
Abstract
Cargo proteins exported from the endoplasmic reticulum to the Golgi apparatus are typically transported in coat protein complex II (COPII)-coated vesicles of 60–90 nm diameter. Several cargo molecules including collagens and chylomicrons form structures that are too large to be accommodated by these vesicles, but their secretion still requires COPII proteins. Here, we first review recent progress on large cargo secretions derived especially from animal models and human diseases, which indicate the importance of COPII proteins. We then discuss the recent isolation of specialized factors that modulate the process of COPII-dependent cargo formation to facilitate the exit of large-sized cargoes from the endoplasmic reticulum. Based on these findings, we propose a model that describes the importance of the GTPase cycle for secretion of oversized cargoes. Next, we summarize reports that describe the structures of COPII proteins and how these results provide insight into the mechanism of assembly of the large cargo carriers. Finally, we discuss what issues remain to be solved in the future.
Similar content being viewed by others
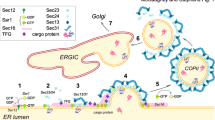

The requirement of COPII proteins for collagen export from the ER
Cargoes exiting the endoplasmic reticulum (ER) to the Golgi apparatus are packaged into coat protein complex II (COPII)-coated vesicles that typically have diameters of 60–90 nm [1]. The formation of COPII vesicles occurs in particular regions of the ER called ER exit sites, also known as transitional ER (tER), which stain as punctuated dots scattered throughout the cytoplasm by immunofluorescence analysis of mammalian cells (Fig. 1). The mechanisms to form transport vesicles is highly conserved from yeast to humans. The small GTPase Sar1 is activated by its guanine-nucleotide exchange factor (GEF), Sec12 [2–5]. After activation, Sar1 is recruited to the ER membrane [6–8] and forms the pre-budding complex [9–12], which consists of the inner coat complex Sec23/Sec24 and Sec24-bound cargo molecules [13–15]. Subsequently, the outer coat complex Sec13/Sec31 binds, and this binding event enhances the GTPase-activating protein activity of Sec23, thereby completing coat assembly [16–19]. Sec16 is the other factor essential in COPII biogenesis and functions as a scaffold to interact with particular coat proteins [20–25]. Recently, Sec16 has also been reported to negatively regulate GTP hydrolysis by Sar1 [26–28]. Additional factors involved in vesicle production have been identified, such as p125, TFG-1, and ALG2 [29–38]. The details of conventional COPII biogenesis have been reviewed extensively in other recently published articles [39–46].

Localization of ER exit sites within HeLa cells stained by antibodies against trans-membrane protein Sec12 (rat monoclonal 6B3) and cytoplasmic Sec31 (BD biosciences mouse monoclonal). HeLa cells were fixed with cold methanol and processed for immunofluorescence staining as described previously [99]. Bar 10 μm
Collagens synthesized in the ER fold into hetero- or homo-trimers, which form >300-nm-long rigid structures that are too large to fit into conventional COPII-coated vesicles [47–50]. However, imaging by fluorescent and electron microscopy indicates that collagen I exits the ER via the COPII-dependent process. Stephens and Pepperkok showed that microinjection of a GTPase-deficient mutant of Sar1a (Sar1a H79G) blocks the secretion of collagen I. Moreover, they showed that collagen I exits the ER in structures labeled with Sec24D, but segregates from vesicular stomatitis virus glycoprotein VSVG-ts045, a model of the conventional cargo proteins. The latter result implies that collagen transport to the Golgi is COPII dependent but distinct from conventional cargo trafficking [51]. Mironov et al. strengthened this finding by electron microscopy analysis, which showed that VSVG and collagen I exit the ER by a COPII-dependent process, but from distinct domains. Moreover, it was observed that protrusions from the ER domains in the vicinity of the ER exit sites form carriers containing either VSVG or collagen I. Interestingly, the formation of carriers is COPII dependent but does not seem to involve budding and fusion of COPII-coated vesicles [52].
Sec23A
The importance of COPII proteins for collagen secretion has also been suggested by analyzing human diseases and animal models (Table 1). Two point mutations in Sec23A genes (F382L and M702V) have been identified as being responsible for cranio-lenticulo-sutural dysplasia (CLSD), an autosomal recessive disorder characterized by late-closing fontanels, facial dysmorphisms, and skeletal defects [53, 54]. Fibroblasts isolated from CLSD patients showed extensive dilation of the ER and accumulation of collagen I within the ER. Both mutations are located close to the binding site of Sec31. Biochemical and structural characterization suggests that Sec23A/F382L cannot recruit Sec13/Sec31 and therefore vesicle budding does not occur [18, 55]. In contrast, Sec23A/M702V is capable of interacting with Sec13/Sec31 and has no appreciable effects on vesicle budding in vitro. Interestingly, the M702V mutation seems to enhance Sar1B GTPase activity through an interaction with Sec13/Sec31 [56]. The zebrafish crusher mutation was obtained through a chemical mutagenesis screen to identify genes involved in craniofacial development [57]. Further analysis revealed that crusher has a nonsense mutation at residue 402 of the Sec23A gene. Crusher chondrocytes have distended ER with accumulated collagen II inside [58], further supporting that Sec23A is required for collagen export from the ER.
Recently, Sec23A has been identified as a target of the ER-resident transcription factor BBF2H7, also known as Creb3L2 [59]. BBF2H7 is expressed in chondrocytes and normally degraded by the ubiquitin-proteasome pathway, but upon ER stress, the transcription factor is stabilized and transported to the Golgi, then activated by proteolysis with Golgi-localized site-1 protease (S1P) and site-2 protease (S2P). The cleaved N-terminus translocates to the nucleus to upregulate the expression of Sec23A [60–62]. BBF2H7 knockout mice were found to show severe chondrodysplasia. Chondrocytes from knockout mice have expanded ER, where collagen II and cartilage oligomeric matrix protein accumulate in large amounts [59]. A zebrafish mutant carrying a missense mutation in BBF2H7 (feelgood) also showed defects in chondrocyte development, and the accumulation of collagen II was observed in distended ER [63]. These results suggest that BBF2H7-mediated transcription activation of Sec23A is necessary for collagen transport in chondrocytes. The requirement of the BBF2H7-Sec23A pathway for collagen transport was also reported for dermal fibroblasts [64].
Sec24D
The vertebrate possesses four isoforms of Sec24, and these isoforms are considered to fulfill the demands to traffic varieties of cargo molecules, although they appear to be partially redundant in cargo recognition [65]. Recent studies in fish indicated that Sec24D is specifically important for collagen secretion from the ER. Mutagenesis screens performed in medaka and zebrafish independently led to the identification of the nonsense mutations named vbi and bulldog, respectively [57]. Both mutants showed craniofacial defects, and chondrocytes from these mutants failed to secrete collagen II and displayed dilated ER [66, 67]. Osteogenesis imperfecta, a disorder associated with reduced bone mass, increased bone fragility, and bone deformity, is caused primarily by heterozygotic mutations in genes encoding collagen I (COL1A1 or COL1A2) [68]. A recent clinical study revealed that mutations of Sec24D are also responsible for the osteogenesis imperfecta phenotype. Affected individuals either possess two missense mutations in each Sec24D allele or one missense and the other nonsense mutation. Fibroblasts from patients showed accumulation of collagen I within the dilated ER [69]. Interestingly, knockout of Sec24D in mice revealed early embryonic lethality [70]. These results imply that Sec24D-dependent cargo transport is required for early stages of development, and truncated or mutated forms of Sec24D from fish mutants and patients have marginal activity required for early development, but not sufficient for secreting collagen I from the ER. Interestingly, the expression pattern of Sec24d has been reported to change during development. It is ubiquitously expressed during the early stages of development, whereas the expression is restricted to craniofacial cartilage during later stages of development [66].
Sec13/31
Sec13 functions by forming individual complexes in different locations within cells. Sec13 is known to constitute the nuclear pore complex (NPC) [71]. However, Sec13 interacts with Sec31 to serve as an outer layer of COPII vesicles. In addition, Sec13 has been recently suggested to form a complex with Sec16, serving as a template for the formation of Sec13/31 outer coats [25].
Townley et al. first reported that zebrafish Sec13 morphants exhibit defects in craniofacial development. In mammalian cells, depletion of Sec13 by siRNA impairs collagen I secretion without affecting conventional cargo transport [72]. Interestingly, a zebrafish mutant originally identified as the small-liver phenotype in a screen was revealed to have a C-terminal truncation of Sec13, which makes it incapable of binding to Sec31 [73]. The mutant fish exhibits a hypoplastic digestive organ and small eyes with disrupted retinal lamination, and chondrocytes of the mutant showed collagen II accumulation in the dilated ER structures [74, 75]. In this context, a Sec31A knockdown by morpholino was performed in fish and showed malformation of the digestive organ, as observed in Sec13 mutants. Moreover, chondrocytes from morphants accumulate collagen II within the dilated ER. These results strongly suggest that defects in digestive organ development and collagen secretion in Sec13 mutant fish are due to the compromised COPII function. The mutation in NPC component Nup107 exhibits failure of retinal lamination, although knockdown of both Sec31A and Sec31B or treatment with brefeldin A, an inhibitor of ER to Golgi trafficking, has no effect on eye development. Thus, the function of Sec13 in the NPC complex appears to be necessary for retinal development [76].
As evidence accumulates, there is no doubt that collagen secretion from the ER requires COPII components. However, these results are not sufficient to conclude whether collagen is directly transported by modified COPII-coated structures, which can accommodate large-sized cargoes, or the COPII requirement for oversized-cargo secretion is limited and indirect. Recently, several molecules associated with ER exit sites have been identified to be specifically required for large cargo secretion and some models have been proposed. We will focus on this topic in the next section.
Components specifically required for collagen secretion
TANGO1
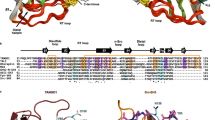
Genome-wide screening in Drosophila S2 cells revealed several genes involved in protein secretion and Golgi morphology [77]. Transport ANd Golgi Organization 1 (TANGO1), also known as Melanoma Inhibitory Activity 3 (MIA3), was isolated in this screening as being involved in the ER to Golgi trafficking. TANGO1, a multi domain-containing protein, with a non-canonical SH3 domain, trans-membrane regions, two coiled-coil domains, and proline-rich domain (PRD), is only conserved through metazoans (Fig. 2). TANGO1 is localized at ER exit sites with the SH3 domain facing into the luminal side and PRD to the cytoplasmic side. Luminal SH3-like folds in MIA-family proteins have unique structural properties when compared with canonical cytoplasmic SH3 domains [78]. PRD of TANGO1 has been shown to interact with Sec23/Sec24, probably in a manner similar to the binding of Sec31 with Sec23/Sec24, because PRD of Sec31 is responsible for the interaction with Sec23/Sec24 [79, 80]. The SH3 domain of TANGO1 is capable of interacting with collagen VII, and a knockdown of TANGO1 impairs collagen VII export from the ER without affecting general transport of proteins. These data suggest that TANGO1 acts as a cargo receptor for collagen VII at ER exit sites [81]. Of note, TANGO1 is not required for collagen I secretion in cultured cells, suggesting that the role of TANGO1 as a cargo receptor is limited to a certain set of molecules and not for all oversized cargoes. In an in vitro vesicle budding assay, TANGO1 was shown to not be exported from the ER along with collagens. In contrast, conventional cargo receptors do exit the ER together with cargo proteins within COPII-coated vesicles. Thus, TANGO1 may employ a unique mechanism for exporting large cargoes from the ER.

The domain organization of MIA2, cTAGE5, and TANGO1
TANGO1 knockout mice have been made and exhibit chondrodysplasia, which leads to dwarfing of the fetus, peripheral edema, and neonatal lethality. These phenotypes are probably due to the intracellular accumulation of collagens and the induction of the strong unfolded protein response, especially in the developing skeleton. The TANGO1 knockout was found to show defects in secretion and part of the maturation of collagen I, II, III, IV, VII, and IX from chondrocytes, fibroblasts, endothelial cells, and mural cells [82]. Unlike TANGO1 depletion by siRNA, the TANGO1 knockout inhibited collagen I and VII export from the ER. It is interesting to note that the transcriptional block of the collagen I gene in mice by retrovirus insertion led to embryonic lethality [83], which is a more severe phenotype than the TANGO1 knockout. Whether TANGO1 has direct roles in the secretion and maturation of a broad range of collagens remains to be investigated; however, we propose that the phenotype of knockout animals is an accumulative outcome because of impaired secretion of a limited number of collagens. Of note, it has been reported that TANGO1 in drosophila is also involved in collagen IV secretion from the ER [84, 85].
cTAGE5
Cutaneous T cell lymphoma-associated antigen 5 (cTAGE5), also known as meningioma-expressed antigen-6 (MGEA6), which was originally isolated as tumor-specific antigens for several types of cancer, is a close homolog to TANGO1 in mammalian cells [86–89]. Although cTAGE5 lacks the N-terminal long luminal stretch when compared with TANGO1, it contained a trans-membrane region, two coiled-coil domains, and a PRD located at the C-terminus, and it localizes to the ER exit sites (Fig. 2). The PRD of cTAGE5 also binds to Sec23/Sec24. cTAGE5 directly interacts with TANGO1 through one of the coiled-coil domains. Cells depleted of cTAGE5 by siRNA accumulate collagen VII within the ER, suggesting that cTAGE5 acts as a co-receptor of TANGO1 at ER exit sites [90].
cTAGE5 is conserved throughout vertebrates and forms a multigene family with nine pseudogenes in humans and possibly in other primates [91]. cTAGE5 expression is fairly ubiquitous, but tissue-specific alternative splicing produces longer forms of cTAGE5, designated as MIA2 (Fig. 2). MIA2 expression is only restricted to hepatocytes, and like TANGO1, it contains an N-terminal SH3-like fold. Mice possessing point mutations in MIA2 have lower circulating VLDL, LDL, HDL, and triglycerides [92]. Further investigation is required to determine whether MIA2 acts as a cargo receptor for oversized cargo. MIA2 is known to have a shorter secreted form, and its role in carcinogenesis has been reported (Fig. 2) [93–97].
Based on the experimental data described above, a model for collagen VII export by the cTAGE5/TANGO1 complex has been proposed (Fig. 3) [81, 90, 98]. The cTAGE5/TANGO1 complex at ER exit sites binds to collagen VII via the luminal SH3-like fold of TANGO1 and Sec23/Sec24 through the PRDs of both cTAGE5 and TANGO1. The interaction of these two PRDs with Sec23/Sec24 inhibits the recruitment of Sec13/Sec31, thereby delaying the Sar1 GTP hydrolysis required for vesicle formation. Once collagen VII is accommodated in a COPII carrier of the right size, collagen VII may dissociate from TANGO1, which would lead to the dissociation of the PRDs from Sec23/Sec24. Sec13/Sec31 may then be recruited and complete the carrier formation. Further experimental validation is required for proving this hypothesis.

Schematic of conventional COPII-vesicle budding and cTAGE5/TANGO1-mediated collagen export. Collagen secretion may require tight regulation of the Sar1 GTPase cycle. The cTAGE5/Sec12 complex efficiently activates Sar1, whereas TANGO1/Sedlin enhances its hydrolysis for collagen export
Sec12
Immunoprecipitation following mass spectrometry analysis revealed Sec12 as a new binding partner of cTAGE5 [99]. Sec12, also known as prolactin regulatory element-binding protein in mammalian cells, is a type II transmembrane protein with WD-40 folds and acts as a GEF for Sar1 [5, 100]. Sec12 binds directly to one of the coiled-coil domains of cTAGE5, and this interaction does not exert any changes to the GEF activity of Sec12 toward Sar1. The interaction, however, is necessary for Sec12 to correctly localize to the ER exit sites, as a knockdown of cTAGE5 leads to the dispersion of Sec12 throughout the ER. Interestingly, the cTAGE5 knockdown inhibits collagen VII secretion, but has no effects on general protein secretion, indicating that conventional cargoes can be secreted as long as Sec12 is present within the ER. Thus, Sec12 recruitment to the ER exit sites by interaction with cTAGE5 appears to be specifically required for collagen VII to exit from the ER [99]. As Sec12 is a GEF for Sar1, these data imply that collagen export from the ER requires high levels of activated Sar1 in the vicinity of ER exit sites (Fig. 3).
Sly1-syntaxin18
Nogueira et al. recently reported that Sly1 interacts with the cytoplasmic domain of TANGO1 in the presence of a crosslinker. Sly1, one of the Sec1/Munc18 (SM) proteins involved in the membrane-fusion reaction, is known to interact with ER-specific target-soluble N-ethylmaleimide-sensitive fusion protein-attachment proteins (t-SNAREs), syntaxin17 and syntaxin18 [101, 102]. A knockdown of Sly1 or syntaxin18 specifically blocks secretion of collagen VII, but not collagen I or other conventional cargoes exported from the ER [103]. Recently, a model of collagen VII transport incorporating these findings was reported, suggesting that sly1-syntaxin18-dependent fusion of recycling membranes such as the ER-Golgi intermediate compartment is responsible for enlarging the COPII-mediated carrier, in which formation would be triggered by the action of cTAGE5/TANGO1 [103, 104].
Cul3-KLHL12
Jin et al. recently reported that ubiquitylation of COPII components is involved in large cargo secretions. Mouse ES cells depleted with ubiquitin ligase CUL3 form tightly packed cell clusters, suggesting the aberrant deposition of the extracellular matrix (ECM). KLHL12 was isolated as an adaptor of CUL3, which binds and shows a similar phenotype to CUL3 when knocked down in ES cells. Interestingly, CUL3-KLHL12 monoubiquitylates Sec31, and this ubiquitylation promotes the formation of enlarged COPII-coated structures (200–500 nm in diameter) where KLHL12 is also present. In addition, the formation of these enlarged COPII structures is required for collagen I and IV transport [105].
Sedlin
Sedlin, also known as TRAPPC2, is a component of the TRAnsport Protein Particle (TRAPP) complex, which is involved in the tethering of vesicles during ER to Golgi and intra-Golgi transport [106]. Sedlin has been identified as a gene mutated in spondyloepiphyseal dysplasia tarda, an X-linked skeletal disorder characterized by disproportionately short stature with a short trunk and degenerative joints, and chondrocytes from patients show impaired secretion of ECM molecules [107]. Venditti et al. recently showed that Sedlin is localized to the ER exit sites by interaction with TANGO1 and seems to directly interact with the GTP-bound form of Sar1. Knockdown of Sedlin leads to the accumulation of an activated form of Sar1 at ER exit sites and specifically blocks the secretion of collagen I and II from chondrocytes and fibroblasts. The authors suggest that Sedlin regulates the Sar1 GTPase cycle for controlling collagen exit from the ER (Fig. 3) [108].
As described above, collagen secretion from the ER appears to be regulated by specialized factors, which would modify the function of conventional COPII proteins. A cargo receptor complex cTAGE5/TANGO1 is proposed to regulate the Sar1 GTPase cycle by interacting with Sec12, in addition to the binding to Sec23/Sec24 for possible competition with Sec13/Sec31; CUL3-KLHL12 monoubiquitylates Sec31 for enlarging the carriers, and molecules engaged in tethering and fusion, Sedlin and Sly1-syntaxin18, are also involved in the secretion of large proteins.
Chylomicron secretion
Sar1B
Chylomicrons synthesized in the enterocyte ER differ in size under different conditions (75–450 nm), but some are considered to be larger than conventional COPII vesicles [109, 110]. Chylomicron retention disease (CMRD), Anderson disease, and CMRD with the neuromuscular disorder Marinesco-Sjogren syndrome (MSS) are all inherited disorders of severe fat malabsorption with impaired chylomicron transport and found to be associated with mutations in Sar1B (Table 1) [111]. The mutations in these diseases mostly lie in the nucleotide binding pockets of Sar1B, indicating the importance of the Sar1 GTPase cycle [111–113]. The zebrafish model of Sar1B deficiency based on a morpholino knockdown showed a similar phenotype, where dietary lipids accumulate in enterocytes of mutant fish [114]. Of note, the fish also exhibit defects in craniofacial cartilage associated with abnormal collagen II secretion, although these defects are not normally seen with CMRD and related diseases.
Interestingly, Sar1B appears to have unique characteristics when compared with the function of Sar1A. Based on the biochemical analysis of a CLSD mutant, Sar1B has been suggested to have a weaker affinity than Sar1A toward Sec13/Sec31. The loose interaction of Sec13/Sec31 with Sar1B may facilitate the formation of a more flexible outer coat that can then accommodate large cargoes [55, 115].
Structural analysis of COPII proteins that provide insight into the assembly of large cargo carriers
Cryo-electron microscopic analysis of purified Sec23/Sec24 and Sec13/Sec31 showed that these two complexes can co-assemble into the cage-like structure from a cuboctahedron with a diameter of 60 nm to an icosidodecahedron with a diameter of 100 nm [116]. However, these structures are apparently not large enough to accommodate large cargoes such as collagens and chylomicrons. O’Donnell et al. recently reported that Sec13/Sec31 could also form tubules with 330-nm-long hollow cylindrical interiors with a diameter of 30 nm [117].
COPII-dependent tubule formation has also been reported in semi-intact cells, which were treated with an activated form of Sar1 (Sar1 H79G) [7, 118]. In addition, several reports indicate that artificial liposomes can be tubulated by incubation with Sar1 H79G or with Sar1 in the presence of non-hydrolyzable GTP analogs such as GMP-PNP and GTPγS [8, 119–122]. These data imply that secretion of large cargoes requires either large amounts or stabilized GTP-bound Sar1, which is consistent with the proposed function of Sec12 in collagen secretion described above (Fig. 3) [99]. By using cryo-electron tomography and subtomogram averaging, Zanetti et al. showed that giant unilamellar vesicles incubated with Sec12, Sar1, Sec23/Sec24, Sec13/Sec31, and GMP-PNP generate tubules coated with COPII proteins. The arrangement of the inner and outer coats into these tubules is structurally connected to but distinct from that of conventional COPII vesicles reported previously [10, 116, 123–127]. It should be noted that the architecture of assembly of these tubules is quite different from the in vitro-assembled Sec13/31 tubules described above [117].
Although yet to be proven, these tubules may be involved in large cargo transport, and it is interesting to speculate that tubulation is dependent on Sec31 monoubiquitylation. In this regard, a recent study of S. cerevisiae showed that the function of Sec13 in COPII vesicle formation may be to rigidify the COPII outer complex for increasing membrane-bending capacity [128]. In addition, phosphorylation of Sec31 by casein kinase II has been reported to reduce its affinity to Sec23, although its involvement in large cargo secretion remains unclear [129]. Future work is expected to reveal whether these modifications of core COPII components are (partly) responsible for the formation of carriers of over-sized cargoes [130, 131].
Future perspectives
Recent studies on human diseases and animal models have revealed that COPII components are crucial not only for conventional cargo export but also for the export of large proteins and protein complexes from the ER. Moreover, several factors specifically involved in the secretion of huge proteins have been identified and found to modify the process of COPII-dependent vesicle formation to enable them to secrete from the ER. However, several issues still need to be addressed. A major issue that remains to be resolved is the identification of the carriers responsible for large protein transport. The existence of megacarriers or tubules, which could accommodate large cargoes, has been proposed as described in this review, but the exact entities of and the mechanisms to form these containers are not fully understood.
A second issue is the relationship between the specialized factors identified. The cTAGE5/TANGO1 complex and sly1-syntaxin18 axis are reported to be rather specific for collagen VII transport, but not for collagen I. However, CUL3-KLHL12 and Sedlin have been implicated in collagen I transport. Whether they can cooperate to accomplish the large cargo export or they individually apply distinct mechanisms requires further investigation. Although there are still unresolved matters, recent identification of specialized factors has certainly provided us with clues to solve these problems.
Abbreviations
- CMRD:
-
Chylomicron retension disease
- COPII:
-
Coat protein complex II
- CLSD:
-
Cranio-lenticulo-sutural dysplasia
- ECM:
-
Extracellular matrix
- ER:
-
Endoplasmic reticulum
- GEF:
-
Guanine-nucleotide exchange factor
- MIA:
-
Melanoma inhibitory activity
- MSS:
-
Marinesco-Sjogren syndrome
- NPC:
-
Nuclear pore complex
- TANGO:
-
Transport ANd Golgi Organization
- TRAPP:
-
TRAnsport Protein Particle
References
Barlowe C, Orci L, Yeung T, Hosobuchi M, Hamamoto S, Salama N, Rexach MF, Ravazzola M, Amherdt M, Schekman R (1994) COPII: a membrane coat formed by Sec proteins that drive vesicle budding from the endoplasmic reticulum. Cell 77(6):895–907
Barlowe C, Schekman R (1993) SEC12 encodes a guanine-nucleotide-exchange factor essential for transport vesicle budding from the ER. Nature 365(6444):347–349. doi:10.1038/365347a0
Nakano A, Muramatsu M (1989) A novel GTP-binding protein, Sar1p, is involved in transport from the endoplasmic reticulum to the Golgi apparatus. J Cell Biol 109(6 Pt 1):2677–2691
Barlowe C, d’Enfert C, Schekman R (1993) Purification and characterization of SAR1p, a small GTP-binding protein required for transport vesicle formation from the endoplasmic reticulum. J Biol Chem 268(2):873–879
Weissman JT, Plutner H, Balch WE (2001) The mammalian guanine nucleotide exchange factor mSec12 is essential for activation of the Sar1 GTPase directing endoplasmic reticulum export. Traffic 2(7):465–475
Huang M, Weissman JT, Beraud-Dufour S, Luan P, Wang C, Chen W, Aridor M, Wilson IA, Balch WE (2001) Crystal structure of Sar1-GDP at 1.7 A resolution and the role of the NH2 terminus in ER export. J Cell Biol 155(6):937–948. doi:10.1083/jcb.200106039
Bielli A, Haney CJ, Gabreski G, Watkins SC, Bannykh SI, Aridor M (2005) Regulation of Sar1 NH2 terminus by GTP binding and hydrolysis promotes membrane deformation to control COPII vesicle fission. J Cell Biol 171(6):919–924. doi:10.1083/jcb.200509095
Lee MC, Orci L, Hamamoto S, Futai E, Ravazzola M, Schekman R (2005) Sar1p N-terminal helix initiates membrane curvature and completes the fission of a COPII vesicle. Cell 122(4):605–617. doi:10.1016/j.cell.2005.07.025
Tabata KV, Sato K, Ide T, Nishizaka T, Nakano A, Noji H (2009) Visualization of cargo concentration by COPII minimal machinery in a planar lipid membrane. EMBO J 28(21):3279–3289. doi:10.1038/emboj.2009.269
Bi X, Corpina RA, Goldberg J (2002) Structure of the Sec23/24-Sar1 pre-budding complex of the COPII vesicle coat. Nature 419(6904):271–277. doi:10.1038/nature01040
Kuehn MJ, Herrmann JM, Schekman R (1998) COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature 391(6663):187–190. doi:10.1038/34438
Aridor M, Weissman J, Bannykh S, Nuoffer C, Balch WE (1998) Cargo selection by the COPII budding machinery during export from the ER. J Cell Biol 141(1):61–70
Miller E, Antonny B, Hamamoto S, Schekman R (2002) Cargo selection into COPII vesicles is driven by the Sec24p subunit. EMBO J 21(22):6105–6113
Miller EA, Beilharz TH, Malkus PN, Lee MC, Hamamoto S, Orci L, Schekman R (2003) Multiple cargo binding sites on the COPII subunit Sec24p ensure capture of diverse membrane proteins into transport vesicles. Cell 114(4):497–509
Pagant S, Wu A, Edwards S, Diehl F, Miller EA (2015) Sec24 is a coincidence detector that simultaneously binds two signals to drive ER export. Curr Biol 25(4):403–412. doi:10.1016/j.cub.2014.11.070
Yoshihisa T, Barlowe C, Schekman R (1993) Requirement for a GTPase-activating protein in vesicle budding from the endoplasmic reticulum. Science 259(5100):1466–1468
Antonny B, Madden D, Hamamoto S, Orci L, Schekman R (2001) Dynamics of the COPII coat with GTP and stable analogues. Nat Cell Biol 3(6):531–537. doi:10.1038/35078500
Bi X, Mancias JD, Goldberg J (2007) Insights into COPII coat nucleation from the structure of Sec23.Sar1 complexed with the active fragment of Sec31. Dev Cell 13(5):635–645. doi:10.1016/j.devcel.2007.10.006
Matsuoka K, Orci L, Amherdt M, Bednarek SY, Hamamoto S, Schekman R, Yeung T (1998) COPII-coated vesicle formation reconstituted with purified coat proteins and chemically defined liposomes. Cell 93(2):263–275
Connerly PL, Esaki M, Montegna EA, Strongin DE, Levi S, Soderholm J, Glick BS (2005) Sec16 is a determinant of transitional ER organization. Curr Biol 15(16):1439–1447. doi:10.1016/j.cub.2005.06.065
Iinuma T, Shiga A, Nakamoto K, O’Brien MB, Aridor M, Arimitsu N, Tagaya M, Tani K (2007) Mammalian Sec16/p250 plays a role in membrane traffic from the endoplasmic reticulum. J Biol Chem 282(24):17632–17639. doi:10.1074/jbc.M611237200
Ivan V, de Voer G, Xanthakis D, Spoorendonk KM, Kondylis V, Rabouille C (2008) Drosophila Sec16 mediates the biogenesis of tER sites upstream of Sar1 through an arginine-rich motif. Mol Biol Cell 19(10):4352–4365. doi:10.1091/mbc.E08-03-0246
Montegna EA, Bhave M, Liu Y, Bhattacharyya D, Glick BS (2012) Sec12 binds to Sec16 at transitional ER sites. PLoS One 7(2):e31156. doi:10.1371/journal.pone.0031156
Watson P, Townley AK, Koka P, Palmer KJ, Stephens DJ (2006) Sec16 defines endoplasmic reticulum exit sites and is required for secretory cargo export in mammalian cells. Traffic 7(12):1678–1687. doi:10.1111/j.1600-0854.2006.00493.x
Whittle JR, Schwartz TU (2010) Structure of the Sec13-Sec16 edge element, a template for assembly of the COPII vesicle coat. J Cell Biol 190(3):347–361. doi:10.1083/jcb.201003092
Bharucha N, Liu Y, Papanikou E, McMahon C, Esaki M, Jeffrey PD, Hughson FM, Glick BS (2013) Sec16 influences transitional ER sites by regulating rather than organizing COPII. Mol Biol Cell 24(21):3406–3419. doi:10.1091/mbc.E13-04-0185
Kung LF, Pagant S, Futai E, D’Arcangelo JG, Buchanan R, Dittmar JC, Reid RJ, Rothstein R, Hamamoto S, Snapp EL, Schekman R, Miller EA (2012) Sec24p and Sec16p cooperate to regulate the GTP cycle of the COPII coat. EMBO J 31(4):1014–1027. doi:10.1038/emboj.2011.444
Yorimitsu T, Sato K (2012) Insights into structural and regulatory roles of Sec16 in COPII vesicle formation at ER exit sites. Mol Biol Cell 23(15):2930–2942. doi:10.1091/mbc.E12-05-0356
Helm JR, Bentley M, Thorsen KD, Wang T, Foltz L, Oorschot V, Klumperman J, Hay JC (2014) Apoptosis-linked gene-2 (ALG-2)/Sec31 interactions regulate endoplasmic reticulum (ER)-to-Golgi transport: a potential effector pathway for luminal calcium. J Biol Chem 289(34):23609–23628. doi:10.1074/jbc.M114.561829
la Cour JM, Schindler AJ, Berchtold MW, Schekman R (2013) ALG-2 attenuates COPII budding in vitro and stabilizes the Sec23/Sec31A complex. PLoS One 8(9):e75309. doi:10.1371/journal.pone.0075309
Shibata H, Inuzuka T, Yoshida H, Sugiura H, Wada I, Maki M (2010) The ALG-2 binding site in Sec31A influences the retention kinetics of Sec31A at the endoplasmic reticulum exit sites as revealed by live-cell time-lapse imaging. Biosci Biotechnol Biochem 74(9):1819–1826. doi:10.1271/bbb.100215
Shibata H, Suzuki H, Yoshida H, Maki M (2007) ALG-2 directly binds Sec31A and localizes at endoplasmic reticulum exit sites in a Ca2+-dependent manner. Biochem Biophys Res Commun 353(3):756–763. doi:10.1016/j.bbrc.2006.12.101
Yamasaki A, Tani K, Yamamoto A, Kitamura N, Komada M (2006) The Ca2+-binding protein ALG-2 is recruited to endoplasmic reticulum exit sites by Sec31A and stabilizes the localization of Sec31A. Mol Biol Cell 17(11):4876–4887. doi:10.1091/mbc.E06-05-0444
Yoshibori M, Yorimitsu T, Sato K (2012) Involvement of the penta-EF-hand protein Pef1p in the Ca2+-dependent regulation of COPII subunit assembly in Saccharomyces cerevisiae. PLoS One 7(7):e40765. doi:10.1371/journal.pone.0040765
Johnson A, Bhattacharya N, Hanna M, Pennington JG, Schuh AL, Wang L, Otegui MS, Stagg SM, Audhya A (2015) TFG clusters COPII-coated transport carriers and promotes early secretory pathway organization. EMBO J. doi:10.15252/embj.201489032
Witte K, Schuh AL, Hegermann J, Sarkeshik A, Mayers JR, Schwarze K, Yates JR 3rd, Eimer S, Audhya A (2011) TFG-1 function in protein secretion and oncogenesis. Nat Cell Biol 13(5):550–558. doi:10.1038/ncb2225
Ong YS, Tang BL, Loo LS, Hong W (2010) p125A exists as part of the mammalian Sec13/Sec31 COPII subcomplex to facilitate ER-Golgi transport. J Cell Biol 190(3):331–345. doi:10.1083/jcb.201003005
Shimoi W, Ezawa I, Nakamoto K, Uesaki S, Gabreski G, Aridor M, Yamamoto A, Nagahama M, Tagaya M, Tani K (2005) p125 is localized in endoplasmic reticulum exit sites and involved in their organization. J Biol Chem 280(11):10141–10148. doi:10.1074/jbc.M409673200
Brandizzi F, Barlowe C (2013) Organization of the ER-Golgi interface for membrane traffic control. Nat Rev Mol Cell Biol 14(6):382–392. doi:10.1038/nrm3588
D’Arcangelo JG, Stahmer KR, Miller EA (2013) Vesicle-mediated export from the ER: COPII coat function and regulation. Biochim Biophys Acta 1833(11):2464–2472. doi:10.1016/j.bbamcr.2013.02.003
Venditti R, Wilson C, De Matteis MA (2014) Exiting the ER: what we know and what we don’t. Trends Cell Biol 24(1):9–18. doi:10.1016/j.tcb.2013.08.005
Barlowe CK, Miller EA (2013) Secretory protein biogenesis and traffic in the early secretory pathway. Genetics 193(2):383–410. doi:10.1534/genetics.112.142810
Faini M, Beck R, Wieland FT, Briggs JA (2013) Vesicle coats: structure, function, and general principles of assembly. Trends Cell Biol 23(6):279–288. doi:10.1016/j.tcb.2013.01.005
Lord C, Ferro-Novick S, Miller EA (2013) The highly conserved COPII coat complex sorts cargo from the endoplasmic reticulum and targets it to the golgi. Cold Spring Harb Perspect Biol. doi:10.1101/cshperspect.a013367
Gillon AD, Latham CF, Miller EA (2012) Vesicle-mediated ER export of proteins and lipids. Biochim Biophys Acta 1821(8):1040–1049. doi:10.1016/j.bbalip.2012.01.005
Zanetti G, Pahuja KB, Studer S, Shim S, Schekman R (2012) COPII and the regulation of protein sorting in mammals. Nat Cell Biol 14(1):20–28. doi:10.1038/ncb2390
Ishikawa Y, Boudko S, Bachinger HP (2015) Ziploc-ing the structure: triple helix formation is coordinated by rough endoplasmic reticulum resident PPIases. Biochim Biophys Acta. doi:10.1016/j.bbagen.2014.12.024
Fromme JC, Schekman R (2005) COPII-coated vesicles: flexible enough for large cargo? Curr Opin Cell Biol 17(4):345–352. doi:10.1016/j.ceb.2005.06.004
Bonfanti L, Mironov AA Jr, Martinez-Menarguez JA, Martella O, Fusella A, Baldassarre M, Buccione R, Geuze HJ, Mironov AA, Luini A (1998) Procollagen traverses the Golgi stack without leaving the lumen of cisternae: evidence for cisternal maturation. Cell 95(7):993–1003
Bachinger HP, Morris NP, Davis JM (1993) Thermal stability and folding of the collagen triple helix and the effects of mutations in osteogenesis imperfecta on the triple helix of type I collagen. Am J Med Genet 45(2):152–162. doi:10.1002/ajmg.1320450204
Stephens DJ, Pepperkok R (2002) Imaging of procollagen transport reveals COPI-dependent cargo sorting during ER-to-Golgi transport in mammalian cells. J Cell Sci 115(Pt 6):1149–1160
Mironov AA, Mironov AA Jr, Beznoussenko GV, Trucco A, Lupetti P, Smith JD, Geerts WJ, Koster AJ, Burger KN, Martone ME, Deerinck TJ, Ellisman MH, Luini A (2003) ER-to-Golgi carriers arise through direct en bloc protrusion and multistage maturation of specialized ER exit domains. Dev Cell 5(4):583–594
Boyadjiev SA, Fromme JC, Ben J, Chong SS, Nauta C, Hur DJ, Zhang G, Hamamoto S, Schekman R, Ravazzola M, Orci L, Eyaid W (2006) Cranio-lenticulo-sutural dysplasia is caused by a SEC23A mutation leading to abnormal endoplasmic-reticulum-to-Golgi trafficking. Nat Genet 38(10):1192–1197. doi:10.1038/ng1876
Boyadjiev SA, Kim SD, Hata A, Haldeman-Englert C, Zackai EH, Naydenov C, Hamamoto S, Schekman RW, Kim J (2011) Cranio-lenticulo-sutural dysplasia associated with defects in collagen secretion. Clin Genet 80(2):169–176. doi:10.1111/j.1399-0004.2010.01550.x
Fromme JC, Ravazzola M, Hamamoto S, Al-Balwi M, Eyaid W, Boyadjiev SA, Cosson P, Schekman R, Orci L (2007) The genetic basis of a craniofacial disease provides insight into COPII coat assembly. Dev Cell 13(5):623–634. doi:10.1016/j.devcel.2007.10.005
Kim SD, Pahuja KB, Ravazzola M, Yoon J, Boyadjiev SA, Hammamoto S, Schekman R, Orci L, Kim J (2012) The [corrected] SEC23-SEC31 [corrected] interface plays critical role for export of procollagen from the endoplasmic reticulum. J Biol Chem 287(13):10134–10144. doi:10.1074/jbc.M111.283382
Neuhauss SC, Solnica-Krezel L, Schier AF, Zwartkruis F, Stemple DL, Malicki J, Abdelilah S, Stainier DY, Driever W (1996) Mutations affecting craniofacial development in zebrafish. Development 123:357–367
Lang MR, Lapierre LA, Frotscher M, Goldenring JR, Knapik EW (2006) Secretory COPII coat component Sec23a is essential for craniofacial chondrocyte maturation. Nat Genet 38(10):1198–1203. doi:10.1038/ng1880
Saito A, Hino S, Murakami T, Kanemoto S, Kondo S, Saitoh M, Nishimura R, Yoneda T, Furuichi T, Ikegawa S, Ikawa M, Okabe M, Imaizumi K (2009) Regulation of endoplasmic reticulum stress response by a BBF2H7-mediated Sec23a pathway is essential for chondrogenesis. Nat Cell Biol 11(10):1197–1204. doi:10.1038/ncb1962
Kondo S, Hino SI, Saito A, Kanemoto S, Kawasaki N, Asada R, Izumi S, Iwamoto H, Oki M, Miyagi H, Kaneko M, Nomura Y, Urano F, Imaizumi K (2012) Activation of OASIS family, ER stress transducers, is dependent on its stabilization. Cell Death Differ 19(12):1939–1949. doi:10.1038/cdd.2012.77
Lui WO, Zeng L, Rehrmann V, Deshpande S, Tretiakova M, Kaplan EL, Leibiger I, Leibiger B, Enberg U, Hoog A, Larsson C, Kroll TG (2008) CREB3L2-PPARgamma fusion mutation identifies a thyroid signaling pathway regulated by intramembrane proteolysis. Cancer Res 68(17):7156–7164. doi:10.1158/0008-5472.CAN-08-1085
Saito A, Kanemoto S, Zhang Y, Asada R, Hino K, Imaizumi K (2014) Chondrocyte proliferation regulated by secreted luminal domain of ER stress transducer BBF2H7/CREB3L2. Mol Cell 53(1):127–139. doi:10.1016/j.molcel.2013.11.008
Melville DB, Montero-Balaguer M, Levic DS, Bradley K, Smith JR, Hatzopoulos AK, Knapik EW (2011) The feelgood mutation in zebrafish dysregulates COPII-dependent secretion of select extracellular matrix proteins in skeletal morphogenesis. Disease models & mechanisms 4(6):763–776. doi:10.1242/dmm.007625
Ishikura-Kinoshita S, Saeki H, Tsuji-Naito K (2012) BBF2H7-mediated Sec23A pathway is required for endoplasmic reticulum-to-Golgi trafficking in dermal fibroblasts to promote collagen synthesis. J Invest Dermatol 132(8):2010–2018. doi:10.1038/jid.2012.103
Wendeler MW, Paccaud JP, Hauri HP (2007) Role of Sec24 isoforms in selective export of membrane proteins from the endoplasmic reticulum. EMBO Rep 8(3):258–264. doi:10.1038/sj.embor.7400893
Sarmah S, Barrallo-Gimeno A, Melville DB, Topczewski J, Solnica-Krezel L, Knapik EW (2010) Sec24D-dependent transport of extracellular matrix proteins is required for zebrafish skeletal morphogenesis. PLoS One 5(4):e10367. doi:10.1371/journal.pone.0010367
Ohisa S, Inohaya K, Takano Y, Kudo A (2010) sec24d encoding a component of COPII is essential for vertebra formation, revealed by the analysis of the medaka mutant, vbi. Dev Biol 342(1):85–95. doi:10.1016/j.ydbio.2010.03.016
Rauch F, Glorieux FH (2004) Osteogenesis imperfecta. Lancet 363(9418):1377–1385. doi:10.1016/S0140-6736(04)16051-0
Garbes L, Kim K, Riess A, Hoyer-Kuhn H, Beleggia F, Bevot A, Kim MJ, Huh YH, Kweon HS, Savarirayan R, Amor D, Kakadia PM, Lindig T, Kagan KO, Becker J, Boyadjiev SA, Wollnik B, Semler O, Bohlander SK, Kim J, Netzer C (2015) Mutations in SEC24D, encoding a component of the COPII machinery, cause a syndromic form of osteogenesis imperfecta. Am J Hum Genet. doi:10.1016/j.ajhg.2015.01.002
Baines AC, Adams EJ, Zhang B, Ginsburg D (2013) Disruption of the Sec24d gene results in early embryonic lethality in the mouse. PLoS One 8(4):e61114. doi:10.1371/journal.pone.0061114
Siniossoglou S, Wimmer C, Rieger M, Doye V, Tekotte H, Weise C, Emig S, Segref A, Hurt EC (1996) A novel complex of nucleoporins, which includes Sec13p and a Sec13p homolog, is essential for normal nuclear pores. Cell 84(2):265–275
Townley AK, Feng Y, Schmidt K, Carter DA, Porter R, Verkade P, Stephens DJ (2008) Efficient coupling of Sec23-Sec24 to Sec13-Sec31 drives COPII-dependent collagen secretion and is essential for normal craniofacial development. J Cell Sci 121(Pt 18):3025–3034. doi:10.1242/jcs.031070
Huang H, Ruan H, Aw MY, Hussain A, Guo L, Gao C, Qian F, Leung T, Song H, Kimelman D, Wen Z, Peng J (2008) Mypt1-mediated spatial positioning of Bmp2-producing cells is essential for liver organogenesis. Development 135(19):3209–3218. doi:10.1242/dev.024406
Niu X, Gao C, Jan Lo L, Luo Y, Meng C, Hong J, Hong W, Peng J (2012) Sec13 safeguards the integrity of the endoplasmic reticulum and organogenesis of the digestive system in zebrafish. Dev Biol 367(2):197–207. doi:10.1016/j.ydbio.2012.05.004
Schmidt K, Cavodeassi F, Feng Y, Stephens DJ (2013) Early stages of retinal development depend on Sec13 function. Biol Open 2(3):256–266. doi:10.1242/bio.20133251
Niu X, Hong J, Zheng X, Melville DB, Knapik EW, Meng A, Peng J (2014) The nuclear pore complex function of Sec13 protein is required for cell survival during retinal development. J Biol Chem 289(17):11971–11985. doi:10.1074/jbc.M114.547190
Bard F, Casano L, Mallabiabarrena A, Wallace E, Saito K, Kitayama H, Guizzunti G, Hu Y, Wendler F, Dasgupta R, Perrimon N, Malhotra V (2006) Functional genomics reveals genes involved in protein secretion and Golgi organization. Nature 439(7076):604–607. doi:10.1038/nature04377
Stoll R, Renner C, Zweckstetter M, Bruggert M, Ambrosius D, Palme S, Engh RA, Golob M, Breibach I, Buettner R, Voelter W, Holak TA, Bosserhoff AK (2001) The extracellular human melanoma inhibitory activity (MIA) protein adopts an SH3 domain-like fold. EMBO J 20(3):340–349. doi:10.1093/emboj/20.3.340
Shaywitz DA, Espenshade PJ, Gimeno RE, Kaiser CA (1997) COPII subunit interactions in the assembly of the vesicle coat. J Biol Chem 272(41):25413–25416
Shugrue CA, Kolen ER, Peters H, Czernik A, Kaiser C, Matovcik L, Hubbard AL, Gorelick F (1999) Identification of the putative mammalian orthologue of Sec31P, a component of the COPII coat. J Cell Sci 112(Pt 24):4547–4556
Saito K, Chen M, Bard F, Chen S, Zhou H, Woodley D, Polischuk R, Schekman R, Malhotra V (2009) TANGO1 facilitates cargo loading at endoplasmic reticulum exit sites. Cell 136(5):891–902. doi:10.1016/j.cell.2008.12.025
Wilson DG, Phamluong K, Li L, Sun M, Cao TC, Liu PS, Modrusan Z, Sandoval WN, Rangell L, Carano RA, Peterson AS, Solloway MJ (2011) Global defects in collagen secretion in a Mia3/TANGO1 knockout mouse. J Cell Biol 193(5):935–951. doi:10.1083/jcb.201007162
Schnieke A, Harbers K, Jaenisch R (1983) Embryonic lethal mutation in mice induced by retrovirus insertion into the alpha 1(I) collagen gene. Nature 304(5924):315–320
Lerner DW, McCoy D, Isabella AJ, Mahowald AP, Gerlach GF, Chaudhry TA, Horne-Badovinac S (2013) A Rab10-dependent mechanism for polarized basement membrane secretion during organ morphogenesis. Dev Cell 24(2):159–168. doi:10.1016/j.devcel.2012.12.005
Pastor-Pareja JC, Xu T (2011) Shaping cells and organs in Drosophila by opposing roles of fat body-secreted collagen IV and perlecan. Dev Cell 21(2):245–256. doi:10.1016/j.devcel.2011.06.026
Heckel D, Brass N, Fischer U, Blin N, Steudel I, Tureci O, Fackler O, Zang KD, Meese E (1997) cDNA cloning and chromosomal mapping of a predicted coiled-coil proline-rich protein immunogenic in meningioma patients. Hum Mol Genet 6(12):2031–2041
Comtesse N, Niedermayer I, Glass B, Heckel D, Maldener E, Nastainczyk W, Feiden W, Meese E (2002) MGEA6 is tumor-specific overexpressed and frequently recognized by patient-serum antibodies. Oncogene 21(2):239–247. doi:10.1038/sj.onc.1205005
Usener D, Schadendorf D, Koch J, Dubel S, Eichmuller S (2003) cTAGE: a cutaneous T cell lymphoma associated antigen family with tumor-specific splicing. J Invest Dermatol 121(1):198–206. doi:10.1046/j.1523-1747.2003.12318.x
Guinn BA, Gilkes AF, Mufti GJ, Burnett AK, Mills KI (2006) The tumour antigens RAGE-1 and MGEA6 are expressed more frequently in the less lineage restricted subgroups of presentation acute myeloid leukaemia. Br J Haematol 134(2):238–239. doi:10.1111/j.1365-2141.2006.06135.x
Saito K, Yamashiro K, Ichikawa Y, Erlmann P, Kontani K, Malhotra V, Katada T (2011) cTAGE5 mediates collagen secretion through interaction with TANGO1 at endoplasmic reticulum exit sites. Mol Biol Cell 22(13):2301–2308. doi:10.1091/mbc.E11-02-0143
Comtesse N, Reus K, Meese E (2001) The MGEA6 multigene family has an active locus on 14q and at least nine pseudogenes on different chromosomes. Genomics 75(1–3):43–48. doi:10.1006/geno.2001.6576
Pitman JL, Bonnet DJ, Curtiss LK, Gekakis N (2011) Reduced cholesterol and triglycerides in mice with a mutation in Mia2, a liver protein that localizes to ER exit sites. J Lipid Res 52(10):1775–1786. doi:10.1194/jlr.M017277
Bosserhoff AK, Moser M, Scholmerich J, Buettner R, Hellerbrand C (2003) Specific expression and regulation of the new melanoma inhibitory activity-related gene MIA2 in hepatocytes. J Biol Chem 278(17):15225–15231. doi:10.1074/jbc.M212639200
Hellerbrand C, Bataille F, Schlegel J, Hartmann A, Muhlbauer M, Scholmerich J, Buttner R, Hofstadter F, Bosserhoff AK (2005) In situ expression patterns of melanoma inhibitory activity 2 in healthy and diseased livers. Liver Int 25(2):357–366. doi:10.1111/j.1478-3231.2005.01099.x
Hellerbrand C, Amann T, Schlegel J, Wild P, Bataille F, Spruss T, Hartmann A, Bosserhoff AK (2008) The novel gene MIA2 acts as a tumour suppressor in hepatocellular carcinoma. Gut 57(2):243–251. doi:10.1136/gut.2007.129544
Kurihara M, Kirita T, Sasahira T, Ohmori H, Matsushima S, Yamamoto K, Bosserhoff AK, Kuniyasu H (2013) Protumoral roles of melanoma inhibitory activity 2 in oral squamous cell carcinoma. Br J Cancer 108(7):1460–1469. doi:10.1038/bjc.2013.27
Kong B, Wu W, Valkovska N, Jager C, Hong X, Nitsche U, Friess H, Esposito I, Erkan M, Kleeff J, Michalski CW (2015) A common genetic variation of melanoma inhibitory activity-2 labels a subtype of pancreatic adenocarcinoma with high endoplasmic reticulum stress levels. Sci Rep 5:8109. doi:10.1038/srep08109
Malhotra V, Erlmann P (2011) Protein export at the ER: loading big collagens into COPII carriers. EMBO J 30(17):3475–3480. doi:10.1038/emboj.2011.255
Saito K, Yamashiro K, Shimazu N, Tanabe T, Kontani K, Katada T (2014) Concentration of Sec12 at ER exit sites via interaction with cTAGE5 is required for collagen export. J Cell Biol 206(6):751–762. doi:10.1083/jcb.201312062
McMahon C, Studer SM, Clendinen C, Dann GP, Jeffrey PD, Hughson FM (2012) The structure of Sec12 implicates potassium ion coordination in Sar1 activation. J Biol Chem 287(52):43599–43606. doi:10.1074/jbc.M112.420141
Yamaguchi T, Dulubova I, Min SW, Chen X, Rizo J, Sudhof TC (2002) Sly1 binds to Golgi and ER syntaxins via a conserved N-terminal peptide motif. Dev Cell 2(3):295–305
Steegmaier M, Oorschot V, Klumperman J, Scheller RH (2000) Syntaxin 17 is abundant in steroidogenic cells and implicated in smooth endoplasmic reticulum membrane dynamics. Mol Biol Cell 11(8):2719–2731
Nogueira C, Erlmann P, Villeneuve J, Santos AJ, Martinez-Alonso E, Martinez-Menarguez JA, Malhotra V (2014) SLY1 and Syntaxin 18 specify a distinct pathway for procollagen VII export from the endoplasmic reticulum. eLife 3:e02784. doi:10.7554/eLife.02784
Malhotra V, Erlmann P, Nogueira C (2015) Procollagen export from the endoplasmic reticulum. Biochem Soc Trans 43(1):104–107. doi:10.1042/BST20140286
Jin L, Pahuja KB, Wickliffe KE, Gorur A, Baumgartel C, Schekman R, Rape M (2012) Ubiquitin-dependent regulation of COPII coat size and function. Nature 482(7386):495–500. doi:10.1038/nature10822
Barrowman J, Bhandari D, Reinisch K, Ferro-Novick S (2010) TRAPP complexes in membrane traffic: convergence through a common Rab. Nat Rev Mol Cell Biol 11(11):759–763. doi:10.1038/nrm2999
Gedeon AK, Colley A, Jamieson R, Thompson EM, Rogers J, Sillence D, Tiller GE, Mulley JC, Gecz J (1999) Identification of the gene (SEDL) causing X-linked spondyloepiphyseal dysplasia tarda. Nat Genet 22(4):400–404. doi:10.1038/11976
Venditti R, Scanu T, Santoro M, Di Tullio G, Spaar A, Gaibisso R, Beznoussenko GV, Mironov AA, Mironov A Jr, Zelante L, Piemontese MR, Notarangelo A, Malhotra V, Vertel BM, Wilson C, De Matteis MA (2012) Sedlin controls the ER export of procollagen by regulating the Sar1 cycle. Science 337(6102):1668–1672. doi:10.1126/science.1224947
Hussain MM (2000) A proposed model for the assembly of chylomicrons. Atherosclerosis 148(1):1–15
Sabesin SM, Frase S (1977) Electron microscopic studies of the assembly, intracellular transport, and secretion of chylomicrons by rat intestine. J Lipid Res 18(4):496–511
Jones B, Jones EL, Bonney SA, Patel HN, Mensenkamp AR, Eichenbaum-Voline S, Rudling M, Myrdal U, Annesi G, Naik S, Meadows N, Quattrone A, Islam SA, Naoumova RP, Angelin B, Infante R, Levy E, Roy CC, Freemont PS, Scott J, Shoulders CC (2003) Mutations in a Sar1 GTPase of COPII vesicles are associated with lipid absorption disorders. Nat Genet 34(1):29–31. doi:10.1038/ng1145
Charcosset M, Sassolas A, Peretti N, Roy CC, Deslandres C, Sinnett D, Levy E, Lachaux A (2008) Anderson or chylomicron retention disease: molecular impact of five mutations in the SAR1B gene on the structure and the functionality of Sar1b protein. Mol Genet Metab 93(1):74–84. doi:10.1016/j.ymgme.2007.08.120
Peretti N, Sassolas A, Roy CC, Deslandres C, Charcosset M, Castagnetti J, Pugnet-Chardon L, Moulin P, Labarge S, Bouthillier L, Lachaux A, Levy E, Department of Nutrition-Hepatogastroenterology HFMEBUL, Department of Pediatrics CHUS-JRCUdM (2010) Guidelines for the diagnosis and management of chylomicron retention disease based on a review of the literature and the experience of two centers. Orphanet J Rare Dis 5:24. doi:10.1186/1750-1172-5-24
Levic DS, Minkel J, Wang W, Rybski WM, Melville DB, Knapik EW (2015) Animal model of Sar1b deficiency presents lipid absorption deficits similar to Anderson disease. J Mol Med 93(2):165–176. doi:10.1007/s00109-014-1247-x
Fromme JC, Orci L, Schekman R (2008) Coordination of COPII vesicle trafficking by Sec23. Trends Cell Biol 18(7):330–336. doi:10.1016/j.tcb.2008.04.006
Stagg SM, LaPointe P, Razvi A, Gurkan C, Potter CS, Carragher B, Balch WE (2008) Structural basis for cargo regulation of COPII coat assembly. Cell 134(3):474–484. doi:10.1016/j.cell.2008.06.024
O’Donnell J, Maddox K, Stagg S (2011) The structure of a COPII tubule. J Struct Biol 173(2):358–364. doi:10.1016/j.jsb.2010.09.002
Aridor M, Fish KN, Bannykh S, Weissman J, Roberts TH, Lippincott-Schwartz J, Balch WE (2001) The Sar1 GTPase coordinates biosynthetic cargo selection with endoplasmic reticulum export site assembly. J Cell Biol 152(1):213–229
Bacia K, Futai E, Prinz S, Meister A, Daum S, Glatte D, Briggs JA, Schekman R (2011) Multibudded tubules formed by COPII on artificial liposomes. Sci Rep 1:17. doi:10.1038/srep00017
Long KR, Yamamoto Y, Baker AL, Watkins SC, Coyne CB, Conway JF, Aridor M (2010) Sar1 assembly regulates membrane constriction and ER export. J Cell Biol 190(1):115–128. doi:10.1083/jcb.201004132
Zanetti G, Prinz S, Daum S, Meister A, Schekman R, Bacia K, Briggs JA (2013) The structure of the COPII transport-vesicle coat assembled on membranes. eLife 2:e00951. doi:10.7554/eLife.00951
Hariri H, Bhattacharya N, Johnson K, Noble AJ, Stagg SM (2014) Insights into the mechanisms of membrane curvature and vesicle scission by the small GTPase Sar1 in the early secretory pathway. J Mol Biol 426(22):3811–3826. doi:10.1016/j.jmb.2014.08.023
Bhattacharya N, O Donnell J, Stagg SM (2012) The structure of the Sec13/31 COPII cage bound to Sec23. J Mol Biol 420(4–5):324–334. doi:10.1016/j.jmb.2012.04.024
Stagg SM, Gurkan C, Fowler DM, LaPointe P, Foss TR, Potter CS, Carragher B, Balch WE (2006) Structure of the Sec13/31 COPII coat cage. Nature 439(7073):234–238. doi:10.1038/nature04339
Noble AJ, Zhang Q, O’Donnell J, Hariri H, Bhattacharya N, Marshall AG, Stagg SM (2013) A pseudoatomic model of the COPII cage obtained from cryo-electron microscopy and mass spectrometry. Nat Struct Mol Biol 20(2):167–173. doi:10.1038/nsmb.2467
Mancias JD, Goldberg J (2008) Structural basis of cargo membrane protein discrimination by the human COPII coat machinery. EMBO J 27(21):2918–2928. doi:10.1038/emboj.2008.208
Fath S, Mancias JD, Bi X, Goldberg J (2007) Structure and organization of coat proteins in the COPII cage. Cell 129(7):1325–1336. doi:10.1016/j.cell.2007.05.036
Copic A, Latham CF, Horlbeck MA, D’Arcangelo JG, Miller EA (2012) ER cargo properties specify a requirement for COPII coat rigidity mediated by Sec13p. Science 335(6074):1359–1362. doi:10.1126/science.1215909
Koreishi M, Yu S, Oda M, Honjo Y, Satoh A (2013) CK2 phosphorylates Sec31 and regulates ER-To-Golgi trafficking. PLoS One 8(1):e54382. doi:10.1371/journal.pone.0054382
Lord C, Bhandari D, Menon S, Ghassemian M, Nycz D, Hay J, Ghosh P, Ferro-Novick S (2011) Sequential interactions with Sec23 control the direction of vesicle traffic. Nature 473(7346):181–186. doi:10.1038/nature09969
Bhandari D, Zhang J, Menon S, Lord C, Chen S, Helm JR, Thorsen K, Corbett KD, Hay JC, Ferro-Novick S (2013) Sit4p/PP6 regulates ER-to-Golgi traffic by controlling the dephosphorylation of COPII coat subunits. Mol Biol Cell 24(17):2727–2738. doi:10.1091/mbc.E13-02-0114
Schwarz K, Iolascon A, Verissimo F, Trede NS, Horsley W, Chen W, Paw BH, Hopfner KP, Holzmann K, Russo R, Esposito MR, Spano D, De Falco L, Heinrich K, Joggerst B, Rojewski MT, Perrotta S, Denecke J, Pannicke U, Delaunay J, Pepperkok R, Heimpel H (2009) Mutations affecting the secretory COPII coat component SEC23B cause congenital dyserythropoietic anemia type II. Nat Genet 41(8):936–940. doi:10.1038/ng.405
Bianchi P, Fermo E, Vercellati C, Boschetti C, Barcellini W, Iurlo A, Marcello AP, Righetti PG, Zanella A (2009) Congenital dyserythropoietic anemia type II (CDAII) is caused by mutations in the SEC23B gene. Hum Mutat 30(9):1292–1298. doi:10.1002/humu.21077
Tao J, Zhu M, Wang H, Afelik S, Vasievich MP, Chen XW, Zhu G, Jensen J, Ginsburg D, Zhang B (2012) SEC23B is required for the maintenance of murine professional secretory tissues. Proc Natl Acad Sci USA 109(29):E2001–E2009. doi:10.1073/pnas.1209207109
Chen XW, Wang H, Bajaj K, Zhang P, Meng ZX, Ma D, Bai Y, Liu HH, Adams E, Baines A, Yu G, Sartor MA, Zhang B, Yi Z, Lin J, Young SG, Schekman R, Ginsburg D (2013) SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. eLife 2:e00444. doi:10.7554/eLife.00444
Wansleeben C, Feitsma H, Montcouquiol M, Kroon C, Cuppen E, Meijlink F (2010) Planar cell polarity defects and defective Vangl2 trafficking in mutants for the COPII gene Sec24b. Development 137(7):1067–1073. doi:10.1242/dev.041434
Merte J, Jensen D, Wright K, Sarsfield S, Wang Y, Schekman R, Ginty DD (2010) Sec24b selectively sorts Vangl2 to regulate planar cell polarity during neural tube closure. Nat Cell Biol 12(1):41–46. doi:10.1038/ncb2002
Adams EJ, Chen XW, O’Shea KS, Ginsburg D (2014) Mammalian COPII coat component SEC24C is required for embryonic development in mice. J Biol Chem 289(30):20858–20870. doi:10.1074/jbc.M114.566687
Acknowledgments
We thank members of the Katada Laboratory for valuable discussions. This work was supported in part by research grants from the Ministry of Education, Culture, Sports, Science, and Technology (MEXT) of Japan (K.S. and T.K.) and Japan Society for the Promotion of Science (JSPS; K.S. and T.K.).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Saito, K., Katada, T. Mechanisms for exporting large-sized cargoes from the endoplasmic reticulum. Cell. Mol. Life Sci. 72, 3709–3720 (2015). https://doi.org/10.1007/s00018-015-1952-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-015-1952-9