S, Bechmann I, Frei UA, Pugh CW, Ratcliffe PJ, Bachmann S, Maxwell PH, Eckardt K-U (2002) Widespread, hypoxia-inducible expression of HIF-2α in distinct cell populations of different organs. FASEB J 17:271–273
Carver LA, Hogenesch JB, Bradfield CA (1994) Tissue specific expression of the rat Ah-receptor and ARNT mRNAs. Nucleic Acids Res 22:3038–3044
Drutel G, Kathmann M, Heron A, Schwartz J-C, Arrang J-M (1996) Cloning and selective expression in brain and kidney of ARNT2 homologous to the Ah receptor nuclear translocator (ARNT). Biochem Biophys Res Commun 225:333–339
Shimba S, Ishii N, Ohta Y, Ohno T, Watabe Y, Hayashi M, Wada T, Aoyagi T, Tezuka M (2005) Brain and muscle Arnt-like protein-1 (BMAL1), a component of the molecular clock, regulates adipogenesis. Proc Natl Acad Sci USA 102:12071–12076
Ryan HE, Lo J, Johnson RS (1998) HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J 17:3005–3015
Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL (1998) Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev 12:149–162
Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL (1998) The hypoxia responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev 12:3320–3324
Peng J, Zhang L, Drysdale L, Fong GH (2000) The transcription factor EPAS-1/hypoxia-inducible factor 2α plays an important role in vascular remodeling. Proc Natl Acad Sci USA 97:8386–8391
Compernolle V, Brusselmans K, Acker T, Hoet P, Tjwa M, Beck H, Plaisance S, Dor Y, Keshet E, Lupu F, Nemery B, Dewerchin M, Van Veldhoven P, Plate K, Moons L, Collen D, Carmeliet P (2002) Loss of HIF-2α and inhibition of VEGF impair fetal lung maturation, whereas treatment with VEGF prevents fatal respiratory distress in premature mice. Nat Med 8:702–710
Scortegagna M, Ding K, Oktay Y, Gaur A, Thurmond F, Yan L-J, Marck BT, Matsumoto AM, Shelton JM, Richardson JA, Bennett MJ, Garcia JA (2003) Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1 −/− mice. Nat Genet 35:331–340
Kozak KR, Abbott B, Hankinson O (1997) ARNT-deficient mice and placental differentiation. Dev Biol 191:247–305
Keith B, Adelman DM, Simon MC (2001) Targeted mutation of the murine arhylhydrocarbon receptor nuclear translocator 2 (Arnt2) gene reveals partial redundancy with Arnt. Proc Natl Acad Sci USA 98:6692–6697
Hu CJ, Sataur A, Wang L, Chen H, Simon MC (2007) The N-terminal transactivation domain confers target gene specificity of hypoxia-inducible factors HIF-1alpha and HIF-2alpha. Mol Biol Cell 18:4528–4542
Sowter HM, Raval RR, Moore J, Ratcliffe PJ, Harris AL (2003) Predominant role of hypoxia-inducible transcription factor (Hif)-1α versus Hif-2α in regulation of the transcription. Cancer Res 63:6130–6134
Raval RR, Lau KW, Tran MG, Sowter HM, Mandriota SJ, Li JL, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ (2005) Contrasting properties of hypoxia-inducible factor 1 (hif-1) and hif-2 in von Hippel-Lindau-associated renal cell carcinoma. Mol Cell Biol 25:5675–5686
Kondo K, Kim WY, Lechpammer M, Kaelin WG Jr (2003) Inhibition of HIF2α is sufficient to suppress pVHL-defective tumor growth. PLoS Biol 1:439–444
Kondo K, Kico J, Nakamura E, Lechpammer M, Kaelin WGJ (2002) Inhibition of HIF is necessary for tumor suppression by the von Hippel-Lindau protein. Cancer Cell 1:237–246
Maranchie JK, Vasselli JR, Riss J, Bonifacino JS, Linehan WM, Klausner RD (2002) The contribution of VHL substrate binding and HIF1-α to the phenotype of VHL loss in renal cell carcinoma. Cancer Cell 1:247–255
Mandriota SJ, Turner KJ, Davies DR, Murray PG, Morgan NV, Sowter HM, Wykoff CC, Maher ER, Harris AL, Ratcliffe PJ, Maxwell PH (2002) HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 1:459–468
Covello KL, Simon MC, Keith B (2005) Targeted replacement of hypoxia-inducible factor-1alpha by a hypoxia-inducible factor-2alpha knock-in allele promotes tumor growth. Cancer Res 65:2277–2286
Brusselmans K, Bono F, Maxwell P, Dor Y, Dewerchin M, Collen D, Herbert JM, Carmeliet P (2001) Hypoxia-inducible factor-2α (HIF-2α) is involved in the apoptotic response to hypoglycemia but not to hypoxia. J Biol Chem 276:39192–391966
Lau KW, Tian YM, Raval RR, Ratcliffe PJ, Pugh CW (2007) Target gene selectivity of hypoxia-inducible factor-alpha in renal cancer cells is conveyed by post-DNA-binding mechanisms. Br J Cancer 96:1284–1292
Dayan F, Roux D, Brahimi-Horn MC, Pouyssegur J, Mazure NM (2006) The oxygen sensor factor-inhibiting hypoxia-inducible factor-1 controls expression of distinct genes through the bifunctional transcriptional character of hypoxia-inducible factor-1alpha. Cancer Res 66:3688–3698
Gordan JD, Bertout JA, Hu CJ, Diehl JA, Simon MC (2007) HIF-2alpha promotes hypoxic cell proliferation by enhancing c-myc transcriptional activity. Cancer Cell 11:335–347
Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA (1998) Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3alpha. Gene Expr 7:205–213
Hara S, Hamada J, Kobayashi C, Kondo Y, Imura N (2001) Expression and characterization of hypoxia-inducible factor (HIF)-3alpha in human kidney: suppression of HIF-mediated gene expression by HIF-3alpha. Biochem Biophys Res Commun 287:808–813
Young RM, Wang SJ, Gordan JD, Ji X, Liebhaber SA, Simon MC (2008) Hypoxia-mediated selective mRNA translation by an internal ribosome entry site-independent mechanism. J Biol Chem 283:16309–16319
Kallio PJ, Okamoto K, O’Brien S, Carrero P, Makino Y, Tanaka H, Poellinger L (1998) Signal transduction in hypoxic cells: inducible nuclear translocation and recruitment of the CBP/p300 coactivator by the hypoxia-inducible factor-1α. EMBO J 17:6573–6586
Salceda S, Caro J (1997) Hypoxia-inducible factor 1α (HIF-1α) protein is rapidly degraded by the ubiquitin–proteasome system under normoxic conditions. J Biol Chem 272:22642–22647
Tanimoto K, Makino Y, Pereira T, Poellinger L (2000) Mechanism of regulation of the hypoxia-inducible factor-1α by the von Hippel-Lindau tumor suppressor protein. EMBO J 19:4298–4309
Paltoglou S, Roberts BJ (2007) HIF-1alpha and EPAS ubiquitination mediated by the VHL tumour suppressor involves flexibility in the ubiquitination mechanism, similar to other RING E3 ligases. Oncogene 26:604–609
Maxwell PH, Wiesener MS, Chang G-W, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399:271–275
Clifford SC, Cockman ME, Smallwood AC, Mole DR, Woodward ER, Maxwell PH, Ratcliffe PJ, Maher ER (2001) Contrasting effects on HIF-1α regulation by disease-causing pVHL mutations correlate with patterns of tumourigenesis in von Hippel-Lindau disease. Hum Mol Genet 10:1029–1038
Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG (2000) Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol 2:423–427
Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH (2000) Hypoxia inducible factor-α binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem 275:25733–25741
Hoffman MA, Ohh M, Yang H, Klco JM, Ivan M, Kaelin WGJ (2001) von Hippel-Lindau protein mutants linked to type 2C VHL disease preserve the ability to downregulate HIF. Hum Mol Genet 10:1019–1027
Ang SO, Chen H, Hirota K, Gordeuk VR, Jelinek J, Guan Y, Liu E, Sergueeva AI, Miasnikova GY, Mole D, Maxwell PH, Stockton DW, Semenza GL, Prchal JT (2002) Disruption of oxygen homeostasis underlies congenital Chuvash polycythemia. Nat Genet 32:614–621
Jaakkola P, Mole DR, Tian Y-M, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, von Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Targeting of HIF-α to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292:468–472
Epstein ACR, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian Y-M, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ (2001) C. elegans EGL-9 and mammalian homologues define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107:43–54
Masson N, Willam C, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Independent function of two destruction domains in hypoxia-inducible factor-α chains activated by prolyl hydroxylation. EMBO J 20:5197–5206
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WGJr (2001) HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292:464–468
Bruick RK, McKnight SL (2001) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294:1337–1340
Hon WC, Wilson MI, Harlos K, Claridge TD, Schofield CJ, Pugh CW, Maxwell PH, Ratcliffe PJ, Stuart DI, Jones EY (2002) Structural basis for the recognition of hydroxyproline in HIF-1α by pVHL. Nature 417:975–978
Lando D, Peet DJ, Whelan DA, Gorman JJ, Whitelaw ML (2002) Asparagine hydroxylation of the HIF transactivation domain: a hypoxic switch. Science 295:858–861
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK (2002) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 16:1466–1471
Hewitson KS, McNeill LA, Riordan MV, Tian Y-M, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, Ratcliffe PJ, Pugh CW, Schofield CJ (2002) Hypoxia inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J Biol Chem 277:26351–26355
Elkins JM, Hewitson KS, McNeill LA, Seibel JF, Schlemminger I, Pugh CW, Ratcliffe PJ, Schofield CJ (2003) Structure of factor-inhibiting hypoxia-inducible factor (HIF) reveals mechanism of oxidative modification of HIF-1α. J Biol Chem 278:1802–1806
McDonough MA, Li V, Flashman E, Chowdhury R, Mohr C, Lienard BM, Zondlo J, Oldham NJ, Clifton IJ, Lewis J, McNeill LA, Kurzeja RJ, Hewitson KS, Yang E, Jordan S, Syed RS, Schofield CJ (2006) Cellular oxygen sensing: crystal structure of hypoxia-inducible factor prolyl hydroxylase (PHD2). Proc Natl Acad Sci USA 103:9814–9819
Wenger RH, Stiehl DP, Camenisch G (2005) Integration of oxygen signaling at the consensus HRE. Science signal 306:re12
Ivan M, Harris AL, Martelli F, Kulshreshtha R (2008) Hypoxia response and microRNAs: no longer two separate worlds. J Cell Mol Med 12:1426–1431
Centanin L, Ratcliffe PJ, Wappner P (2005) Reversion of lethality and growth defects in fatiga oxygen-sensor mutant flies by loss of hypoxia-inducible factor-alpha/sima. EMBO Rep 6:1070–1075
Koivunen P, Tiainen P, Hyvarinen J, Williams KE, Sormunen R, Klaus SJ, Kivirikko KI, Myllyharju J (2007) An endoplasmic reticulum transmembrane prolyl 4-hydroxylase is induced by hypoxia and acts on hypoxia-inducible factor alpha. J Biol Chem 282:30544–30552
Lancaster DE, McNeill LA, McDonough MA, Aplin RT, Hewitson KS, Pugh CW, Ratcliffe PJ, Schofield CJ (2004) Disruption of dimerization and substrate phosphorylation inhibit factor inhibiting hypoxia-inducible factor (FIH) activity. Biochem J 383:429–437
Hirsila M, Koivunen P, Gunzler V, Kivirikko KI, Myllyharju J (2003) Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor HIF. J Biol Chem 278:30772–30780
Koivunen P, Hirsila M, Kivirikko KI, Myllyharju J (2006) The length of peptide substrates has a marked effect on hydroxylation by the hypoxia-inducible factor prolyl 4 hydroxylases. J Biol Chem 281:28712–28720
Ehrismann D, Flashman E, Genn DN, Mathioudakis N, Hewitson KS, Ratcliffe PJ, Schofield CJ (2007) Studies on the activity of the hypoxia-inducible factor hydroxylases using an oxygen consumption assay. Biochem J 401:227–234
Stolze IP, Tian YM, Appelhoff RJ, Turley H, Wykoff CC, Gleadle JM, Ratcliffe PJ (2004) Genetic analysis of the role of the asparaginyl hydroxylase FIH in regulating HIF transcriptional target genes. J Biol Chem 279:42719–42725
Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J (2003) HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J 22:4082–4090
Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM (2004) Differential function of the prolyl hydroxylases PHD1, PHD2 and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem 279:38458–38465
Tian YM, Mole DR, Ratcliffe PJ, Gleadle JM (2006) Characterization of different isoforms of the HIF prolyl hydroxylase PHD1 generated by alternative initiation. Biochem J 397:179–186
Habelhah H, Laine A, Erdjument-Bromage H, Tempst P, Gershwin ME, Bowtell DD, Ronai Z (2004) Regulation of 2-oxoglutarate (alpha-ketoglutarate) dehydrogenase stability by the RING finger ubiquitin ligase Siah. J Biol Chem 279:53782–53788
Nakayama K, Frew IJ, Hagensen M, Skals M, Habelhah H, Bhoumik A, Kadoya T, Erdjument-Bromage H, Tempst P, Frappell PB, Bowtell DD, Ronai Z (2004) Siah2 regulates stability of prolyl-hydroxylases, controls HIF1α abundance, and modulates physiological responses to hypoxia. Cell 117:941–952
Fukuba H, Yamashita H, Nagano Y, Jin HG, Hiji M, Ohtsuki T, Takahashi T, Kohriyama T, Matsumoto M (2007) Siah-1 facilitates ubiquitination and degradation of factor inhibiting HIF-1alpha (FIH). Biochem Biophys Res Commun 353:324–329
Barth S, Nesper J, Hasgall PA, Wirthner R, Nytko KJ, Edlich F, Katschinski DM, Stiehl DP, Wenger RH, Camenisch G (2007) The peptidyl prolyl cis/trans isomerase FKBP38 determines hypoxia-inducible transcription factor prolyl-4-hydroxylase PHD2 protein stability. Mol Cell Biol 27:3758–3768
Knowles HJ, Raval RR, Harris AL, Ratcliffe PJ (2003) Effect of ascorbate on the activity of hypoxia inducible factor (HIF) in cancer cells. Cancer Res 63:1764–1768
Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Merino M, Trepel J, Zbar B, Toro J, Ratcliffe PJ, Lineham M, Neckers L (2005) HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 8:143–153
Ginouves A, Ilc K, Macias N, Pouyssegur J, Berra E (2008) PHDs overactivation during chronic hypoxia “desensitizes” HIFalpha and protects cells from necrosis. Proc Natl Acad Sci USA 105:4745–4750
Wax SD, Tsao L, Lieb ME, Fallon JT, Taubman MB (1996) SM-20 is a novel 40-kd protein whose expression in the arterial wall is restricted to smooth muscle. Lab Invest 74:797–808
Willam C, Maxwell PH, Nichols L, Lygate C, Tian YM, Bernhardt W, Wiesener M, Ratcliffe PJ, Eckardt KU, Pugh CW (2006) HIF prolyl hydroxylases in the rat; organ distribution and changes in expression following hypoxia and coronary artery ligation. J Mol Cell Cardiol 41:68–77
Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C, Hellwig-Burgel T, Jelkmann W, Acker H, Fandrey J (2002) Intracellular localisation of human HIF-1α hydroylases: implications for oxygen sensing. J Cell Sci 116:1319–1326
Percy MJ, Zhao Q, Flores A, Harrison C, Lappin TR, Maxwell PH, McMullin MF, Lee FS (2006) A family with erythrocytosis establishes a role for prolyl hydroxylase domain protein 2 in oxygen homeostasis. Proc Natl Acad Sci USA 103:654–659
Percy MJ, Furlow PW, Beer PA, Lappin TR, McMullin MF, Lee FS (2007) A novel erythrocytosis-associated PHD2 mutation suggests the location of a HIF binding groove. Blood 110:2193–2196
Takeda K, Ho V, Takeda H, Duan LJ, Nagy A, Fong GH (2006) Placental but not heart defect is associated with elevated hif{alpha} levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol 26:8336–8346
Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG Jr (2008) Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood 111:3236–3244
Aragones J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, Lambrechts D, Bishop T, Lafuste P, Diez-Juan A, Harten SK, Van Noten P, De Bock K, Willam C, Tjwa M, Grosfeld A, Navet R, Moons L, Vandendriessche T, Deroose C, Wijeyekoon B, Nuyts J, Jordan B, Silasi-Mansat R, Lupu F, Dewerchin M, Pugh C, Salmon P, Mortelmans L, Gallez B, Gorus F, Buyse J, Sluse F, Harris RA, Gnaiger E, Hespel P, Van Hecke P, Schuit F, Van Veldhoven P, Ratcliffe P, Baes M, Maxwell P, Carmeliet P (2008) Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet 40:170–180
Bishop T, Gallagher D, Pascual A, Lygate CA, de Bono JP, Nicholls LG, Ortega-Saenz P, Oster H, Wijeyekoon B, Sutherland AI, Grosfeld A, Aragones J, Schneider M, van Geyte K, Teixeira D, Diez-Juan A, Lopez-Barneo J, Channon KM, Maxwell PH, Pugh CW, Davies AM, Carmeliet P, Ratcliffe PJ (2008) Abnormal sympathoadrenal development and systemic hypotension in PHD3−/− mice. Mol Cell Biol 28:3386–3400
Kuznetsova AV, Meller J, Schnell PO, Nash JA, Ignacak ML, Sanchez Y, Conaway JW, Conaway RC, Czyzyk-Krzeska MF (2003) von Hippel-Lindau protein binds hyperphosphorylated large subunit of RNA polymerase II through a proline hydroxylation motif and targets it for ubiquitination. Proc Natl Acad Sci USA 100:2706–2711
Mikhaylova O, Ignacak ML, Barankiewicz TJ, Harbaugh SV, Yi Y, Maxwell PH, Schneider M, Van Geyte K, Carmeliet P, Revelo MP, Wyder M, Greis KD, Meller J, Czyzyk-Krzeska MF (2008) The von Hippel-Lindau tumor suppressor protein and Egl-9-Type proline hydroxylases regulate the large subunit of RNA polymerase II in response to oxidative stress. Mol Cell Biol 28:2701–2717
Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, Godson C, Nielsen JE, Moynagh P, Pouyssegur J, Taylor CT (2006) Prolyl hydroxylase-1 negatively regulates I{kappa}B kinase-beta, giving insight into hypoxia-induced NF{kappa}B activity. Proc Natl Acad Sci USA 103:18154–18159
Xie L, Xiao K, Whalen EJ, Forrester MT, Freeman RS, Fong G, Gygi SP, Lefkowitz RJ, Stamler JS (2009) Oxygen-regulated beta(2)-adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by VHL. Sci Signal 2:ra33
Cockman ME, Lancaster DE, Stolze IP, Hewitson KS, McDonough MA, Coleman ML, Coles CH, Yu X, Hay RT, Ley SC, Pugh CW, Oldham NJ, Masson N, Schofield CJ, Ratcliffe PJ (2006) Posttranslational hydroxylation of ankyrin repeats in I{kappa}B proteins by the hypoxia-inducible factor (HIF) asparaginyl hydroxylase, factor inhibiting HIF (FIH). Proc Natl Acad Sci USA 103:14767–14772
Coleman ML, McDonough MA, Hewitson KS, Coles C, Mecinovic J, Edelmann M, Cook KM, Cockman ME, Lancaster DE, Kessler BM, Oldham NJ, Ratcliffe PJ, Schofield CJ (2007) Asparaginyl hydroxylation of the Notch ankyrin repeat domain by factor inhibiting hypoxia-inducible factor. J Biol Chem 282:24027–24038
Ferguson JE 3rd, Wu Y, Smith K, Charles P, Powers K, Wang H, Patterson C (2007) ASB4 is a hydroxylation substrate of FIH and promotes vascular differentiation via an oxygen-dependent mechanism. Mol Cell Biol 27:6407–6419
Zheng X, Linke S, Dias JM, Zheng X, Gradin K, Wallis TP, Hamilton BR, Gustafsson M, Ruas JL, Wilkins S, Bilton RL, Brismar K, Whitelaw ML, Pereira T, Gorman JJ, Ericson J, Peet DJ, Lendahl U, Poellinger L (2008) Interaction with factor inhibiting HIF-1 defines an additional mode of cross-coupling between the Notch and hypoxia signaling pathways. Proc Natl Acad Sci USA 105:3368–3373
Gradin K, Takasaki C, Fujii-Kuriyama Y, Sogawa K (2002) The transcriptional activation function of the HIF-like factor requires phosphorylation at a conserved threonine. J Biol Chem 277:23508–23514
Sang N, Stiehl DP, Bohensky J, Leshchinsky I, Srinivas V, Caro J (2003) Mitogen-activated protein kinase (MAPK) signaling up-regulates the activity of hypoxia-inducible factors by its effects on p300. J Biol Chem 278:14013–14019
Mylonis I, Chachami G, Samiotaki M, Panayotou G, Paraskeva E, Kalousi A, Georgatsou E, Bonanou S, Simos G (2006) Identification of MAPK phosphorylation sites and their role in the localization and activity of hypoxia-inducible factor-1alpha. J Biol Chem 281:33095–33106
Mylonis I, Chachami G, Paraskeva E, Simos G (2008) An atypical CRM1-dependent nuclear export signal mediates regulation of hypoxia-inducible factor HIF-1alpha by MAPK. J Biol Chem 283:27620–27627
Brugarolas JB, Vazquez F, Reddy A, Sellers WR, Kalin WGJ (2003) TSC2 regulates VEGF through mTOR-dependent and -independent pathways. Cancer Cell 4:147–157
Brugarolas J, Kaelin WG Jr (2004) Dysregulation of HIF and VEGF is a unifying feature of the familial hamartoma syndromes. Cancer Cell 6:7–10
Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WG Jr (2004) Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev 18:2893–2904
Jeong J-W, Bae M–K, Ahn M-Y, Kim S–H, Sohn T-K, Bae M-H, Yoo M-A, Song EJ, Lee K-J, Kim K-W (2002) Regulation and destabilization of HIF-1α by ARD1-mediated acetylation. Cell 111:709–720
Bae SH, Jeong JW, Park JA, Kim SH, Bae MK, Choi SJ, Kim KW (2004) Sumoylation increases HIF-1alpha stability and its transcriptional activity. Biochem Biophys Res Commun 324:394–400
Fisher TS, Etages SD, Hayes L, Crimin K, Li B (2005) Analysis of ARD1 function in hypoxia response using retroviral RNA interference. J Biol Chem 280:17749–17757
Bilton R, Mazure N, Trottier E, Hattab M, Dery MA, Richard DE, Pouyssegur J, Brahimi-Horn MC (2005) ARD1, an acetyltransferase, does not alter stability of hypoxia-inducible factor-1alpha and is not induced by hypoxia or HIF. J Biol Chem 280:31132–31140
Arnesen T, Kong X, Evjenth R, Gromyko D, Varhaug JE, Lin Z, Sang N, Caro J, Lillehaug JR (2005) Interaction between HIF-1 alpha (ODD) and hARD1 does not induce acetylation and destabilization of HIF-1 alpha. FEBS Lett 579:6428–6432
Berta MA MN, Hattab M, Pouysségur J, Brahimi-Horn MC (2007) SUMOylation of hypoxia-inducible factor-1alpha reduces its transcriptional activity. Biochem Biophys Res Commun 360:646–652
Cash TP, Pan Y, Simon MC (2007) Reactive oxygen species and cellular oxygen sensing. Free Radic Biol Med 43:1219–1225
Sogawa K, Numayama-Tsuruta K, Ema M, Abe M, Abe H, Fujii-Kuriyama Y (1998) Inhibition of hypoxia-inducible factor 1 activity by nitric oxide donors in hypoxia. Proc Natl Acad Sci USA 95:7368–7373
Sandau KB, Zhou J, Kietzmann T, Brune B (2001) Regulation of the hypoxia-inducible factor 1α by the inflammatory mediators nitric oxide and tumor necrosis factor-α in contrast to desferroxamine and phenylarsine oxide. J Biol Chem 276:39805–39811
Wang F, Sekine H, Kikuchi Y, Takasaki C, Miura C, Heiwa O, Shuin T, Fujii-Kuriyama Y, Sogawa K (2002) HIF-1a-prolyl hydroxylase: molecular target of nitric oxide in the hypoxic signal transduction pathway. Biochem Biophys Res Commun 295:657–662
Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B (2003) Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell 14:3470–3481
Berchner-Pfannschmidt U, Yamac H, Trinidad B, Fandrey J (2007) Nitric oxide modulates oxygen sensing by hypoxia-inducible factor 1-dependent induction of prolyl hydroxylase 2. J Biol Chem 282:1788–1796
Hagen T, Taylor CT, Lam F, Moncada S (2003) Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF-1α. Science 302:1975–1978
Baek JH, Mahon PC, Oh J, Kelly B, Krishnamachary B, Pearson M, Chan DA, Giaccia AJ, Semenza GL (2005) OS-9 interacts with hypoxia-inducible factor 1alpha and prolyl hydroxylases to promote oxygen-dependent degradation of HIF-1alpha. Molecular Cell 17:503–512
Ozer A, Wu LC, Bruick RK (2005) The candidate tumor suppressor ING4 represses activation of the hypoxia inducible factor (HIF). Proc Natl Acad Sci USA 102:7481–7486
Hopfer U, Hopfer H, Jablonski K, Stahl RA, Wolf G (2006) The novel WD-repeat protein Morg1 acts as a molecular scaffold for hypoxia-inducible factor prolyl hydroxylase 3 (PHD3). J Biol Chem 281:8645–8655
Masson N, Appelhoff RJ, Tuckerman JR, Tian YM, Demol H, Puype M, Vandekerckhove J, Ratcliffe PJ, Pugh CW (2004) The HIF prolyl hydroxylase PHD3 is a potential substrate of the TRiC chaperonin. FEBS Lett 570:166–170
Bracken CP, Whitelaw ML, Peet DJ (2005) Activity of hypoxia-inducible factor 2 alpha (HIF-2 alpha) is regulated by association with the NF-kappa B essential modulator (NEMO/IKK gamma). J Biol Chem 280:14240–14251
Bhattacharya S, Michels CL, Leung M–K, Arany ZP, Kung AL, Livingston DM (1999) Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev 13:64–75
Alam NA, Rowan AJ, Wortham NC, Pollard PJ, Mitchell M, Tyrer JP, Barclay E, Calonje E, Manek S, Adams SJ, Bowers PW, Burrows NP, Charles-Holmes R, Cook LJ, Daly BM, Ford GP, Fuller LC, Hadfield-Jones SE, Hardwick N, Highet AS, Keefe M, MacDonald-Hull SP, Potts EDA, Crone M, Wilkinson S, Camacho-Martinez F, Jablonska S, Ratnavel R, MacDonald A, Mann RJ, Grice K, Guillet G, Lewis-Jones MS, McGrath H, Seukeran DC, Morrison PJ, Fleming S, Rahman S, Kelsell D, Leigh I, Olpin S, Tomlinson IPM (2003) Genetic and functional analyses of FH mutations in multiple cutaneous and uterine leiomyomatosis, heritary leiomyomatosis and renal cancer, and fumarate hydratase deficiency. Hum Mol Genet 12:1241–1252
Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, Tomlinson IP (2005) Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum Mol Genet 14:2231–2239
Pollard PJ, Spencer-Dene B, Shukla D, Howarth K, Nye E, El-Bahrawy M, Deheragoda M, Joannou M, MacDonald S, Martin A, Igarashi P, Varsani-Brown S, Rosewell I, Poulsom R, Maxwell PH, Stamp GW, Tomlinson IP (2007) Targeted inactivation of Fh1 causes proliferative renal cyst development and activation of the hypoxia pathway. Cancer Cell 11:311–319
Thrash-Bingham CA, Tartof KD (1999) aHIF: a natural antisense transcript overexpressed in human renal cancer and during hypoxia. J Natl Cancer Inst 91:143–151
Semenza GL (2001) Hypoxia-inducible factor 1: control of oxygen homeostasis in health and disease. Pediatr Res 49:614–617
Semenza GL (2001) Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med 7:345–350
Vincent KA, Shyu KG, Luo Y, Magner M, Tio RA, Jiang C, Goldberg MA, Akita GY, Gregory RJ, Isner JM (2000) Angiogenesis is induced in a rabbit model of hindlimb ischemia by naked DNA encoding an HIF-1a/VP16 hybrid transcription factor. Circulation 102:2255–22561
Shyu KG, Wang MT, Wang BW, Chang CC, Leu JG, Kuan P, Chang H (2002) Intramyocardial injection of naked DNA encoding HIF-1α/VP16 hybrid to enhance angiogenesis in an acute myocardial infarction model in the rat. Cardiovasc Res 54:576–583
Philipp S, Cui L, Ludolph B, Kelm M, Schulz R, Cohen MV, Downey JM (2006) Desferoxamine and ethyl-3, 4-dihydroxybenzoate protect myocardium by activating NOS and generating mitochondrial ROS. Am J Physiol Heart Circ Physiol 290:H450–H457
Xi L, Taher M, Yin C, Salloum F, Kukreja RC (2004) Cobalt chloride induces delayed cardiac preconditioning in mice through selective activation of HIF-1alpha and AP-1 and iNOS signaling. Am J Physiol Heart Circ Physiol 287:H2369–H2375
Nwogu NI, Greenen D, Bean M, Brenner MC, Huang X, Buttrick PM (2001) Inhibition of collagen synthesis with prolyl 4-hydroxylase inhibitor improves left ventricular function and alters the pattern of left ventricular dilatation after myocardial infarction. Circulation 104:2216–2221
Ivan M, Haberberger T, Gervasi DC, Michelson KS, Gunzler V, Kondo K, Yang H, Sorokina I, Conaway RC, Conaway JW, Kaelin WG Jr (2002) Biochemical purification and pharmacological inhibition of a mammalian prolyl hydroxylase acting on hypoxia-inducible factor. Proc Natl Acad Sci USA 99:13459–13464
Warnecke C, Griethe W, Weidemann A, Jurgensen JS, Willam C, Bachmann S, Ivashchenko Y, Wagner I, Frei U, Wiesener M, Eckardt KU (2003) Activation of the hypoxia-inducible factor-pathway and stimulation of angiogenesis by application of prolyl hydroxylase inhibitors. FASEB J 17:1186–1188
Milkiewicz M, Pugh CW, Egginton S (2004) Inhibition of endogenous HIF inactivation induces angiogenesis in ischaemic skeletal muscles of mice. J Physiol 560:21–26
Philipp S, Jurgensen JS, Fielitz J, Bernhardt WM, Weidemann A, Schiche A, Pilz B, Dietz R, Regitz-Zagrosek V, Eckardt KU, Willenbrock R (2006) Stabilization of hypoxia inducible factor rather than modulation of collagen metabolism improves cardiac function after acute myocardial infarction in rats. Eur J Heart Fail 8:347–354
Bernhardt WM, Campean V, Kany S, Jurgensen JS, Weidemann A, Warnecke C, Arend M, Klaus S, Gunzler V, Amann K, Willam C, Wiesener MS, Eckardt KU (2006) Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol 17:1970–1978
Kung AL, Wang S, Klco JM, Kaelin WG, Livingston DM (2000) Suppression of tumor growth through disruption of hypoxia-inducible transcription. Nat Med 6:1335–1340
Kung AL, Zabludoff SD, France DS, Freedman SJ, Tanner EA, Vieira A, Cornell-Kennon S, Lee J, Wang B, Wang J, Memmert K, Naegeli HU, Petersen F, Eck MJ, Bair KW, Wood AW, Livingston DM (2004) Small molecule blockade of transcriptional coactivation of the hypoxia-inducible factor pathway. Cancer Cell 6:33–43
Carroll VA, Ashcroft M (2006) Role of hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res 66:6264–6270
Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI, Dang CV, Semenza GL (2007) HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell 11:407–420
Aravind L, Koonin EV (2001) The DNA-repair protein AlkB, EGL-9, and leprecan define new families of 2-oxoglutarate- and iron-dependent dioxygenases. Genome Biol 2:RESEARCH0007
Hewitson KS, Granatino N, Welford RW, McDonough MA, Schofield CJ (2005) Oxidation by 2-oxoglutarate oxygenases: non-haem iron systems in catalysis and signalling. Philos Transact A Math Phys Eng Sci 363:807–828
Duncan T, Trewick SC, Koivisto P, Bates PA, Lindahl T, Sedgwick B (2002) Reversal of DNA alkylation damage by two human dioxygenases. Proc Natl Acad Sci USA 99:16660–16665
Trewick SC, McLaughlin PJ, Allshire RC (2005) Methylation: lost in hydroxylation? EMBO Rep 6:315–320
Valegard K, van Scheltinga AC, Lloyd MD, Hara T, Ramaswamy S, Perrakis A, Thompson A, Lee HJ, Baldwin JE, Schofield CJ, Hajdu J, Andersson I (1998) Structure of a cephalosporin synthase. Nature 394:805–809
Kivirikko KI, Myllyla R (1980) The hydroxylation of prolyl and lysyl residues. In: Freeman RB, Hawkins HC (eds) The enzymology of post-translational modification of proteins. Academic Press, London, pp 53–104
Gerken T, Girard CA, Tung YC, Webby CJ, Saudek V, Hewitson KS, Yeo GS, McDonough MA, Cunliffe S, McNeill LA, Galvanovskis J, Rorsman P, Robins P, Prieur X, Coll AP, Ma M, Jovanovic Z, Farooqi IS, Sedgwick B, Barroso I, Lindahl T, Ponting CP, Ashcroft FM, O’Rahilly S, Schofield CJ (2007) The obesity-associated FTO gene encodes a 2-oxoglutarate-dependent nucleic acid demethylase. Sciencexpress 318:1469–1472
Mukherji M, Chien W, Kershaw NJ, Clifton IJ, Schofield CJ, Wierzbicki AS, Lloyd MD (2001) Structure–function analysis of phytanoyl-CoA 2-hydroxylase mutations causing Refsum’s disease. Hum Mol Genet 10:1971–1982
Wierzbicki AS, Mitchell J, Lambert-Hammill M, Hancock M, Greenwood J, Sidey MC, de Belleroche J, Gibberd FB (2000) Identification of genetic heterogeneity in Refsum’s disease. Eur J Hum Genet 8:649–651
Whetstine JR, Nottke A, Lan F, Huarte M, Smolikov S, Chen Z, Spooner E, Li E, Zhang G, Colaiacovo M, Shi Y (2006) Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 125:467–481
Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, Zhang Y (2006) Histone demethylation by a family of JmjC domain-containing proteins. Nature 439:811–816
Frescas D, Guardavaccaro D, Bassermann F, Koyama-Nasu R, Pagano M (2007) JHDM1B/FBXL10 is a nucleolar protein that represses transcription of ribosomal RNA genes. Nature 450:309–313
Hulse JD, Ellis SR, Henderson LM (1978) Carnitine biosynthesis beta-hydroxylation of trimethyllysine by an alpha-ketoglutarate-dependent mitochondrial dioxygenase. J Biol Chem 253:1654–1659
Lindblad B, Lindstedt G, Tofft M, Lindstedt S (1969) The mechanism of alpha-ketoglutarate oxidation in coupled enzymatic reactions. J Am Chem Soc 91:4604–4606
Lindstedt G, Lindstedt S (1970) Co-factor requirements of gamma-butyrobetaine hydroxylase from rat liver. J Biol Chem 245:4178–4186
Vaz FM, Wanders RJ (2002) Carnitine biosynthesis in mammals. Biochem J 361:417–429
Hanson ES, Rawlins ML, Leibold EA (2003) Oxygen and iron regulation of iron regulatory protein 2. J Biol Chem 278:40337–40342
Acknowledgments
The authors acknowledge many uncited contributions from co-workers in the field and helpful discussions with those involved now, and in the past, with oxygen-sensing research in Oxford. Work in the authors’ laboratory has been funded by the Wellcome Trust, MRC, CRUK, BHF, European Commission and a Fellowship to MC from Jesus College, Oxford. CWP is a scientific co-founder of ReOx Ltd.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Webb, J.D., Coleman, M.L. & Pugh, C.W. Hypoxia, hypoxia-inducible factors (HIF), HIF hydroxylases and oxygen sensing. Cell. Mol. Life Sci. 66, 3539–3554 (2009). https://doi.org/10.1007/s00018-009-0147-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-009-0147-7