
Abstract
Chromatin organization is central to many conserved biological processes, but it is generally unknown how the underlying nucleosomes are arranged in situ. Here, we have used electron cryotomography to study chromatin in the picoplankton Ostreococcus tauri, the smallest known free-living eukaryote. By visualizing the nucleosome densities directly, we find that O. tauri chromosomes do not arrange into discrete, compact bodies or any other higher level of order. In contrast to the textbook 30-nm fiber model, O. tauri chromatin resembles a disordered assemblage of nucleosomes akin to the polymer melt model. This disorganized nucleosome arrangement has important implications for potentially conserved functions in tiny eukaryotes such as the clustering of nonhomologous chromosomes at the kinetochore during mitosis and the independent regulation of closely positioned adjacent genes.
Similar content being viewed by others


Introduction
The nucleosome hypothesis (Kornberg 1974) defined a fundamental unit of eukaryotic chromosome organization. While the structure of the nucleosome core particle is now known, it is still unclear how nucleosomes pack into the higher order chromatin structures that influence transcription, replication, and mitosis (Luger et al. 1997; van Steensel 2011). An electron microscopy study of purified chromatin lead to the 30-nm fiber model of chromatin organization, which proposed that nucleosomes pack into ordered helical fibers (Finch and Klug 1976). Two later electron cryomicroscopy (cryo-EM) studies of frozen-hydrated sections (cryosections) showed that chromatin organization can differ substantially depending on the cell type. One study did not find 30-nm fibers in mitotic Chinese hamster ovary cells (McDowall et al. 1986), but the other study did observe 30-nm chromatin fibers in starfish sperm, sea cucumber sperm, and purified chicken erythrocyte nuclei (Woodcock 1994). Recently, two groups searched for 30-nm fibers using 2D Fourier analysis of HeLa cell cryosections and 3D electron cryotomography (cryo-ET) of cryosections of purified chicken erythrocyte nuclei (Eltsov et al. 2008; Scheffer et al. 2011). They showed that HeLa cells do not have 30-nm fibers, but chicken erythrocytes do (though they appear short). The absence of 30-nm fibers has further been supported by groups using small-angle X-ray scattering and electron spectroscopic imaging of mammalian chromosomes and cells, respectively (Fussner et al. 2011, 2012; Joti et al. 2012; Maeshima et al. 2010; Nishino et al. 2012).
We study the smallest known free-living eukaryote, the picoplankton Ostreococcus tauri, as a model cell-biology system. O. tauri is a unicellular organism of the green lineage and has just one chloroplast, one mitochondrion, and a tiny nucleus that contains 20 linear interphase chromosomes (Courties et al. 1994; Derelle et al. 2006). Using cryo-ET of intact plunge-frozen cells, we found that each cell typically contained just one cytoplasmic microtubule (Henderson et al. 2007). This minimalistic ultrastructure suggested that further studies of O. tauri might reveal new principles of conserved cell-biological processes. For instance, when we imaged mitotic O. tauri cells by both cryo-ET of cryosections and room-temperature electron tomography of serial plastic sections, we found that each cell had only ~10 spindle microtubules, which was significantly fewer than the minimum 40 expected from textbook models (Gan et al. 2011). We therefore proposed that O. tauri might cluster kinetochores together to allow spindle microtubules to segregate more than one chromosome at a time.
To gain insights into how O. tauri chromatin is organized, here we have further analyzed our tomograms of interphase and mitotic O. tauri cryosections. Thirty-nanometer fibers were not seen: instead, in both interphase and mitotic cells, the nucleosome packing was patternless. Using a template-matching approach, we found that there is no large-scale reorganization indicative of condensation. O. tauri chromatin is therefore organized as a “polymer melt”—a disordered configuration with great flexibility (Eltsov et al. 2008; Maeshima et al. 2010). This chromatin model could explain how centromeres from multiple nonhomologous chromosomes, for instance, could cluster kinetochores and enable segregation by a smaller number of spindle microtubules, or how closely positioned adjacent genes could be independently regulated.
Results
O. tauri chromatin is not organized as 30-nm fibers
We first checked whether O. tauri chromatin is organized as 30-nm fibers by inspecting the chromatin densities in tomograms of cryosections of both the interphase and mitotic cells prepared in our previous study (Gan et al. 2011). We did not see any 30-nm fibers, which, if present, would have been even more easily resolved than the 25-nm, ribosome-like particles (Fig. 1a, b). To test for the 30-nm fiber in a different way, we searched for a characteristic peak at 30 nm (1/30 nm−1) in rotationally averaged amplitudes of the Fourier transforms of chromatin densities. We did not see a peak at 30 nm (1/30 nm−1), but we did see a broad peak centered at 10 nm (1/10 nm−1), most likely corresponding to the disordered nucleosome assemblages (Fig. 1c). Since ribosomes are abundant and approximate the size of a 30-nm fiber, they serve as a good positive control (Nishino et al. 2012). We analyzed the Fourier amplitudes of a cluster of cytoplasmic ribosomes and found a broad peak at approximately 30 nm (1/30 nm−1) as expected for such large complexes. The absence of 30-nm fibers is neither a sample-preparation nor an imaging artifact because 30-nm fibers were clearly seen in cryo-EM images of cryosectioned starfish sperm and chicken erythrocyte nuclei (Scheffer et al. 2011; Woodcock 1994), and even thinner helical protein assemblies such as bacterial type VI secretion systems (15 nm), bacterial microtubules (8 nm), and FtsZ filaments (5 nm) have been seen previously in the same electron cryomicroscope used here (Basler et al. 2012; Li et al. 2007; Pilhofer et al. 2011). We therefore conclude that O. tauri chromatin is not organized as 30-nm fibers.

O. tauri chromatin is not organized as 30-nm fibers. a Tomographic slice, 60 nm thick, through a cryosection of a mitotic O. tauri cell. The intranuclear spindle microtubules are not located in this slice. The nucleus (Nuc), chloroplast (Chl), mitochondrion (Mito), a granule (gr), and gold fiducials (Au) are labeled. A position including either chromatin (blue) or cytoplasmic ribosomes (red) was selected for Fourier analysis. The semiperiodic horizontal structures (most visible in the left side of the chloroplast) are crevasses from cryomicrotomy. b A 10-nm-thick tomographic slice corresponding to the black/white box in a, enlarged 3-fold and rotated 90° counterclockwise. A cytoplasmic ribosome is indicated by the arrow and an intranuclear nucleosome-sized density is indicated by the arrowhead. c Rotationally averaged amplitudes (log scale, arbitrary units) of the Fourier transform of the two color-coded positions boxed in a. Arrows point to the ~30-nm (1/30 nm−1) (left) and 10-nm (1/10 nm−1) (right) spatial frequencies. A 10-nm-thick tomographic slice corresponding to the blue box in a is shown, enlarged 3-fold, either d unmodified or e with a Gaussian-shaped bandpass filter centered at 10 nm (1/10 nm−1). The arrowhead points to an example nucleosome-sized density. Note that image compression artifacts make the chromatin densities look smaller than in the original, uncompressed image
Even though O. tauri does not have 30-nm chromatin fibers, we could not rule out the possibility that the nucleosomes may exist in previously uncharacterized ordered oligomers. This hypothesis could be tested using our best tomograms, in which we could resolve approximately 10-nm-wide, 6-nm-thick granular intranuclear particles (Fig. 1d). We suspected that these intranuclear particles were nucleosomes, since these particles were plentiful and nucleosomes are the most abundant macromolecular complexes in nuclei. To improve visualization of these particles, we bandpass-filtered the tomograms to enhance the structural details broadly centered at 10 nm (1/10 nm−1) spatial frequency (Fig. 1e). Bandpass filtering enhanced only Fourier components at spacings larger than the first CTF zero (~4 to 4.5 nm) and did not produce any spurious densities. Since similar nucleosome-like particles were exceedingly rare in the extracellular space and were also only infrequently seen in the cytoplasm (where other similarly sized complexes reside), they were not the result of random noise or imaging artifacts. Some of the nucleosome-like particles were arranged in short chains reminiscent of beads-on-a-string (Olins and Olins 1974), but none were found in longer or thicker arrays (Fig. 2).

Some nucleosomes appear to form small clusters. Tomographic slices (10 nm) showing examples of small nucleosome clusters (indicated by arrows) in an interphase cell (a–d) and a mitotic cell (e–h). Both the bandpass-filtered images (a, e) and the original lowpass-filtered images (b, f) are shown. The template-matching result for the 10-nm-thick volume is indicated by green circles overlaid on the tomographic densities (c, g) and alone (d, h). The diameter of the circle is related to the nucleosome’s “z” position within the subvolume: the largest circles denote nucleosomes centered within the subvolume, while the smallest circles denote nucleosomes centered above or below the subvolume. Some densities appear to be smaller than the nominal ~10-nm nucleosome diameter; these smaller densities may be the “tops” or “bottoms” of nucleosomes that are just within the 10-nm-thick volume. They may also result from the noise in the cryotomogram that produces some false-positive hits. The presence of some false-positives and false-negatives does not affect, however, the main conclusion that there are no discernable higher-order structures present
O. tauri chromatin is disorganized
Next we used a template-matching search to more objectively locate nucleosome-like particles in 3D (Förster et al. 2010; Huiskonen et al. 2010). While template matching cannot accurately discriminate between different small (<250 kDa) macromolecular complexes (Förster et al. 2010) or determine the exact nucleosome orientations, it can identify strong candidates as long as they are not packed close together face-to-face (Fig. S1). We therefore searched our tomograms with an 8-nm-diameter spherical template, which is a compromise of the nucleosome’s dimensions. Template matching ranks candidate densities according to how well they match the template. By taking into account the nuclear volumes from whole-cell tomograms (Henderson et al. 2007), the number of bases in the genome (Derelle et al. 2006), and the nucleosome-repeat length from a new micrococcal nuclease digestion experiment (198 bp, Fig. S2), we estimated the interphase nucleosome concentration to be approximately 700 μM. This estimate is comparable to the lower local nucleosome concentration values that can be calculated for both interphase (820 μM) and metaphase (980 μM) chromosomes in other species (Daban 2003). To further check for ordered nucleosome arrays, we filtered the template-matching results to include 0.66× and 1.5× the nominal number of hits (based on the nominal 700 μM concentration); these results (together with the nominal hits) are presented in single tomographic slices in both Figs. 2c, d, g, and h and 3a–d and in full 3D in Fig. 3e. No ordered nucleosome arrays were seen, regardless of the correlation cutoff used. Ordered nucleosome arrays were also absent in mitotic chromatin (Fig. 3f–j); not even discrete chromatids were seen. In our published serial tomograms of plastic-sectioned O. tauri (Gan et al. 2011), we observed a lightly stained zone surrounding the spindle microtubules that we called the “spindle tunnel.” In the cryosections analyzed here, nucleosomes were rarely seen in this tunnel, and no ordered nucleosome arrangements were seen in the surrounding chromatin either (Fig. 4). We conclude that O. tauri chromatin is a disordered assemblage of nucleosomes and that no large-scale condensation-like reorganizations occur during mitosis.

O. tauri nucleosomes do not undergo large-scale reorganization in mitosis. Tomographic slices (1.3 nm thick) through the nucleus of an interphase (a–e) and a mitotic (f–j) O. tauri cell. To enhance the visualization of nucleosome-sized densities in such thin tomographic slices, the tomograms were bandpass-filtered as in Fig. 1d. Template-matching results showing 0.66× (b, g), 1× (c, h), and 1.5× (d, i) the nominal number of hits are shown as green circles superposed on the tomographic densities. Only those hits that are centered in the particular tomographic slice are circled; hits in slices “above” and “below” are not circled for clarity. Many of the ~10-nm densities are nucleosomes centered just below or above the current slice and therefore are not circled; in contrast, see Fig. 2. e, j 3D model of the centers of masses of all the nucleosome-like densities. Each sphere is ~8 nm wide

Spindle microtubules reside in a nucleosome-depleted zone. a, b Tomographic slices (10 nm) of two mitotic O. tauri cells, taken at the spindle tunnel. The chromatin (Chr), mitochondrion (Mito), and chloroplast (Chl) are indicated. Each tomogram was rotated to a view along the longitudinal axis of the spindle microtubules (arrowhead), one of which is incomplete (b). As a result of the image rotation, crevasses (arrows) are visible in the right-hand side of b. Subvolumes containing the spindle microtubule(s) and the spindle tunnel are boxed and enlarged in c and d, corresponding to the cells (a) and (b), respectively. The boundary surrounding the nucleosome-depleted zone is delineated by the blue dotted line
The 30-nm fiber model has been central to our understanding of eukaryotic chromosome biology (Alberts 2008; Lodish 2013). There are many variants of the 30-nm fiber model (Grigoryev and Woodcock 2012), but they all suggest that nucleosomes are packed in ordered arrays with fibers 25–40 nm wide (Dorigo et al. 2004; Robinson et al. 2006). Under special experimental conditions, chromatin fibers can be assembled with well-defined dimensions. These fibers have often been used in studies of chromatin structure and function at the first level beyond the nucleosome (Robinson and Rhodes 2006). Larger-scale chromatin organization is typically studied in the context of chromosomes or cells. A recent ultra-low angle X-ray scattering study of purified HeLa chromosomes found evidence of a “fractal” organization (Nishino et al. 2012), in agreement with earlier chromatin conformation capture and light microscopy studies of mammalian cells (Bancaud et al. 2009; Lieberman-Aiden et al. 2009). Fractal structures do not have a characteristic length scale; examples of fractal structures include self-similar motifs that span different length scales such as the textbook 10-nm, 30-nm, and 100 + −nm “chromonema” fiber-folding hierarchy (Belmont and Bruce 1994) as well as a polymer with a random walk path (Mirny 2011). While we did not see any structural evidence of self-similarity of higher order structures in our tomograms of either interphase or mitotic cells, they might be “fractal” in that the path of individual nucleosome strings might be random walks.
Discussion
O. tauri chromatin has characteristics of a polymer melt
Few methods can parse chromatin organization in situ at “molecular” resolution, i.e., sufficiently to resolve nucleosomes. Electron microscopy has sufficient resolution, but the samples have usually been prepared with aldehyde fixation, solvent dehydration, plastic embedment, and heavy-metal staining, which can all introduce artifacts to native structure. To reliably resolve individual macromolecular complexes in the native state within the crowded milieu of the nucleus, cell imaging must be done by cryo-ET (Gan and Jensen 2012). Using cryo-ET of cryosections, we have now shown that O. tauri chromatin resembles a “polymer melt” chromatin proposed for HeLa cells (Eltsov et al. 2008). The polymer melt model evolved from the “liquid” model that Dubochet and colleagues proposed after imaging frozen-hydrated CHO cells with cryo-EM (McDowall et al. 1986). O. tauri is now the first nonmammalian cell known to have such a disorganized nucleosome arrangement (Fig. 5a, b).

O. tauri chromatin is disorganized. Cartoon models of nucleosomes (blue disks) in O. tauri chromatin in a interphase and b mitosis. In textbook models (c), chromatin is universally presented using the 30-nm fiber; modeled from Scheffer et al. (2011). d Hypothetical model of O. tauri polymer melt chromatin in a mitotic cell, viewed along the spindle axis. Canonical nucleosomes (light blue spheres) and centromeric nucleosomes from nonhomologous chromosomes (multicolored spheres) are positioned around the spindle. Kinetochore protein complexes (lilac rods) connect the centromere to the spindle microtubules (green rings)
Polymer melt chromatin may facilitate conserved nuclear functions
Compared to 30-nm fiber chromatin (Fig. 5c), polymer melt-like chromatin is more compatible with key cell-biological processes in tiny nuclei. For example, we previously proposed that O. tauri might cluster mitotic chromosomes together at their kinetochores so mitosis could be completed in a single round of anaphase (Gan et al. 2011). Since kinetochores are nucleated by centromeric chromatin and positioned by pericentromeric chromatin (Blower et al. 2002; Marshall et al. 2008; Zinkowski et al. 1991), the chromatin path must make tight turns in order to cluster 20 kinetochores in a <1-μm nucleus. We speculate one possible configuration in which centromeric nucleosomes could cluster in a ring surrounding the spindle microtubules (Fig. 5d). Polymer melt chromatin could be flexible enough to make such turns because a loosely ordered beads-on-a-string nucleosome arrangement is likely to have a persistence length similar to naked dsDNA (<50 nm) (Brinkers et al. 2009). The polymer melt could therefore be instrumental to chromosome and kinetochore organization in some Trypanosome species, which may also have fewer kinetochores than chromosomes (Solari 1995). It is currently unknown if 30-nm fibers also exhibit such flexibility because their reported persistence length ranges from 30 to 220 nm (Bystricky et al. 2004; Cui and Bustamante 2000; Dekker et al. 2002; Kepper et al. 2008; Wedemann and Langowski 2002). While we have not yet visualized kinetochore distribution in O. tauri, kinetochore clustering has indeed been shown in yeasts (Appelgren et al. 2003; Jin et al. 2000). Genome-wide chemical mapping and two-color fluorescence light microscopy studies have argued, however, that budding yeast chromatin organizes as 30-nm fibers (Brogaard et al. 2012; Bystricky et al. 2004). It is therefore important to confirm whether or not 30-nm fibers do in fact exist in budding yeast and determine how interchromosomal interactions are mediated.
Chromatin organization also plays a role in transcriptional initiation by modulating the accessibility and positioning of both cis- and trans-regulatory elements. Transcriptional regulation models in humans must therefore take into account both the gene density and long intergenic sequences. In chromosome 11, for example, there are 10.6 genes/megabase and genes are separated by an average of 86 kb (Taylor et al. 2006). As illustrated in a compelling model of the β-globin locus (in human chromosome 11), an extended 30-nm fiber could act as a mechanical scaffold that loops in order to position an array of RNA polymerase II complexes (a “reading head”) on the coding sequences located several kilobases away (Wong et al. 2009). Such a transcriptional-regulation mechanism does not appear plausible in O. tauri and possibly other eukaryotes due to their much higher gene density (Derelle et al. 2006). Since 80 % of the O. tauri genome codes for genes and these are separated on average by less than 200 bp, the gene density is 100-fold larger than the human average. The typical gene would therefore span 1.3 kb—just 6.5 nucleosomes, with a single additional nucleosome separating adjacent genes. In the context of the 30-nm fiber, the average O. tauri gene would span less than one helical turn! According to the β-globin model, activating just one O. tauri gene could then force the shutdown of tens of other genes, which we find unlikely. Polymer melt chromatin would allow independent regulation of each gene in O. tauri and therefore finer control of transcriptional programs. A recent study furthermore showed that due to its flexible nature, polymer melt chromatin can even facilitate chromatin accessibility in both interphase and mitotic chromosomes (Hihara et al. 2012). Other gene-dense organisms may use polymer melt chromatin for similar purposes.
It remains unknown just how common 30-nm fiber-like chromatin is among eukaryotes. There is now an incipient consensus that somatic mammalian cells (Chinese hamster ovary; HeLa; mouse embryonic fibroblasts, spleen, and liver) do not have 30-nm fibers; instead they pack chromatin either as a polymer melt or as 10-nm fibers (Eltsov et al. 2008; Fussner et al. 2012; McDowall et al. 1986; Nishino et al. 2012). In contrast, specialized transcriptionally silent eukaryotic cells (chicken erythrocyte, starfish, and sea cucumber sperm) have 30-nm fibers as the predominant form of chromatin (Scheffer et al. 2011; Woodcock 1994). Here we have presented evidence that a unicellular picoeukaryote also packs chromatin as a polymer melt. Since the O. tauri cells analyzed here were isolated from both interphase and mitotic cultures, they are most analogous to somatic cells in higher eukaryotes. Cells with substantially differing gene densities and spatial and evolutionary constraints can therefore package chromatin without using 30-nm fibers. Unlike higher eukaryotes, however, O. tauri does not undergo large-scale chromosome condensation during mitosis. In other words, O. tauri chromatin does not reorganize into discrete chromatids separated by large cytoplasmic spaces; for an example of condensed mitotic chromatin, see Fig. 2 in Eltsov et al. (2008). It is possible that this picoplankton does not have the genes needed to condense chromosomes, which may also be needed to achieve 30-nm fibers, a form of local chromatin condensation. To test if the features of O. tauri chromatin are unique to this “untypical” organism or an adaptation, our analyses should be applied to other tiny eukaryotes that also have high gene density and small nuclear size.
Materials and methods
Cell preparation and electron cryotomography
Details of cell culture, synchronization, freezing, cryosectioning, imaging, and tomographic reconstruction are described (Gan et al. 2011). In summary, cell cultures of strain RCC745 were grown in artificial seawater and were naturally synchronized to a 12-h light/12-h dark cycle. To enrich for mitotic cells, cultures were arrested sequentially with hydroxyurea and propyzamide. Cells released from propyzamide were able to complete mitosis, progressing through prometaphase, metaphase, and anaphase. These cells could also be arrested in metaphase by treatment with MG132. The mitotic cells analyzed in this study were isolated after release from propyzamide treatment. Cell cultures were then mixed with 22 % dextran (an extracellular cryoprotectant) and 10-nm colloidal gold (fiducial markers for tomographic image alignment) and then rapidly frozen in an HPM-010 high-pressure freezer (Leica Microsystems). Cryosections were cut using an EM-UC6/FC6 cryoultramicrotome (Leica Microsystems) at −145 °C with a nominal feed of 130–150 nm. Ribbons of cryosections were controlled with a micromanipulator (Leitz model “M,” Leica Microsystems) and secured onto a C-flat CF422C-T grid (Protochips, Inc.). Tomographic imaging was done on a FEI “Polara” electron cryomicroscope, operated at 300 kV and −193 °C. Tilt series images were recorded using UCSF Tomo or Leginon (Suloway et al. 2009; Zheng et al. 2004) at a magnification of 18,000 or 22,500, corresponding respectively to 1.26 or 0.96 nm pixels at the specimen level. Nominal underfocus values ranged from 8 to 10 μm, which places the first CTF zero between 4 and 4.5 nm resolution. The nominal tilt range was ±66°, tilt increment was 1.5° or 2°, and cumulative dose was 100–140 electrons/Å2. Tomograms were reconstructed using IMOD (Kremer et al. 1996; Mastronarde 1997).
Fourier analysis
Tomographic slices were generated using IMOD (Kremer et al. 1996; Mastronarde 1997) and both Fourier transforms and rotational averaging of the amplitudes was done using ImageJ (Schneider et al. 2012). Since knife marks and crevasses at the cryosection surfaces could interfere with this analysis (Dubochet et al. 2007), we used subvolumes containing only the interior of the cryosection.
Template matching
Nucleosomes were located automatically using the template-matching function in Jsubtomo (www.opic.ox.ac.uk/jsubtomo) (Huiskonen et al. 2010). An initial search was done using a low correlation cutoff—typically less than 0.05. Hit lists having higher correlation cutoffs were then created manually. For example, this list can be filtered so that the number of hits is less than, equal to, or greater than the number of nucleosomes calculated from other types of data (see “Nucleosome concentration calculations” section). Positions corresponding to each hit were mapped to a 3D volume using the IMOD program “point2model.” The hits were then visualized with the tomographic densities using the IMOD programs “3dmod” and “slicer.”
Simulations
Atomic models of polynucleosomes were adjusted manually using UCSF Chimera (Pettersen et al. 2004) and tomograms were simulated using Bsoft (Heymann and Belnap 2007) following our previous protocol (Gan et al. 2008).
Micrococcal nuclease digests
O. tauri nucleosome-repeat length was determined using the classic micrococcal nuclease digestion experiment (Hewish and Burgoyne 1973). O. tauri cells were pelleted and then resuspended with digestion buffer (50 mM Tris, pH 8, 5 mM CaCl2, 0.1 % saponin, 25 μg/ml RNase A). Next, micrococcal nuclease (Roche Applied Science) was added and incubated at room temperature for 5 min. The reaction was stopped by the addition of and incubation with 0.2 mg/ml Proteinase K for 2 min, followed by the addition of one volume of 2× Stop Buffer (20 mM Tris, pH 7.5, 20 mM EDTA, and 20 mM EGTA). Digested DNA was purified using the DNeasy blood & tissue kit (Qiagen) according to the manufacturer’s instructions. DNA bands were then resolved in a 1.5 % agarose/0.5× Tris–borate–EDTA gel, electrophoresed at 50 V for 3 h, and visualized with FloroSafe stain (1st BASE). DNA bands were photographed in a G:BOX gel imager (Synoptics, Ltd.). Since limited micrococcal nuclease digestion products migrate as a DNA ladder corresponding to oligonucleosomes, the nucleosome-repeat length was determined by least-squares linear-regression analysis of log(band size) versus nucleosome number. The mono- and dinucleosomes were excluded from the analysis because they undergo more extensive digestion, producing shorter DNA fragments that, when included in the linear fit, result in a longer apparent nucleosome-repeat length.
Nucleosome concentration calculations
To calculate the nucleosome concentration, we used the haploid nuclear genome size (12.56 Mb) known from genome sequencing (Derelle et al. 2006; Grimsley et al. 2010). We then used the O. tauri nucleosome-repeat length (198 ± 6 bp, Fig. S2) to convert the genome size to nucleosomes (63,000). Finally, we used the volume of early interphase nuclei (0.155 ± 0.02 femtoliter), which we previously measured in our tomograms of intact O. tauri cells (Henderson et al. 2007). From these data, we estimate that early interphase cells pack nucleosomes at 680 ± 90 μM average concentration. We could not estimate the nucleosome concentration of mitotic cells because the contrast of these large plunge-frozen cells was too low to permit accurate segmentation of the nuclei. To calculate the local nucleosome concentration from Daban’s DNA concentration estimates, we assumed that the average base pair has a molecular weight MWbp = 610 Da and that a typical nucleosome wraps 200 bp of DNA. The local DNA concentration, ρ DNA, was defined by Daban as “the mass of DNA per unit volume of the structure that contains it” (Daban 2003). For interphase chromosomes, ρ DNA = 100 g/l (0.1 g/ml). Based on these values, the molar concentration per base pair is C bp = ρ DNA/MWbp or 0.16 M. The local nucleosome concentration is therefore 0.16/200 = 8.2 × 10−4 mol/l or 820 μM.
Figure preparation
Figures were created using 3dmod (Figs. 1, 2, 3, 4, and S1), UCSF Chimera (Figs. 5 and S1), Adobe Illustrator (Fig. 5), Microsoft Excel (Figs. 1 and S2), and Syngene (Fig. S2) and arranged using Adobe Photoshop.
References
Alberts B (2008) Molecular biology of the cell. Garland Science, New York
Appelgren H, Kniola B, Ekwall K (2003) Distinct centromere domain structures with separate functions demonstrated in live fission yeast cells. J Cell Sci 116:4035–4042
Bancaud A, Huet S, Daigle N, Mozziconacci J, Beaudouin J, Ellenberg J (2009) Molecular crowding affects diffusion and binding of nuclear proteins in heterochromatin and reveals the fractal organization of chromatin. EMBO J 28:3785–3798
Basler M, Pilhofer M, Henderson GP, Jensen GJ, Mekalanos JJ (2012) Type VI secretion requires a dynamic contractile phage tail-like structure. Nature 483:182–186
Belmont AS, Bruce K (1994) Visualization of G1 chromosomes: a folded, twisted, supercoiled chromonema model of interphase chromatid structure. J Cell Biol 127:287–302
Blower MD, Sullivan BA, Karpen GH (2002) Conserved organization of centromeric chromatin in flies and humans. Dev Cell 2:319–330
Brinkers S, Dietrich HR, de Groote FH, Young IT, Rieger B (2009) The persistence length of double stranded DNA determined using dark field tethered particle motion. J Chem Phys 130:215105
Brogaard K, Xi L, Wang JP, Widom J (2012) A map of nucleosome positions in yeast at base-pair resolution. Nature 486:496–501
Bystricky K, Heun P, Gehlen L, Langowski J, Gasser SM (2004) Long-range compaction and flexibility of interphase chromatin in budding yeast analyzed by high-resolution imaging techniques. Proc Natl Acad Sci U S A 101:16495–16500
Courties C, Vaquer A, Troussellier M, Lautier J (1994) Smallest eukaryotic organism. Nature 370:255
Cui Y, Bustamante C (2000) Pulling a single chromatin fiber reveals the forces that maintain its higher-order structure. Proc Natl Acad Sci U S A 97:127–132
Daban J-R (2003) High concentration of DNA in condensed chromatin. Biochem Cell Biol 81:91–99
Dekker J, Rippe K, Dekker M, Kleckner N (2002) Capturing chromosome conformation. Science 295:1306–1311
Derelle E, Ferraz C, Rombauts S, Rouzé P, Worden AZ, Robbens S, Partensky F, Degroeve S, Echeynié S, Cooke R, Saeys Y, Wuyts J, Jabbari K, Bowler C, Panaud O, Piégu B, Ball SG, Ral J-P, Bouget F-Y, Piganeau G, De Baets B, Picard A, Delseny M, Demaille J, Van de Peer Y, Moreau H (2006) Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc Natl Acad Sci USA 103:11647–11652
Dorigo B, Schalch T, Kulangara A, Duda S, Schroeder RR, Richmond TJ (2004) Nucleosome arrays reveal the two-start organization of the chromatin fiber. Science 306:1571–1573
Dubochet J, Zuber B, Eltsov M, Bouchet-Marquis C, Al-Amoudi A, Livolant F (2007) How to “read” a vitreous section. Methods Cell Biol 79:385–406
Eltsov M, Maclellan KM, Maeshima K, Frangakis AS, Dubochet J (2008) Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc Natl Acad Sci USA 105:19732–19737
Finch JT, Klug A (1976) Solenoidal model for superstructure in chromatin. Proc Natl Acad Sci U S A 73:1897–1901
Förster F, Han B-G, Beck M (2010) Visual proteomics. Meth Enzymol 483:215–243
Fussner E, Ching RW, Bazett-Jones DP (2011) Living without 30 nm chromatin fibers. Trends Biochem Sci 36:1–6
Fussner E, Strauss M, Djuric U, Li R, Ahmed K, Hart M, Ellis J, Bazett-Jones DP (2012) Open and closed domains in the mouse genome are configured as 10-nm chromatin fibres. EMBO reports
Gan L, Chen S, Jensen GJ (2008) Molecular organization of Gram-negative peptidoglycan. Proc Natl Acad Sci USA 105:18953–18957
Gan L, Jensen GJ (2012) Electron tomography of cells. Q Rev Biophys 45:27–56
Gan L, Ladinsky MS, Jensen GJ (2011) Organization of the smallest eukaryotic spindle. Curr Biol 21:1578–1583
Grigoryev SA, Woodcock CL (2012) Chromatin organization—the 30 nm fiber. Exp Cell Res 318:1448–1455
Grimsley N, Péquin B, Bachy C, Moreau H, Piganeau G (2010) Cryptic sex in the smallest eukaryotic marine green alga. Mol Biol Evol 27:47–54
Henderson GP, Gan L, Jensen GJ (2007) 3-D ultrastructure of O. tauri: electron cryotomography of an entire eukaryotic cell. PLoS One 2:e749
Hewish DR, Burgoyne LA (1973) Chromatin sub-structure. The digestion of chromatin DNA at regularly spaced sites by a nuclear deoxyribonuclease. Biochem Biophys Res Commun 52:504–510
Heymann JB, Belnap DM (2007) Bsoft: image processing and molecular modeling for electron microscopy. J Struct Biol 157:3–18
Hihara S, Pack CG, Kaizu K, Tani T, Hanafusa T, Nozaki T, Takemoto S, Yoshimi T, Yokota H, Imamoto N, Sako Y, Kinjo M, Takahashi K, Nagai T, Maeshima K (2012) Local nucleosome dynamics facilitate chromatin accessibility in living mammalian cells. Cell Rep 2:1645–1656
Huiskonen JT, Hepojoki J, Laurinmaki P, Vaheri A, Lankinen H, Butcher SJ, Grunewald K (2010) Electron cryotomography of Tula hantavirus suggests a unique assembly paradigm for enveloped viruses. J Virol 84:4889–4897
Jin QW, Fuchs J, Loidl J (2000) Centromere clustering is a major determinant of yeast interphase nuclear organization. J Cell Sci 113(Pt 11):1903–1912
Joti Y, Hikima T, Nishino Y, Kamada F, Hihara S, Takata H, Ishikawa T, Maeshima K (2012) Chromosomes without a 30-nm chromatin fiber. Nucleus 3:404–410
Kepper N, Foethke D, Stehr R, Wedemann G, Rippe K (2008) Nucleosome geometry and internucleosomal interactions control the chromatin fiber conformation. Biophys J 95:3692–3705
Kornberg RD (1974) Chromatin structure: a repeating unit of histones and DNA. Science 184:868–871
Kremer JR, Mastronarde DN, McIntosh JR (1996) Computer visualization of three-dimensional image data using IMOD. J Struct Biol 116:71–76
Li Z, Trimble MJ, Brun YV, Jensen GJ (2007) The structure of FtsZ filaments in vivo suggests a force-generating role in cell division. EMBO J 26:4694–4708
Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 326:289–293
Lodish HF (2013) Molecular cell biology. Freeman, New York
Luger K, Mäder AW, Richmond RK, Sargent DF, Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389:251–260
Maeshima K, Hihara S, Eltsov M (2010) Chromatin structure: does the 30-nm fibre exist in vivo? Current Opinion in Cell Biology
Marshall OJ, Marshall AT, Choo KHA (2008) Three-dimensional localization of CENP-A suggests a complex higher order structure of centromeric chromatin. J Cell Biol 183:1193–1202
Mastronarde DN (1997) Dual-axis tomography: an approach with alignment methods that preserve resolution. J Struct Biol 120:343–352
McDowall AW, Smith JM, Dubochet J (1986) Cryo-electron microscopy of vitrified chromosomes in situ. EMBO J 5:1395–1402
Mirny LA (2011) The fractal globule as a model of chromatin architecture in the cell. Chromosome Res: Int J Mol Supramol Evol Asp Chromosome Biol 19:37–51
Nishino Y, Eltsov M, Joti Y, Ito K, Takata H, Takahashi Y, Hihara S, Frangakis AS, Imamoto N, Ishikawa T, Maeshima K (2012) Human mitotic chromosomes consist predominantly of irregularly folded nucleosome fibres without a 30-nm chromatin structure. EMBO J 31:1644–1653
Olins AL, Olins DE (1974) Spheroid chromatin units (v bodies). Science 183:330–332
Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612
Pilhofer M, Ladinsky MS, McDowall AW, Petroni G, Jensen GJ (2011) Microtubules in bacteria: ancient tubulins build a five-protofilament homolog of the eukaryotic cytoskeleton. PLoS biology 9:e1001213
Robinson PJ, Rhodes D (2006) Structure of the '30 nm' chromatin fibre: a key role for the linker histone. Curr Opin Struct Biol 16:336–343
Robinson PJJ, Fairall L, Huynh VAT, Rhodes D (2006) EM measurements define the dimensions of the "30-nm" chromatin fiber: evidence for a compact, interdigitated structure. Proc Natl Acad Sci USA 103:6506–6511
Scheffer MP, Eltsov M, Frangakis AS (2011) Evidence for short-range helical order in the 30-nm chromatin fibers of erythrocyte nuclei. Proc Natl Acad Sci U S A 108:16992–16997
Schneider CA, Rasband WS, Eliceiri KW (2012) NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675
Solari AJ (1995) Mitosis and genome partition in trypanosomes. Biocell 19:65–84
Suloway C, Shi J, Cheng A, Pulokas J, Carragher B, Potter CS, Zheng SQ, Agard DA, Jensen GJ (2009) Fully automated, sequential tilt-series acquisition with Leginon. J Struct Biol 167:11–18
Taylor TD, Noguchi H, Totoki Y, Toyoda A, Kuroki Y, Dewar K, Lloyd C, Itoh T, Takeda T, Kim DW, She X, Barlow KF, Bloom T, Bruford E, Chang JL, Cuomo CA, Eichler E, FitzGerald MG, Jaffe DB, LaButti K, Nicol R, Park HS, Seaman C, Sougnez C, Yang X, Zimmer AR, Zody MC, Birren BW, Nusbaum C, Fujiyama A, Hattori M, Rogers J, Lander ES, Sakaki Y (2006) Human chromosome 11 DNA sequence and analysis including novel gene identification. Nature 440:497–500
van Steensel B (2011) Chromatin: constructing the big picture. EMBO J 30:1885–1895
Wedemann G, Langowski J (2002) Computer simulation of the 30-nanometer chromatin fiber. Biophys J 82:2847–2859
Wong H, Winn PJ, Mozziconacci J (2009) A molecular model of chromatin organisation and transcription: how a multi-RNA polymerase II machine transcribes and remodels the beta-globin locus during development. Bioessays: News Rev Mol Cel Dev Biol 31:1357–1366
Woodcock CL (1994) Chromatin fibers observed in situ in frozen hydrated sections. Native fiber diameter is not correlated with nucleosome repeat length. J Cell Biol 125:11–19
Zheng QS, Braunfeld MB, Sedat JW, Agard DA (2004) An improved strategy for automated electron microscopic tomography. J Struct Biol 147:91–101
Zinkowski RP, Meyne J, Brinkley BR (1991) The centromere–kinetochore complex: a repeat subunit model. J Cell Biol 113:1091–1110
Acknowledgments
We thank Drs. J. Huiskonen for advice on Jsubtomo, M. Swulius for discussions on template matching, H. Wong and J. Mozziconacci for sharing their 30-nm fiber model, and Drs. A. McDowall and D. Rhodes for discussions on chromatin. This work was supported by the Howard Hughes Medical Institute and the Gordon and Betty Moore Center for Integrative Study of Cell Regulation. MSL was supported by NIH grant 2 R37 AI041239-06A1 to P. Björkman. LG was supported by a fellowship from the Damon Runyon Cancer Research Foundation (DRG-1940-07) and startup funds from NUS.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Fig. S1
Tests of template-matching performance using simulated tomograms. Simulated tomograms of linear arrays of 5 nucleosome core particles without linker DNA. In (A) and (B), the tilt axis points up (arrow), while in (C), the tilt axis points out of the plane of the image (bulls-eye symbol). The center-to-center distance between nucleosomes is indicated at the bottom right of each square in the top row. The middle row shows the simulated tomograms. In the bottom row, simulated nucleosome densities are oriented according to the top hits of a template-matching search using a nucleosome as the template. Searches that produced the wrong number of hits or with grossly mis-oriented nucleosomes are highlighted in red; there were typically 1 or 2 false positives or negatives. Panel Cii is rotated 90° around the vertical axis relative to panel Ci (curved arrow symbol) to better show that the lower-most nucleosome is mis-oriented relative to the upper four nucleosomes. Searches using a sphere as a template yielded comparable error rates (JPEG 61 kb)
Fig. S2
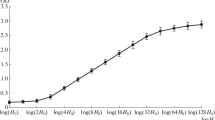
The O. tauri nucleosome-repeat length is 198 base pairs. (A) Agarose gel showing O. tauri DNA from an MNase digestion. Peripheral lanes show markers (base pairs) and central lanes show DNA digested with 0, 0.1, 0.2, 0.4, 0.8, and 1.6 units MNase in 110 μl reaction volume. (B) Linear fit of DNA size vs. nucleosome oligomers, ranging from trimers to octamers. The individual bands are plotted as squares (JPEG 17 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Gan, L., Ladinsky, M.S. & Jensen, G.J. Chromatin in a marine picoeukaryote is a disordered assemblage of nucleosomes. Chromosoma 122, 377–386 (2013). https://doi.org/10.1007/s00412-013-0423-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00412-013-0423-z