
Abstract
Aims/hypothesis
The molecular mechanisms of insulin release are only partially known. Among putative factors for coupling glucose metabolism to insulin secretion, anaplerosis has lately received strong support. The anaplerotic enzyme pyruvate carboxylase is highly expressed in beta cells, and anaplerosis influences insulin secretion in beta cells. By inhibiting pyruvate carboxylase in rat islets, we aimed to clarify the hitherto unknown metabolic events underlying anaplerotic regulation of insulin secretion.
Methods
Phenylacetic acid (5 mmol/l) was used to inhibit pyruvate carboxylase in isolated rat islets, which were then assessed for insulin secretion, fuel oxidation, ATP:ADP ratio, respiration, mitochondrial membrane potential, exocytosis and ATP-sensitive K+ channel (KATP-channel) conductance.
Results
We found that the glucose-provoked rise in ATP:ADP ratio was suppressed by inhibition of pyruvate carboxylase. In contrast, fuel oxidation, respiration and mitochondrial membrane potential, as well as Ca2+-induced exocytosis and KATP-channel conductance in single cells, were unaffected. Insulin secretion induced by α-ketoisocaproic acid was suppressed, whereas methyl-succinate-stimulated secretion remained unchanged. Perifusion of rat islets revealed that inhibition of anaplerosis decreased both the second phase of insulin secretion, during which KATP-independent actions of fuel secretagogues are operational, as well as the first and KATP-dependent phase.
Conclusions/interpretation
Our results are consistent with the concept that anaplerosis via pyruvate carboxylase determines pyruvate cycling, which has previously been shown to correlate with glucose responsiveness in clonal beta cells. These processes, controlled by pyruvate carboxylase, seem crucial for generation of an appropriate ATP:ADP ratio, which may regulate both phases of fuel-induced insulin secretion.
Similar content being viewed by others
Introduction
Type 2 diabetes is characterised by dysregulated insulin secretion [1], which is manifested as an inadequate secretory response to glucose. The stimulus-secretion coupling of glucose in beta cells involves at least two different pathways. In the well-defined triggering or KATP-dependent pathway, glucose metabolism increases the ATP:ADP ratio, closing the ATP-sensitive K+ (KATP) channels. This depolarises the plasma membrane, voltage-dependent Ca2+-channels open, and cytosolic Ca2+ concentrations increase [2], triggering exocytosis. The less well-characterised amplifying or KATP-independent pathway is also dependent on the rise in cytosolic Ca2+, but does not necessarily involve the KATP-channels, since glucose also stimulates insulin secretion in a dose-dependent manner when these channels are bypassed [3, 4]. The KATP-dependent pathway has been suggested to account for the rapid first phase, in which a readily releasable pool of insulin granules is exocytosed. The slower, sustained, second phase requires metabolism of glucose and, presumably, the KATP-independent pathway acts on another pool of granules, converting them to a state in which they become readily releasable [5, 6].
The mechanisms underlying the KATP-independent pathway are poorly defined, and there are several hypotheses regarding the identity of the factors coupling glucose metabolism to insulin secretion. Glutamate [7], malonyl-CoA [8], ATP [9] and NADPH [10] are among the proposed signals, all of which, however, are the subjects of considerable controversy. Nonetheless, a unifying aspect of these candidates for metabolic coupling in beta cells is anaplerosis, i.e. the filling-up of carbon intermediates into the tricarboxylic acid cycle [10, 11]. The anaplerotic enzyme pyruvate carboxylase is highly expressed in beta cells as compared with islet non-beta-cells [11], and ∼40% of pyruvate entering the tricarboxylic acid cycle during glucose stimulation is carboxylated by pyruvate carboxylase [12], indicating a critical role of the enzyme. Indeed, inhibition of pyruvate carboxylase with phenylacetic acid (PAA) [13] decreases insulin secretion in INS-1 cells, 832/13 cells and rat islets [14–16]. PAA is metabolised to phenylacetyl-CoA intracellularly, and inhibits pyruvate carboxylase by competing with acetyl-CoA, a key regulator of the enzyme [17].
The aim of the present study was to investigate the metabolic events behind these observations, addressing the role of pyruvate-carboxylase-mediated anaplerosis for insulin secretion. We used PAA as a tool to inhibit pyruvate carboxylase and anaplerosis in rat islets, and examined the consequences for insulin secretion and islet metabolism. We found that pyruvate carboxylase, and thereby anaplerosis, is crucial for an appropriate rise in the ATP:ADP ratio in response to fuel metabolism, a rise thought to trigger and sustain insulin secretion.
Materials and methods
Reagents and animals
All chemicals were from Sigma (St Louis, MO, USA) if not stated otherwise. Female Sprague–Dawley rats (180–250 g; B&K, Stockholm, Sweden) were used for the experiments. The studies were approved by the Regional Animal Ethics Committee in Lund, Sweden.
Islet isolations, batch incubations and islet perifusions
Pancreatic islets from fed rats were isolated by standard collagenase digestion [18], and subsequently handpicked under a stereo microscope. All experiments were conducted on freshly isolated islets if not stated otherwise. For culture overnight, the isolated islets were kept in RPMI-1640 medium, containing 11.1 mmol/l glucose, 10% fetal calf serum, 100 U/ml penicillin and 100 μg/ml streptomycin, at 37°C in 95% air and 5% CO2. For secretion studies, batches (n=8) of three islets for each condition were kept in HEPES balanced salt solution (HBSS; in mmol/l: 125 NaCl, 5.9 KCl, 1.2 MgCl2, 20 HEPES, 1.3 CaCl2, 0.1% BSA; pH 7.35) containing 2.8 mmol/l glucose for 60 min at 37°C. The islets were then incubated in 200 μl buffer combined with the respective secretagogue and inhibitor at 37°C for an additional hour. Afterwards, a sample from the buffer was measured for insulin by ELISA (Mercodia, Uppsala, Sweden). Perifusion of islets was performed as previously described [19]. In brief, islets were pre-incubated in HBSS at 2.8 mmol/l glucose for 60 min. Then, batches of 20 islets were sandwiched between two layers of gel (Bio-Gel P4; Bio-Rad, Richmond, CA, USA). The HBSS was warmed to 37°C. Islets were perifused at a flow rate of 0.5 ml/min, fractions were collected at 1-min intervals, and insulin in the perifusate was measured by ELISA.
ATP and ADP determinations
Isolated islets were cultured overnight followed by a 2-h incubation in HBSS containing 16.7 mmol/l glucose, 2.5 μmol/l forskolin and 35 mmol/l KCl. These measures were taken to degranulate beta cells, depleting them of ATP contained in insulin granules, which obscures changes in ATP generated by metabolism [20]. Islets were washed in HBSS, followed by preincubation for 1 h in HBSS at 2.8 mmol/l glucose; the buffer was then replaced by HBSS containing 2.8 or 16.7 mmol/l glucose, or 2.8 mmol/l glucose and 10 mmol/l methyl succinate. After an additional 5 or 30 min, trichloroacetic acid was added at a final concentration of 2% to the islets, which were kept on ice for 20 min. After centrifugation (13,000 g), the supernatant was assayed for ATP content using bioluminescence (BioThema, Haninge, Sweden). ADP was measured after ATP had been depleted by ATP sulphurylase followed by conversion of ADP to ATP by pyruvate kinase, as described in detail [21].
Fuel oxidation
Isolated islets were kept in HBSS containing 2.8 mmol/l glucose for 60 min at 37°C. Batches of islets (n=20) were transferred to a cup (Kimble-Kontes, Vineland, NJ, USA) suspended from a rubber sleeve stopper (Fisher, Pittsburgh, PA, USA), which was inserted into a glass scintillation vial. For glucose oxidation, a 50 μl reaction mixture containing 13 kBq d-[14C(U)]glucose (NEN Life Science Products, Boston, MA, USA) and glucose at a final concentration of 2.8 or 16.7 mmol/l was added to the cups and the vials were sealed. For palmitate oxidation, a 100 μl reaction mixture consisting of 0.5 mmol/l palmitic acid complexed to 1% BSA (essentially fatty acid-free), with approximately 9.2 kBq [1-14C]palmitic acid (NEN) as tracer, 0.8 mmol/l l-carnitine, and glucose, at a final concentration of 2.8 or 16.7 mmol/l, was added. For glutamine oxidation, a 50 μl reaction mixture containing 3.7 kBq l-[14C(U)]glutamine (PerkinElmer, Life Sciences, Boston, MA, USA), 10 mmol/l glutamine and 10 mmol/l 2-amino-2-norbornane-carboxylic acid was added. The reaction was terminated after 1 h by injection of 100 μl 10% trichloroacetic acid into the suspended cup. The oxidation rate was measured as 14CO2 trapped in 300 μl benzethonium hydroxide added to the bottom of the sealed vials, followed by an additional incubation overnight at room temperature. 14CO2 was determined by scintillation counting.
MTS assay
Isolated islets were kept in HBSS containing 2.8 mmol/l glucose for 60 min at 37°C. Batches of islets (n=10) were transferred to a 96-well plate containing 100 μl HBSS and 20 μl 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H-(MTS) tetrazolium compound (Promega, Madison, WI, USA) and 2.8 or 16.7 mmol/l glucose. After incubation for 2 h at 37°C, the absorbance was measured at 490 nm.
Oxygen consumption
Oxygen consumption rates were monitored with an oxygen electrode (Hansatech, Cambridge, UK). Islets were kept in HBSS containing 2.8 mmol/l glucose for 30 min at 37°C. Thereafter 100 islets were added to a magnetically stirred oxygen electrode chamber thermostated to 37°C containing 300 μl HBSS. Oxygen consumption was measured during 4 min at 2.8, 16.7 and 16.7 mmol/l glucose with 5 mmol/l PAA.
Mitochondrial membrane potential
Mitochondrial membrane potential was measured by Rh123 fluorescence [22]. Isolated islets were incubated with 13 μmol/l Rh123 (Sigma–Aldrich, Sweden) for 30 to 60 min in Hanks’ balanced salt solution (Sigma–Aldrich), supplemented with 4.1 mmol/l NaHCO3. Upon investigation, a single islet was transferred to a separate glass-bottomed dish containing 1,000 μl Hanks’ balanced salt solution and placed on a heated and gas-equilibrated (37°C, 95% air–5% CO2) microscope stage (Carl Zeiss, Göttingen, Germany). Time-lapse images were recorded every 20 s. Rh123 fluorescence was excited using the 488 nm line of a krypton–argon laser of a Zeiss LSM 510 META confocal microscope, and emitted light was collected via a Zeiss Plan-APOCHROMAT ×10 objective using a band-pass 526–537 nm filter. The pinhole was maximised, resulting in a confocal section thickness of 140 μm. Rh123 fluorescence intensity was measured from the entire islet for each image frame. The osmolarity of the solutions were kept constant throughout the experiment.
Electrophysiology
Measurements in dispersed islet cells were conducted using an EPC-10 patch-clamp amplifier in conjunction with the Pulse software suite (version 8.53; HEKA Elektronik, Lambrecht/Pfalz, Germany). Exocytosis was monitored as increases in cell capacitance using the sine+DC mode of the lock-in amplifier included in the Pulse software suite. The experiments were made in the standard whole-cell configuration patch in which the pipette solution dialyses the cell and replaces the cytosol. The pipette solution consisted of (in mmol/l) 125 K-glutamate, 10 KCl, 10 NaCl, 1 MgCl2, 5 HEPES, 3 Mg-ATP, 0.1 cAMP, 10 EGTA and 9 CaCl2 (pH 7.2 with KOH). The resulting free intracellular Ca2+ concentration of this Ca2+/EGTA buffer was estimated as 1.5 μmol/l using the binding constants of Martell and Smith [23]. The extracellular solution contained (in mmol/l) 138 NaCl, 5.6 KCl, 2.6 CaCl2, 1.2 MgCl2, 5 glucose and 5 HEPES with 5 PAA added as indicated (pH 7.4 with NaOH). The bath (∼1.5 ml) was continuously perfused (6 ml/min) and the temperature maintained at ∼34°C. Effects of PAA on KATP-channel conductance were measured using the standard and perforated-patch whole-cell configuration of the patch-clamp technique. The same extracellular buffer as above was used. In standard whole-cell experiments the pipette solution (=intracellular) consisted of (in mmol/l) 125 K-glutamate, 10 KCl, 10 NaCl, 1 MgCl2, 5 HEPES, 1 Mg-ATP, 3 Mg-ADP with pH 7.2 (KOH). In the perforated-patch mode the pipette solution contained 76 mmol/l K2SO4.
Statistical analysis
Student’s t-test was used for comparison of two groups. When multiple groups were compared, ANOVA was performed followed by Bonferroni’s test post hoc.
Results
Static incubation of isolated islets
It has previously been shown that PAA decreases insulin secretion in INS-1 cells and rat islets [14]. To confirm this model for inhibition of pyruvate carboxylase and insulin secretion, we performed secretion studies in static incubations with or without PAA (Fig. 1). Upon an increase in glucose from 2.8 to 16.7 mmol/l, insulin secretion rose ninefold. In the presence of 5 mmol/l PAA insulin secretion at 16.7 mmol/l glucose was decreased by 50%.

PAA inhibits insulin secretion under both KATP-dependent and -independent conditions. Static incubations of islets at 2.8 and 16.7 mmol/l glucose in the presence (grey bars) or absence (black bars) of 5 mmol/l PAA, and in combination with 35 mmol/l K+ (K) and 250 mmol/l diazoxide (dz). Values are means±SEM for three independent experiments. *p<0.05
To further define the role of pyruvate carboxylase in glucose-stimulated insulin secretion (GSIS), we studied secretion under conditions where the KATP-dependent pathway was bypassed using 35 mmol/l KCl and 250 mmol/l diazoxide. Here, insulin secretion was stimulated 11-fold by the rise in glucose; again, insulin secretion at 16.7 mmol/l glucose was decreased by 50% when PAA was added. These results show that the actions of pyruvate carboxylase and anaplerosis involve KATP-independent processes.
According to a recent study, activation of glutamate dehydrogenase (GDH), is sufficient to fully activate the KATP-independent pathway of insulin secretion [24]. Therefore we also investigated the effect of PAA on insulin secretion stimulated by non-glucidic fuels. Stimulation of islets with 10 mmol/l α-ketoisocaproic acid (α-KIC), which is transaminated to leucine activating GDH, increased insulin secretion 14-fold at 2.8 mmol/l glucose compared with 2.8 mmol/l glucose alone (Fig. 2a). Addition of PAA decreased α-KIC-induced insulin secretion by 47% (p<0.01). Similar results were obtained when leucine was used (data not shown). Succinate is another widely used anaplerotic insulin secretagogue. Mono-methyl succinate, which is converted into succinate intracellularly, at 10 mmol/l, provoked a sixfold increase in insulin secretion (Fig. 2b). Notably, this increase remained unaffected by PAA.

Effects of PAA on non-glucidic fuel-induced insulin secretion. Islets were incubated for 1 h in 2.8 mmol/l glucose combined with 10 mmol/l α-ketoisocaproic acid (a) or 10 mmol/l methyl succinate (b) in the presence (chequered bars) or absence (white bars) of 5 mmol/l PAA. 16.7 mmol/l glucose (grey bars) was used as a stimulatory control. Values are means±SEM for six (a) or three (b) independent experiments. **p<0.01
Perifusion of islets
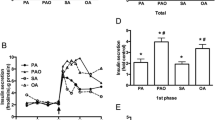
To obtain information about the dynamics of insulin secretion regarding the two phases, we analysed insulin secretion of islets in a perifusion system. Islets were perifused for 10 min with 2.8 mmol/l, followed by 30 min with 16.7 mmol/l glucose, in the presence or absence of PAA. Then the glucose concentration was lowered to 2.8 mmol/l. As shown in Fig. 3a, this stimulated a clear biphasic secretion pattern in the control islets with a rapid increase in insulin secretion followed by a nadir and then a gradually increasing rate of secretion reaching a plateau after 20 min. When PAA was added, insulin secretion rose to a level that was markedly lower, and no clear first or second phase was distinguished. PAA reduced insulin secretion significantly during both the first phase (1–10 min after elevated glucose) (p<0.01) and the second phase (11–30 min) (p<0.01).

PAA inhibits insulin secretion during both the first and the second phase of insulin secretion. a Islets were perifused for 10 min with 2.8 mmol/l glucose (glc), followed by 30 min with 16.7 mmol/l glucose, in the presence (open squares) or absence (closed diamonds) of PAA. Then the glucose concentration was lowered to 2.8 mmol/l. b Total amount of insulin released, where the first and seconds phases are defined as 1–10 and 11–30 min, respectively, after the rise in glucose. Grey bars PAA; black bars no PAA. Values are means±SEM for five independent experiments. **p<0.01
ATP:ADP measurement
Since the increase in ATP:ADP ratio is the generally accepted trigger of GSIS and responsible for initiation of the first phase of insulin secretion, we next examined the effects of PAA on adenine nucleotide levels. Measurements of ATP and ADP were performed after a 5- or 30-min incubation in 16.7 mmol/l glucose (Tables 1, 2 and 3). After 5 min, the ATP:ADP ratio increased significantly (p<0.01) when the concentration of glucose was elevated from 2.8 to 16.7 mmol/l; this increase was substantially decreased by PAA (p<0.05). ATP levels increased significantly when glucose concentrations were elevated (p<0.05), whereas ADP levels decreased (p<0.01). In PAA-treated islets, ATP levels were not significantly increased in response to glucose, and ADP levels were consistently higher than in the control islets (p<0.05). Similar changes were observed after 30 min (Table 2). Thus, anaplerosis appears to be essential for a sustained glucose-induced rise in the ATP:ADP ratio that may both trigger and amplify insulin secretion.
Since PAA had no effect on succinate-stimulated insulin secretion, we also investigated the impact of PAA on nucleotide levels in succinate-stimulated islets. Like glucose, methyl succinate induced an increase in the ATP:ADP ratio (Table 3). Again, this increase was abolished by PAA.
Fuel oxidation studies
A low ATP:ADP ratio could reflect an overall reduction of beta cell metabolism and a disturbance of the tricarboxylic acid cycle. Thus, to investigate the influence of pyruvate carboxylase inhibition on beta cell metabolism, we performed oxidation measurements in the presence and absence of PAA. Glucose oxidation increased sixfold when the glucose concentration was increased from 2.8 to 16.7 mmol/l (0.68±0.14 vs 4.5±0.76 pmol·islet−1·h−1). Addition of PAA did not affect the oxidation of glucose at either 2.8 or 16.7 mmol/l (1.22±0.55 vs 3.83±0.83 pmol· islet−1·h−1). This indicates that the replenishment of metabolites via pyruvate carboxylase into the tricarboxylic acid cycle and the oxidation of acetyl-CoA to CO2 are separated events. Also, the oxidation of glutamine was unaffected by PAA (6.32±0.96 pmol·islet−1·h−1 in control islets vs 6.76±0.87 pmol·islet−1·h−1 in PAA-treated islets). In both control and PAA-treated islets, palmitate oxidation was reduced by an increase in glucose from 2.8 to 16.7 mmol/l. However, at neither low nor high glucose was there any significant difference in the rate of palmitate oxidation between the two groups (3.42±0.57 vs 2.55± 0.58 pmol·islet−1·h−1 at 2.8 mmol/l glucose and 1.85±0.37 vs 1.52±0.36 pmol·islet−1·h−1 at 16.7 mmol/l glucose). Together, these data indicate that fuel oxidation in beta cell mitochondria is undisturbed when anaplerosis is inhibited. Furthermore, PAA has not depleted the cell of CoA, since oxidation of the lipid which requires this co-factor was unaffected.
MTS assay
To further verify that the treatment with PAA has no adverse effects on cell metabolism, we incubated islets in the presence of MTS. This agent is reduced to coloured formazan, requiring NADPH or NADH produced by dehydrogenase enzymes in metabolically active cells. It is thus an indicator of overall mitochondrial metabolism. Again, no difference was seen between PAA-treated and control islets, either at low or high glucose concentrations (101 vs 90% of control), ensuring that the cells were viable with metabolism intact.
Oxygen consumption measurements
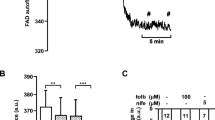
To ascertain whether PAA has any influence on the electron transport chain per se, we performed oxygen consumption measurements (Fig. 4). Oxygen consumption increased by 43% (p<0.01) when the glucose concentration was changed from 2.8 to 16.7 mmol/l. When PAA was added to the buffer, there was no change in respiration. Hence, we can conclude that while PAA specifically inhibits pyruvate carboxylase and consequently anaplerosis, there appears to be no general effect on metabolism in beta cells.

Oxygen consumption in rat islets is not affected by PAA. Oxygen consumption was measured in islets by an oxygraph, where consumption was calculated as pmol O2 consumed·islet−1·min−1 (a). O2 consumption increased significantly when the glucose concentration was increased from 2.8 (white bar) to 16.7 mmol/l (black bar), whereas no effect was seen when PAA was added (grey bar). ***p<0.001. b A representative experiment where arrows indicate addition of glucose (glc) and PAA. Values are means±SEM for six independent experiments
Mitochondrial membrane potential
If PAA acts as an uncoupler, then hyperpolarisation of the mitochondrial membrane will be reduced. To address this possibility, the mitochondrial membrane potential was assessed using the potentiometric dye Rh123 (13 μmol/l). Raising the glucose concentration from 1 to 20 mmol/l resulted in mitochondrial hyperpolarisation, as evidenced by the average 5.8±0.4% decrease in Rh123 fluorescence measured after 1–2 min at steady-state. When 5 mmol/l PAA was added to the islets either 5 min before, or concomitantly with, the sugar, Rh123 fluorescence decreased by 5.9±1.1% (ns; n=10 islets for both conditions).
Electrophysiology
To verify that PAA has no effect on the exocytotic machinery per se, we performed single-cell capacitance measurements where ATP was added in the pipette solution. Ca2+-elicited exocytosis in beta cells was monitored as increases in whole-cell membrane capacitance in the whole-cell configuration. Exocytosis commenced immediately after establishment of the whole-cell configuration at t=0, and remained unaffected in the presence of PAA (5 mmol/l) in the extracellular solution (25±7 and 27±7 fF/s, n=10 and 11, with or without PAA, respectively; Fig. 5a,b). Thus, PAA appears to exert no direct effects on exocytosis.

Effects of PAA on single beta cell electrophysiology. a Increases in cell capacitance (ΔC) evoked by intracellular dialysis of a Ca2+-containing patch electrode solution (free intracellular Ca2+ ∼1.5 μmol/l; Mg-ATP 3 mmol/l) in rat beta cells. Data represent average values±SEM every 10 s after establishing the standard whole-cell configuration (at t=0), and are from 11 cells when 5 mmol/l PAA was present in the extracellular medium (open circles), or from ten control cells (closed circles). b In intact cells using the perforated-patch configuration, the addition of 5 mmol/l PAA, increased the KATP-channel conductance. ctrl control. This effect was reversible upon wash-out. Data represent responses in seven different beta cells, summarised in (c). G M , membrane conductance; nS, nanoseconds. Left column control, middle column PAA, right column control. **p<0.01
Next, measurements of KATP-channel conductance assessed the effect of PAA on the KATP-channel activity. In intact cells (perforated-patch configuration) the addition of 5 mmol/l PAA increased the KATP-channel conductance (G) from 5.3±0.3 to 7.8±0.3 nS (5 min after addition; p<0.01, n=4). The effect was reversible as G returned to 5.1±0.2 nS upon wash-out (p<0.01). In the standard whole-cell experiments, where the intracellular ATP:ADP ratio was clamped at 1:3, G remained unaffected and averaged 12.7±2.0, 12.2±1.2 and 12.6±1.8 nS, before, 3 min after addition and after the wash-out of the acid, respectively. Taken together, the data indicate that PAA exerts no direct effect on KATP-channel activity, and that the observed effect of PAA requires a process in intact cells.
Discussion
It has been recognised that anaplerosis plays an important role in the regulation of insulin secretion. Despite this realisation, little is known about the precise mechanisms of this control. We show here that anaplerosis via pyruvate carboxylase is required for an appropriate rise in the ATP:ADP ratio to be induced by glucose, a rise thought to trigger insulin secretion.
Previous studies have shown that glucose-induced insulin secretion is decreased when the anaplerotic key enzyme pyruvate carboxylase is inhibited by PAA [14]. This was believed mainly to affect the second phase of insulin secretion, which requires activation of the KATP-independent pathway in addition to KATP-channel closure. Here, we have extended these studies and found that inhibition of pyruvate carboxylase decreased both the KATP-dependent and KATP-independent components of insulin secretion. In agreement with these observations, when examining insulin secretion in a dynamic perifusion system, we found that both phases of insulin secretion were inhibited by PAA. This indicates that anaplerosis is essential not only for sustained insulin secretion during the second phase, but also for initiating the KATP-dependent first phase of secretion. Our investigations of the effect of PAA on the KATP-channels agree with this suggestion. In the perforated-patch configuration, where metabolism presumably is unperturbed, PAA increased KATP-channel activity. In contrast, KATP-channel conductance in disrupted cells (standard whole-cell configuration), with a clamped ATP:ADP ratio, was unaffected by PAA. Collectively, this indicates that inhibition of pyruvate carboxylase influences metabolism, where the most apparent candidate is the ATP:ADP ratio. An increased KATP-channel activity could be a cause of the lacking first phase, because the closure of the KATP-channels is the generally accepted trigger of first-phase insulin secretion. Increased KATP-channel activity is not a likely explanation of the abrogated second phase, because PAA decreased insulin secretion also under KATP-independent conditions. However, adenine nucleotides may play a critical role in the second and KATP-independent phase via other mechanisms. This is supported by our findings that the ATP:ADP ratio was decreased also 30 min after a rise in glucose concentration, when the KATP-independent pathway is thought to be operating. The hypothesis was originally based on the ability of glucose to increase the ATP:ADP ratio in a concentration-dependent manner [9, 25]. Moreover, the priming of insulin granules, which modifies them into a readily releasable state, is ATP-dependent, whereas exocytosis of the immediately releasable pool is not [5]. While priming mechanisms are still to be elucidated, they may be attributed to the KATP-independent pathway.
An increased ATP:ADP ratio depends on glucose metabolism [26], and anaplerosis may be the pivotal regulator. We found that the glucose-induced rise in ATP was absent when anaplerosis was inhibited; hence our results show that influx of carbon intermediates into the tricarboxylic acid cycle is critical for appropriate ATP production in beta cells. Although the underlying mechanisms have yet to be resolved, pyruvate cycling may explain the regulatory role of anaplerosis via pyruvate carboxylase [16]. The model is based on the existence of two pools of pyruvate in beta cells, one feeding acetyl-CoA into the tricarboxylic acid cycle through pyruvate dehydrogenase, and one exchanging with tricarboxylic acid cycle intermediates via pyruvate carboxylase [16]. In clonal beta cells, only the latter correlates with glucose responsiveness and insulin secretion [16]. Pyruvate cycling will generate flux through either the pyruvate–malate [10] or pyruvate–citrate shuttles [14, 27], both of which have been proposed to generate coupling signals. This hypothesis is supported by the fact that inhibition of pyruvate carboxylase by PAA decreases pyruvate cycling and GSIS in parallel [16]. This agrees with the fact that we observed no effect of pyruvate carboxylase inhibition on glucose oxidation, while insulin secretion was markedly decreased. This model and our current results indicate that pyruvate carboxylase and pyruvate dehydrogenase act on different pools of pyruvate, and that these processes are separate from each other. The decrease of α-KIC-stimulated insulin secretion when pyruvate carboxylase was inhibited suggests that α-KIC or leucine exerted some of its effects on insulin secretion via pyruvate carboxylase. In line with this, leucine provokes an increased pyruvate carboxylase-mediated anaplerotic flux in beta cells, whereas the flux through GDH is unchanged [28]. Collectively, these observations further support the view that pyruvate cycling with flux via pyruvate carboxylase is critical and the most relevant anaplerotic pathway for regulation of insulin secretion. The stimulatory effect on insulin secretion by succinate, another anaplerotic fuel, was not affected by PAA, indicating that succinate mediates its effect through other pathways, independently of pyruvate carboxylase and pyruvate cycling. The ATP:ADP measurements, where succinate was used as secretagogue and which showed a similar decrease in ATP:ADP ratio in the presence of PAA as with glucose, suggest that succinate stimulates insulin secretion in an ATP-independent way.
Throughout the study, we have used PAA, the most widely used inhibitor of pyruvate carboxylase; so far, effects of PAA other than on metabolism and insulin secretion have not been reported in beta cells [13, 14]. However, we acknowledge that pharmacological inhibitors must be used with great caution, since non-specific effects can never be entirely excluded. To address this concern, as well as to investigate the mechanisms of anaplerosis-regulated insulin secretion, we examined relevant metabolic and functional parameters, such as fuel oxidation, respiration, mitochondrial membrane potential and exocytosis, and found that none was changed, with the exception of the ATP:ADP ratio. Many of the effects attributed to PAA when used in other systems involve more long-term alterations in function, presumably via changes in gene expression [29]. We believe that they may be of less relevance in our experiments, where acute effects were assessed.
It remains to be elucidated whether the regulation of adenine nucleotides is the sole effect of flux through pyruvate carboxylase and pyruvate cycling, or whether other molecules or signals involved in insulin secretion also are generated. NADPH is a candidate for such a messenger [10], and its role has recently received additional support [30]. Nevertheless, we show that anaplerosis via pyruvate carboxylase is a key regulator of fuel-induced insulin secretion, essential for both phases of insulin secretion. We suggest that this regulation is exerted via generation of an increase in the ATP:ADP ratio, for which pyruvate cycling plays an important role.
Abbreviations
- G:
-
conductance
- GDH:
-
glutamate dehydrogenase
- GSIS:
-
glucose-stimulated insulin secretion
- HBSS:
-
HEPES balanced salt solution
- KATP-channels:
-
ATP-sensitive K+ channels
- α-KIC:
-
α-ketoisocaproic acid
- MTS:
-
3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulphophenyl)-2H tetrazolium
- PAA:
-
phenylacetic acid
References
Porte D Jr (2001) Clinical importance of insulin secretion and its interaction with insulin resistance in the treatment of type 2 diabetes mellitus and its complications. Diabetes Metab Res Rev 17:181–188
Ashcroft FM, Harrison DE, Ashcroft SJ (1984) Glucose induces closure of single potassium channels in isolated rat pancreatic beta-cells. Nature 312:446–448
Gembal M, Gilon P, Henquin JC (1992) Evidence that glucose can control insulin release independently from its action on ATP-sensitive K+ channels in mouse B cells. J Clin Invest 89:1288–1295
Henquin JC (2000) Triggering and amplifying pathways of regulation of insulin secretion by glucose. Diabetes 49:1751–1760
Eliasson L, Renstrom E, Ding WG, Proks P, Rorsman P (1997) Rapid ATP-dependent priming of secretory granules precedes Ca(2+)-induced exocytosis in mouse pancreatic B-cells. J Physiol 503:399–412
Straub SG, Sharp GW (2002) Glucose-stimulated signaling pathways in biphasic insulin secretion. Diabetes Metab Res Rev 18:451–463
Maechler P, Wollheim CB (1999) Mitochondrial glutamate acts as a messenger in glucose-induced insulin exocytosis. Nature 402:685–689
Prentki M, Vischer S, Glennon MC, Regazzi R, Deeney JT, Corkey BE (1992) Malonyl-CoA and long chain acyl-CoA esters as metabolic coupling factors in nutrient-induced insulin secretion. J Biol Chem 267:5802–5810
Sato Y, Henquin JC (1998) The K+-ATP channel-independent pathway of regulation of insulin secretion by glucose: in search of the underlying mechanism. Diabetes 47:1713–1721
MacDonald MJ (1995) Feasibility of a mitochondrial pyruvate malate shuttle in pancreatic islets. Further implication of cytosolic NADPH in insulin secretion. J Biol Chem 270:20051–20058
Schuit F, De Vos A, Farfari S et al (1997) Metabolic fate of glucose in purified islet cells. Glucose-regulated anaplerosis in beta cells. J Biol Chem 272:18572–18579
Khan A, Ling ZC, Landau BR (1996) Quantifying the carboxylation of pyruvate in pancreatic islets. J Biol Chem 271:2539–2542
Bahl JJ, Matsuda M, DeFronzo RA, Bressler R (1997) In vitro and in vivo suppression of gluconeogenesis by inhibition of pyruvate carboxylase. Biochem Pharmacol 53:67–74
Farfari S, Schulz V, Corkey B, Prentki M (2000) Glucose-regulated anaplerosis and cataplerosis in pancreatic beta-cells: possible implication of a pyruvate/citrate shuttle in insulin secretion. Diabetes 49:718–726
Liu YQ, Jetton TL, Leahy JL (2002) Beta-cell adaptation to insulin resistance. Increased pyruvate carboxylase and malate-pyruvate shuttle activity in islets of nondiabetic Zucker fatty rats. J Biol Chem 277:39163–39168
Lu D, Mulder H, Zhao P et al (2002) 13C NMR isotopomer analysis reveals a connection between pyruvate cycling and glucose-stimulated insulin secretion (GSIS). Proc Natl Acad Sci USA 99:2708–2713
Jitrapakdee S, Wallace JC (1999) Structure, function and regulation of pyruvate carboxylase. Biochem J 340:1–16
Lacy PE, Kostianovsky M (1967) Method for the isolation of intact islets of Langerhans from the rat pancreas. Diabetes 16:35–39
Mulder H, Yang S, Winzell MS, Holm C, Ahren B (2004) Inhibition of lipase activity and lipolysis in rat islets reduces insulin secretion. Diabetes 53:122–128
Detimary P, Jonas JC, Henquin JC (1995) Possible links between glucose-induced changes in the energy state of pancreatic B cells and insulin release. Unmasking by decreasing a stable pool of adenine nucleotides in mouse islets. J Clin Invest 96:1738–1745
Schultz V, Sussman I, Bokvist K, Tornheim K (1993) Bioluminometric assay of ADP and ATP at high ATP/ADP ratios: assay of ADP after enzymatic removal of ATP. Anal Biochem 215:302–304
Duchen MR, Smith PA, Ashcroft FM (1993) Substrate-dependent changes in mitochondrial function, intracellular free calcium concentration and membrane channels in pancreatic beta-cells. Biochem J 294:35–42
Martell AE, Smith RM (1971) Critical stability constants. Plenum, New York
Liu YJ, Cheng H, Drought H, MacDonald MJ, Sharp GW, Straub SG (2003) Activation of the KATP channel-independent signaling pathway by the nonhydrolyzable analog of leucine, BCH. Am J Physiol Endocrinol Metab 285:E380–E389
Detimary P, Van den Berghe G, Henquin JC (1996) Concentration dependence and time course of the effects of glucose on adenine and guanine nucleotides in mouse pancreatic islets. J Biol Chem 271:20559–20565
Deeney JT, Prentki M, Corkey BE (2000) Metabolic control of beta-cell function. Semin Cell Dev Biol 11:267–275
Flamez D, Berger V, Kruhoffer M, Orntoft T, Pipeleers D, Schuit FC (2002) Critical role for cataplerosis via citrate in glucose-regulated insulin release. Diabetes 51:2018–2024
Cline GW, Lepine RL, Papas KK, Kibbey RG, Shulman GI (2004) 13C NMR isotopomer analysis of anaplerotic pathways in INS-1 cells. J Biol Chem 279:44370–44375
Hudgins WR, Shack S, Myers CE, Samid D (1995) Cytostatic activity of phenylacetate and derivatives against tumor cells. Correlation with lipophilicity and inhibition of protein prenylation. Biochem Pharmacol 50:1273–1279
Ivarsson R, Quintens R, Dejonghe S et al (2005) Redox control of exocytosis: regulatory role of NADPH, thioredoxin, and glutaredoxin. Diabetes 54:2132–2142
Acknowledgements
We thank K. Andersson for technical assistance with perifusion experiments and S. Lucas for aid with O2 consumption experiments. These studies were supported by grants from the Swedish Research Council (14196-03A: H.M.), the Swedish Diabetes Association (to H. Mulder and E. Renström), the Albert Påhlsson, Åke Wiberg, Ingrid and Fredrik Thuring foundations, and the Faculty of Medicine, Lund.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fransson, U., Rosengren, A.H., Schuit, F.C. et al. Anaplerosis via pyruvate carboxylase is required for the fuel-induced rise in the ATP:ADP ratio in rat pancreatic islets. Diabetologia 49, 1578–1586 (2006). https://doi.org/10.1007/s00125-006-0263-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-006-0263-y