
Abstract
Cortical microinfarcts (CMIs) observed in brains of patients with Alzheimer’s disease tend to be located close to vessels afflicted with cerebral amyloid angiopathy (CAA). CMIs in Alzheimer’s disease are preferentially distributed in the arterial borderzone, an area most vulnerable to hypoperfusion. However, the causal association between CAA and CMIs remains to be elucidated. This study consists of two parts: (1) an observational study using postmortem human brains (n = 31) to determine the association between CAA and CMIs, and (2) an experimental study to determine whether hypoperfusion worsens CAA and induces CMIs in a CAA mouse model. In postmortem human brains, the density of CMIs was 0.113/cm2 in mild, 0.584/cm2 in moderate, and 4.370/cm2 in severe CAA groups with a positive linear correlation (r = 0.6736, p < 0.0001). Multivariate analysis revealed that, among seven variables (age, disease, senile plaques, neurofibrillary tangles, CAA, atherosclerosis and white matter damage), only the severity of CAA was a significant multivariate predictor of CMIs (p = 0.0022). Consistent with the data from human brains, CAA model mice following chronic cerebral hypoperfusion due to bilateral common carotid artery stenosis induced with 0.18-mm diameter microcoils showed accelerated deposition of leptomeningeal amyloid β (Aβ) with a subset of them developing microinfarcts. In contrast, the CAA mice without hypoperfusion exhibited very few leptomeningeal Aβ depositions and no microinfarcts by 32 weeks of age. Following 12 weeks of hypoperfusion, cerebral blood flow decreased by 26% in CAA mice and by 15% in wild-type mice, suggesting impaired microvascular function due to perivascular Aβ accumulation after hypoperfusion. Our results suggest that cerebral hypoperfusion accelerates CAA, and thus promotes CMIs.
Similar content being viewed by others


Introduction
Cortical microinfarcts (CMIs) are frequently observed in the brains of Alzheimer’s disease (AD) patients [33, 41], and tend to be located in the vascular territory of leptomeningeal arteries or cortical arterioles exhibiting cerebral amyloid angiopathy (CAA), a pathological hallmark of AD [20, 33]. Furthermore, CMIs in AD are preferentially distributed in the arterial borderzone, an area particularly vulnerable to hypoperfusion [41], suggesting a causal relationship between CAA and CMIs, with hypoperfusion serving as a mediating factor. Indeed, our neuropathological study has shown that CMIs are present predominantly in the arterial borderzone between the middle and posterior cerebral artery territories in AD patients [33], which may coincide with the relative predilection of CAA pathology in posterior brain regions [42].
Nevertheless, the previous reports on the association between CAA and CMIs in AD brains have been conflicting [12, 24, 34, 40]; some studies have reported an association [34, 40] while others have found no such link [12, 24] (Supplementary Table 1). One of the plausible explanations for the disparity in the previous reports is the difficulty in assessing how the various AD-related pathologies, such as senile plaques (SPs), neurofibrillary tangles (NFTs), and CAA contribute to CMI development. Another complication arising from the findings is that these pathological changes can also be interdependent on concomitant atherosclerosis or arteriolosclerosis [34, 46]. Such difficulties are exemplified by the fact that SPs and CAA appear in close proximity in AD patients [48], though on a case-by-case level, an inverse correlation between CAA and plaque density is apparent [50].
The purpose of this study was to elucidate the possible association between the burden of CMIs and CAA by investigating postmortem brains exhibiting CAA where the final neuropathological diagnoses included not only AD but also other neurodegenerative disorders and vascular cognitive impairment. By avoiding a biased selection of postmortem brains, we anticipated this study would enable comparison of AD and non-AD patients that are accompanied by pathologically proven CAA to investigate the association between CMIs and CAA.
To explore whether chronic cerebral hypoperfusion is associated with the relationship between CMIs and CAA, we used a transgenic mouse CAA model that expresses human vasculotropic Swedish/Dutch/Iowa mutant amyloid precursor protein (APP) (Tg-SwDI mice) [11] and subjected them to bilateral common carotid artery stenosis (BCAS) to mimic chronic cerebral hypoperfusion [37]. By combining the animal investigations with postmortem human work, we aimed to reveal the underlying mechanisms in the relationship between CMIs and CAA.
Materials and methods
Postmortem human brain material
Two hundred seventy-five autopsied brains were obtained from Kyoto University Hospital and Osaka Saiseikai Nakatsu Hospital from 1992 to 2009 through a process approved by an institutional research committee. As we described previously [1, 21], neuropathological diagnoses were made by thorough histopathological examination of extensively sampled brain sections (Supplementary Fig. 1). In brief, in all brains at least 20 different samples, including anteroinferior frontal region, anterior cingulate region, middle frontal gyrus, superior frontal gyrus, precentral gyrus, superior and middle temporal gyri, amygdala, hippocampus, entorhinal cortex, supramarginal sulcus, occipital lobe, basal ganglia, thalamus, cerebellum, and at least 3 levels of the brainstem, were systematically taken from the formalin-fixed brains (Supplementary Fig. 1). Among the 275 patients, 31 patients were pathologically proved to have CAA by hematoxylin and eosin (H&E) staining and then by β-amyloid (Aβ) immunostaining, all of which were included in this study. Autopsies were performed at 12.9 ± 11.9 h (mean ± SD) (range 1.5–45.5 h) after death. The average fixation time was 37 ± 57 days (range 6–330 days) (Supplementary Table 2).
Clinical and pathological diagnosis
The 31 patients consisted of 14 AD (mean ± SD, 81 ± 8-year old) and 17 non-AD patients (78 ± 8-year old). The breakdown of the 17 non-AD patients is listed in Supplementary Table 2. The premortem clinical diagnoses, causes of death, vascular risk factors, postmortem pathological diagnoses and other demographic and pathological data of the 31 patients are also shown in Supplementary Table 2.
The clinical diagnosis of dementia met the criteria of the Diagnostic and Statistical Manual of Mental Disorders IV [3]. The neuropathological diagnoses of AD were made if the postmortem brains revealed the presence of frequent neuritic plaques in the neocortex (Consortium to Establish a Registry for Alzheimer’s Disease, CERAD) [29], and NFT stage was no less than IV, according to the Braak and Braak neuropathological staging of Alzheimer-related changes [7, 8] as assessed with modified Bielschowsky staining. Two observers (T.Y. and Y.O.) assessed SP and NFT stage individually, and if required, a joint assessment was scrutinized under a two headed microscope. The diagnosis of diffuse Lewy body disease was also made by thorough histopathological examination of extensively sampled brain sections [27]. The diagnosis of subcortical ischemic vascular dementia was made clinically [5], and was retrospectively found to meet the pathological criteria outlined by Kalaria et al. [19]: (1) the presence of bilateral diffuse white matter lesions, (2) the presence of lacunar infarctions in the perforator territory, and (3) the presence of arteriolosclerosis.
Grading of atherosclerosis
At autopsy, the atherosclerosis stage was consistently graded by one of the authors (T.Y.). The degree of atherosclerosis at skull base was classified into four grades: ‘normal’ (no atherosclerosis), ‘mild’ (the presence of patchy atheroma), ‘moderate’ (a severity that is intermediate between mild and severe), or ‘severe’ (the presence of atheroma along the entire length of the vessels). The above staging was re-examined and confirmed by another author (Y.O.) using the autopsy report and macroscopic images taken at autopsy.
Tinctorial and immunohistochemical staining
Tissue blocks were obtained from the frontal, temporal, parietal, and occipital lobes (Supplementary Fig. 1). The blocks were embedded in paraffin and sectioned at 12 μm thickness for Congo Red staining, and 6 μm thickness for other staining on a microtome. To minimize variability in staining intensity, tissue sections were prepared by the same technician and stained with or using freshly prepared tinctorial and buffer solutions. Routine histological assessment was carried out with Congo Red, H&E, Klüver-Barrera (KB), modified Bielschowsky, and Gallyas staining. Pearls-Stieda staining was added as it was needed. The rest of the blocks were used for immunohistochemistry, involving sequential incubation with primary antibody, appropriate biotinylated secondary antibody (diluted 1:200, Vector Laboratories, Burlingame, CA, USA), and avidin–biotin–peroxidase complex (1:200, Vector Laboratories) in 0.1 M phosphate-buffered saline (PBS, pH 7.4). The sections were visualized with 0.01% diaminobenzidine tetrahydrochloride and 0.005% H2O2 in 50 mM Tris–HCl (pH 7.6). The primary antibodies were mouse anti-Aβ8–17 (6F/3D) (1:100, Novocastra, Newcastle, UK), rabbit anti-cow glial fibrillary acidic protein (GFAP) (1:200, DAKO, Glostrup, Denmark), mouse anti-human paired helical filament-tau (AT8; 1:200, Thermo Scientific, Rockford, IL, USA), and mouse anti-cluster of differentiation 68 (CD68) (1:100, DAKO) antibodies.
Senile plaque and neurofibrillary tangle burden
The burden of neuritic plaques was classified into ‘none’, ‘sparse’, ‘moderate’, and ‘frequent’ categories in the cortical sections stained with the modified Bielschowsky staining according to CERAD protocol [29]. The stage of NFTs was assessed according to the Braak and Braak neuropathological staging of Alzheimer-related changes [7, 8]. In this study, we used 6 μm thick paraffin sections using the modified Bielschowsky method as we have previously reported [52]; because, the modified Bielschowsky method is far more effective over other silver staining methods in detecting NFTs.
Staging of CAA
For staging of CAA, 12 μm thick sections were stained with Congo Red and viewed with polarized light [13, 42]. CAA was classified into CAA Type 1 (affected capillaries with or without larger cerebral vessel involvement) and CAA Type 2 (affected leptomeningeal arteries, cortical arteries/arterioles, or rarely veins), as proposed by Attems et al. [4]. CAA Type 2 was further analyzed, and was divided into three grades proposed by Vonsattel et al. [44]: those with ‘mild’ (focal Aβ deposits in the smooth muscle layer of the vessel walls), ‘moderate’ (circumferential Aβ deposits in the smooth muscle layer of the vessel walls), and ‘severe’ (extensive Aβ deposition with morphological changes such as microaneurysms, fibrinoid necrosis, double barreling, inflammation, thrombus, or hemorrhage) (Fig. 1). When several grades were observed in one case, the dominant grade represented the case.

Representative photomicrographs of various grades of CAA. Congo Red staining showing mild CAA (a), moderate CAA (b), and severe CAA associated with double barreling (c). Bars indicate 100 μm in a and c, and 50 μm in b
Definition and quantitative analysis of CMI
CMIs were analyzed in the same sections that were used for pathological confirmation of CAA. CMIs were defined as cerebral cortex lesions visible only microscopically [19] and usually accompanied by reactive glial proliferation. Regions of interest with evidence of expanded Virchow-Robin space or microabscess as well as those accompanied by hemorrhagic changes or cortical laminar necrosis on H&E staining were excluded from analysis. Following further confirmation of CMIs with immunohistochemistry for GFAP and CD68, we determined the density of CMIs using a method reported previously [33]. In brief, the number of CMIs in each lobe was counted in sections stained with H&E. Images of the H&E stained slides were scanned (GT-X770 EPSON, Nagano, Japan). The cerebral cortices were outlined on each slide and the areas were measured using the ImageJ software package (image processing and analysis in JAVA, ImageJ bundled with JAVA 1.43, NIH, USA). The number of CMIs per cm2 of the cortex was calculated as a measure of CMI density. The CMI density in the frontal cortex was the mean of the values obtained from the five areas (anteroinferior frontal region, anterior cingulate region, middle frontal gyrus, superior frontal gyrus, precentral gyrus), and that in the temporal cortex was the mean of the values obtained from the two areas (lateral and medial temporal). The CMI density in the parietal cortex was obtained from the parietal supramarginal gyrus, and that in the occipital cortex was obtained from the occipital calcarine cortex (Supplementary Fig. 1).
Assessment of white matter changes
Using H&E- and KB-stained slides cut coronally at the level of mid-hippocampus, parietal, and occipital lobes, we classified white matter lesions into four grades as reported previously [9, 17]: those with ‘normal’ (normal white matter), ‘mild’ (no appreciable reduction in axonal meshwork density, and a slightly increased number of reactive astrocytes), ‘moderate’ (a slight reduction of axonal meshwork density, a reduction of oligodendroglial cell nuclei, and a further increased number of reactive astrocytes), and ‘severe’ (a marked reduction of myelin, axons and oligodendroglial cell nuclei with relatively marked astrocytic reaction, loosely scattered macrophages but no complete cerebral infarction).
Experimental animals
We used transgenic mice, C57BL/6-Tg(Thy1-APPSwDutIowa)BWevn/J [11] (Jackson Laboratory, Bar Harbor, ME, USA), which overexpress the neuronally derived human APP gene, encoding the Swedish p.K670N/M671L, Dutch p.E693Q and Iowa p.D694N mutations, under the control of the mouse thymus cell antigen 1 (Thy1) promoter. Generally, Thy1-driven exogenous gene expression is not altered by hypoxic/ischemic condition [26, 43]. The mice were screened for transgene expression by polymerase chain reaction, and heterozygous mice were mated with non-transgenic C57BL/6J mice (Japan SLC, Hamamatsu, Japan). All mice were given free access to food and water.
Surgical procedures and rearing methods
Male heterozygous mice were subjected to either sham or BCAS operation using microcoils [22, 30, 32, 37, 38]. Body weight and rectal temperature were measured, and blood pressure was monitored from the tail artery of the sham- or BCAS-operated mice. Under anesthesia with 1.5% isoflurane, the common carotid arteries were exposed through a midline cervical incision, and a microcoil with an internal diameter of 0.18 mm was applied to the bilateral common carotid arteries. Sham-operated animals underwent bilateral exposure of the common carotid arteries without inserting the microcoil. Rectal temperature was maintained between 36.5 and 37.5°C, and body weight monitored until the animals were euthanized. After the operation, the mice were housed in cages with a 12-h light/dark cycle (lights on at 7:00 a.m.). Three animal groups were prepared in this experiment (6 groups, n = 4–7 per group, n = 31 in total; detailed information given in Supplementary Table 3). The timing of sham/BCAS and the duration of cerebral hypoperfusion are described in Supplementary Fig. 2. In brief, Tg-SwDI mice were subjected to sham or BCAS operation at 10, 16, and 20 weeks of age, and they were subsequently killed at 18, 24, and 32 weeks of age, respectively, to assess histology. Wild-type age-matched C57BL/6J mice (n = 5 per group) were also subjected to BCAS operation. The number of mice used in this study was minimized for ethical reasons, and all procedures were performed in accordance with the guidelines for animal experimentation from the ethical committee of Kyoto University.
Systolic blood pressure and cerebral blood flow measurements
Mice were thermostatically controlled at 37°C on a warming pad and cerebral blood flow (CBF) and systolic blood pressure (SBP) recorded preoperatively and postoperatively at 32 weeks of age (12 weeks after BCAS or sham operation). Mean value of ten replicate measurements of CBF or SBP was determined for each mouse. The SBP was monitored in conscious mice by the tail-cuff method (MK-2000ST; Muromachi Co., Kyoto, Japan). The CBF was measured in identically sized regions of interest (900 pixels) located 1 mm posterior and 2 mm lateral from the bregma by Laser speckle blood flow imager (Omega Zone; Omegawave, Tokyo, Japan) under anesthesia with 1.5% isoflurane after the periosteum was widely removed with fine-tip forceps and calibration was carried out with a calibration reference device (Calibrator S/N 080715-5, Omegawave, Inc., Tokyo, Japan). CBF values were expressed as a percentage of the preoperative value. Since repetitive CBF measurement leads to fibrous scar tissue build up and bone opacification, CBF was measured only at two time points.
Histological investigation in mice
Mice were anesthetized with sodium pentobarbital (50 mg/kg, intraperitoneal) and transcardially perfused with 0.01 M phosphate buffer (PB) in normal saline. The removed brains were immersion-fixed in 4% paraformaldehyde in 0.1 M PB, and embedded in paraffin. The brains were then sliced into 6 μm thick sagittal sections at 1, 2, 3, and 4 mm lateral from the midline, and subjected to H&E and modified Bielschowsky staining. Immunohistochemical staining was performed according to the same protocol as human tissue. Mouse anti-Aβ1–40 (BA27) (Wako amyloid kit, Wako, Osaka, Japan), mouse anti-Aβ1–42 (BC05) (Wako amyloid kit, Wako), and mouse anti-Aβ5–10 (6E10) (1:500, Covance, Princeton, NJ, USA) antibodies were used.
Densitometric analysis of mouse brains
The Aβ-stained slides were captured with a digital camera (BZ-9000 KEYENCE, Osaka, Japan). Then, using the ImageJ software, the densitometric analysis of Aβ was performed blindly to animal groups by setting regions of interest in the cerebral cortex, the hippocampus, and the leptomeninges with the identical threshold in the Aβ (6E10)-immunostained sections. Leptomeningeal vascular, as well as pial, Aβ accumulations were jointly analyzed as ‘leptomeningeal Aβ’.
Statistical analysis
For human samples, numerical scores were computed from the data analysis as follows: age, 65- to 93-year old; disease group, AD = 1 or non-AD = 0; the grade of atherosclerosis, 0–3; the severity of CAA, 1–3; SP burden, 0–3; NFT stage, 0–6; and the grade of WML, 0–3. We first performed univariate analysis to determine whether age, disease type, or the above pathological changes were predictive of CMI formation using Fisher’s exact test. Next, multivariate analysis was performed while taking into account the effects of all variables on the parameters measured, including CMI formation.
In mice, Student’s t test was used to evaluate possible differences between the sham- and BCAS-operated mice groups at each time point, and two-way ANOVA was used to test for the effect of age and operation on Aβ deposition in the hippocampus, cerebral cortex, and leptomeninges. Differences with p < 0.05 were considered statistically significant in all analyses used.
Results
The grade of CAA and atherosclerosis, SP and NFT stage, the degree of WML, and the CMI densities in the autopsy series are shown in Table 1.
Capillary CAA
Capillary CAA was found in only one patient (Table 1, Patient no. 26). In this patient, findings of Aβ depositions in the leptomeningeal arteries and cortical arterioles were also prominent. In the remaining 30 patients, capillary CAA was undetectable with H&E staining and Aβ immunohistochemistry. In the following analyses, therefore, only CAA Type 2 was graded.
Cortical microinfarcts
CMIs were present along the sulcus in the superficial cortex with variable frequency among the postmortem brains (Fig. 2a, b). The average microinfarct diameter was approximately 200 μm (range 100–500 μm). Some lesions were recently formed, with the presence of GFAP-positive gemistocytic astroglial cells and CD68-positive microglia/macrophages (Fig. 2c–e), while others were relatively old with induction of fibrillary astrocyte gliosis. Thrombus formation was found in one patient with severe CAA (Table 1, Patient no. 13).

Representative photomicrographs of CMIs from patients with CAA. a CMIs (arrows) were present in the temporal cortex in a patient with CAA-related inflammation (H&E staining). Vascular Aβ depositions (brown) and marked vascular/perivascular infiltration of inflammatory cells are shown in insets (left, anti-Aβ immunostaining with hematoxylin counterstain; right, H&E staining). b, A CMI (arrows) in the occipital cortex in a patient with AD. c–e A CMI was accompanied with GFAP-positive reactive astroglial cells and CD68-positive macrophage/microglial proliferation (adjacent sections; c H&E; d GFAP; and e CD68). Bars indicate 300 μm in a, 50 μm in b, and 100 μm in c–e and insets in a
Most CMIs (>90%) were located in close proximity (~1 cm) to Aβ-deposited vessels (Fig. 3a–c). Around ischemic foci, such as CMIs or necrosis, SPs were sometimes not present (Fig. 3c). Furthermore, in and around ischemic foci, vascular Aβ deposition appeared to increase in intensity (Fig. 3a–c).

Representative photomicrographs of CMIs stained with H&E and Aβ from patients with CAA. a, b CMIs were located in close proximity to Aβ-deposited vessels. a and b are adjacent sections (a H&E; b Aβ). c Aβ-positive senile plaques were not apparent inside a CMI (arrows). The arrowhead indicates an Aβ-positive arteriole within a CMI. Bars indicate 500 μm in a and b, and 100 μm in c
CMI density in AD and non-AD patients
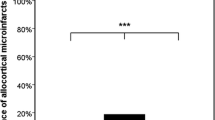
The results of the univariate analyses are shown in Table 2. The mean CMI density within the four cortical lobes was not significantly different (r = −0.1007, p = 0.5900) in AD group (0.71/cm2) and non-AD group (1.12/cm2) (Fig. 4a). When AD and non-AD groups were combined, CMI density in the frontal, temporal, parietal, and occipital lobes was 1.02, 0.65, 1.18, and 0.93/cm2, respectively (Fig. 4b). There were no significant differences in the CMI density within the four cortical lobes. CMI density in the frontal, temporal, parietal, and occipital lobes was 1.32, 0.29, 1.03, and 0.30/cm2, respectively, in the AD group, while in the non-AD group the CMI density was 0.76, 0.97, 1.29, and 1.44/cm2, respectively (Fig. 4c).

Correlation of CMI density with CAA severity. a A diagram showing no significant difference in CMI density between AD and non-AD groups (*p = 0.5900). b A diagram showing CMI density in each lobe of AD and non-AD groups combined. c Diagram showing CMI density in each lobe of AD and non-AD groups. d Significant correlation of CMI density with CAA severity (*p < 0.0001). e CAA-CMI correlation was observed in each lobe. f CAA–CMI correlation was observed in both AD and non-AD group. g CMI density was not associated with SP burden (*p = 0.2204). h CMI density was not associated with NFT stage (*p = 0.4597). i CMI density was not associated with atherosclerosis grade (*p = 0.6312)
CMI density was 0.11/cm2 in mild, 0.58/cm2 in moderate, and 4.37/cm2 in severe CAA groups, meaning it became greater as CAA severity increased (r = 0.6736, p < 0.0001) (Fig. 4d). This CAA–CMI correlation was observed in all lobes (Fig. 4e); the r value was 0.6377 in the frontal lobe (p = 0.0014), 0.5676 in the temporal lobe (p = 0.0055), 0.5355 in the parietal lobe (p = 0.0078), and 0.5652 in the occipital lobe (p = 0.0033). Such CAA–CMI correlation was also observed in both disease groups (AD or non-AD) (Fig. 4f). CMI density was not associated with SP burden (r = −0.2265, p = 0.2204) (Fig. 4g), or NFT stage (r = −0.1378, p = 0.4597) (Fig. 4h). Thus, CMI density did not change with greater burden of SP or NFT. CMI density was also not associated with the grade of atherosclerosis (r = −0.0913, p = 0.6312) (Fig. 4i).
The results of the multivariate analysis are shown in Table 3. The severity of CAA was a significant multivariate predictor (p = 0.0022), with a standard partial regression coefficient of 0.6395. Similar trends were evident in each lobe (frontal, p = 0.0014; temporal, p = 0.0055; parietal, p = 0.0078; occipital, p = 0.0033). However, age (p = 0.5979), the grade of atherosclerosis (p = 0.3973), AD or non-AD (p = 0.8364), SP burden (p = 0.5164), NFT stage (p = 0.3870), or the grade of WML (p = 0.6931) were not significant multivariate predictors.
Cerebral blood flow in mice after BCAS
The BCAS operation reduced CBF by 15% in 20-week-old wild-type mice and 26% in Tg-SwDI mice of the same age after 12 weeks with a statistically significant difference (p = 0.0412) (Supplementary Fig. 3).
Histological findings in Tg-SwDI mice
We found that microinfarcts developed in a subset of BCAS-operated Tg-SwDI mice (Fig. 5). The percentage of BCAS-operated Tg-SwDI mice (n = 17) exhibiting microinfarcts was 33.3 (2 out of 6), 42.9 (3 out of 7) and 25.0% (1 out of 4) at 18, 24, and 32 weeks of age. However, microinfarcts were not found in sham-operated Tg-SwDI mice (n = 14) or BCAS-operated wild-type mice (n = 15) up to 32 weeks of age. The number of microinfarcts in BCAS-operated Tg-SwDI mice, sham-operated Tg-SwDI mice, and BCAS-operated wild-type mouse was listed in Supplementary Table 3. Immunohistochemical analysis showed that ischemic insults accelerated perivascular and leptomeningeal Aβ accumulations (Fig. 6a, b). Generally, the immunostained patterns of Aβ1–40 and Aβ1–42 were quite similar, although Aβ40 immunostaining was more intense than Aβ1–42, in accordance with a previous biochemical study [11]. Aβ accumulation was evident in and around microvessels as well as larger cerebral vessels (approximately 10–15 μm in diameter), in the leptomeningeal arteries of BCAS-operated Tg-SwDI mice at 18 weeks of age; however, Aβ accumulation in the leptomeningeal arteries were less apparent in sham-operated Tg-SwDI mice at 18 or 24 weeks of age (Fig. 6b). As a result, two-way ANOVA showed that BCAS had a significant [F (1, 25) = 18.5, p = 0.0002] effect on leptomeningeal Aβ accumulation. However, there was no effect of age on leptomeningeal Aβ accumulation [F (2, 25) = 1.73, p = 0.20] (Fig. 6f), suggesting a ceiling effect of age on leptomeningeal Aβ accumulation.

Appearance of microinfarcts and microhemorrhages in Tg-SwDI mice after cerebral hypoperfusion. a–e Representative photomicrographs of H&E staining showing that following BCAS-induced cerebral hypoperfusion, a cortical microinfarct developed at 18 weeks of age in three of six Tg-SwDI mice (b), a hippocampal microinfarct at 24 weeks of age in three of seven Tg-SwDI mice (d), and a striatal microhemorrhage at 32 weeks of age in one of four Tg-SwDI mice (e), although such changes were not found in sham-operated Tg-SwDI mice at indicated time points (a, c). f Perls-Stieda’s iron staining showing the striatal microhemorrhage. Bars indicate 200 μm in a–d and 40 μm in e and f

Enhanced Aβ deposition in Tg-SwDI mice after cerebral hypoperfusion. a Aβ immunohistochemical staining showing that following cerebral hypoperfusion, Tg-SwDI mice exhibited cortical Aβ deposition at 24 weeks of age (right middle panel) and more extensively at 32 weeks of age (right lower panel). Sham-operated Tg-SwDI mice showed cortical Aβ deposition at 32 weeks of age (left lower panel). Insets show representative images of vascular and perivascular Aβ deposition. b Aβ immunohistochemical staining showing that following cerebral hypoperfusion, Tg-SwDI mice exhibited Aβ deposition in the leptomeninges containing leptomeningeal arteries (arrows) at 18 weeks of age (right upper panel) and more extensively at 24 and 32 weeks of age (right middle and lower panels), although sham-operated Tg-SwDI mice began to show such changes at 32 weeks of age (left lower panel). Insets show representative images of vascular and perivascular Aβ deposition in the leptomeningeal artery. In a and b, the mouse anti-Aβ1–40 (BA27) antibody was used. c The schematic illustration of the regions of interest depicted in the cerebral cortex (size, 500 × 300 μm each), the hippocampus (500 × 300 μm each), and the leptomeninges including the pia and arteries (300 × 50 μm each) of the parasagittal section of the brain. d–f Densitometric analyses of temporal profiles of Aβ depositions in the cerebral cortex (d), the hippocampus (e), and the leptomeninges (f). Leptomeningeal vascular, as well as pial, Aβ accumulation was jointly analyzed as ‘leptomeningeal Aβ’. There seems to be a ceiling effect of age on leptomeningeal Aβ accumulation. CPu caudoputamen, Cx cortex, Hip hippocampus, LM leptomeninge, LV lateral ventricle, Thal thalamus. Bars indicate 40 μm in a and b and 8 μm in insets in a and b
By contrast, in the hippocampus and the cerebral cortex, two-way ANOVA showed a significant effect of age [hippocampus, F (2, 25) = 15.8, p < 0.0001; cortex, F (2, 25) = 7.02, p = 0.0038] but no effect of the operation [hippocampus, F (1, 25) = 2.79, p = 0.11; cortex, F (1, 25) = 0.73, p = 0.40] on Aβ accumulation (Fig. 6a, d, e).
In all Tg-SwDI mice combined, the amount of leptomeningeal Aβ was significantly greater in mice with microinfarcts (3.4-fold), compared to those without microinfarcts (p = 0.0004). Most microinfarcts were seen in the cortex (Fig. 5b) and close to the Aβ-deposited vessels; however, some microinfarcts were also seen in the hippocampus CA1 area in the 18, 24, and 32-week-old BCAS-operated Tg-SwDI mice (Fig. 5d). A small number of microhemorrhages were also detected in the ventral part of the thalamus in the 32-week-old BCAS-operated Tg-SwDI mice (Fig. 5e, f), although such changes were not found in sham-operated Tg-SwDI mice or BCAS-operated wild-type mice up to 32 weeks of age (Fig. 5a, c). Thrombus formation was not apparent in any sections assessed.
Discussion
We found that CMI density is related to CAA severity, but not to SP or NFT burden severity, suggesting that the presence of Aβ causes loss of vascular autoregulation associated with rigidity of arterioles, leading to infarction in the territory of their branching vessels. Thrombus formation induced by endothelial damage in CAA may have also been involved in the CMI development although the thrombus was found only in one patient with CAA but not in any Tg-SwDI mice. The severity of white matter changes was not associated with the CMI density, which may raise the possibility that the CMIs and white matter changes have different formation processes as white matter damage is reported to reflect a progressive microangiopathy due to CAA [10]. Furthermore, our study showed that CMI density is no greater in AD than in other disorders accompanied with CAA. Given that CMI is also a strong determinant for dementia [23, 49], the observed correlation between CMI and CAA strengthens the notion that, although underestimated, these two pathologies are important substrates of cognitive decline in the elderly [15, 16].
Previous studies have reported an association between CMI and CAA in AD [15, 16, 34, 40], but others have found no such link [12, 24]. Inconsistencies between different studies might be attributable to the heterogeneity of underlying vascular pathology in AD as cerebrovascular changes can occur concomitantly with AD pathology, and also because CAA may contribute to the development of various types of cerebrovascular diseases such as lobar hemorrhage, microbleeds, and white matter lesions [39]. The issue is further complicated by the fact that CAA usually accompanies AD, but does not exclusively result from AD. Thus, accordingly, a significant correlation has been reported between the severity of CAA and the amount of CMIs in only one other study using postmortem brains of vascular dementia [16]. The current study, which used an unbiased collection of brains including non-AD samples, confirms the previous report and suggests that CAA along with chronic cerebral hypoperfusion may give rise to CMIs, which appear to be true substrates of cognitive impairment [14]. This may explain the previous findings, which have indicated that severity of CAA is associated with increased frequency of antemortem cognitive decline [25, 31]. Our findings, which show a strong association between CMIs and CAA, are supported by additional animal studies using the Tg-SwDI mice subjected to chronic cerebral hypoperfusion; when combined these findings have demonstrated that cerebral hypoperfusion induces: (1) accumulation of CAA-like vascular Aβ and resultant vascular compromise and (2) development of microinfarcts and even microhemorrhages in a subset of animals. Since microinfarcts did not develop in wild-type mice after BCAS, the data suggest that both CAA and hypoperfusion are required for microinfarct development. Tg-SwDI mice may have morphological changes in the endothelium and in the basement membranes of capillaries and arteries, leading to a failure of the mechanisms of clearance of Aβ and other metabolites. Hypoperfusion may interfere with the arterial pulsations and with the interstitial fluid pressure, leading to reduced perivascular clearance of Aβ as manifested by CAA associated with microinfarcts.
We have previously shown that chronic cerebral hypoperfusion increased the amount of filter-trap Aβ in the extracellular-enriched soluble brain fraction of mice overexpressing a mutant form of the human APP [22, 51]; in this mouse model exhibiting senile plaques, the neuronal loss and cognitive impairment that were both accelerated by hypoperfusion was explained by the disturbed Aβ metabolism. In the current study using mice overexpressing vasculotropic mutant Aβ, the hypoperfusion-induced aggravation of CAA may then result from increased synthesis of Aβ [45]. Alternatively, cerebral hypoperfusion and reduced vascular pulsation may impede clearance of Aβ as it is hypothesized that motive force of vessel pulsation is required for perivascular clearance of interstitial fluid containing Aβ [47]. This mechanism may temporarily aggravate CAA, which may compromise cerebral perfusion and ultimately lead to development of microinfarcts. Thus, the impaired microvascular function observed may impede perivascular drainage pathway of Aβ, forming a cycle of vascular Aβ deposition.
Our findings may also have implications in the understanding of the pathological complications of Aβ immunotherapy. The hypoperfusion-induced abnormalities seen in Tg-SwDI mice may be similar to the findings in AD patients who have received Aβ immunotherapy; in immunized AD group, microvascular lesions, such as CMIs or microhemorrhages, occurred with a higher density than non-immunized AD control group; this may have been because soluble Aβ mobilized from the amyloid deposits had not cleared efficiently from AD brains [6, 35]. It is plausible that the aging- and AD-associated microvascular changes [18] cannot cope with overwhelming amount of Aβ mobilized from the Aβ deposits at least partially due to existing reduction of vessel pulsation and/or hypoperfusion, leading to formation of CMIs or microhemorrhages in immunized AD patients. The current study indicates that such mechanisms underlie the formation of CMIs, even in non-immunized patients with CAA. The degree of hypoperfusion and microvascular changes may predict the response rate of Aβ immunotherapy and the emergence rate of ischemic complications. However, it remains to be determined whether chronic cerebral hypoperfusion hampers the Aβ drainage pathway directly through reduced arterial pulsation or indirectly through upregulated Aβ synthesis.
The main limitation of this study is the lack of clinical and neuropsychometric information on the postmortem brains used. This meant that we could not directly relate our pathological findings to antemortem hypoperfusive and cognitive status. Another limitation is that CAA or CMI severity could not be related to the apolipoprotein E genotype [36], due to ethical and legal constraints of genotype extraction. In addition, the use of the CERAD criteria for assessing the burden of neuritic plaques and the Braak staging for the burden of NFTs both based on the modified Bielschowsky staining may be the third limitation, considering that the CERAD is semiquantitative rather than quantitative measures of neuropathology [28] and the Braak staging criteria for tau pathology have been recently modified [2], although the two neuropathologists agreed on their final grading of the 31 CAA patients enrolled in the current study.
In summary, using postmortem brain analysis of relevant patients and models of transgenic mice, our study provides strong evidence for the relationship between CAA and microinfarcts with cerebral hypoperfusion as the mediating factor. Our observations support the notion that CAA is one of the pathological substrates that links neurodegenerative and vascular processes.
References
Akiguchi I, Tomimoto H, Suenaga T, Wakita H, Budka H (1998) Blood–brain barrier dysfunction in Binswanger’s disease; an immunohistochemical study. Acta Neuropathol 95:78–84
Alafuzoff I, Arzberger T, Al-Sarraj S et al (2008) Staging of neurofibrillary pathology in Alzheimer’s disease: a study of the BrainNet Europe Consortium. Brain Pathol 18:484–496
American Psychiatric Association (1994) Diagnostic and statistical manual of mental disorders, 4th edn. American Psychiatric Press, Washington, DC
Attems J, Jellinger K, Thal DR, Van Nostrand W (2011) Review: sporadic cerebral amyloid angiopathy. Neuropathol Appl Neurobiol 37:75–93
Bennett DA, Wilson RS, Gilley DW, Fox JH (1990) Clinical diagnosis of Binswanger’s disease. J Neurol Neurosurg Psychiatry 53:961–965
Boche D, Zotova E, Weller RO et al (2008) Consequence of Abeta immunization on the vasculature of human Alzheimer’s disease brain. Brain 131:3299–3310
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K (2006) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112:389–404
Brun A, Englund E (1986) A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol 19:253–262
Chen YW, Gurol ME, Rosand J et al (2006) Progression of white matter lesions and hemorrhages in cerebral amyloid angiopathy. Neurology 67:83–87
Davis J, Xu F, Deane R et al (2004) Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem 279:20296–20306
Ellis RJ, Olichney JM, Thal LJ et al (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer’s disease: the CERAD experience, part XV. Neurology 46:1592–1596
Grinberg LT, Thal DR (2010) Vascular pathology in the aged human brain. Acta Neuropathol 119:277–290
Hachinski V, Iadecola C, Petersen RC et al (2006) National Institute of Neurological Disorders and Stroke–Canadian Stroke Network vascular cognitive impairment harmonization standards. Stroke 37:2220–2241
Haglund M, Sjobeck M, Englund E (2004) Severe cerebral amyloid angiopathy characterizes an underestimated variant of vascular dementia. Dement Geriatr Cogn Disord 18:132–137
Haglund M, Passant U, Sjobeck M, Ghebremedhin E, Englund E (2006) Cerebral amyloid angiopathy and cortical microinfarcts as putative substrates of vascular dementia. Int J Geriatr Psychiatry 21:681–687
Ihara M, Polvikoski TM, Hall R et al (2010) Quantification of myelin loss in frontal lobe white matter in vascular dementia, Alzheimer’s disease, and dementia with Lewy bodies. Acta Neuropathol 119:579–589
Kalaria RN (1996) Cerebral vessels in ageing and Alzheimer’s disease. Pharmacol Ther 72:193–214
Kalaria RN, Kenny RA, Ballard CG, Perry R, Ince P, Polvikoski T (2004) Towards defining the neuropathological substrates of vascular dementia. J Neurol Sci 226:75–80
Kalaria RN, Thomas A, Oakley A et al (2003) Cerebrovascular amyloidosis and dementia. Curr Med Chem Immunol Endocr Metab Agents 3:317–327
Kawamoto Y, Akiguchi I, Nakamura S, Budka H (2002) Accumulation of 14-3-3 proteins in glial cytoplasmic inclusions in multiple system atrophy. Ann Neurol 52:722–731
Kitaguchi H, Tomimoto H, Ihara M et al (2009) Chronic cerebral hypoperfusion accelerates amyloid beta deposition in APPSwInd transgenic mice. Brain Res 1294:202–210
Kovari E, Gold G, Herrmann FR et al (2004) Cortical microinfarcts and demyelination significantly affect cognition in brain aging. Stroke 35:410–414
Launer LJ, Petrovitch H, Ross GW, Markesbery W, White LR (2008) AD brain pathology: vascular origins? Results from the HAAS autopsy study. Neurobiol Aging 29:1587–1590
Matthews FE, Brayne C, Lowe J, McKeith I, Wharton SB, Ince P (2009) Epidemiological pathology of dementia: attributable-risks at death in the Medical Research Council Cognitive Function and Ageing Study. PLoS Med 6:e1000180
McCarran WJ, Goldberg MP (2007) White matter axon vulnerability to AMPA/kainate receptor-mediated ischemic injury is developmentally regulated. J Neurosci 27:4220–4229
McKeith IG, Dickson DW, Lowe J et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872
Mirra SS, Gearing M, McKeel DW Jr et al (1994) Interlaboratory comparison of neuropathology assessments in Alzheimer’s disease: a study of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). J Neuropathol Exp Neurol 53:303–315
Mirra SS, Heyman A, McKeel D et al (1991) The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology 41:479–486
Nakaji K, Ihara M, Takahashi C et al (2006) Matrix metalloproteinase-2 plays a critical role in the pathogenesis of white matter lesions after chronic cerebral hypoperfusion in rodents. Stroke 37:2816–2823
Neuropathology Group. Medical Research Council Cognitive Function Aging Study (2001) Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS). Lancet 357:169–175
Nishio K, Ihara M, Yamasaki N et al (2010) A mouse model characterizing features of vascular dementia with hippocampal atrophy. Stroke 41:1278–1284
Okamoto Y, Ihara M, Fujita Y, Ito H, Takahashi R, Tomimoto H (2009) Cortical microinfarcts in Alzheimer’s disease and subcortical vascular dementia. Neuroreport 20:990–996
Olichney JM, Hansen LA, Hofstetter CR, Grundman M, Katzman R, Thal LJ (1995) Cerebral infarction in Alzheimer’s disease is associated with severe amyloid angiopathy and hypertension. Arch Neurol 52:702–708
Patton RL, Kalback WM, Esh CL et al (2006) Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: a biochemical analysis. Am J Pathol 169:1048–1063
Premkumar DR, Cohen DL, Hedera P, Friedland RP, Kalaria RN (1996) Apolipoprotein E-epsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer’s disease. Am J Pathol 148:2083–2095
Shibata M, Ohtani R, Ihara M, Tomimoto H (2004) White matter lesions and glial activation in a novel mouse model of chronic cerebral hypoperfusion. Stroke 35:2598–2603
Shibata M, Yamasaki N, Miyakawa T et al (2007) Selective impairment of working memory in a mouse model of chronic cerebral hypoperfusion. Stroke 38:2826–2832
Smith EE, Greenberg SM (2009) Beta-amyloid, blood vessels, and brain function. Stroke 40:2601–2606
Soontornniyomkij V, Lynch MD, Mermash S et al (2010) Cerebral microinfarcts associated with severe cerebral beta-amyloid angiopathy. Brain Pathol 20:459–467
Suter OC, Sunthorn T, Kraftsik R et al (2002) Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke 33:1986–1992
Thal DR, Griffin WS, de Vos RA, Ghebremedhin E (2008) Cerebral amyloid angiopathy and its relationship to Alzheimer’s disease. Acta Neuropathol 115:599–609
Underhill SM, Goldberg MP (2007) Hypoxic injury of isolated axons is independent of ionotropic glutamate receptors. Neurobiol Dis 25:284–290
Vonsattel JP, Myers RH, Hedley-Whyte ET, Ropper AH, Bird ED, Richardson EP Jr (1991) Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann Neurol 30:637–649
Wang Z, Wu D, Vinters HV (2002) Hypoxia and reoxygenation of brain microvascular smooth muscle cells in vitro: cellular responses and expression of cerebral amyloid angiopathy-associated proteins. APMIS 110:423–434
Weller RO, Boche D, Nicoll JA (2009) Microvasculature changes and cerebral amyloid angiopathy in Alzheimer’s disease and their potential impact on therapy. Acta Neuropathol 118:87–102
Weller RO, Djuanda E, Yow HY, Carare RO (2009) Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol 117:1–14
Weller RO, Massey A, Newman TA, Hutchings M, Kuo YM, Roher AE (1998) Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer’s disease. Am J Pathol 153:725–733
White L, Petrovitch H, Hardman J et al (2002) Cerebrovascular pathology and dementia in autopsied Honolulu-Asia Aging Study participants. Ann N Y Acad Sci 977:9–23
Wyss-Coray T, Lin C, Yan F et al (2001) TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med 7:612–618
Yamada M, Ihara M, Okamoto Y et al (2011) The influence of chronic cerebral hypoperfusion on cognitive function and amyloid beta metabolism in APP overexpressing mice. PLoS One 6:e16567
Yamamoto T, Hirano A (1986) A comparative study of modified Bielschowsky, Bodian and thioflavin S stains on Alzheimer’s neurofibrillary tangles. Neuropathol Appl Neurobiol 12:3–9
Acknowledgments
We would like to express our cordial gratitude to Ms. Takako Kawada and Ms. Mayumi Ohara for their excellent technical assistance, and Dr. Ahmad Khundakar for his critical reading of the manuscript. We gratefully acknowledge grant support from the Lifelong Health & Wellbeing programme supported by the Research Councils UK Newcastle Centre for Brain Ageing and Vitality (R.N.K.), Alzheimer’s Research UK (R.N.K.), the UK NIHR Biomedical Research Centre for Ageing and Age Related Diseases award to the Newcastle upon Tyne Hospitals NHS Foundation Trust (R.N.K.), the Bayer Scholarship for Cardiovascular Research (M.I.), the Univers Foundation (M.I.), and the Grants-in-Aid for Scientific Research (B) (M.I. No. 23390233).
Conflict of interest
The authors have no conflict of interest to declare.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Okamoto, Y., Yamamoto, T., Kalaria, R.N. et al. Cerebral hypoperfusion accelerates cerebral amyloid angiopathy and promotes cortical microinfarcts. Acta Neuropathol 123, 381–394 (2012). https://doi.org/10.1007/s00401-011-0925-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-011-0925-9