
Abstract
Many central nervous system diseases currently lack effective treatment and are often associated with defects in microvascular function, including a failure to match the energy supplied by the blood to the energy used on neuronal computation, or a breakdown of the blood–brain barrier. Pericytes, an under-studied cell type located on capillaries, are of crucial importance in regulating diverse microvascular functions, such as angiogenesis, the blood–brain barrier, capillary blood flow and the movement of immune cells into the brain. They also form part of the “glial” scar isolating damaged parts of the CNS, and may have stem cell-like properties. Recent studies have suggested that pericytes play a crucial role in neurological diseases, and are thus a therapeutic target in disorders as diverse as stroke, traumatic brain injury, migraine, epilepsy, spinal cord injury, diabetes, Huntington’s disease, Alzheimer’s disease, diabetes, multiple sclerosis, glioma, radiation necrosis and amyotrophic lateral sclerosis. Here we report recent advances in our understanding of pericyte biology and discuss how pericytes could be targeted to develop novel therapeutic approaches to neurological disorders, by increasing blood flow, preserving blood–brain barrier function, regulating immune cell entry to the CNS, and modulating formation of blood vessels in, and the glial scar around, damaged regions.
Similar content being viewed by others

Introduction
Capillary pericytes have important roles in blood vessel formation and stabilization [5, 149], blood–brain barrier (BBB) formation and maintenance [6, 11, 29], control of capillary diameter and cerebral blood flow (CBF) regulation [9, 54, 55, 116], amyloid β clearance [124], mediation of neuroinflammation [67, 122, 135], glial scar formation [46], and in some circumstances they exhibit properties of stem cells [33, 98, 109]. Recent studies have revealed that pericytes have important roles in numerous CNS disorders including ischaemic stroke [36, 54, 155], epilepsy [84], spinal cord injury (SCI) [46, 89], diabetes [43], Huntington’s disease [35], Alzheimer’s disease (AD) [55, 124], multiple sclerosis (MS) [122], glioma [20, 53, 138, 148], radiation necrosis [87] and amyotrophic lateral sclerosis (ALS) [150]. In these disorders, pericyte malfunction often leads to BBB disruption and/or a decrease of blood flow, thus causing secondary neurological damage. In this review, we will initially introduce the biological characteristics of pericytes and then discuss how they act either protectively or to promote damage in the progression of CNS disorders. We will focus in particular on the possible mechanisms and consequences for disease of pericyte contraction, pericyte death, abnormal angiogenesis, immunological derangement and scar formation, which will shed light on possible therapies for CNS diseases. We will first consider how pericytes are defined and the heterogeneity of their properties, then review their main physiological functions, before describing how their malfunction contributes to different CNS diseases and suggesting therapeutic approaches that are based on targeting pericytes.
Defining pericytes and their heterogeneity
Pericytes, also known as mural cells (a classification which also includes vascular smooth muscle cells around arterioles) or Rouget cells, were first described by the German scientist Ebert in 1871 and the French scientist Rouget in 1873. They were named as “pericytes” by Zimmermann in 1923 [81, 166] due to their location enveloping the endothelium, and being embedded in the basement membrane outside brain vessels including capillaries, post-capillary venules and terminal arterioles (Figs. 1, 2) [5].

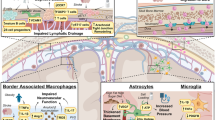
Functions of CNS pericytes in health. Pericytes form a chain contacting endothelial cells [14], interacting physically with them via a peg and socket structure. Several functions of CNS pericytes are illustrated. (1) BBB formation and maintenance, by regulating tight and adherens junctions, and transcytosis across endothelial cells. (2) Immunoregulation by pericytes regulating the entrance and movement of immune cells such as neutrophils. (3) Capillary diameter (arrows) and hence CBF are regulated by α smooth muscle actin-expressing circumferential processes of pericytes on at least the first four branching order vessels of the capillary bed. (4) Angiogenesis and vessel stabilization are mediated by pericytes during the development and repair of the vasculature. (5) Pericytes can proliferate after conditions like ischaemia, and may also be able to differentiate into other cell types

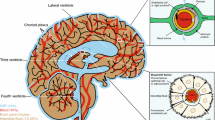
Morphology of, and common labelling methods for, pericytes. a Human cortical pericytes, in healthy tissue removed to allow glioma removal. Isolectin B4 tagged with a green dye is used to label the basement membrane, which extends along capillaries and around pericytes. Pericytes can be seen on straight parts of the capillary and at junctions [white and yellow arrow-heads, respectively, here and in c]. b Human cortical pericyte as in a, labelled for IB4 (green) and the pericyte marker PDGFRβ. c Cortical capillaries labelled for IB4 (green) in an NG2-DsRed mouse in which pericytes are red. d Larger views of the top left pericyte show circumferential DsRed-labelled processes (arrows) that will adjust capillary diameter when they contract
Even today, correct identification of pericytes is challenging because of their heterogeneity [5, 49, 59], an understanding of which is likely to be important for understanding their role in disease. For example, there are more contractile pericytes expressing α smooth muscle actin (αSMA) at the arteriole end of the capillary bed [9, 64] (out to the 4th branching order of the capillary bed), there is probably differential regulation of immune cell migration by pericytes at different locations along the capillary bed [135], and a subset of pericytes can proliferate after CNS injury and contribute to the scar which isolates damaged tissue from surrounding healthy tissue [32, 46, 63]. Furthermore, pericytes need to be distinguished from a population of fibroblast-like cells that are present on CNS blood vessels other than capillaries [144].
Nevertheless, platelet-derived growth factor receptor β (PDGFRβ) [90], alanyl aminopeptidase (CD13) [82], the proteoglycan neuron-glial antigen 2 (NG2) [110] and desmin [99] are markers often used to identify pericytes (Fig. 2), with α smooth muscle actin being used to define a contractile sub-class of pericytes [100]. Other markers, such as regulator of G-protein signalling protein 5 (RGS5) [18], SUR2 (ATP-binding cassette transporter subfamily C member 9 or ABCC9) [17], Kir6.1 (potassium inwardly rectifying channel subfamily J member 8) [17], delta-like non-canonical Notch ligand 1 (DLK1) [17], T-box transcription factor 18 (Tbx18) [50], GLAST [46] and endosialin [23], also label pericytes, but in addition label other cells. The expression of all these markers changes during growth and development, and may be up- or downregulated in pathological conditions [5]. Therefore, cell morphology, anatomical position, and the absence of endothelial and glial cell markers, should also be taken into consideration to reduce misidentification of pericytes. In particular, a standard anatomical criterion for defining pericytes on capillaries is that they have spatially isolated nuclei, with processes running along the capillary that separate them from the next pericyte soma along the vessel. This distinguishes them unambiguously from vascular smooth muscle cells (vSMCs), which abut each other when forming the smooth muscle around arterioles. Ignoring this fundamental distinction has led to a recent misidentification [64] of contractile pericytes, which express αSMA and extend processes wrapping around capillaries, as being vSMCs (see discussion in Ref. [9]).
The use of genetic mouse models such as NG2-dsRed [126], NG2-EYFP [160], RGS5-GFP [109], NG2-eGFP [64] and NG2/PDGFRβ-tdTomato [59] mice, which label (at least some classes of) pericytes and their progeny, has paved the way for studying the fate of pericytes in physiology and pathology using intravital imaging. Additionally, the development of NG2- and PDGFRβ-driven Cre expression (constitutive or inducible) which can be crossed with specific floxed mice lines to delete genes of interest, may aid the field in gaining a deeper understanding of the role of pericytes in physiological and pathological settings [25]. Some pericyte-deficient mice, such as Pdgfbret/ret mice in which the PDGF-B retention motif is depleted to disrupt its binding to heparan sulphate proteoglycans [91] and Pdgfrβ+/− or Pdgf hypomorph mice, which have a 20–50% reduction of pericytes [6, 11, 29], have also been created to study the effect of pericyte degeneration on neurovascular function. However, the use of some mouse lines requires care. For example, along with being expressed in pericytes, NG2 is also expressed in oligodendrocyte progenitor cells and PDGFRβ is reported to be expressed in some neurons, so understanding the effects of Cre driven by the promoters for these proteins requires control experiments to be sure that the effects seen are mediated by pericytes.
It has recently been reported that a subset of pericytes specifically take up the fluorescent Nissl dye NeuroTrace 500/525 so that they can be distinguished from other brain cells, which may also open a window for studying pericyte behaviour in physical and pathological states [28]. Why this label preferentially enters pericytes is unclear, and it is still uncertain whether this dye labels all pericytes or just the non-contractile pericytes in the middle of the capillary bed.
Transcriptome studies of pericyte heterogeneity
Ultimately we can hope that transcriptome and proteome studies will define precisely the mRNA and protein expression of pericytes at different positions along the capillary bed, and how they change in disease. However, for “-omics” studies, it is essential to have a marker for the cells to be studied (to isolate and sort the cells), and choosing one particular marker to define pericytes may well lead to the exclusion of certain subclasses of the cells. Indeed, studies to date have given very different results when characterising pericyte mRNA expression.
For example, comparison of five different mural cell (pericyte plus vascular smooth muscle cell) transcriptomes by the Betsholtz group [62] revealed a “surprisingly limited overlap” of the main genes expressed, with the five studies reporting only three “core” transcripts in common. Two of these transcripts were related to actomyosin contraction, yet a subsequent study by the same group [144] reported that pericytes express almost no α smooth muscle actin. This is very surprising, since pericytes are visibly observed to contract and relax in videos provided by several studies [54, 116], and α smooth muscle actin has been observed in immunohistochemical studies of pericytes by numerous groups [2, 9, 10, 64, 156] (even in mid-capillary bed pericytes when actin depolymerisation is inhibited [2]). At present it is unclear whether these discrepancies reflect differences in the method of selecting pericytes for transcriptome analysis, a rapid down-regulation of αSMA expression during processing for the transcriptomic analysis, or mediation of contraction by a previously unappreciated process (perhaps involving γ actin [49]).
Normal functions of pericytes underlying their role in CNS disease
Below we will describe recent data showing how pericytes contribute to CNS disorders. To understand their role in disease, it is essential to understand the normal functions of pericytes and how they may vary between pericytes at different locations on the capillary bed. Here, therefore, we will review the main functions of pericytes, before describing how deficits in these pericyte functions contribute to neuronal damage in disease.
Blood vessel formation
Pericytes play a key role in the generation of new blood vessels. A complex web of bidirectional signalling pathways mediating interactions between endothelial cells and pericytes is essential for forming new blood vessels and stabilising existing ones (clinical disorders resulting from defects in the operation of these pathways are discussed below). Briefly (for reviews see [134, 139]), endothelial cells release PDGF-BB which binds to PDGFRβ on pericytes, enhancing their proliferation and recruiting them to the endothelial tube. An association of pericytes with capillaries is essential for a capillary to be stable [90], and in the brain it is also a prerequisite for a properly functioning blood–brain barrier (see below). Signalling mediated by pericyte-derived angiopoietin-1 (Ang-1) binding to Tie-2 tyrosine kinase receptors mainly on endothelial cells (but also on pericytes [141]) promotes vessel formation by increasing endothelial cell proliferation, migration and survival [1], while a related ligand, Ang-2 (expressed in developing blood vessels), inhibits the effects of Ang-1. TGF-β1, which is produced by endothelial cells and pericytes, induces the formation of pericytes and inhibits endothelial cell proliferation [5, 13], while in hypoxic conditions VEGF is produced and induces proliferation and migration of pericytes [68, 159]. Together, these pathways, with additional signalling via Notch-3 [146] and NG2, lead to the formation of a fairly regularly spaced array of capillaries (with pericytes located at intervals along them) that deliver adequate oxygen and glucose to the CNS tissue. Interestingly, the ratio of pericytes to endothelial cells is somewhat higher in CNS tissue than in most other vascular beds and varies between CNS locations [5, 38], presumably reflecting some quantitative difference in the strength of signalling by all these pathways that results in an adequate number of pericytes to maintain an adequate vessel density, blood flow and BBB function. Table 1 summarizes the functions and disorders in which these signalling pathways are involved.
Constriction and dilation of capillaries
Calcium-dependent contraction of pericyte processes that run around capillaries (Fig. 2d) evokes capillary constriction. This occurs both in response to pericyte depolarization produced by a microelectrode (which is presumed to open voltage-gated Ca2+ channels in the pericytes, as will a rise of [K+]o in pathology), and when a rise in [Ca2+]i is evoked by a range of neurotransmitters and other vasoactive molecules including noradrenaline, ACh, ATP, angiotensin II, endothelin-1 and lactate [54, 72, 73, 83, 89, 116, 142, 151, 153]. A claim that these constrictions are mediated by vSMCs [64] has been explained to reflect an erroneous definition of pericytes [9]: any contractile cell on vessel walls with circumferential processes was defined in Ref. [64] to be a vSMC, ignoring the conventional (nearly 100 year old) definition of spatially isolated mural cells (even those with circumferential processes) as being pericytes [81, 166]. Noradrenaline release from locus coeruleus neurons confers a contractile tone to pericytes. This allows pericyte relaxation, capillary dilation and an increase in capillary blood flow [15, 54, 79] when neuronal activity releases dilating factors such as glutamate (which evokes ATP release from neurons, thus raising astrocyte [Ca2+]i and generating prostaglandin E2 release), adenosine or lactate, or when NO is released from endothelial cells [27, 42, 54, 116, 152, 153]. When neurons are active, pericytes relax and increase the diameter of capillaries faster than vSMCs relax to dilate penetrating arterioles [54, 79]. Since most of the vascular resistance within the brain parenchyma is located in capillaries [47], and the magnitude of the dilation that pericytes produce of capillaries is similar to that which occurs in penetrating arterioles when neurons are active [54], it follows that pericyte-mediated capillary dilation contributes significantly to the increase of blood flow that is triggered by neuronal activity [15, 54, 79]. Interestingly, even mid-capillary bed pericytes (which mainly lack circumferential processes that could contract to provide tone) may regulate blood flow by adjusting capillary diameter, perhaps by altering growth of the endothelial tube [14] or by relaxing and decreasing vessel wall stiffness when neurons are active to allow easier passive dilation [121].
Blood–brain barrier maintenance
The BBB is conferred by tight junctions between endothelial cells, and low rates of transcellular vesicular transport (transcytosis) across CNS endothelial cells. Transgenic experiments in which the level of PDGFRβ is reduced, thus reducing the number of pericytes present by up to 50%, have revealed that the presence of pericytes is essential to maintain the BBB [6, 11, 29]. Pericytes achieve this by: (1) increasing endothelial expression of Ang1 and decreasing expression of Ang2, which leads to a suppression of vascular permeability [6]; (2) promoting expression of the major facilitator superfamily domain containing 2a (Mfsd2a) transporter protein in endothelial cells, which in turn suppresses vesicle-mediated transcytosis across the endothelial layer [6, 12, 22, 29]; and (3) maintaining tight junction protein expression in older animals [11]. Loss of pericyte-induced BBB function leads to influx into the brain parenchyma of molecules with a molecular weight up to 500 kDa, including serum proteins (such as thrombin and fibrinogen) and perhaps other toxic molecules (such as glutamate and ATP) that cause neuronal and vascular damage and lead to microglial activation [11]. Pericyte-deficient mice have reduced cerebral blood flow resulting in neurovascular uncoupling, reduced oxygen supply to brain and metabolic stress [79].
Regulation of immune cell entry
In pathology, immune cells enter the brain. This process is regulated by pericytes. Transgenic deletion of pericytes leads to an upregulation of leukocyte adhesion molecules and plasmalemma vesicle-associated protein (PLVAP) in endothelial cells [29]. PLVAP regulates leukocyte migration both in blood vessels and lymphatic vessels [52, 118]. In muscle it has been shown that leukocytes cross the venule endothelial cell layer and then migrate along pericyte processes before exiting into the tissue at gaps between pericytes [117], while work in the placenta has shown that, having entered the tissue, they are then attracted to capillary pericytes by release of the chemoattractant macrophage migration–inhibitory factor (MIF) [135]. Loss of pericytes leads to leukocytes entering the brain, and may modulate the inflammatory response [29, 122].
Proliferation and migration in response to injury
CNS injury evokes a recruitment of immune cells to the injury site, but also astrogliosis which leads to the formation of a so-called ‘glial scar’ around the injury site, forming a barrier between the injured and the non-injured tissue which may reduce further neuronal loss, at the possible expense of hindering the regeneration of axons through the lesion [3, 147, 165]. Some cells within the glial scar express NG2, which may contribute to the hindrance of axon regrowth [96]. Although some of these cells are oligodendrocyte precursor cells, a significant fraction of cells within the glial scar are apparently derived by proliferation and migration of a subset of NG2-expressing pericytes that express the glutamate transporter GLAST [32, 46, 63].
Role in CNS disorders
Given the important roles of pericytes in normal CNS function described above, it is unsurprising that they play a major role in disease. In general, such contributions include alterations in vasculogenesis, capillary diameter, BBB function, immune cell entry and glial scar formation (Fig. 3). In this section, we will review the evidence for these actions in a series of neurological diseases, before considering what drug treatments might be used to target pericytes therapeutically. We deal first with disorders involving energetic challenge and cell depolarization (stroke, the spreading depression occurring in traumatic brain injury and migraine, epilepsy and spinal cord injury), then a situation of energy-oversupply (diabetes), before discussing protein malfunction diseases (Huntington’s and Alzheimer’s), and finally diseases that cannot easily be grouped with others including multiple sclerosis, glioma, radiation necrosis and amyotrophic lateral sclerosis. In general, the evidence cited comes from animal models of disease, but where possible we describe human patient or post-mortem data.

Pericyte responses to brain injury. Injury can: (1) induce pericyte (green) mediated constriction of capillaries; (2) evoke pericyte-mediated regulation of immune function by recruitment of immune cells (leukocytes, blue) to the brain parenchyma, phagocytosis (of green circles) and release of factors (blue open circles) that modulate microglial and macrophage function; and (3) cause loss of BBB function with detachment of pericytes from capillaries and apoptosis, and vessel leakage. Later after injury, pericytes proliferate and migrate to contribute to the scar around the damaged area, and promote blood vessel formation to re-supply the damaged area with nutrients
Stroke
During ischaemia, for example caused by block of an upstream artery, pericytes constrict capillaries both in vitro and in vivo [54, 116, see also 64 but note that this paper mis-named pericytes as smooth muscle cells)]. This is presumably because the fall of ATP levels in pericytes leads to less Ca2+ extrusion and a rise of [Ca2+]i—a process facilitated by the large rise of [K+]o and concomitant depolarization of all cells that occurs during the anoxic depolarization which occurs after a few minutes of ischaemia [57]. In profound ischaemia (“chemical ischaemia”, which prevents synthesis of ATP from either glycolysis or oxidative phosphorylation), this constriction occurs over 15–30 min [54], and a similar pericyte-mediated constriction of coronary capillaries occurs within 45 min when the heart experiences ischaemia in vivo [103]. In the brain this constriction, which will reduce blood flow, is followed by the pericytes dying [36, 54]. This death is thus expected to occur with the pericytes in rigor, constricting the capillaries [54], suggesting that, even after the upstream artery is unblocked by administration of tissue plasminogen activator or use of a stent retriever, a long-lasting decrease of blood flow will occur (until the dead pericytes are removed by microglia). In animal experiments, this so-called no-reflow phenomenon leads to blood flow being reduced by ~ 45% when the upstream artery is unblocked [61], which presumably contributes to continuing generation of neuronal damage. In addition to the loss of blood flow, neuronal damage will also be promoted by any loss of blood–brain barrier function [6, 11, 29] which results from pericyte death.
These data suggest that better restoration of cerebral blood flow and maintenance of BBB function after ischaemia might be achieved by the development of therapies targeted at preventing capillary constriction by pericytes, and preventing pericyte death. Below, we will consider strategies to achieve this. In the longer term after ischaemia, as described below for spinal cord injury, pericytes also proliferate and migrate to contribute to the scar that forms around damaged tissue [36].
Spreading depression
Cortical spreading depression (SD) is a wave of profound neuronal depolarization triggered either by brain trauma, epilepsy (discussed below) or during migraine attacks. After traumatic brain injury in human patients, the occurrence of SD waves correlates with long-term brain damage [58], probably at least in part due to the reduction of blood flow that occurs. SD is associated with a rise of [K+]o to ~ 25 mM, and a prolonged decrease of cerebral blood flow that is generated by release of the vasoconstricting arachidonic acid derivative 20-HETE [37]. Although the involvement of pericytes in the reduction of blood flow remains to be shown, pericyte-mediated capillary constriction by 20-HETE may occur in these conditions, since 20-HETE is known to constrict pericytes [54], and much of the adjustable vascular resistance within the brain parenchyma is located in capillaries [47].
Epilepsy
In animal models of epilepsy, it has recently been shown that focal capillary constrictions occur in close spatial association (< 3 μm) with NG2-expressing mural cells (pericytes in this case), and that these constrictions are surrounded by regions of neuronal damage [84]. Although the stimulus for the constriction was not studied, a rise of [K+]o during the seizure might lead to pericyte depolarization and activation of voltage-gated calcium channels, evoking Ca2+ entry and pericyte myofilament contraction. Alternatively, the seizure-evoked [K+]o rise could evoke local release of noradrenaline from locus coeruleus axon terminals, or the seizure-associated rise of [Ca2+]i might evoke the release of arachidonic acid from astrocytes and generation of vasoconstrictive 20-HETE [8], as occurs in spreading depression [37]. In addition, status epilepticus is associated with increased turnover of pericytes, associated with vessel leakage and a decreased responsivity to glutamate and endothelin [4, 93].
Spinal cord injury
Spinal cord injury (SCI) often leads to a crushing of blood vessels, generating ischaemia, which may result in pericytes constricting capillaries and dying, as described above. Recently, however, another pericyte-mediated constriction mechanism that decreases spinal blood flow, and produces hypoxia below the lesion, has been revealed [89]. Below the lesion, pericyte 5-HT1 and α2 adrenergic receptors become activated, evoking capillary constriction, despite the fact that the lesion often leads to a loss of descending monoaminergic neurons. Activation of these receptors results from the production of trace amines (e.g., tryptamine and tyramine) by pericytes that ectopically express the enzyme aromatic l-amino acid decarboxylase (AADC), which synthesizes trace amines from dietary amino acids such as tryptophan [48]. The resulting contraction of pericytes locally constricts capillaries, thus reducing blood flow and causing a chronic state of hypoxia in the spinal cord below the injured site for months in a rat model of SCI. Blocking these amine receptors or AADC was found to restore blood flow and return the tissue oxygen level to normal below the lesion [89], and this improved the motor function and locomotion. Remarkably, inspiring a higher than normal oxygen concentration also raised the oxygen level below the lesion for a prolonged period (~ 20 min), perhaps by increasing neuronal activity and thus evoking the release of vasodilating factors that increased blood flow [89].
Suppression of trace amine generation or blocking the downstream pericyte constriction therefore seem to be promising approaches for partial restoration of function after SCI. The long-lasting effect of raising the local oxygen level by transiently breathing hyperoxic air also suggests the presence of a positive feedback loop, whereby an increase of local blood flow produces a further increase of flow, which might potentiate the effect of therapeutic interventions.
A further aspect of pericyte function after spinal cord injury is that a subset of pericytes (possibly pericytes with properties different from those releasing the trace amines) migrates to form the fibrotic core of the glial scar around the spinal lesion [46]. There is controversy over the functional consequences of the pericyte contribution to the scar, with one study claiming it is needed for revascularisation of the damaged area [63] and another claiming that it reduces axon regrowth through the lesion [32].
Diabetes
Pericyte loss from retinal capillaries is an early symptom of diabetic retinopathy. The high blood glucose concentration occurring in diabetes activates a pathway, involving protein kinase C δ (PKC-δ) and MAP kinase, which induces a tyrosine phosphatase (SHP-1) to dephosphorylate PDGFRβ and thus inhibit endothelial PDGF-BB signalling to pericytes via this receptor [43]. This leads to pericyte apoptosis, and a decrease in the number of pericytes on capillaries [13, 19]. P2X7 receptor activation [137], and increased secretion of Ang2 from endothelial cells acting via the α3β1 integrin-p53 pathway [56, 114], can also contribute to pericyte apoptosis in diabetes. The loss of capillary pericytes causes loss of blood–retina barrier function. This can be reversed by applying β adrenergic agonists, which may act via Akt [158]. Pericyte loss also leads to microaneurysms [143], which can leak and cause local oedema. Diabetes may similarly induce pericyte loss from brain capillaries [131, 133], although this has been less studied. Pericyte-targeted therapy may thus be useful to protect the retina and brain in diabetes.
Huntington’s disease
Huntington’s disease (HD) is caused by a mutation (a trinucleotide repeat) in the first exon of the huntingtin gene, which results in a loss of medium spiny neurons in the striatum. HD in humans is also associated with a loss of BBB function, resulting from a decrease in tight junction expression and an increase in endothelial transcytosis, which may reflect a decrease in PDGFRβ expression in pericytes [35]. There is an increase of vessel density, possibly due to more VEGF release from reactive astrocytes, and pericyte density may increase early in the disease but decrease later on [65, 112]. The pericyte changes early in the disease precede neuronal loss [112], and may therefore contribute to that loss.
Alzheimer’s disease
In Alzheimer’s disease (AD), amyloid β accumulates around the walls of arterioles and capillaries, a condition termed cerebral amyloid angiopathy (CAA). Capillaries in the brains of AD patients also show an abnormal focally constricted morphology [60, 77] with some resemblance to that produced by pericyte contraction [54, 116]. This presumably contributes to the decrease of cerebral blood flow seen in human AD, which is one of the first changes to occur and which can be greater than 40% [7, 66]. This CBF reduction may also, in part, reflect a decrease of the coupling between neuronal activity and blood flow [102], which is partly mediated by pericytes [54, 78, 94]. In human AD and its mouse models, a loss of pericytes from the capillary wall coincides with a loss of blood–brain barrier function [124, 130], consistent with the role of pericytes in maintaining the BBB that was discussed above. Loss of pericytes also appears to promote amyloid β accumulation, tau pathology and early neuronal loss [124]. These changes suggest that therapies aimed at maintenance of normal pericyte function in AD may, by preventing the decrease of CBF and loss of BBB function, serve to preserve neuronal function longer.
Multiple sclerosis
In multiple sclerosis (MS), peripheral lymphocytes are believed to enter the CNS (a process which may be regulated by pericytes: [29, 52, 117, 118, 135]) and damage myelin and neurons, although there may also be a hypoxic component to the disease [30] which could theoretically induce pericyte loss as in brain ischaemia. Indeed, a loss of BBB function associated with pericyte degeneration is an early feature of human MS [24]. In transgenic mice with low pericyte numbers [31], differentiation of oligodendrocyte precursor cells is slowed during remyelination after a demyelinating insult. Based on culture experiments, this was suggested to reflect laminin 2 derived from pericytes promoting differentiation (and pericytes may also secrete other pro-regenerative molecules [41]), however, interpretation is complicated by the loss of BBB function that occurs in vivo when pericyte number is decreased.
Glioma
Growing tumours require a supply of energy and carbon skeletons and thus need to become vascularised. In the hypoxic tumour environment, vascular endothelial growth factor (VEGF) is released by the hypoxia and acts on endothelial cells to promote angiogenesis; consequently antibody (bevacizumab) to VEGF has been used clinically to try to suppress blood vessel formation, but patients become resistant to this treatment. Another signalling mechanism that may be worthy of therapeutic attention is the PDGF-BB—PDGFRβ pathway since, to become vascularised, glioma cells express PDGF-BB to attract pericytes to newly formed vessels [53, 138, 148]. Some of the pericytes mediating this function may differentiate from tumour stem cells [20]. In addition, PDGFRβ signalling promotes expression of IL-33 by pericytes, which recruits tumour-associated macrophages that promote tumour metastasis [154]. Therapeutically targeting pericytes to prevent angiogenesis and IL-33 production might thus be used to restrict tumour growth.
Radiation necrosis
Treatment of tumours with radiation, within or outside the brain, is associated with a loss of pericytes and endothelial cells from the capillaries nearby [87, 127], causing, in the case of the human brain, a leaky BBB and neuronal damage.
Amyotrophic lateral sclerosis
In human ALS, pericyte loss occurs from spinal cord capillaries, and the magnitude of the loss is correlated with breakdown of the blood–spinal cord barrier and accumulation of blood proteins in the parenchyma [150]. This suggests that prevention of pericyte loss might help to ameliorate the progression of ALS.
Targeting pericytes to ameliorate brain disorders
Common themes recur in the roles of pericytes in the disorders considered above. In disease, pericytes can:
-
1.
constrict capillaries and reduce cerebral blood flow (epilepsy, stroke, spinal cord injury, and possibly spreading depression and Alzheimer’s disease);
-
2.
cause a loss of BBB function by dying or decreasing PDGFRβ expression (stroke, epilepsy, Huntington’s disease, Alzheimer’s disease, diabetes, multiple sclerosis, radiation necrosis, ALS);
-
3.
migrate to a damaged area to isolate it but may thereby prevent neuronal regeneration through the damage site (spinal cord injury, stroke);
-
4.
be involved in angiogenesis in any insult that involves blood vessel loss or tissue growth (spinal cord injury, traumatic brain injury, stroke, glioma); and
-
5.
probably regulate immune cell entry in the majority of disorders (stroke, spinal cord injury, epilepsy, Alzheimer’s disease, multiple sclerosis).
How can the negative aspects of pericyte function in disease be targeted therapeutically, while promoting the beneficial functions?
Targeting drugs to CNS pericytes
Two generic issues of developing therapies for CNS pericyte malfunction are: (1) how to get the drug across the BBB, and (2) how to make it act specifically on pericytes.
In some diseases, the BBB will already be defective at sites where pericytes are malfunctioning, perhaps providing an automatic specificity in the brain region where peripherally administered drug action will occur. Alternatively, new methods for achieving penetration of the BBB may be used, such as encapsulating drugs in liposomes or other types of nano-carriers [40, 42, 164].
Some proteins that may be attractive therapeutic targets may happen to be expressed only on pericytes or their interacting endothelial cells (such as Tie2: [161]), while others may be also expressed on other cell types (such as PDGFRβ [161]). For the less selectively expressed targets, it will be necessary to devise drugs that, in addition to recognising their therapeutic target, also bind to another molecule that is selectively expressed on pericytes (or interacting cells).
Preventing pericyte-mediated constriction of capillaries and pericyte death
Since pericyte-mediated constriction and the pericyte death which occurs in ischaemia are both mediated by Ca2+ entry [54, 116], a potential therapeutic approach would be to apply blockers of pericyte voltage-gated Ca2+ channels as early as possible after an ischaemic event, for example, when removing an arterial thrombus with a stent retriever or with tissue plasminogen activator. Indeed, voltage-gated Ca2+ channel blockers slow ischaemia-evoked capillary constriction in brain slices [104], and also reduce pericyte death evoked by ATP [136] or ischaemia (R. Nortley, F. O’Farrell and D. Attwell, unpublished). Consistent with this, an unpublished study by A. Neuhaus, Y. Couch, B. Sutherland and A. Buchan has found that administering nimodipine at the end of a period of middle cerebral artery occlusion in rats leads to improved blood flow after the simulated stroke and a better behavioural outcome. Similarly, administering nano-carrier-attached adenosine (which may decrease calcium channel activity) maintains capillary dilation after ischaemia and improves behavioural outcome [42]. These approaches may also be beneficial in migraine [75], and after traumatic brain injury or subarachnoid haemorrhage, when pericyte constriction of some capillaries can induce heterogeneity of the transit time for blood flow through the capillaries which decreases oxygen supply to the tissue [107, 108].
The rise of [Ca2+]i that evokes pericyte constriction of capillaries might be significantly potentiated by pericyte depolarization generated by Ca2+-activated chloride channels [151], as in smooth muscle [119]. Block of these channels would offer another possible target for reducing capillary constriction in pathology.
The constriction of pericytes that occurs below spinal cord injuries and leads to local hypoxia, may be relieved by blocking the AADC enzyme that produces trace amines that constrict the pericytes, or blocking the receptors that these amines act on [89]. It is likely that, in other disorders (e.g., spreading depression, epilepsy and Alzheimer’s disease), signalling pathways upstream of pericyte Ca2+, or operating in parallel with pericyte Ca2+ (such as oxidative stress in ischaemia, which promotes occlusion of vessels by pericytes [156]), will be found that can be blocked to relieve pericyte-mediated capillary constriction.
Prevention of loss of BBB function
BBB function may be maintained by preventing pericyte death, either as described above for conditions involving a rise of pericyte [Ca2+]i, or by targeting specific death-inducing pathways in other disorders, such as PKC-δ in diabetes [43]. In conditions more mild than those involving pericyte death, BBB function can be improved by promoting interactions between pericytes and endothelial cells, to preserve the activity of Mfsd2a and suppression of transcytosis that are essential for normal BBB function [12, 22]. This can be achieved by increasing PDGF-BB signalling from endothelial cells to PDGFRβ receptors on pericytes (mirroring the loss of BBB function which occurs when PDGFRβ signalling is reduced transgenically [6, 11, 29]), by increasing TGFβ signalling to increase pericyte number, or by modulating Ang2 and Tie2 function [56, 113,114,115]. Intracerebroventricular administration of exogenous PDGF-BB has entered human clinical trials and appears to be well-tolerated and safe [115]. Thus, in a cell culture model, BBB function is better maintained in hypoxia when PDGF-BB or TGFβ is administered [132]; in status epilepticus, intravenous administration of PDGF-BB reduces blood vessel leakage and normalises blood flow [4]; and in an animal model of Parkinson’s disease, PDGF-BB may restore neurovascular function by rescuing PDGFRβ signalling [111].
Despite its toxic reputation, thalidomide, an immunomodulating agent that is applied in cancer and rheumatic disease [39], can induce pericyte proliferation, recruit pericytes to capillaries, and thus induce vessel maturation, mainly by increasing PDGF-BB expression in endothelial cells [86]. An increased density of pericytes on capillaries would be expected to promote the integrity of the BBB and indeed, in an animal model of AD, administering thalidomide decreases BBB leakiness [123]. Thalidomide has been successfully used to treat hereditary haemorrhagic telangiectasia in humans [86], and has been patented for use to prevent loss of BBB function after radiation therapy [85]. A beneficial effect of thalidomide on pericyte survival has also been confirmed in sunitinib-induced cardiotoxicity (caused by a decrease of PDGFRβ signalling) [21] and radiation-induced kidney injury [127].
Another therapeutic approach to maintaining BBB function is to apply the inhibitor of phosphodiesterase type 3, cilostazol, or the prostacyclin analogue iloprost which activates Gs-coupled IP receptors, both of which are expected to raise the level of cyclic AMP in pericytes. These agents have been shown to reduce the detachment of pericytes and astrocyte endfeet from endothelial cells in stroke-prone rats [105], to preserve BBB function in white matter subjected to demyelination with lysophosphatidylcholine [97], and (in a cell culture model of the BBB) to preserve BBB function in the face of oxygen–glucose deprivation by upregulating tight junctions between endothelial cells [140], apparently by inhibiting TGFβ signalling. This suggests further therapeutic approaches could be targeted to TGFβ signalling.
The importance of PDGFRβ signalling for maintaining pericyte number and BBB function has been exploited to provide a biomarker for AD progression. Loss of pericytes and degradation of BBB function has been shown to correlate with human cognitive decline and the appearance of soluble PDGFRβ in the cerebrospinal fluid [95]. In the long term, it will highly desirable to develop similar assays for other aspects of pericyte function, including TGFβ and Ang-Tie signalling, and ideally to extend such an approach to allow simple blood tests to be used to assess CNS pericyte function.
Control of immune cell entry in pathology
Numerous neurological conditions are associated with recruitment of immune cells from the blood to the brain. Whether modulating this entry by targeting pericyte functions (such as MIF release [135]) could provide beneficial therapies will depend on whether the net effect of immune cell recruitment is damaging or positive (e.g., releasing anti-inflammatory factors that suppress deleterious microglial actions [129]).
Controlling pericyte migration into the glial scar
At present, little is known about the factors stimulating pericytes to proliferate and move to damaged areas to contribute to the glial scar. However, blocking proliferation can lessen the pericyte contribution to the scar, which may promote axon regrowth [32] or alternatively hinder revascularisation [63]. Periostin expressed by pericytes is a key molecule involved in regulating pericyte movement into the scar [157], and genetic or pharmacological inhibition of its function decreases pericyte proliferation and scar formation, and improves long-term outcome after spinal cord injury [157].
Regulating glioma growth
Pericytes contribute to tumour growth both by promoting angiogenesis and by releasing IL-33 to promote metastasis (see above). Both of these actions are driven by PDGF-BB—PDGFRβ signalling, implying that tumour growth may be limited by agents blocking this signalling, such as imatinib and sunitinib (although these drugs also block other tyrosine kinases) [120].
Pericytes may also be a useful target in facilitating access of drugs to tumours, by disrupting the blood–tumour barrier, while leaving the blood–brain barrier less affected. By inhibiting the tyrosine kinase BMX found in stem cell-derived pericytes [20] with ibrutinib, it has recently been shown [163] that chemotherapy agents can have better access to the brain tumours.
Pericytes as stem cells
A growing body of evidence indicates that pericytes can become multipotential stem cells [33]. Pericytes have been suggested to acquire the ability to differentiate into neuronal, microglial and vascular lineage cells after brain pathology, in conditions such as ischaemic diseases and hypoxia [70, 71, 98, 109]. Thus, reprogramming of pericytes might be employed to promote neurogenesis and vasculogenesis at sites of brain injury. However, all these studies employed ex vivo culture of pericytes to reprogramme their fate, and the idea of pericytes or vascular smooth muscle becoming stem cells has been challenged by a study [50] showing that they do not intrinsically exhibit differentiation potential in vivo during ageing or in pathology.
Conclusions
Despite being relatively neglected components of the CNS, the data reviewed in this article demonstrate that pericytes, located at the interface between CNS cells and the blood supply coming from the periphery, play numerous crucial roles in the healthy CNS. As a result, they offer many opportunities for therapeutic intervention in a broad range of neurological disorders. We predict the widespread development of pericyte-targeted therapies in the next 10 years.
References
Abdel-Malak NA, Srikant CB, Kristof AS, Magder SA, Di Battista JA, Hussain SN (2008) Angiopoietin-1 promotes endothelial cell proliferation and migration through AP-1-dependent autocrine production of interleukin-8. Blood 111:4145–4154
Alarcon-Martinez L, Yilmaz-Ozcan S, Yemisci M, Schallek J, Kılıç K, Can A, Di Polo A, Dalkara T (2018) Capillary pericytes express α-smooth muscle actin, which requires prevention of filamentous-actin depolymerization for detection. Elife 7:e34861
Anderson MA, Burda JE, Ren Y, Ao Y, O’Shea TM, Kawaguchi R, Coppola G, Khakh BS, Deming TJ, Sofroniew MV (2016) Astrocyte scar formation aids central nervous system axon regeneration. Nature 532:195–200
Arango-Lievano M, Boussadia B, De Terdonck LDT, Gault C, Fontanaud PP, Lafont C, Mollard P, Marchi N, Jeanneteau F (2018) Topographic reorganization of cerebrovascular mural cells under seizure conditions. Cell Rep 23:1045–1059
Armulik A, Genové G, Betsholtz C (2011) Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev Cell 21:193–215
Armulik A, Genove G, Mae M, Nisancioglu MH, Wallgard E, Niaudet C, He L, Norlin J, Lindblom P, Strittmatter K, Johansson BR, Betsholtz C (2010) Pericytes regulate the blood-brain barrier. Nature 468:557–561
Asllani I, Habeck C, Scarmeas N, Borogovac A, Brown TR, Stern Y (2008) Multivariate and univariate analysis of continuous arterial spin labeling perfusion MRI in Alzheimer’s disease. J Cereb Blood Flow Metab 28:725–736
Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA (2010) Glial and neuronal control of brain blood flow. Nature 468:232–243
Attwell D, Mishra A, Hall CN, O’Farrell FM, Dalkara T (2016) What is a pericyte? J Cereb Blood Flow Metab 36:451–455
Bandopadhyay R, Orte C, Lawrenson JG, Reid AR, De Silva S, Allt G (2001) Contractile proteins in pericytes at the blood-brain and blood-retinal barriers. J Neurocytol 30:35–44
Bell RD, Winkler EA, Sagare AP, Singh I, LaRue B, Deane R, Zlokovic BV (2010) Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 68:409–427
Ben-Zvi A, Lacoste B, Kur E, Andreone BJ, Mayshar Y, Yan H, Gu C (2014) Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 509:507–511
Bergers G, Song S (2005) The role of pericytes in blood-vessel formation and maintenance. Neuro-Oncology 7:452–464
Berthiaume AA, Grant RI, McDowell KP, Underly RG, Hartmann DA, Levy M, Bhat NR, Shih AY (2018) Dynamic remodeling of pericytes in vivo maintains capillary coverage in the adult mouse brain. Cell Rep 22:8–16
Biesecker KR, Srienc AI, Shimoda AM, Agarwal A, Bergles DE, Kofuji P, Newman EA (2016) Glial cell calcium signaling mediates capillary regulation of blood flow in the retina. J Neurosci 36:9435–9445
Bjorkqvist M, Ohlsson M, Minthon L, Hansson O (2012) Evaluation of a previously suggested plasma biomarker panel to identify Alzheimer’s disease. PLoS One 7:e29868
Bondjers C, He L, Takemoto M, Norlin J, Asker N, Hellstrom M, Lindahl P, Betsholtz C (2006) Microarray analysis of blood microvessels from PDGF-B and PDGF-Rbeta mutant mice identifies novel markers for brain pericytes. FASEB J 20:1703–1705
Bondjers C, Kalen M, Hellstrom M, Scheidl SJ, Abramsson A, Renner O, Lindahl P, Cho H, Kehrl J, Betsholtz C (2003) Transcription profiling of platelet-derived growth factor-B-deficient mouse embryos identifies RGS5 as a novel marker for pericytes and vascular smooth muscle cells. Am J Pathol 162:721–729
Cai J, Boulton M (2002) The pathogenesis of diabetic retinopathy: old concepts and new questions. Eye 16:242–260
Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, Min W, McLendon RE, Rich JN, Bao S (2013) Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 153:139–152
Chintalgattu V, Rees ML, Culver JC, Goel A, Jiffar T, Zhang J, Dunner K Jr, Pati S, Bankson JA, Pasqualini R, Arap W, Bryan NS, Taegtmeyer H, Langley RR, Yao H, Kupferman ME, Entman ML, Dickinson ME, Khakoo AY (2013) Coronary microvascular pericytes are the cellular target of sunitinib malate-induced cardiotoxicity. Sci Transl Med 5:187ra69
Chow BW, Gu C (2017) Gradual suppression of transcytosis governs functional blood-retinal barrier formation. Neuron 93:1325–1333
Christian S, Winkler R, Helfrich I, Boos AM, Besemfelder E, Schadendorf D, Augustin HG (2008) Endosialin (Tem1) is a marker of tumor-associated myofibroblasts and tumor vessel-associated mural cells. Am J Pathol 172:486–494
Claudio L, Raine CS, Brosnan CF (1995) Evidence of persistent blood-brain barrier abnormalities in chronic-progressive multiple sclerosis. Acta Neuropathol 90:228–238
Cuervo H, Pereira B, Nadeem T, Lin M, Lee F, Kitajewski J, Lin CS (2017) PDGFRbeta-P2A-CreERT2 mice: a genetic tool to target pericytes in angiogenesis. Angiogenesis 20:655–662
Cui X, Chopp M, Zacharek A, Ye X, Roberts C, Chen J (2011) Angiopoietin/Tie2 pathway mediates type 2 diabetes induced vascular damage after cerebral stroke. Neurobiol Dis 43:285–292
Dai M, Yang Y, Shi X (2011) Lactate dilates cochlear capillaries via type V fibrocyte-vessel coupling signaled by nNOS. Am J Physiol Heart Circ Physiol 301:H1248–H1254
Damisah EC, Hill RA, Tong L, Murray KN, Grutzendler J (2017) A fluoro-Nissl dye identifies pericytes as distinct vascular mural cells during in vivo brain imaging. Nat Neurosci 20:1023–1032
Daneman R, Zhou L, Kebede AA, Barres BA (2010) Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 468:562–566
Davies AL, Desai RA, Bloomfield PS, McIntosh PR, Chapple KJ, Linington C, Fairless R, Diem R, Kasti M, Murphy MP, Smith KJ (2013) Neurological deficits caused by tissue hypoxia in neuroinflammatory disease. Ann Neurol 74:815–825
De La Fuente AG, Lange S, Silva ME, Gonzalez GA, Tempfer H, van Wijngaarden P, Zhao C, Di Canio L, Trost A, Bieler L, Zaunmair P, Rotheneichner P, O’Sullivan A, Couillard-Despres S, Errea O, Mäe MA, Andrae J, He L, Keller A, Bátiz LF, Betsholtz C, Aigner L, Franklin RJM, Rivera FJ (2017) Pericytes stimulate oligodendrocyte progenitor cell differentiation during CNS remyelination. Cell Rep 20:1755–1764
Dias DO, Kim H, Holl D, Werne Solnestam B, Lundeberg J, Carlén M, Göritz C, Frisén J (2018) Reducing pericyte-derived scarring promotes recovery after spinal cord injury. Cell 173:153–165
Dore-Duffy P (2008) Pericytes: pluripotent cells of the blood brain barrier. Curr Pharm Des 14:1581–1593
Dore-Duffy P, Wang X, Mehedi A, Kreipke CW, Rafols JA (2007) Differential expression of capillary VEGF isoforms following traumatic brain injury. Neurol Res 29:395–403
Drouin-Ouellet J, Sawiak SJ, Cisbani G, Lagacé M, Kuan WL, Saint-Pierre M, Dury RJ, Alata W, St-Amour I, Mason SL, Calon F, Lacroix S, Gowland PA, Francis ST, Barker RA, Cicchetti F (2015) Cerebrovascular and blood-brain barrier impairments in Huntington’s disease: potential implications for its pathophysiology. Ann Neurol 78:160–177
Fernandez-Klett F, Potas JR, Hilpert D, Blazej K, Radke J, Huck J, Engel O, Stenzel W, Genove G, Priller J (2013) Early loss of pericytes and perivascular stromal cell-induced scar formation after stroke. J Cereb Blood Flow Metab 33:428–439
Fordsmann JC, Ko RW, Choi HB, Thomsen K, Witgen BM, Mathiesen C, Lønstrup M, Piilgaard H, MacVicar BA, Lauritzen M (2013) Increased 20-HETE synthesis explains reduced cerebral blood flow but not impaired neurovascular coupling after cortical spreading depression in rat cerebral cortex. J Neurosci 33:2562–2570
Frank RN, Dutta S, Mancini MA (1987) Pericyte coverage is greater in the retinal than in the cerebral capillaries of the rat. Investig Ophthalmol Vis Sci 28:1086–1091
Franks ME, Macpherson GR, Figg WD (2004) Thalidomide. Lancet 363:1802–1811
Fullstone G, Nyberg S, Tian X, Battaglia G (2016) From the blood to the central nervous system: a nanoparticle’s journey through the blood–brain barrier by transcytosis. Int Rev Neurobiol 130:41–72
Gaceb A, Özen I, Padel T, Barbariga M, Paul G (2018) Pericytes secrete pro-regenerative molecules in response to platelet-derived growth factor-BB. J Cereb Blood Flow Metab 38:45–57
Gaudin A, Yemisci M, Eroglu H, Lepetre-Mouelhi S, Turkoglu OF, Donmez-Demir B, Caban S, Sargon MF, Garcia-Argote S, Pieters G, Loreau O, Rousseau B, Tagit O, Hildebrandt N, Le Dantec Y, Mougin J, Valetti S, Chacun H, Nicolas V, Desmaele D, Andrieux K, Capan Y, Dalkara T, Couvreur P (2014) Squalenoyl adenosine nanoparticles provide neuroprotection after stroke and spinal cord injury. Nat Nanotechnol 9:1054–1062
Geraldes P, Hiraoka-Yamamoto J, Matsumoto M, Clermont A, Leitges M, Marette A, Aiello LP, Kern TS, King GL (2009) Activation of PKC-delta and SHP-1 by hyperglycemia causes vascular cell apoptosis and diabetic retinopathy. Nat Med 15:1298–1306
Ghosh M, Balbi M, Hellal F, Dichgans M, Lindauer U, Plesnila N (2015) Pericytes are involved in the pathogenesis of cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy. Ann Neurol 78:887–900
Gonul E, Duz B, Kahraman S, Kayali H, Kubar A, Timurkaynak E (2002) Early pericyte response to brain hypoxia in cats: an ultrastructural study. Microvasc Res 64:116–119
Goritz C, Dias DO, Tomilin N, Barbacid M, Shupliakov O, Frisen J (2011) A pericyte origin of spinal cord scar tissue. Science 333:238–242
Gould IG, Tsai P, Kleinfeld D, Linninger A (2017) The capillary bed offers the largest hemodynamic resistance to the cortical blood supply. J Cereb Blood Flow Metab 37:52–68
Gozal EA, O’Neill BE, Sawchuk MA, Zhu H, Halder M, Chou CC, Hochman S (2014) Anatomical and functional evidence for trace amines as unique modulators of locomotor function in the mammalian spinal cord. Front Neural Circuits 8:134
Grant RI, Hartmann DA, Underly RG, Berthiaume AA, Bhat NR, Shih AY (2017) Organizational hierarchy and structural diversity of microvascular pericytes in adult mouse cortex. J Cereb Blood Flow Metab. https://doi.org/10.1177/0271678X17732229
Guimaraes-Camboa N, Cattaneo P, Sun Y, Moore-Morris T, Gu Y, Dalton ND, Rockenstein E, Masliah E, Peterson KL, Stallcup WB, Chen J, Evans SM (2017) Pericytes of multiple organs do not behave as mesenchymal stem cells in vivo. Cell Stem Cell 20:345–359
Gunsilius E, Petzer AL, Stockhammer G, Kahler CM, Gastl G (2001) Serial measurement of vascular endothelial growth factor and transforming growth factor-beta1 in serum of patients with acute ischemic stroke. Stroke 32:275–278
Guo L, Zhang H, Hou Y, Wei T, Liu J (2016) Plasmalemma vesicle-associated protein: a crucial component of vascular homeostasis. Exp Ther Med 12:1639–1644
Guo P, Hu B, Gu W, Xu L, Wang D, Huang HJ, Cavenee WK, Cheng SY (2003) Platelet-derived growth factor-B enhances glioma angiogenesis by stimulating vascular endothelial growth factor expression in tumor endothelia and by promoting pericyte recruitment. Am J Pathol 162:1083–1093
Hall CN, Reynell C, Gesslein B, Hamilton NB, Mishra A, Sutherland BA, O’Farrell FM, Buchan AM, Lauritzen M, Attwell D (2014) Capillary pericytes regulate cerebral blood flow in health and disease. Nature 508:55–60
Hamilton NB, Attwell D, Hall CN (2010) Pericyte-mediated regulation of capillary diameter: a component of neurovascular coupling in health and disease. Front Neuroenerg 2010:2
Hammes HP, Lin J, Wagner P, Feng Y, Vom Hagen F, Krzizok T, Renner O, Breier G, Brownlee M, Deutsch U (2004) Angiopoietin-2 causes pericyte dropout in the normal retina: evidence for involvement in diabetic retinopathy. Diabetes 53:1104–1110
Hansen AJ (1985) Effect of anoxia on ion distribution in the brain. Physiol Rev 65:101–148
Hartings JA, Watanabe T, Bullock MR, Okonkwo DO, Fabricius M, Woitzik J, Dreier JP, Puccio A, Shutter LA, Pahl C, Strong AJ, Co-Operative Study on Brain Injury Depolarizations (2011) Spreading depolarizations have prolonged direct current shifts and are associated with poor outcome in brain trauma. Brain 134:1529–1540
Hartmann DA, Underly RG, Grant RI, Watson AN, Lindner V, Shih AY (2015) Pericyte structure and distribution in the cerebral cortex revealed by high-resolution imaging of transgenic mice. Neurophotonics 2:041402
Hashimura T, Kimura T, Miyakawa T (1991) Morphological changes of blood vessels in the brain with Alzheimer’s disease. Jpn J Psychiatry Neurol 45:661–665
Hauck EF, Apostel S, Hoffmann JF, Heimann A, Kempski O (2004) Capillary flow and diameter changes during reperfusion after global cerebral ischemia studied by intravital video microscopy. J Cereb Blood Flow Metab 24:383–391
He L, Vanlandewijck M, Raschperger E, Andaloussi Mae M, Jung B, Lebouvier T, Ando K, Hofmann J, Keller A, Betsholtz C (2016) Analysis of the brain mural cell transcriptome. Sci Rep 6:35108
Hesp ZC, Yoseph RY, Suzuki R, Jukkola P, Wilson C, Nishiyama A, McTigue DM (2018) Proliferating NG2-cell-dependent angiogenesis and scar formation alter axon growth and functional recovery after spinal cord injury in mice. J Neurosci 38:1366–1382
Hill RA, Tong L, Yuan P, Murikinati S, Gupta S, Grutzendler J (2015) Regional blood flow in the normal and ischemic brain Is controlled by arteriolar smooth muscle cell contractility and not by capillary pericytes. Neuron 87:95–110
Hsiao HY, Chen YC, Huang CH, Chen CC, Hsu YH, Chen HM, Chiu FL, Kuo HC, Chang C, Chern Y (2015) Aberrant astrocytes impair vascular reactivity in Huntington disease. Ann Neurol 78:178–192
Iturria-Medina Y, Sotero RC, Toussaint PJ, Mateos-Pérez JM, Evans AC, Alzheimer’s Disease Neuroimaging Initiative (2016) Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun 7:11934
Jansson D, Rustenhoven J, Feng S, Hurley D, Oldfield RL, Bergin PS, Mee EW, Faull RL, Dragunow M (2014) A role for human brain pericytes in neuroinflammation. J Neuroinflamm 11:104
Jean LeBlanc N, Guruswamy R, ElAli A (2018) Vascular endothelial growth factor isoform-B stimulates neurovascular repair after ischemic stroke by promoting the function of pericytes via vascular endothelial growth factor receptor-1. Mol Neurobiol 55:3611–3626
Joutel A, Corpechot C, Ducros A, Vahedi K, Chabriat H, Mouton P, Alamowitch S, Domenga V, Cécillion M, Marechal E, Maciazek J, Vayssiere C, Cruaud C, Cabanis EA, Ruchoux MM, Weissenbach J, Bach JF, Bousser MG, Tournier-Lasserve E (1996) Notch3 mutations in CADASIL, a hereditary adult-onset condition causing stroke and dementia. Nature 383:707–710
Karow M, Sanchez R, Schichor C, Masserdotti G, Ortega F, Heinrich C, Gascon S, Khan MA, Lie DC, Dellavalle A, Cossu G, Goldbrunner R, Gotz M, Berninger B (2012) Reprogramming of pericyte-derived cells of the adult human brain into induced neuronal cells. Cell Stem Cell 11:471–476
Karow M, Schichor C, Beckervordersandforth R, Berninger B (2014) Lineage-reprogramming of pericyte-derived cells of the adult human brain into induced neurons. J Vis Exp 12:51433
Kawamura H, Kobayashi M, Li Q, Yamanishi S, Katsumura K, Minami M, Wu DM, Puro DG (2004) Effects of angiotensin II on the pericyte-containing microvasculature of the rat retina. J Physiol 561:671–683
Kawamura H, Sugiyama T, Wu DM, Kobayashi M, Yamanishi S, Katsumura K, Puro DG (2003) ATP: a vasoactive signal in the pericyte-containing microvasculature of the rat retina. J Physiol 551:787–799
Keller A, Westenberger A, Sobrido MJ, García-Murias M, Domingo A, Sears RL, Lemos RR, Ordoñez-Ugalde A, Nicolas G, da Cunha JE, Rushing EJ, Hugelshofer M, Wurnig MC, Kaech A, Reimann R, Lohmann K, Dobričić V, Carracedo A, Petrović I, Miyasaki JM, Abakumova I, Mäe MA, Raschperger E, Zatz M, Zschiedrich K, Klepper J, Spiteri E, Prieto JM, Navas I, Preuss M, Dering C, Janković M, Paucar M, Svenningsson P, Saliminejad K, Khorshid HR, Novaković I, Aguzzi A, Boss A, Le Ber I, Defer G, Hannequin D, Kostić VS, Campion D, Geschwind DH, Coppola G, Betsholtz C, Klein C, Oliveira JR (2013) Mutations in the gene encoding PDGF-B cause brain calcifications in humans and mice. Nat Genet 45:1077–1082
Khennouf L, Gesslein B, Brazhe A, Octeau JC, Kutuzov N, Khakh BS, Lauritzen M (2018) Active role of capillary pericytes during stimulation-induced activity and spreading depolarization. Brain 141:2032–2046
Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N (1993) Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 362:841–844
Kimura T, Hashimura T, Miyakawa T (1991) Observations of microvessels in the brain with Alzheimer’s disease by the scanning electron microscope. Jpn J Psychiatry Neurol 45:671–676
Kisler K, Nelson AR, Montagne A, Zlokovic BV (2017) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 18:419–434
Kisler K, Nelson AR, Rege SV, Ramanathan A, Wang Y, Ahuja A, Lazic D, Tsai PS, Zhao Z, Zhou Y, Boas DA, Sakadžić S, Zlokovic BV (2017) Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat Neurosci 20:406–416
Kofler NM, Cuervo H, Uh MK, Murtomaki A, Kitajewski J (2015) Combined deficiency of Notch1 and Notch3 causes pericyte dysfunction, models CADASIL, and results in arteriovenous malformations. Sci Rep 5:16449
Krueger M, Bechmann I (2010) CNS pericytes: concepts, misconceptions, and a way out. Glia 58:1–10
Kunz J, Krause D, Kremer M, Dermietzel R (1994) The 140-kDa protein of blood-brain barrier-associated pericytes is identical to aminopeptidase N. J Neurochem 62:2375–2386
Lacar B, Herman P, Platel JC, Kubera C, Hyder F, Bordey A (2012) Neural progenitor cells regulate capillary blood flow in the postnatal subventricular zone. J Neurosci 32:16435–16448
Leal-Campanario R, Alarcon-Martinez L, Rieiro H, Martinez-Conde S, Alarcon-Martinez T, Zhao X, LaMee J, Popp PJ, Calhoun ME, Arribas JI, Schlegel AA, Stasi LL, Rho JM, Inge L, Otero-Millan J, Treiman DM, Macknik SL (2017) Abnormal capillary vasodynamics contribute to ictal neurodegeneration in epilepsy. Sci Rep 7:43276
Lebrin F, Soussain C, Thalgott J (2015) https://patents.google.com/patent/WO2015107196A1/en. Accessed 6 Aug 2018
Lebrin F, Srun S, Raymond K, Martin S, van den Brink S, Freitas C, Breant C, Mathivet T, Larrivee B, Thomas JL, Arthur HM, Westermann CJ, Disch F, Mager JJ, Snijder RJ, Eichmann A, Mummery CL (2010) Thalidomide stimulates vessel maturation and reduces epistaxis in individuals with hereditary hemorrhagic telangiectasia. Nat Med 16:420–428
Lee ST, Seo Y, Bae JY, Chu K, Kim JW, Choi SH, Kim TM, Kim IH, Park SH, Park CK (2017) Loss of pericytes in radiation necrosis after glioblastoma treatments. Mol Neurobiol 55:4918–4926
Li F, Lan Y, Wang Y, Wang J, Yang G, Meng F, Han H, Meng A, Wang Y, Yang X (2011) Endothelial Smad4 maintains cerebrovascular integrity by activating N-cadherin through cooperation with Notch. Dev Cell 20:291–302
Li Y, Lucas-Osma AM, Black S, Bandet MV, Stephens MJ, Vavrek R, Sanelli L, Fenrich KK, Di Narzo AF, Dracheva S, Winship IR, Fouad K, Bennett DJ (2017) Pericytes impair capillary blood flow and motor function after chronic spinal cord injury. Nat Med 23:733–741
Lindahl P, Johansson BR, Leveen P, Betsholtz C (1997) Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277:242–245
Lindblom P, Gerhardt H, Liebner S, Abramsson A, Enge M, Hellstrom M, Backstrom G, Fredriksson S, Landegren U, Nystrom HC, Bergstrom G, Dejana E, Ostman A, Lindahl P, Betsholtz C (2003) Endothelial PDGF-B retention is required for proper investment of pericytes in the microvessel wall. Genes Dev 17:1835–1840
Maddaluno L, Rudini N, Cuttano R, Bravi L, Giampietro C, Corada M, Ferrarini L, Orsenigo F, Papa E, Boulday G, Tournier-Lasserve E, Chapon F, Richichi C, Retta SF, Lampugnani MG, Dejana E (2013) EndMT contributes to the onset and progression of cerebral cavernous malformations. Nature 498:492–496
Milesi S, Boussadia B, Plaud C, Catteau M, Rousset MC, De Bock F, Schaeffer M, Lerner-Natoli M, Rigau V, Marchi N (2014) Redistribution of PDGFRbeta cells and NG2DsRed pericytes at the cerebrovasculature after status epilepticus. Neurobiol Dis 71:151–158
Mishra A, Reynolds JP, Chen Y, Gourine AV, Rusakov DA, Attwell D (2016) Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat Neurosci 19:1619–1627
Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV (2015) Blood-brain barrier breakdown in the aging human hippocampus. Neuron 85:296–302
Morgenstern DA, Asher RA, Fawcett JW (2002) Chondroitin sulphate proteoglycans in the CNS injury response. Prog Brain Res 137:313–332
Muramatsu R, Kuroda M, Matoba K, Lin H, Takahashi C, Koyama Y, Yamashita T (2015) Prostacyclin prevents pericyte loss and demyelination induced by lysophosphatidylcholine in the central nervous system. J Biol Chem 290:11515–11525
Nakagomi T, Kubo S, Nakano-Doi A, Sakuma R, Lu S, Narita A, Kawahara M, Taguchi A, Matsuyama T (2015) Brain vascular pericytes following ischemia have multipotential stem cell activity to differentiate into neural and vascular lineage cells. Stem Cells 33:1962–1974
Nehls V, Denzer K, Drenckhahn D (1992) Pericyte involvement in capillary sprouting during angiogenesis in situ. Cell Tissue Res 270:469–474
Nehls V, Drenckhahn D (1993) The versatility of microvascular pericytes: from mesenchyme to smooth muscle? Histochemistry 99:1–12
Nicolas G, Pottier C, Maltête D, Coutant S, Rovelet-Lecrux A, Legallic S, Rousseau S, Vaschalde Y, Guyant-Maréchal L, Augustin J, Martinaud O, Defebvre L, Krystkowiak P, Pariente J, Clanet M, Labauge P, Ayrignac X, Lefaucheur R, Le Ber I, Frébourg T, Hannequin D, Campion D (2013) Mutation of the PDGFRB gene as a cause of idiopathic basal ganglia calcification. Neurology 80:181–187
Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S, Ashe KH, Carlson GA, Iadecola C (2000) Abeta 1–40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci USA 97:9735–9740
O’Farrell FM, Mastitskaya S, Hammond-Haley M, Freitas F, Wah WR, Attwell D (2017) Capillary pericytes mediate coronary no-reflow after myocardial ischaemia. Elife 6:e29280
O’Farrell FM, Reynell C, Mishra A, Nortley R, Papoulia M, Sutherland BA, Buchan AM, Hall CN, Attwell D (2015) Neurovascular physiology and pathophysiology of brain pericytes. Proc Physiol Soc 34, C32. www.physoc.org/proceedings/abstract/Proc%20Physiol%20Soc%2034C32. Accessed 6 Aug 2018
Omote Y, Deguchi K, Kono S, Liu N, Liu W, Kurata T, Yamashita T, Ikeda Y, Abe K (2014) Neurovascular protection of cilostazol in stroke-prone spontaneous hypertensive rats associated with angiogenesis and pericyte proliferation. J Neurosci Res 92:369–374
Oon CE, Bridges E, Sheldon H, Sainson RCA, Jubb A, Turley H, Leek R, Buffa F, Harris AL, Li JL (2017) Role of delta-like 4 in Jagged1-induced tumour angiogenesis and tumour growth. Oncotarget 8:40115–40131
Østergaard L, Aamand R, Karabegovic S, Tietze A, Blicher JU, Mikkelsen IK, Iversen NK, Secher N, Engedal TS, Anzabi M, Jimenez EG, Cai C, Koch KU, Naess-Schmidt ET, Obel A, Juul N, Rasmussen M, Sørensen JC (2013) The role of the microcirculation in delayed cerebral ischemia and chronic degenerative changes after subarachnoid hemorrhage. J Cereb Blood Flow Metab 33:1825–1837
Østergaard L, Engedal TS, Aamand R, Mikkelsen R, Iversen NK, Anzabi M, Næss-Schmidt ET, Drasbek KR, Bay V, Blicher JU, Tietze A, Mikkelsen IK, Hansen B, Jespersen SN, Juul N, Sørensen JC, Rasmussen M (2014) Capillary transit time heterogeneity and flow-metabolism coupling after traumatic brain injury. J Cereb Blood Flow Metab 34:1585–1598
Ozen I, Deierborg T, Miharada K, Padel T, Englund E, Genove G, Paul G (2014) Brain pericytes acquire a microglial phenotype after stroke. Acta Neuropathol 128:381–396
Ozerdem U, Grako KA, Dahlin-Huppe K, Monosov E, Stallcup WB (2001) NG2 proteoglycan is expressed exclusively by mural cells during vascular morphogenesis. Dev Dyn 222:218–227
Padel T, Ozen I, Boix J, Barbariga M, Gaceb A, Roth M, Paul G (2016) Platelet-derived growth factor-BB has neurorestorative effects and modulates the pericyte response in a partial 6-hydroxydopamine lesion mouse model of Parkinson’s disease. Neurobiol Dis 94:95–105
Padel T, Roth M, Gaceb A, Li JY, Björkqvist M, Paul G (2018) Brain pericyte activation occurs early in Huntington’s disease. Exp Neurol 305:139–150
Park DY, Lee J, Kim J, Kim K, Hong S, Han S, Kubota Y, Augustin HG, Ding L, Kim JW, Kim H, He Y, Adams RH, Koh GY (2017) Plastic roles of pericytes in the blood–retinal barrier. Nat Commun 8:15296
Park SW, Yun JH, Kim JH, Kim KW, Cho CH, Kim JH (2014) Angiopoietin 2 induces pericyte apoptosis via alpha3beta1 integrin signaling in diabetic retinopathy. Diabetes 63:3057–3068
Paul G, Zachrisson O, Varrone A, Almqvist P, Jerling M, Lind G, Rehncrona S, Linderoth B, Bjartmarz H, Shafer LL, Coffey R, Svensson M, Mercer KJ, Forsberg A, Halldin C, Svenningsson P, Widner H, Frisen J, Palhagen S, Haegerstrand A (2015) Safety and tolerability of intracerebroventricular PDGF-BB in Parkinson’s disease patients. J Clin Investig 125:1339–1346
Peppiatt CM, Howarth C, Mobbs P, Attwell D (2006) Bidirectional control of CNS capillary diameter by pericytes. Nature 443:700–704
Proebstl D, Voisin MB, Woodfin A, Whiteford J, D’Acquisto F, Jones GE, Rowe D, Nourshargh S (2012) Pericytes support neutrophil subendothelial cell crawling and breaching of venular walls in vivo. J Exp Med 209:1219–1234
Rantakari P, Auvinen K, Jäppinen N, Kapraali M, Valtonen J, Karikoski M, Gerke H, Iftakhar-E-Khuda I, Keuschnigg J, Umemoto E, Tohya K, Miyasaka M, Elima K, Jalkanen S, Salmi M (2015) The endothelial protein PLVAP in lymphatics controls the entry of lymphocytes and antigens into lymph nodes. Nat Immunol 16:386–396
Rorsman NJG, Ta CM, Garnett H, Swietach P, Tammaro P (2018) Defining the ionic mechanisms of optogenetic control of vascular tone by channelrhodopsin-2. Br J Pharmacol 175:2028–2045
Ruan J, Luo M, Wang C, Fan L, Yang SN, Cardenas M, Geng H, Leonard JP, Melnick A, Cerchietti L, Hajjar KA (2013) Imatinib disrupts lymphoma angiogenesis by targeting vascular pericytes. Blood 121:5192–5202
Rungta RL, Chaigneau E, Osmanski BF, Charpak S (2018) Vascular compartmentalization of functional hyperemia from the synapse to the pia. Neuron 99:362–375
Rustenhoven J, Jansson D, Smyth LC, Dragunow M (2017) Brain pericytes as mediators of neuroinflammation. Trends Pharmacol Sci 38:291–304
Ryu JK, McLarnon JG (2008) Thalidomide inhibition of perturbed vasculature and glial-derived tumor necrosis factor-alpha in an animal model of inflamed Alzheimer’s disease brain. Neurobiol Dis 29:254–266
Sagare AP, Bell RD, Zhao Z, Ma Q, Winkler EA, Ramanathan A, Zlokovic BV (2013) Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat Commun 4:2932
Sagare AP, Sweeney MD, Makshanoff J, Zlokovic BV (2015) Shedding of soluble platelet-derived growth factor receptor-beta from human brain pericytes. Neurosci Lett 607:97–101
Schallek J, Geng Y, Nguyen H, Williams DR (2013) Morphology and topography of retinal pericytes in the living mouse retina using in vivo adaptive optics imaging and ex vivo characterization. Investig Ophthalmol Vis Sci 54:8237–8250
Scharpfenecker M, Floot B, Russell NS, Coppes RP, Stewart FA (2014) Thalidomide ameliorates inflammation and vascular injury but aggravates tubular damage in the irradiated mouse kidney. Int J Radiat Oncol Biol Phys 89:599–606
Schulz GB, Wieland E, Wüstehube-Lausch J, Boulday G, Moll I, Tournier-Lasserve E, Fischer A (2015) Cerebral cavernous malformation-1 protein controls DLL4-Notch3 signaling between the endothelium and pericytes. Stroke 46:1337–1343
Schwartz M, Kipnis J, Rivest S, Prat A (2013) How do immune cells support and shape the brain in health, disease, and aging? J Neurosci 33:17587–17596
Sengillo JD, Winkler EA, Walker CT, Sullivan JS, Johnson M, Zlokovic BV (2013) Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease. Brain Pathol 23:303–310
Shao B, Bayraktutan U (2013) Hyperglycaemia promotes cerebral barrier dysfunction through activation of protein kinase C-β. Diabetes Obes Metab 15:993–999
Shen J, Xu G, Zhu R, Yuan J, Ishii Y, Hamashima T, Matsushima T, Yamamoto S, Takatsuru Y, Nabekura J, Sasahara M (2018) PDGFR-β restores blood–brain barrier functions in a mouse model of focal cerebral ischemia. J Cereb Blood Flow Metab. https://doi.org/10.1177/0271678x18769515 [Epub ahead of print]
Shimizu F, Sano Y, Tominaga O, Maeda T, Abe MA, Kanda T (2013) Advanced glycation end-products disrupt the blood-brain barrier by stimulating the release of transforming growth factor-β by pericytes and vascular endothelial growth factor and matrix metalloproteinase-2 by endothelial cells in vitro. Neurobiol Aging 34:1902–1912
Stapor PC, Sweat RS, Dashti DC, Betancourt AM, Murfee WL (2014) Pericyte dynamics during angiogenesis: new Insights from new identities. J Vasc Res 51:163–174
Stark K, Eckart A, Haidari S, Tirniceriu A, Lorenz M, von Bruhl ML, Gartner F, Khandoga AG, Legate KR, Pless R, Hepper I, Lauber K, Walzog B, Massberg S (2013) Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and ‘instruct’ them with pattern-recognition and motility programs. Nat Immunol 14:41–51
Sugiyama T, Kawamura H, Yamanishi S, Kobayashi M, Katsumura K, Puro DG (2005) Regulation of P2X7-induced pore formation and cell death in pericyte-containing retinal microvessels. Am J Physiol Cell Physiol 288:C568–C576
Sugiyama T, Kobayashi M, Kawamura H, Li Q, Puro DG (2004) Enhancement of P2X(7)-induced pore formation and apoptosis: an early effect of diabetes on the retinal microvasculature. Investig Ophthalmol Vis Sci 45:1026–1032
Svensson A, Özen I, Genové G, Paul G, Bengzon J (2015) Endogenous brain pericytes are widely activated and contribute to mouse glioma microvasculature. PLoS One 10:e0123553
Sweeney MD, Ayyadurai S, Zlokovic BV (2016) Pericytes of the neurovascular unit: key functions and signaling pathways. Nat Neurosci 19:771–783
Takeshita T, Nakagawa S, Tatsumi R, So G, Hayashi K, Tanaka K, Deli MA, Nagata I, Niwa M (2014) Cilostazol attenuates ischemia-reperfusion-induced blood-brain barrier dysfunction enhanced by advanced glycation endproducts via transforming growth factor-beta1 signaling. Mol Cell Neurosci 60:1–9
Teichert M, Milde L, Holm A, Stanicek L, Gengenbacher N, Savant S, Ruckdeschel T, Hasanov Z, Srivastava K, Hu J, Hertel S, Bartol A, Schlereth K, Augustin HG (2017) Pericyte-expressed Tie2 controls angiogenesis and vessel maturation. Nat Commun 8:16106
Toribatake Y, Tomita K, Kawahara N, Baba H, Ohnari H, Tanaka S (1997) Regulation of vasomotion of arterioles and capillaries in the cat spinal cord: role of alpha actin and endothelin-1. Spinal Cord 35:26–32
Valdez CN, Arboleda-Velasquez JF, Amarnani DS, Kim LA, D’Amore PA (2014) Retinal microangiopathy in a mouse model of inducible mural cell loss. Am J Pathol 184:2618–2626
Vanlandewijck M, He L, Mäe MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Laviña B, Gouveia L, Sun Y, Raschperger E, Räsänen M, Zarb Y, Mochizuki N, Keller A, Lendahl U, Betsholtz C (2018) A molecular atlas of cell types and zonation in the brain vasculature. Nature 554:475–480
Vinukonda G, Dummula K, Malik S, Hu F, Thompson CI, Csiszar A, Ungvari Z, Ballabh P (2010) Effect of prenatal glucocorticoids on cerebral vasculature of the developing brain. Stroke 41:1766–1773
Wang Y, Pan L, Moens CB, Appel B (2014) Notch3 establishes brain vascular integrity by regulating pericyte number. Development 141:307–317
Wanner IB, Anderson MA, Song B, Levine J, Fernandez A, Gray-Thompson Z, Ao Y, Sofroniew MV (2013) Glial scar borders are formed by newly proliferated, elongated astrocytes that interact to corral inflammatory and fibrotic cells via STAT3-dependent mechanisms after spinal cord injury. J Neurosci 33:12870–12886
Wesseling P, Schlingemann RO, Rietveld FJ, Link M, Burger PC, Ruiter DJ (1995) Early and extensive contribution of pericytes/vascular smooth muscle cells to microvascular proliferation in glioblastoma multiforme: an immuno-light and immuno-electron microscopic study. J Neuropathol Exp Neurol 54:304–310
Winkler EA, Bell RD, Zlokovic BV (2011) Central nervous system pericytes in health and disease. Nat Neurosci 14:1398–1405
Winkler EA, Sengillo JD, Sullivan JS, Henkel JS, Appel SH, Zlokovic BV (2013) Blood-spinal cord barrier breakdown and pericyte reductions in amyotrophic lateral sclerosis. Acta Neuropathol 125:111–120
Wu DM, Kawamura H, Sakagami K, Kobayashi M, Puro DG (2003) Cholinergic regulation of pericyte-containing retinal microvessels. Am J Physiol Heart Circ Physiol 284:H2083–H2090
Wu T, Dai M, Shi XR, Jiang ZG, Nuttall AL (2011) Functional expression of P2X4 receptor in capillary endothelial cells of the cochlear spiral ligament and its role in regulating the capillary diameter. Am J Physiol Heart Circ Physiol 301:H69–H78
Yamanishi S, Katsumura K, Kobayashi T, Puro DG (2006) Extracellular lactate as a dynamic vasoactive signal in the rat retinal microvasculature. Am J Physiol Heart Circ Physiol 290:H925–H934
Yang Y, Andersson P, Hosaka K, Zhang Y, Cao R, Iwamoto H, Yang X, Nakamura M, Wang J, Zhuang R, Morikawa H, Xue Y, Braun H, Beyaert R, Samani N, Nakae S, Hams E, Dissing S, Fallon PG, Langer R, Cao Y (2016) The PDGF-BB-SOX7 axis-modulated IL-33 in pericytes and stromal cells promotes metastasis through tumour-associated macrophages. Nat Commun 7:11385
Yang S, Jin H, Zhu Y, Wan Y, Zhu L, Hu B (2017) Diverse functions and mechanisms of pericytes in ischemic stroke. Curr Neuropharmacol 15:892–905
Yemisci M, Gursoy-Ozdemir Y, Vural A, Can A, Topalkara K, Dalkara T (2009) Pericyte contraction induced by oxidative-nitrative stress impairs capillary reflow despite successful opening of an occluded cerebral artery. Nat Med 15:1031–1037
Yokota K, Kobayakawa K, Saito T, Hara M, Kijima K, Ohkawa Y, Harada A, Okazaki K, Ishihara K, Yoshida S, Kudo A, Iwamoto Y, Okada S (2017) Periostin promotes scar formation through the interaction between pericytes and infiltrating monocytes/macrophages after spinal cord injury. Am J Pathol 187:639–653
Yun JH, Jeong HS, Kim KJ, Han MH, Lee EH, Lee K, Cho CH (2018) β-Adrenergic receptor agonists attenuate pericyte loss in diabetic retinas through Akt activation. FASEB J 32:2324–2338
Zechariah A, ElAli A, Doeppner TR, Jin F, Hasan MR, Helfrich I, Mies G, Hermann DM (2013) Vascular endothelial growth factor promotes pericyte coverage of brain capillaries, improves cerebral blood flow during subsequent focal cerebral ischemia, and preserves the metabolic penumbra. Stroke 44:1690–1697
Zehendner CM, Wedler HE, Luhmann HJ (2013) A novel in vitro model to study pericytes in the neurovascular unit of the developing cortex. PLoS One 8:e81637
Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, Phatnani HP, Guarnieri P, Caneda C, Ruderisch N, Deng S, Liddelow SA, Zhang C, Daneman R, Maniatis T, Barres BA, Wu JQ (2014) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34:11929–11947
Zhou HJ, Qin L, Zhang H, Tang W, Ji W, He Y, Liang X, Wang Z, Yuan Q, Vortmeyer A, Toomre D, Fuh G, Yan M, Kluger MS, Wu D, Min W (2016) Endothelial exocytosis of angiopoietin-2 resulting from CCM3 deficiency contributes to cerebral cavernous malformation. Nat Med 22:1033–1042
Zhou W, Chen C, Shi Y, Wu Q, Gimple RC, Fang X, Huang Z, Zhai K, Ke SQ, Ping YF, Feng H, Rich JN, Yu JS, Bao S, Bian XW (2017) Targeting glioma stem cell-derived pericytes disrupts the blood-tumor barrier and improves chemotherapeutic efficacy. Cell Stem Cell 21:591–603
Zhou Y, Peng Z, Seven ES, Leblanc RM (2018) Crossing the blood-brain barrier with nanoparticles. J Control Release 270:290–303
Zhu Y, Soderblom C, Krishnan V, Ashbaugh J, Bethea JR, Lee JK (2015) Hematogenous macrophage depletion reduces the fibrotic scar and increases axonal growth after spinal cord injury. Neurobiol Dis 74:114–125
Zimmermann KW (1923) Der feinere Bau der Blutkapillaren. Z Anat Entwicklungsgesch 68:29–109
Acknowledgements
Supported by the European Research Council (BrainEnergy), Wellcome Trust (099222/Z/12/Z), BBSRC (LIDo PhD studentship to NK), Leonard Wolfson Experimental Neurology Centre (PhD studentship to RN), Rosetrees Trust, National Natural Science Foundation of China (81622041) and National Key R&D Program of China (2017YFC1307500, 2017YFC1307504).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Cheng, J., Korte, N., Nortley, R. et al. Targeting pericytes for therapeutic approaches to neurological disorders. Acta Neuropathol 136, 507–523 (2018). https://doi.org/10.1007/s00401-018-1893-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-018-1893-0