
Abstract
Facioscapulohumeral muscular dystrophy (FSHD) is the third most common muscular dystrophy after the dystrophinopathies and myotonic dystrophy and is associated with a typical pattern of muscle weakness. Most patients with FSHD carry a large deletion in the polymorphic D4Z4 macrosatellite repeat array at 4q35 and present with 1–10 repeats whereas non-affected individuals possess 11–150 repeats. An almost identical repeat array is present at 10q26 and the high sequence identity between these two arrays can cause difficulties in molecular diagnosis. Each 3.3-kb D4Z4 unit contains a DUX4 (double homeobox 4) gene that, among others, is activated upon contraction of the 4q35 repeat array due to the induction of chromatin remodelling of the 4qter region. A number of 4q subtelomeric sequence variants are now recognised, although FSHD only occurs in association with three ‘permissive’ haplotypes, each of which is associated with a polyadenylation signal located immediately distal of the last D4Z4 unit. The resulting poly-A tail appears to stabilise DUX4 mRNAs transcribed from this most distal D4Z4 unit in FSHD muscle cells. Synthesis of both the DUX4 transcripts and protein in FSHD muscle cells induces significant cell toxicity. DUX4 is a transcription factor that may target several genes which results in a deregulation cascade which inhibits myogenesis, sensitises cells to oxidative stress and induces muscle atrophy, thus recapitulating many of the key molecular features of FSHD.
Similar content being viewed by others

Clinical presentation
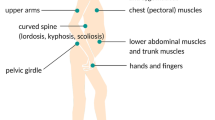
Facioscapulohumeral muscular dystrophy (FSHD) is the third most common autosomal dominant muscular dystrophy after the dystrophinopathies and myotonic dystrophy, affecting approximately 1 in 20,000 individuals worldwide (Padberg 2004). Clinical symptoms usually appear during the second decade and are characterised by progressive muscle weakness, initially of the facial, scapular and humeral muscles, which often show marked asymmetry, and later involving the abdominal muscles and the musculature of the lower limbs and feet (Upadhyaya and Cooper 2004; Padberg 2004). A significant variability in clinical expression is often observed, even among affected family members. The initial facial muscle weakness often goes unreported, with problems only first noted following involvement of the scapular musculature, when patients experience difficulty in raising their arms. Indeed, disease progression is usually relatively slow, with affected individuals often experiencing quite long periods of remission interspersed by sudden and painful periods of muscular deterioration. Several other, non-muscle tissues are also frequently affected in FSHD, with high frequency hearing loss (~75% patients) and retinal telangiectasia (60% patients) reported (Padberg et al. 1995). Central nervous system defects may also occur, with learning difficulties and epilepsy evident in some severely affected children (Saito et al. 2007). Other, less frequent clinical manifestations include respiratory insufficiency (Wohlgemuth et al. 2004) and cardiac conduction defects which may occur in severely affected individuals (Laforêt et al. 1998). The degree of disease severity in FSHD is associated with several factors. For example, affected females are typically less symptomatic than males and older patients are usually more severely affected. The size of the disease-associated 4q35-D4Z4 repeat array is also reported to influence disease severity; with FSHD patients carrying a smaller number of repeats (1–3 units) often being much more severely affected (Tawil et al. 1996). There are, however, significant difficulties in establishing any genotype–phenotype associations in FSHD, mainly due to the significant variability in the degree of muscle weakness observed in patients, even from the same family. Consequently, approximately 15–20% of patients eventually require wheelchair support, while others may remain essentially asymptomatic, exhibiting few, if any, clinical features. Despite the evident morbidity of this disorder, there is surprisingly little evidence for a reduced life expectancy.
Genetics of FSHD
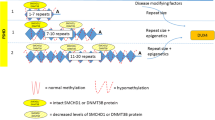
FSHD was first reported to be linked to the distal end of the 4q arm, at the 4q35 region, in 1990 (Wijmenga et al. 1990; Upadhyaya et al. 1990), where it was found to be associated with a polymorphic macrosatellite repeat array. The 3.3-kb repeat unit was named D4Z4, and variable copy numbers are repeated in tandem in a head-to-tail orientation at the 4q35 locus. A number of D4Z4-like sequences are found throughout the human genome, most of which are associated with acrocentric chromosomes, however, only the D4Z4 repeat array situated at 10q26 exhibits almost complete sequence identity (~99%) to the 4q35 array (Lyle et al. 1995; Winokur et al. 1996; Beckers et al. 2001; Deidda et al. 1995). Each 3.3-kb repeat unit has a complex sequence structure, with several GC-rich repeat sequences and an open reading frame containing two homeobox sequences designated as DUX4 (double homeobox 4) (Hewitt et al. 1994; Gabriëls et al. 1999; Ding et al. 1998) (Fig. 1). Both the 4q35 and 10q26 D4Z4 repeat arrays are highly polymorphic and exhibit extensive size variation in the normal population ranging from 38 > 350 kb, corresponding to 11 ≥ 150 3.3 kb repeats. A highlight for FSHD research was recognition that most affected individuals carried at least one much smaller (10–38 kb) 4q35-D4Z4 array containing only 1–10 repeats (Wijmenga et al. 1992). Similar large contractions of the 10q26-D4Z4 array, found in some 10% of the normal population, are however not associated with FSHD (Wijmenga et al. 1992; Lemmers et al. 2001; Zhang et al. 2001) (Fig. 2). The few sequence differences identified between the highly homologous 4q35- and 10q26-D4Z4 arrays can, however, be used to help define their chromosomal origin, with such sequence variations now routinely used as part of an FSHD molecular diagnostic test. Digestion with either EcoRI and BlnI, or, EcoRI and XapI (Fig. 1), separation by pulse-field gel electrophoresis, and hybridization to the p13-E11 probe proximal to the array permits differentiation of the 4q35-derived D4Z4 repeats from the 10q26-derived D4Z4 repeats, with most affected FSHD patients exhibiting a 4q35-derived D4Z4 band of <38 kb (Lunt 1998). The complete loss of the 4q subtelomeric region is however not associated with FSHD, as individuals monosomic for this region, from 4q35-4qter, do not manifest the disorder (Tupler et al. 1996) (Fig. 2b). The finding that at least one 4q35-D4Z4 repeat was required for FSHD expression indicated the possibility that D4Z4 associated gene(s), or other regulatory sequences located at 4q35, have a direct disease-causing effect.

The D4Z4 repeat array contains 11–100 units in normal individuals and is present on both chromosomes 4q35 and 10q26. The repeat array is highly homologous up to 45-kb proximal and differs only by a small number of polymorphisms, one of which creates sites for XapI (X) and BlnI (B) on chromosomes 4q and 10q respectively, with the latter, along with EcoR1 (E), allowing identification of contractions in 4q. Intra-chromosomal variations also exist giving several different haplotypes which are specified both by a proximal simple sequence length polymorphism (SSLP) and a distal region giving variants A, B and C. Each D4Z4 repeat unit contains a DUX4 ORF (yellow rectangles). Several other genes (FRG2, DUX4c, FRG1 and SLC25A4 (ANT1)) were, previously considered to be FSHD candidates, and are situated proximal of the D4Z4 array, from which they are separated by a nuclear matrix attachment site (FR/MAR) located within the SSLP

Only D4Z4 contractions associated with permissive chromosomes result in FSHD. a, c FSHD may only develop in individuals who possess a contraction on permissive alleles containing the poly-A signal ATTAAA in the pLAM region, however, deletion of the entire D4Z4 repeat array (b), which may be accompanied by deletion of other proximal markers (represented by dashed boxes) is not permissive of FSHD and it was suggested that the FSHD gene was included in this array or in the proximal region deleted. d A unique case of FSHD linked to contraction on chromosome 10 was reported suggesting that contractions on non-permissive chromosome may be permissive for FSHD if the poly-A signal is translocated from a permissive chromosome. In this example only the distal end of the D4Z4 array, along with the pLAM region was transferred from a permissive 4q to a non-permissive 10q chromosome and precludes a key role for proximal 4q genes in the pathogenesis of FSHD (Lemmers et al. 2010b)
The sequence complexity of the entire 4q subtelomeric region, coupled with the almost identical sequences at 10q26, has greatly hampered FSHD research and diagnosis. Indeed, a comparative sequence analysis of D4Z4-like sequences from multiple organisms demonstrated that the 10q26 array had initially formed following duplication of an original 4q35 D4Z4 array which was subsequently transfered to 10q26, gaining just a few sequence variations in the process (van Geel et al. 2002). This high sequence identity has also meant that inter- and intra-chromosomal D4Z4 repeat transfers still occur in approximately 10% of normal individuals and FSHD patients, with the resulting hybrid D4Z4 arrays composed of a mixture of both type 4 and type 10 repeats (van Deutekom et al. 1996; Matsumura et al. 2002; Lemmers et al. 2004). Such hybrid arrays further confound molecular analysis, often making it difficult to differentiate standard arrays, containing either 4q or 10q repeats, from the non-standard (hybrid) arrays, composed of a mixture 4q and 10q repeats (Fig. 2). This is particularly problematical as deleted hybrid D4Z4 alleles may also be associated with FSHD.
Diagnostic complexity is further increased with the identification of two additional 4qter variants, designated as 4qA and 4qB (Fig. 1), which differ only by the presence of a 6.2-kb β-satellite sequence in the 4qA allele (van Geel et al. 2002). These sequence variants are located immediately distal of the D4Z4 array and while they are equally frequent in the population, FSHD is exclusively associated with the 4qA variant (Lemmers et al. 2002; Thomas et al. 2007). A third rare sequence variant, 4qC, was recently identified but has not been linked with FSHD (Lemmers et al. 2010a). Additional 4qter sequences have been identified which further subdivide this subtelomeric region into a number of variants, with FSHD found to be associated with only a few permissive haplotypes. A simple sequence length polymorphic site (SSLP) located proximal of the 4q35-D4Z4 array (Fig. 1) identifies three haplotypes, 4A161, 4A159 and 4A168, which, in conjunction with large 4q35-D4Z4 deletions, are specifically associated with FSHD pathogenicity (Lemmers et al. 2010b; Spurlock et al. 2010). Further sequence analysis of permissive and non-permissive FSHD haplotypes has identified a variant C/T SNP associated with the pLAM sequence situated immediately distal of the 4q35 D4Z4 array that can create a consensus polyadenylation signal ATTAAA, but only in FSHD permissive haplotypes. All non-permissive haplotypes, however, either possess a non-functional sequence, ATCAAA, or the sequence is absent due to deletion of the pLAM region (Lemmers et al. 2010b) (Fig. 2). This polyadenylation signal had been previously functionally mapped and found to be involved in the expression of stable DUX4 mRNAs in FSHD primary myoblasts (Dixit et al. 2007). The DUX4 transcript was found to initiate from a transcription start site located in the most distal D4Z4 unit but unexpectedly terminated in the flanking pLAM region, which provided both an intronic sequence and a functional polyadenylation signal (Dixit et al. 2007). Confirmation that this poly-A signal was required to stabilise the pathogenic DUX4 transcript came from cell transfection studies which introduced the T version of this C/T SNP into several non-permissive alleles and resulted in transcription of a stable DUX4 mRNA, while its removal from permissive alleles resulted in the failure of DUX4 detection (Lemmers et al. 2010b). These studies give a clear indication that the complete DUX4 mRNA, and/or the encoded DUX4 protein, directly influences FSHD pathogenesis. Further such evidence came from an FSHD family study in which disease was associated with small 4/10 hybrid D4Z4 alleles (Lemmers et al. 2010b). In one family, the disease-associated D4Z4 allele was found on chromosome 10q26 where a normally non pathological short repeat array had been extended by the translocation of a 4q35 fragment comprising a part of the distal D4Z4 unit with the adjacent pLAM region and its poly-A signal, thus allowing stable DUX4 mRNA expression (Fig. 2). As only a small part of the 4q subtelomeric region was transferred to generate this pathogenic 10q26 array, without direct association with most FSHD candidate genes (FRG1, SLC25A4 (ANT1) and DUX4c), these various genes are clearly not involved in the initiation of FSHD pathogenesis. A study in another FSHD family in which disease was associated with an unusually large deletion extending from the proximal end of the D4Z4 repeat array, deleting both the DUX4c and FRG2 genes, indicated that neither gene is causative of FSHD (Deak et al. 2007). These various findings reinforce the idea that FSHD expression is directly linked to the DUX4 gene with the distal 4q35-D4Z4 repeat unit being juxtaposed to a functional polyadenylation signal in the pLAM sequence, providing compelling evidence that DUX4 is indeed the FSHD gene.
The question remains as to why only certain 4q haplotypes are associated with the T SNP that creates this functional poly-A signal? A possible explanation comes from an evolutionary study of the 4q35 region, which indicates that the present 4q and 10q sequences originated from an initial 4q-located haplotype (Lemmers et al. 2010a). An evolutionary network analysis found that this ancestral D4Z4 region, now found only in chimps, diverged to give the 4A168 and 4A159 haplotypes, with 4A159 then evolving to form 4A161. Interestingly, as these represent the only three FSHD permissive haplotypes, ancestral chimps must have also carried a permissive haplotype, which, having diverged into the 4A168/159/161 haplotypes, subsequently underwent rearrangement deleting the pLAM region and/or its polyadenylation sequence (Lemmers et al. 2010a). This confirms that the terminal 4q35-D4Z4 unit, the enclosed DUX4 gene, and the adjacent pLAM region with a functional poly-A site are essential for the development of FSHD by allowing expression of a stable DUX4 mRNA.
FSHD2 (referred in OMIM as FSHD type 1B)
An intriguing puzzle is the observation that some 5% of the patients exhibit the full clinical spectrum of FSHD without either a contracted 4q35 or 10q26 D4Z4 repeat array (Gilbert et al. 1993). Whilst the apparently different biological mechanism that underlies this form of the disease named FSHD2 is not yet understood, it does share several similarities with FSHD1 (OMIM: FSHD type 1A). This type of disease also only occurs in individuals with at least one permissive 4q haplotype and who exhibit some of the 4q35 epigenetic changes involved in FSHD1 pathogenesis, although in FSHD2 hypomethylation of both the 4q35 and 10q26 D4Z4 repeat arrays occurs (de Greef et al. 2007). Furthermore, DUX4 transcripts that utilise the pLAM-associated poly-A signal have also been identified in FSHD2 muscle cells (Lemmers et al. 2010b).
Epigenetic mechanisms involved in FSHD
The direct association between the 4q35-D4Z4 array and FSHD, led to the search for potential causative genes across the 4qter region and the subsequent identification of at least three possible genes, including the two FSHD region genes, FRG1 and FRG2, and the more centromeric SLC25A4 (ANT1) gene. Initial studies using small sample numbers indicated that these three genes were up-regulated in FSHD muscle in a distance-dependant manner, with FRG2, the gene most proximal to D4Z4, exhibiting the largest increase in expression (Gabellini et al. 2002). It was suggested that normal large-sized 4q35-D4Z4 arrays, with at least 11 repeats, are usually completely heterochromatic resulting in the repression of any associated genes in normal individuals. Meanwhile D4Z4 arrays that contract below the 11-repeat threshold would exhibit a more ‘open’ euchromatic-like state, resulting in the derepression of any associated genes (Hewitt et al. 1994; Winokur et al. 1994). This is supported by the presence of an FSHD-related nuclear matrix attachment site (FR-MAR), which was also mapped in 4q35 and creates two chromatin loops, one containing the D4Z4 array and a second, more proximal loop, encompassing the FRG1, FRG2 and DUX4c genes (Petrov et al. 2006). A potent transcriptional enhancer was also recognised at the 5′-end of the D4Z4 array that is effectively blocked by the FR-MAR in both normal human myoblasts and other non-muscle cells. The FR-MAR was found to be weakened in FSHD muscle cells, thus allowing the D4Z4 array and the 4q35 genes to be contained within a single chromatin loop. This may then permit the D4Z4-associated enhancer to up-regulate transcription of several genes in the vicinity (Petrov et al. 2008).
Much controversy is found in the literature about which genes (or gene) are the causative agents in FSHD. While recent studies indicate a direct link between FSHD and the distal DUX4 ORF (Dixit et al. 2007; Lemmers et al. 2010b), many other studies have reported FSHD-associated changes in expression of various 4q35-located genes. Consistent with a putative role in FSHD, several functional studies of the FRG1 protein have shown it to have a role in both angiogenesis and muscle development by utilising quite different biochemical functions, i.e. alternative RNA splicing and actin bundling (Gabellini et al. 2006; van Koningsbruggen et al. 2007; Hanel et al. 2009; Wuebbles et al. 2009; Liu et al. 2010; Sun et al. 2011). Furthermore, a recent analysis of myoblasts isolated from affected muscle of a transgenic mouse overexpressing FRG1 reported a significant loss of cell proliferation and an increased doubling time not observed in unaffected muscle (Chen et al. 2011). However, no disturbed proliferation rate was reported for FSHD versus control myoblasts (Barro et al. 2010) and while initial reports suggested FRG1 expression levels were elevated in patient muscle cells (Gabellini et al. 2002), later studies have found no alteration in FRG1 expression (Klooster et al. 2009; Arashiro et al. 2009; Masny et al. 2010). There have also been suggestions that the DUX4c gene (Fig. 1), located within the inverted partial D4Z4 unit (D4S2463) 42-kb proximal of the D4Z4 repeat array, which is up-regulated in FSHD muscle cells and probably involved in myoblast proliferation, could contribute to FSHD pathogenesis (Ansseau et al. 2009; Bosnakovski et al. 2008a). However, as mentioned above two unusual FSHD genetic profiles have ruled out a causative role for DUX4c in the disease. These are a 75-kb deletion that removed the FRG2 and DUX4c genes on the 4q35 pathogenic allele (Deak et al. 2007), and the 4q35/10q26 translocation in which the 10q26 pathogenic allele carries none of the genes proximal to the 4q35 D4Z4 array (Lemmers et al. 2010b). Again these studies are pointing towards the D4Z4-associated DUX4 gene being ‘the FSHD gene’.
Each repeat unit in the 4q35 and 10q26 D4Z4 arrays contains GC-rich hhspm3 and LSau repetitive sequences that are predominantly associated with heterochromatin (Hewitt et al. 1994). Such sequences are usually heavily methylated and indeed, all normal sized D4Z4 repeat arrays are hypermethylated, while contracted D4Z4 arrays exhibit variable levels of hypomethylation. Furthermore, DNA hypermethylation is associated with the recruitment of various methylated DNA binding proteins, as well as histone-modifying deacetylases and methyltransferases, all of which are involved in chromatin condensation (Ballestar and Wolffe 2001). Thus, hypomethylation of the contracted 4q D4Z4 alleles in FSHD1, and the hypomethylation of both the 4q35 and 10q26 non-contracted D4Z4 alleles in FSHD2, indicates the significance of these epigenetic changes which induce a more euchromatic-like D4Z4 array that is directly associated with FSHD expression (de Greef et al. 2007). Although it remains a puzzle as to why the widespread D4Z4 hypomethylation coupled with the absence of large 4q D4Z4 deletions, results in FSHD2. However, the recent finding of stable DUX4 mRNA expression in FSHD2 patient muscle cells involving the use of the pLAM polyadenylation signal distal of the 4q35 array, does seem to fit with the general pathogenic mechanism proposed for FSHD1 (Lemmers et al. 2010b). This is further supported by the similar clinical presentation of both disorders (de Greef et al. 2010). The variable level of D4Z4-associated hypomethylation observed in FSHD has been found to correlate to repeat size, with 4q D4Z4 arrays containing only 1–3 repeats generally showing pronounced hypomethylation, while 4q D4Z4 arrays with 4–10 repeats exhibit a far greater variation in the level of methylation (van Overveld et al. 2005; Lunt et al. 1995). Hypomethylation of contracted D4Z4 arrays has also been observed in rare asymptomatic individuals carrying permissive haplotypes, perhaps an indication that while D4Z4 hypomethylation is necessary for FSHD, it is not fully responsible for disease onset (van Overveld et al. 2003). However, a limitation of most such DNA methylation studies is that only the proximal or internal D4Z4 repeats were assessed for hypomethylation and hence, it cannot be excluded that the most distal D4Z4 repeat was differently methylated. Indeed, methylation levels of internal D4Z4 repeats are reported to be 20–25% higher than proximal repeats (de Greef et al. 2009).
Histone modifications of the D4Z4 arrays have also been assessed for features associated with heterochromatin. These modifications can affect transcription and chromatin structure in various ways, such as by the inhibition of transcription factor binding and alteration of interactions between modified histones and DNA that often result in chromatin structural changes (Peterson and Laniel 2004). Specific histone modifications, such as dimethylation at lysine 4 on histone 3 (H3K4me2), are usually associated with transcriptional activation, while trimethylation at lysine 9 (H3K9me3) and 27 (H3K27me3) are normally associated with transcriptional repression, due to the induction of a more heterochromatic state (Lachner et al. 2003). D4Z4 arrays were initially reported to present with an unexpressed euchromatin conformation (Jiang et al. 2003; Yang et al. 2004), although both heterochromatic regions, with H3K9 and H3K27 trimethylation, and euchromatic regions, with H3K4 dimethylation, were subsequently recognised (Zeng et al. 2009). Further study, however found significantly decreased levels of H3K9me3 on D4Z4 arrays in both FSHD1 and FSHD2, whilst the levels of H3K27me3 and H3K4me2 were unaltered, indicating a more euchromatic-like state across the D4Z4 region in FSHD (Zeng et al. 2009). However, it should be noted that loss of H3K9me3 affects not only the 4q contracted allele but also the non-contracted 4q alleles and both 10q D4Z4 alleles, whereas DNA hypomethylation is specific to the contracted 4q D4Z4 allele, an indication perhaps that DNA methylation changes may have a greater influence on disease onset and progression (van Overveld et al. 2003; Zeng et al. 2009).
However, more severe DNA hypomethylation is seen at D4Z4 in another disorder, immunodeficiency, centromere instability and facial anomalies (ICF) syndrome (van Overveld et al. 2003; Kondo et al. 2000), which shows no similarity in clinical presentation to FSHD (de Greef et al. 2007). Meanwhile, H3K9me3 has been found to be preserved at normal levels at D4Z4 in ICF cells (Zeng et al. 2009). As such, it is unclear what relevance DNA hypomethylation and histone methylation at D4Z4 has in FSHD due to their non-specificity to the disease, as D4Z4 hypomethylation is found in both FSHD and ICF, and to the disease chromosome, with a decrease in H3K9me3 identified on both chromosomes 4q and 10q in FSHD, respectively.
A binding of regulatory proteins within D4Z4 has been suggested as a possible contributing factor in FSHD pathogenesis. A multi-protein repressor complex that binds to a site in the D4Z4 repeat has been identified which consists of YY1, a transcriptional activator or repressor according to the promoter context, HMGB2, an architectural protein, and nucleolin. Their depletion resulted in the transcriptional up-regulation of FRG2 (Gabellini et al. 2002). Contracted 4q D4Z4 arrays were postulated to have far fewer binding sites for this repressive complex which might explain both the ‘disease threshold effect’ attributed to D4Z4 arrays with <11 repeats, and the observed transcriptional up-regulation of several 4q35 genes (Gabellini et al. 2002).
Other experiments suggest that the D4Z4 array possesses an insulator element that has both enhancer-blocking and barrier activities and which may be involved in displacing the 4q telomere towards the nuclear periphery (Ottaviani et al. 2009). This positioning activity lies within a short sequence that interacts with the CCCTC-binding factor (CTCF), a protein involved in long-range chromosomal interactions, and with A-type lamins which are integral proteins of the nuclear lamina. CTCF binding is lost in healthy individuals due to D4Z4 multimerization in myoblasts, which is in keeping with its known lack of affinity for methylated DNA present in normal sized D4Z4 arrays (Ottaviani et al. 2010). Conversely, in reporter gene constructs containing just a single D4Z4 unit, CTCF is able to bind to D4Z4 and activate an insulator function which may protect genes in the FSHD region from repressive structures (Ottaviani et al. 2009). Whether such CTCF binding also occurs on the long hypomethylated D4Z4 arrays in FSHD2 myoblasts is, however, unknown.
The overall role of such epigenetic mechanisms in FSHD is still unclear, as is whether D4Z4 methylation changes and histone modifications represent specific causative disease factors, or are simply a secondary response to the disease. Although, the likelihood is that such epigenetic changes are indeed disease-related and somehow influence the functionality of the 4qter region, most probably by up-regulation of its associated genes (Figs. 1, 3). The precise chromatin structure and DNA methylation of the most distal D4Z4 unit, however, is yet to be determined. In fact, it has been shown that during muscle development and adult myogenesis different epigenetic components can co-operate to establish short open chromatin domains with active gene transcription, which is surrounded by large regions of restrictive chromatin which prevent gene expression (Saccone and Puri 2010).

Epigenetic modifications of D4Z4 arrays have a major influence on chromatin status and the 4q35 transcriptional profile. a Normal DNA methylation (red circles) and H3K9 trimethylation (green triangles) lead to a ‘closed’ chromatin configuration previously thought to result in transcriptional inhibition. A contraction of the D4Z4 array results in both decreased DNA methylation and reduced H3K9 trimethylation which induce a more ‘open’ chromatin structure and transcription of DUX4. Non-permissive haplotypes lack any functional polyadenylation signal resulting in the degradation of any DUX4 mRNA (b), whereas an efficiently polyadenylated transcript, derived from a permissive chromosomal haplotype, produces a protein with 2 homeodomains (HD1/2), and a long C-terminal domain, that perturbs normal muscle biology, along with the transcript (c). d A more recent model suggests that normal sized D4Z4 arrays contain domains exhibiting both hetero- and euchromatic-like features that produce a shorter transcript (DUX4-s) by utilising a cryptic splice site. While contracted D4Z4 arrays are hypomethylated and exhibit a more ‘open’ chromatin configuration which permits transcription of a full length mRNA (DUX4-fl)
DUX4: the gene, transcripts and protein
Initial sequence analysis of the D4Z4 repeats defined a large open reading frame (ORF) coding for a double homeodomain-containing protein but which lacked a promoter (Hewitt et al. 1994). A functional promoter was subsequently recognised within this large ORF which defined a potential shorter double homeobox DUX4 gene (Ding et al. 1998; Gabriëls et al. 1999). The homeodomain which allows DNA binding is typically found in transcription factors involved in embryonic development (Gehring 1993) and such a candidate gene for FSHD might have explained the large number of genes already found deregulated in patient muscles at the time (Tupler et al. 1999). However, the lack of any introns or discernible polyadenylation site within the D4Z4 repeat, coupled with the complete failure to detect any specific DUX4 transcripts, suggested that it was likely to be a non-functional retro-transposed pseudogene (Hewitt et al. 1994; Lyle et al. 1995; Winokur et al. 2003a; Osborne et al. 2007; Bickmore and van der Maarel 2003; Yip and Picketts 2003; Alexiadis et al. 2007).
Specific DUX4 mRNA detection was a major technical challenge because of its very low abundance, high GC content and similarity to transcripts of hundreds homologous DUX genes dispersed in the human genome (Beckers et al. 2001). When it could finally be detected by RT- PCR in FSHD myoblasts, the DUX4 mRNA was found to derive from the distal D4Z4 unit and extend to the flanking pLAM sequence which provided both an intron and a polyadenylation signal (Kowaljow et al. 2007, Dixit et al. 2007). In the latter publication, a table was provided as supporting information with the detailed optimized RT-PCR conditions compared to the ones that had previously failed, thus helping other researchers to confirm that the DUX4 gene was effectively transcribed (Snider et al. 2009; Lemmers et al. 2010b). One additional difficulty in detecting DUX4 expression became clear as an elegant serial dilution experiment indicated that only 1/1,000 FSHD myoblasts express DUX4 in cell culture (Snider et al. 2010).
DUX4 mRNA appears to be induced upon myoblast differentiation since it was detected in all FSHD myotube cultures tested, but only in some proliferating myoblast cultures (Kowaljow et al. 2007; Dixit et al. 2007; Snider et al. 2009; 2010). However, the DUX4 mRNA could not always be detected in FSHD muscle biopsies (Snider et al. 2010), again suggesting a very low expression level. An explanation could be that DUX4 is preferentially expressed upon regeneration, in activated satellite cells and their progeny which correspond to the myoblast culture model, but only represent a very low fraction of a total muscle extract. It might, thus, be much more difficult to detect DUX4 in adult FSHD muscle biopsy than in foetal or strongly regenerating (pathological) muscles. The trigger leading to DUX4 expression in some of the activated satellite cells and their progeny most probably relates to the disruption of nucleosomes that takes place during DNA replication and might define a chromatin structure that allows DUX4 transcription in some FSHD cells.
A further level of complexity was suggested by a report that the D4Z4 repeat array exhibits bi-directional transcription which creates sense and anti-sense DUX4 transcripts, as well as several other smaller RNAs (Snider et al. 2009). It was suggested that such transcripts might be involved in recruiting other factors, such as the Heterochromatin Protein 1 (HP1), which might help preserve the heterochromatic nature of the D4Z4 repeat array (Snider et al. 2009). Interestingly, bi-directional transcription was also observed for the mouse DUX paralogue repeat array (Clapp et al. 2007). Besides these different splice forms of the DUX4 transcript, a shorter DUX4 RNA form was reported which utilises a cryptic splice site in the first exon and the poly-A site in pLAM. This might lead to the synthesis of a putative truncated DUX4 protein lacking the C-terminal region similar to DUX4c (Fig. 3) (Snider et al. 2010). The transcript encompassing the complete DUX4 ORF was named DUX4-fl and the shorter one DUX4-s. Several groups could detect long DUX4 RNAs but only a single report mentioned the short one which is yet to be confirmed (Ansseau et al. 2009).
The DUX4-fl transcripts are able to produce a DUX4 protein of about 55 kDa. This was confirmed by immunodetection on a western blot from human testis extracts (Snider et al. 2010). However, detection of the DUX4 protein in FSHD muscle cells again proved controversial. Following the development of a specific monoclonal antibody and optimized immunodetection on western blot, Dixit et al. (2007, supporting information) identified the protein in FSHD but not control myoblast extracts. With new antibodies a nuclear DUX4 protein was stained by immunofluorescence in an estimated 1/1,000 myoblasts (Snider et al. 2010), thus in agreement with the RNA data. Additional issues currently include DUX4 turn over and toxicity. DUX4 appears to be unstable as treatment of myoblast cultures with a proteasome inhibitor (MG-132) strongly improved its detection (Dixit et al. 2007; A Tassin, Unpublished data). Moreover, DUX4 mediated cell death appears more frequent in myoblasts than in myotubes, thus restricting DUX4 detection to few surviving myoblasts while more DUX4 positive nuclei can be found after differentiation to myotubes (Bosnakovski et al. 2008b). No specific antibody allowing immunohistochemical detection of DUX4 in muscle sections has been published and so investigating whether DUX4 expression is restricted to satellite cells or regenerating fibres is not yet possible.
Much current FSHD research is focused on elucidation of the biological functions of the DUX4 transcripts, its encoded protein, and their myopathic potential. Initial transfection studies have identified DUX4 as a critical transcription factor which probably targets multiple genes. For example, it was shown to up-regulate the PITX1 (paired-like homeodomain transcription factor 1) gene by binding to a specific site in its promoter (Dixit et al. 2007). However, PITX1 is itself a transcription factor that controls hind limb development, and is also associated with regulating right/left symmetry, which is often affected in FSHD. Additionally, over-expression of DUX4 results in significant cell toxicity in cultures with DUX4 protein localizing to the nucleus where it is involved in emerin relocalization and caspase 3 and/or 7 induction, resulting in increased cell death (Kowaljow et al. 2007). Indeed, of all the proposed FSHD candidate genes, only DUX4 displays overt cell toxicity and induces an FSHD-like pathology in C2C12 myoblasts, increased apoptosis and a reduction in MyoD expression levels and its target genes, resulting in muscle differentiation defects (Bosnakovski et al. 2008b). Interestingly, this DUX4 myopathogenecity could be relieved by increasing PAX3 and PAX7 expression, two homologous genes which encode similar paired class homeoproteins with major transcriptional roles in myogenesis and muscle regeneration (Buckingham et al. 2003; Bosnakovski et al. 2008b). It is possible that DUX4 is a potential competitor of PAX3 and PAX7 and its toxicity involves interference with PAX3/7-regulated genes, such as the viability-associated genes and the myogenic regulatory factors, MyoD and MYF5, thereby preventing normal cell signalling. Many genes were also found to be deregulated 4 h after induction of DUX4 expression in C2C12 cells, suggesting there may be multiple direct DUX4 target genes (Bosnakovski et al. 2008b). It is possible that some of these deregulated genes encode other transcription factors such as PITX1, thus affecting their target genes in turn, leading to a large deregulation cascade (Dixit et al. 2007).
While no full DUX4 transgenic mouse has yet been published, animal models with local injection of a DUX4 expression vector have proved useful in defining its myotoxicity, with DUX4 over-expression in both zebrafish and mice inducing abnormal muscle histology and degeneration (Snider et al. 2009; Wuebbles et al. 2010; Wallace et al. 2010). This DUX4 mediated myotoxicity can, however, be suppressed either by the introduction of mutations within the homeodomain regions, or when the DUX4 protein is over-expressed in the muscle of Tp53 knock-out mice (Wallace et al. 2010). This latter experiment further defined a role for p53 in DUX4 toxicity, confirming earlier studies in which p53 pathway activation was noted in FSHD muscle cells (Winokur et al. 2003b; Laoudj-Chenivesse et al. 2005; Sandri et al. 2001). It is possible that the Tp53 gene may be activated in the DUX4-induced cascade as it is a direct target of PITX1 in cancer cells (Liu and Lobie 2007) which was recently confirmed in human primary myoblasts (C Vanderplanck, Unpublished data). Besides its role in cell cycle and apoptosis control, p53 is also emerging as a critical regulator of both metabolic homeostasis and muscle atrophy (Maddocks and Vousden 2011; Schwarzkopf et al. 2008; Dirks-Naylor and Lennon-Edwards 2011). This evidence, therefore, defines DUX4 as an important transcription factor with a central role in signalling pathways and which controls myogenesis, induces apoptosis, and probably regulates many other cellular processes (Belayew 2010).
Furthermore, transfection studies have shown that expression of the full size DUX4 protein, but not the DUX4c protein, which has a shorter C-terminal region, in a manner similar to DUX4-s, resulted in nuclear foci formation and increased cell death, demonstrating that toxicity is associated with the presence of the full C-terminal region of the protein, therefore highlighting the importance of the splicing event (Snider et al. 2009; Ansseau et al. 2009; Bosnakovski et al. 2008a). However, DUX4 toxicity appeared much milder when its expression was induced in C2C12 cells after their switch to differentiation medium or in myotubes (Bosnakovski et al. 2008a).
It is unclear at what stage (proliferation, confluence, differentiation) the endogenous DUX4 gene is expressed in FSHD muscle cells and at what level. The published RNA and protein data suggest that DUX4 is mostly expressed in FSHD myotubes but clearly larger scale studies are required. Detection of a short DUX4 RNA form in control tissues suggests that the DUX4 gene might be expressed normally in muscle (Snider et al. 2010). Since the DUX4 promoter has different consensus binding sites (E-box, YY1, Sp1 and SRF) that are characteristic of muscle genes induced during terminal differentiation such as the dystrophin gene, a search for DUX4 expression at the RNA and/or protein level should be performed in a large set of controls as well as FSHD muscle samples. Moreover, the study could be extended to include myotubes formed after a long differentiation period in culture, a condition that was never investigated and which could favour DUX4 expression.
It should be noted that all published studies on DUX4-fl function are based on cell culture transfections with potent expression vectors and nothing is yet known about the role of the endogenous protein. The only data about the function of an expressed endogenous part of DUX4 comes from a study of sarcomas resulting from a chromosome translocation that fused the end of the DUX4 gene to part the CIC gene encoding the DNA binding domain of a transcription factor. The resulting CIC-DUX4 fusion protein could induce about 300-fold the transcription level of CIC-target genes (Kawamura-Saito et al. 2006). This very potent transcriptional activity associated with the DUX4 carboxyl-terminal domain suggests that even low expression of an unstable DUX4 protein might still be sufficient to activate target genes. Indeed, the significance of the DUX4 carboxyl-terminal region was further demonstrated by finding that a putative protein which initiated at an internal translation start site of the DUX4 transcript, and corresponded to just the DUX4 carboxyl-terminal region, was by itself sufficient to suppress myogenesis, by inhibiting MyoD transcription and activating several of its target genes, including myogenin and myosin light chain (Snider et al. 2009). Although, inhibition of myogenesis seems to involve both the N- and C-terminal domains of DUX4 and so may be attributed to either the DUX4-fl or the DUX4-s transcripts (Snider et al. 2009).
The normal DUX4 gene demonstrates complex transcription in certain tissues, notably the human testis. Intriguingly, the heterochromatin associated hhspm3 repeat, which is found in the part of D4Z4 units corresponding to the DUX4 promoter (Hewitt et al. 1994, Gabriëls et al. 1999), had been found to be hypomethylated in testis nearly three decades ago (Zhang et al. 1985) suggesting DUX4 could be expressed in that tissue. In germ line cells DUX4 is transcribed from both 4qA and 4qB arrays, as well as from the 10q D4Z4 arrays, although the 10q DUX4 transcript extends over 4 additional non-coding exons and utilises a polyadenylation site that is situated some 6.5-kb distal of the pLAM region (Snider et al. 2010). Similar DUX4 transcripts are expressed in induced pleuripotent stem (iPS) cells from normal individuals, again demonstrating a likely role for DUX4 in human development. Furthermore, while polyadenylated DUX4 mRNAs are transcribed from both 4q and 10q arrays in germ line cells, following their differentiation into somatic tissue only DUX4 transcripts derived from the 4qA-associated D4Z4 array are able to utilise the poly-A signal in the pLAM region. However, why only germ cells are able to express DUX4 from 4qB and 10q arrays is uncertain, although these germ cells may exhibit epigenetic alterations, such as hypomethylated hhspm3 sequences (Zhang et al. 1985) which may permit production of the full length transcript. This then raises a question relating to the likely pathogenesis of FSHD2, in which somatic DUX4 transcription may be comparable to germ line cells due to the absence of 4q D4Z4 contraction. Might this indicate an abnormal developmental status for the 4q35 region in FSHD2, resulting in a decrease in the normal epigenetic changes during differentiation, the maintenance of an open chromatin conformation and a lack of transcriptional silencing? This could then lead to stable DUX4 mRNA expression through the polyadenylation signal on 4qA permissive haplotypes (Lemmers et al. 2010b). What still has to be determined, however, is whether DUX4 transcription also occurs on both 4qB and 10q arrays in FSHD2, and also whether the DNA methylation patterns and histone modifications differ at 4q35 during the differentiation of FSHD2 cells, as compared to FSHD1 and normal cells.
We are currently unaware just which splice forms are transcribed in different cell types, although the majority of cells with a contracted 4q D4Z4 array appear to produce DUX4-fl, while cells with non-contracted arrays only synthesise DUX4-s transcripts. Alterations in DUX4 splice form also occur during embryonic development, with high DUX4-fl levels found in developing human testis, but only DUX4-s found in a subset of differentiated tissues. Such changes in DUX4 splice form between germ line to somatic cells indicates a probable developmental control, which was assessed using iPS cells developed from normal and FSHD fibroblasts (Snider et al. 2010). It was found that both FSHD and control iPS cells produced the normal DUX4-fl transcript prior to differentiation, possibly indicating a more open chromatin structure (Fig. 3). However, following differentiation into embryoid bodies, normal cells switched exclusively to DUX4-s expression while FSHD cells continued to transcribe DUX4-fl, therefore indicating that the full length transcript, and/or the DUX4 protein, is a key factor in FSHD pathology. Additional evidence for a specific biological role for DUX4 transcripts came from a study that found inhibition of myogenesis by DUX4 was retained even following the introduction of stop codons into the DUX4 ORF which eliminated DUX4 protein production (Snider et al. 2009). If confirmed, this unexpected role of the DUX4 transcript in addition to all the published data about the DUX4 protein suggests that DUX4 involvement in FSHD occurs at both the RNA and protein levels.
Treatment
With our current lack of understanding of the pathological mechanisms involved in FSHD, it is perhaps not surprising that few, if any, rational disease-specific therapies have yet been developed. Indeed, to date, most FSHD treatments involve attempts to physically improve functional impairment, with surgery used to alleviate both scapular fixation and ‘foot drop’ in patients (Tawil 2008). An increasing knowledge of at least some aspects of the pathology involved is, however, leading to more directed FSHD therapies. In contrast to Duchenne muscular dystrophy which results from a loss of function mutation, the change in chromatin structure at the FSHD locus causes a gain of function, and a logical therapeutic approach is thus to employ antisense strategies against specifically activated target genes. In the absence of a DUX4 over-expression mouse model researchers have recently assessed RNA interference against the FRG1 mRNA (Wallace et al. 2011; Bortolanza et al. 2011). These studies examined the potential benefits of using FRG1-specific miRNAs and shRNAs in the myopathy mouse model that over expresses the FRG1 protein. This ‘proof-of-principle’ methodology to specifically ‘knockdown’ the FRG1 mRNA was reported to significantly improve muscle histology, with increased muscle mass, reduced fat deposition and fibrosis, a decline in myofibre degeneration, and an overall improvement of muscle function and strength. Similarly, the potential benefits of using siRNA and antisense oligomers to ‘knockdown’ DUX4 mRNA have also been explored with the reported decrease in DUX4 expression found to normalise several FSHD deregulated genes, (C Vanderplanck, Unpublished data).
A major issue in strategies targeting the DUX4 gene is the reported very low abundance of DUX4 expressing cells in culture (1/1,000 myoblasts) (Snider et al. 2010). This number has not been investigated in muscle tissues where it might be larger since DUX4 expression appears to increase with differentiation (Dixit et al. 2007; Snider et al. 2009). However, such a low number of DUX4 positive cells could still be pathogenic during muscle regeneration in vivo. Indeed, the chromatin remodelling processes occurring in the heterogeneous population of muscle progenitors/satellite cells (Saccone and Puri 2010) could favour DUX4 gene expression in several cell types. With DUX4 being a potent transcription factor it could induce a deregulation cascade that may propagate by the proliferation/expansion of these cells, and by their fusion to existing myofibers. The DUX4 protein expressed from a single recently fused nucleus could spread in the fibre cytoplasm and reach neighbouring nuclei where it could also deregulate gene expression. This process may explain the slow disease progression and large phenotype variability. Thus, targeting DUX4 even in 0.1% of muscle cells would be essential in blocking the spreading of the deregulation cascade from cells newly produced by the regeneration to myofibers.
Conclusion
The genetic and biological events that result in FSHD pathogenesis are complex and while not yet fully understood, recent studies have significantly increased our knowledge of this enigmatic and multifaceted disorder (Fig. 4). While large deletions of the 4q35-D4Z4 repeat array were recognised to be disease-associated in most FSHD patients more than 2 decades ago, a complete understanding of the molecular mechanisms leading to the deregulation of multiple genes in this disease is only beginning to be unravelled.

The pathogenic mechanisms involved in FSHD are complex. D4Z4 contractions on both permissive and non-permissive alleles result in DNA hypomethylation and loss of H3K9me3, suggesting the formation of a more ‘open’ chromatin structure. While transcription of DUX4 can now occur, the absence of a recognisable polyadenylation site on non-permissive backgrounds results in transcript degradation. Permissive alleles are associated with a functional poly-A signal and permit transcription of stableDUX4-s and DUX4-fl mRNAs, with DUX4-fl protein synthesis resulting in pathogenic biological events. In the absence of any contraction, D4Z4 has a closed chromatin configuration resulting in production of DUX4-s which is degraded if produced on a non-permissive background. In FSHD2 patients, hypomethylation and reduced levels of H3K9me3 are present on non-contracted chromosomes, and results in FSHD suggesting they favour the expression of the DUX4-fl transcript
Part of the difficulty is caused by the publication of divergent data by different laboratories most probably caused by low sample numbers and the heterogeneity of individuals and sample/model systems. Individual genetic polymorphisms could occur not only in the 4q35 region where they might affect DUX4 expression but also in any of the many genes shown to be deregulated in FSHD muscles. SNPs have been well studied in 4q35 but large polymorphisms have only started to be investigated by a combination of fluorescent in situ hybridization and DNA combing demonstrating unexpected rearrangements in this subtelomeric region (Nguyen et al. 2011). Another source of heterogeneity is the biopsy: i.e. is the selected muscle always, often or never affected in the pathology, where is the biopsy site (e.g. as compared to tendon location), what were the physiological conditions of the patient when this muscle sample was taken (e.g. diet, fasting status, physical activity, cigarette smoking) since this tissue presents a high plasticity (e.g. changing fibre type according to exercise, or exhibiting a small level of atrophy after a night fasting)? For studies performed with myoblast cultures, if the cells are indeed mostly derived from muscle stem cells, i.e. satellite cells, other cell populations inside the muscle biopsy (i.e. fibroblasts, pericytes, and inflammatory cells) can contribute to the culture in varying degrees unless myoblasts were selected based on their CD56 surface marker. Moreover, satellite cells in the muscle itself are a heterogeneous population with distinct embryological origin and multiple levels of biochemical and functional diversity (Biressi and Rando 2010). This heterogeneity might be altered during cell plating and culture in various media chosen for proliferation or differentiation. Moreover passage number and proximity to senescence, cell density, and differentiation level differently affect muscle cells at the molecular level. An additional factor is the recent development of immortalized FSHD myoblast lines in which addition of an activated CDK4 gene is expected to affect the differentiation process (Stadler et al. 2011).
In FSHD, a first level of gene deregulation might be considered to result from alterations in overall chromosome structures (DNA loops, DNA methylation changes, and chromatin marks at both the DNA and protein level) caused by large contractions of the 4q35 D4Z4 array; resulting in the involvement of additional 4q35 genes, besides FRG2, FRG1, DUX4c, and SLC25A4 (ANT1), and possibly genes on other chromosomes that localize to the same peripheral nuclear domain. A second level of gene deregulation results from the production of a complete and stable DUX4 transcript from permissive alleles containing a functional pLAM-associated polyadenylation site, either from small 4q35-D4Z4 arrays in FSHD1, or from normal sized hypomethylated D4Z4 arrays in FSHD2. The resulting DUX4 protein is a transcription factor that may regulate the expression of multiple genes, several of which encode other transcription factors, such as MyoD, which is inhibited, and PITX1 that is activated. These in turn target many other genes, some of which encode other transcription factors, for example, myogenin, a MyoD target, or TP53 which is activated by PITX1, resulting in a large deregulation cascade (Fig. 5). Given that many sequence variants might be expected in such a large cohort of genes in the normal population, this may partially explain the clinical heterogeneity often observed in FSHD patients.

A transcription dysregulation cascade in FSHD. The DUX4 gene mapped in the D4Z4 repeated element at 4q35 is activated either by the pathogenic deletion that contracts the repeat array, or by another uncharacterized mutation that leads to chromatin opening of normal sized repeat arrays. The chromatin changes allow transcription of the DUX4 gene. On permissive alleles that carry the poly-A signal in the pLAM region this results in a stable mRNA that can be translated. The expressed DUX4 protein is a transcription factor that may directly or indirectly interact with a set of target genes. Among those, DUX4 expression results in the inhibition of the MyoD gene which encodes the transcription master switch of muscle differentiation thus causing inhibition of the MyoD target genes in FSHD. DUX4 over-expression also inhibits the expression of genes involved in response to oxidative stress, and probably inducing the μcrystallin (CRYM) gene whose promoter carries a DUX4 binding site. A direct DUX4 target gene is PITX1 at 5q31 which encodes a transcription factor that is the master switch for hindlimb development in embryogenesis. PITX1 is specifically induced in FSHD muscles as compared to 11 neuromuscular disorders; it induces E3 ubiquitin ligase which is linked to atrophy in adult skeletal muscles and is involved in inflammation. Among the PITX1 target genes is TP53 which has major roles in the control of DNA repair, cell cycling and apoptosis as well as in multiple levels of cell metabolism and muscle atrophy. Scheme based on data from Bosnakovski et al. 2008b; Dixit et al. 2007; Liu and Lobie 2007; Reed et al. 2007; C Vanderplanck, Unpublished data; Maddocks and Vousden 2011; Schwarzkopf et al. 2008; Dirks-Naylor and Lennon-Edwards 2011
Questions still remain as to the physiological function of DUX4 which has yet to be identified. Although only ~75% of individuals possessing 4qA permissive chromosomes are able to produce a DUX4-fl or DUX4-s transcript using the poly-A addition signal in exon 3, a DUX4-fl mRNA could also derive from non-permissive 4qA, 4qB and 10q alleles. Detection of these transcripts in germline and iPS cells strongly suggests a role for either the RNA or protein in early embryogenesis (Snider et al. 2010). Meanwhile, it may be possible that the DUX4-s transcript, which is produced in the majority of cells possessing no 4q D4Z4 contraction, is redundant and has little or no function. Another puzzle relates to the putative role of the various DUX4-related transcripts, and other associated RNA sequences in the pathogenesis of FSHD. Are some of these RNA species involved in chromatin structure alterations at 4q35 or elsewhere? Furthermore, why is synthesis of these DUX4 transcripts apparently confined to just a few myonuclei in FSHD muscles, do these rare cells exhibit a critical difference in their ability to alter chromatin loops or process DUX4 transcripts? And how does FSHD develop, if only such a small proportion of muscle cells are likely to be affected by abnormal myogenesis and increased cell death? A ‘spreading’ model has been proposed in which DUX4 gene activation in just a few myonuclei leads to a diffusion of these expressed mRNAs, and their translated proteins, throughout the cytoplasm of the myofibre, with the subsequent importation of these proteins, via nuclear location signals, into multiple nuclei throughout the myofibre (A Tassin, Unpublished data).
From the evidence presented in this review, we can look forward with confidence that future FSHD research will provide answers to these many questions, and, in doing so, provide further insight into this elusive and enigmatic disease. The better understanding of the pathogenic mechanisms underlying both FSHD1 and FSHD2 will then hopefully allow us to develop the specifically targeted therapies needed by our many long-suffering FSHD patients.
References
Alexiadis V, Ballestas ME, Sanchez C, Winokur S, Vedanarayanan V, Warren M, Ehrlich M (2007) RNAPol-ChIP analysis of transcription from FSHD-linked tandem repeats and satellite DNA. Biochim Biophys Acta 1769:29–40
Ansseau E, Laoudj-Chenivesse D, Marcowycz A, Tassin A, Vanderplanck C, Sauvage S, Barro M, Mahieu I, Leroy A, Leclercq I, Mainfroid V, Figlewicz D, Mouly V, Butler-Browne G, Belayew A, Coppée F (2009) DUX4c is up-regulated in FSHD. It induces the MYF5 protein and human myoblast proliferation. PLoS One 4:e7482
Arashiro P, Eisenberg I, Kho AT, Cerqueira AM, Canovas M, Silva HC, Pavanello RC, Verjovski-Almeida S, Kunkel LM, Zatz M (2009) Transcriptional regulation differs in affected facioscapulohumeral muscular dystrophy patients compared to asymptomatic related carriers. Proc Natl Acad Sci USA 106:6220–6225
Ballestar E, Wolffe AP (2001) Methyl-CpG-binding proteins: targeting specific gene repression. Eur J Biochem 268:1–6
Barro M, Carnac G, Flavier S, Mercier J, Vassetzky Y, Laoudj-Chenivesse D (2010) Myoblasts from affected and non-affected FSHD muscles exhibit morphological differentiation defects. J Cell Mol Med 14:275–289
Beckers M, Gabriëls J, van der Maarel S, De Vriese A, Frants RR, Collen D, Belayew A (2001) Active genes in junk DNA? Characterization of DUX genes embedded within 3.3 kb repeated elements. Gene 264:51–57
Belayew A (2010) Cascade of gene activation in Landouzy Dejerine muscular dystrophy. Bull Mem Acad R Med Belg 165:87–97
Bickmore WA, van der Maarel SM (2003) Perturbations of chromatin structure in human genetic disease: recent advances. Hum Mol Genet 12:R207–R213
Biressi S, Rando TA (2010) Heterogeneity in the muscle satellite cell population. Semin Cell Dev Biol 21:845–854
Bortolanza S, Nonis A, Sanvito F, Maciotta S, Sitia G, Wei J, Torrente Y, Di Serio C, Chamberlain JR, Gabellini D (2011) AAV6-mediated systemic shRNA delivery reverses disease in a mouse model of facioscapulohumeral muscular dystrophy. Mol Ther [Epub ahead of print]. doi:10.1038/mt.2011.153
Bosnakovski D, Lamb S, Simsek T, Xu Z, Belayew A, Perlingeiro R, Kyba M (2008a) DUX4c, an FSHD candidate gene, interferes with myogenic regulators and abolishes myoblast differentiation. Exp Neurol 214:87–96
Bosnakovski D, Xu Z, Gang EJ, Galindo CL, Liu M, Simsek T, Garner HR, Agha-Mohammadi S, Tassin A, Coppée F, Belayew A, Perlingeiro RR, Kyba M (2008b) An isogenetic myoblast expression screen identifies DUX4-mediated FSHD-associated molecular pathologies. EMBO J 27:2766–2779
Buckingham M, Bajard L, Chang T, Daubas P, Hadchouel J, Meilhac S, Montarras D, Rocancourt D, Relaix F (2003) The formation of skeletal muscle: from somite to limb. J Anat 202:59–68
Chen SC, Frett E, Marx J, Bosnakovski D, Reed X, Kyba M, Kennedy BK (2011) Decreased proliferation kinetics of mouse myoblasts overexpressing FRG1. PLoS One 6:e19780
Clapp J, Mitchell LM, Bolland DJ, Fantes J, Corcoran AE, Scotting PJ, Armour JA, Hewitt JE (2007) Evolutionary conservation of a coding function for D4Z4, the tandem DNA repeat mutated in facioscapulohumeral muscular dystrophy. Am J Hum Genet 81:264–279
de Greef JC, Wohlgemuth M, Chan OA, Hansson KB, Smeets D, Frants RR, Weemaes CM, Padberg GW, van der Maarel SM (2007) Hypomethylation is restricted to the D4Z4 repeat array in phenotypic FSHD. Neurology 69:1018–1026
de Greef JC, Lemmers RJ, van Engelen BG, Sacconi S, Venance SL, Frants RR, Tawil R, van der Maarel SM (2009) Common epigenetic changes of D4Z4 in contraction-dependent and contraction-independent FSHD. Hum Mutat 30:1449–1459
de Greef JC, Lemmers RJ, Camaño P, Day JW, Sacconi S, Dunand M, van Engelen BG, Kiuru-Enari S, Padberg GW, Rosa AL, Desnuelle C, Spuler S, Tarnopolsky M, Venance SL, Frants RR, van der Maarel SM, Tawil R (2010) Clinical features of facioscapulohumeral muscular dystrophy 2. Neurology 75:1548–1554
Deak KL, Lemmers RJ, Stajich JM, Klooster R, Tawil R, Frants RR, Speer MC, van der Maarel SM, Gilbert JR (2007) Genotype-phenotype study in an FSHD family with a proximal deletion encompassing p13E−11 and D4Z4. Neurology 68:578–582
Deidda G, Cacurri S, Grisanti P, Vigneti E, Piazzo N, Felicetti L (1995) Physical mapping evidence for a duplicated region on chromosome 10qter showing high homology with the facioscapulohumeral muscular dystrophy locus on chromosome 4qter. Eur J Hum Genet 3:155–167
Ding H, Beckers MC, Plaisance S, Marynen P, Collen D, Belayew A (1998) Characterization of a double homeodomain protein (DUX1) Encoded by a cDNA Homologous to 3.3 Kb dispersed repeated elements. Hum Mol Genet 7:1681–1694
Dirks-Naylor AJ, Lennon-Edwards S (2011) Cellular and molecular mechanisms of apoptosis in age-related muscle atrophy. Curr Aging Sci [Epub ahead of print]
Dixit M, Ansseau E, Tassin A, Winokur S, Shi R, Qian H, Sauvage S, Mattéotti C, van Acker AM, Leo O, Figlewicz D, Barro M, Laoudj-Chenivesse D, Belayew A, Coppée F, Chen YW (2007) DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc Natl Acad Sci USA 104:18157–18162
Gabellini D, Green MR, Tupler R (2002) Inappropriate gene activation in FSHD: a repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell 110:339–348
Gabellini D, D’Antona G, Moggio M, Prelle A, Zecca C, Adami R, Angeletti B, Ciscato P, Pellegrino MA, Bottinelli R, Green MR, Tupler R (2006) Facioscapulohumeral muscular dystrophy in mice overexpressing FRG1. Nature 439:973–977
Gabriëls J, Beckers MC, Ding H, De Vriese A, Plaisance S, van der Maarel SM, Padberg GW, Frants RR, Hewitt JE, Collen D, Belayew A (1999) Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 236:25–32
Gehring WJ (1993) Exploring the homeobox. Gene 135:215–221
Gilbert JR, Stajich JM, Wall S, Carter SC, Qiu H, Vance JM, Stewart CS, Speer MC, Pufky J, Yamaoka LH, Rozear M, Samson F, Fardeau M, Roses AD, Pericak-Vance MA (1993) Evidence for heterogeneity in facioscapulohumeral muscular dystrophy (FSHD). Am J Hum Genet 53:401–408
Hanel ML, Wuebbles RD, Jones PL (2009) Muscular dystrophy candidate gene FRG1 is critical for muscle development. Dev Dyn 238:1502–1512
Hewitt JE, Lyle R, Clark LN, Valleley EM, Wright TJ, Wijmenga C, van Deutekom J, Francis F, Sharpe PT, Hofker M, Frants RR, Williamson R (1994) Analysis of the tandem repeat locus D4Z4 associated with facioscapulohumeral muscular dystrophy. Hum Mol Genet 3:1287–1295
Jiang G, Yang F, van Overveld PG, Vedanarayanan V, van der Maarel S, Ehrlich M (2003) Testing the position-effect variegation hypothesis for facioscapulohumeral muscular dystrophy by analysis of histone modification and gene expression in subtelomeric 4q. Hum Mol Genet 12:2909–2921
Kawamura-Saito M, Yamazaki Y, Kaneko K, Kawaguchi N, Kanda H, Mukai H, Gotoh T, Motoi T, Fukayama M, Aburatani H, Takizawa T, Nakamura T (2006) Fusion between CIC and DUX4 up-regulates PEA3 family genes in Ewing-like sarcomas with t(4;19)(q35;q13) translocation. Hum Mol Genet 15:2125–2137
Klooster R, Straasheijm K, Shah B, Sowden J, Frants R, Thornton C, Tawil R, van der Maarel S (2009) Comprehensive expression analysis of FSHD candidate genes at the mRNA and protein level. Eur J Hum Genet 17:1615–1624
Kondo T, Bobek MP, Kuick R, Lamb B, Zhu X, Narayan A, Bourc’his D, Viegas-Pequignot E, Ehrlich M, Hanash SM (2000) Whole-genome methylation scan in ICF syndrome: hypomethylation of non-satellite DNA repeats D4Z4 and NBL2. Hum Mol Genet 9:597–604
Kowaljow V, Marcowycz A, Ansseau E, Conde CB, Sauvage S, Mattéotti C, Arias C, Corona ED, Nuñez NG, Leo O, Wattiez R, Figlewicz D, Laoudj-Chenivesse D, Belayew A, Coppée F, Rosa AL (2007) The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul Disord 17:611–623
Lachner M, O’Sullivan RJ, Jenuwein T (2003) An epigenetic road map for histone lysine methylation. J Cell Sci 116:2117–2124
Laforêt P, de Toma C, Eymard B, Becane HM, Jeanpierre M, Fardeau M, Duboc D (1998) Cardiac involvement in genetically confirmed facioscapulohumeral muscular dystrophy. Neurology 51:1454–1456
Laoudj-Chenivesse D, Carnac G, Bisbal C, Hugon G, Bouillot S, Desnuelle C, Vassetzky Y, Fernandez A (2005) Increased levels of adenine nucleotide translocator 1 protein and response to oxidative stress are early events in facioscapulohumeral muscular dystrophy muscle. J Mol Med 83:216–224
Lemmers RJL, de Kievit P, van Geel M, van der Wielen MJ, Bakker E, Padberg GW, Frants RR, van der Maarel SM (2001) Complete allele information in the diagnosis of facioscapulohumeral muscular dystrophy by triple DNA analysis. Ann Neurol 50:816–819
Lemmers RJ, de Kievit P, Sandkuijl L, Padberg GW, van Ommen GJ, Frants RR, van der Maarel SM (2002) Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat Genet 32:235–236
Lemmers RJ, Van Overveld PG, Sandkuijl LA, Vrieling H, Padberg GW, Frants RR, van der Maarel SM (2004) Mechanism and timing of mitotic rearrangements in the subtelomeric D4Z4 repeat involved in facioscapulohumeral muscular dystrophy. Am J Hum Genet 75:44–53
Lemmers RJ, van der Vliet PJ, van der Gaag KJ, Zuniga S, Frants RR, de Knijff P, van der Maarel SM (2010a) Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete interchromosomal sequence transfers in human evolution. Am J Hum Genet 86:364–377
Lemmers RJ, van der Vliet PJ, Klooster R, Sacconi S, Camaño P, Dauwerse JG, Snider L, Straasheijm KR, van Ommen GJ, Padberg GW, Miller DG, Tapscott SJ, Tawil R, Frants RR, van der Maarel SM (2010b) A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 329:1650–1653
Liu DX, Lobie PE (2007) Transcriptional activation of p53 by Pitx1. Cell Death Differ 14:1893–1907
Liu Q, Jones TI, Tang VW, Brieher WM, Jones PL (2010) Facioscapulohumeral muscular dystrophy region gene-1 (FRG-1) is an actin-bundling protein associated with muscle-attachment sites. J Cell Sci 123:1116–1123
Lunt PW (1998) 44th ENMC international workshop: facioscapulohumeral muscular dystrophy: molecular studies 19–21 July 1996, Naarden, The Netherlands. Neuromuscul Disord 8:126–30
Lunt PW, Jardine PE, Koch MC, Maynard J, Osborn M, Williams M, Harper PS, Upadhyaya M (1995) Correlation between fragment size at D4F104S1 and age at onset or at wheelchair use, with a possible generational effect, accounts for much phenotypic variation in 4q35-facioscapulohumeral muscular dystrophy (FSHD). Hum Mol Genet 4:951–958
Lyle R, Wright TJ, Clark LN, Hewitt JE (1995) The FSHD-associated repeat, D4Z4, is a member of a dispersed family of homeobox-containing repeats, subsets of which are clustered on the short arms of the acrocentric chromosomes. Genomics 28:389–397
Maddocks OD, Vousden KH (2011) Erratum to: Metabolic regulation by p53. J Mol Med 89:531
Masny PS, Chan OY, de Greef JC, Bengtsson U, Ehrlich M, Tawil R, Lock LF, Hewitt JE, Stocksdale J, Martin JH, van der Maarel SM, Winokur ST (2010) Analysis of allele-specific RNA transcription in FSHD by RNA-DNA FISH in single myonuclei. Eur J Hum Genet 18:448–456
Matsumura T, Goto K, Yamanaka G, Lee JH, Zhang C, Hayashi YK, Arahata K (2002) Chromosome 4q:10q translocations; comparison with different ethnic populations and FSHD patients. BMC Neurol 2:7
Nguyen K, Walrafen P, Bernard R, Attarian S, Chaix C, Vovan C, Renard E, Dufrane N, Pouget J, Vannier A, Bensimon A, Lévy N (2011) Molecular combing reveals allelic combinations in facioscapulohumeral dystrophy. Ann Neurolog. Accepted manuscript online: 7 JUN 2011 01:19PM EST | (in press). doi:10.1002/ana.22513
Osborne RJ, Welle S, Venance SL, Thornton CA, Tawil R (2007) Expression profile of FSHD supports a link between retinal vasculopathy and muscular dystrophy. Neurology 68:569–577
Ottaviani A, Rival-Gervier S, Boussouar A, Foerster AM, Rondier D, Sacconi S, Desnuelle C, Gilson E, Magdinier F (2009) The D4Z4 macrosatellite repeat acts as a CTCF and A-type lamins-dependent insulator in facio-scapulo-humeral dystrophy. PLoS Genet 5:e1000394
Ottaviani A, Schluth-Bolard C, Gilson E, Magdinier F (2010) D4Z4 as a prototype of CTCF and lamins-dependent insulator in human cells. Nucleus 1:30–36
Padberg GW (2004) Facioscapulohumeral muscular dystrophy: a clinician’s experience. In: Upadhyaya M, Cooper DN (eds) Facioscapulohumeral muscular dystrophy: clinical medicine and molecular cell biology. Garland Science/BIOS Scientific, Abingdon, pp 41–54
Padberg GW, Brouwer OF, de Keizer RJ, Dijkman G, Wijmenga C, Grote JJ, Frants RR (1995) On the significance of retinal vascular disease and hearing loss in facioscapulohumeral muscular dystrophy. Muscle Nerve 2:S73–S80
Peterson CL, Laniel MA (2004) Histones and histone modifications. Curr Biol 14:R546–R551
Petrov A, Pirozhkova I, Carnac G, Laoudj D, Lipinski M, Vassetzky YS (2006) Chromatin loop domain organization within the 4q35 locus in facioscapulohumeral dystrophy patients versus normal human myoblasts. Proc Natl Acad Sci 103:6982–6987
Petrov A, Allinne J, Pirozhkova I, Laoudj D, Lipinski M, Vassetzky YS (2008) A nuclear matrix attachment site in the 4q35 locus has an enhancer-blocking activity in vivo: implications for facioscapulohumeral dystrophy. Genome Res 18:39–45
Reed PW, Corse AM, Porter NC, Flanigan KM, Bloch RJ (2007) Abnormal expression of mu-crystallin in facioscapulohumeral muscular dystrophy. Exp Neurol 205:583–586
Saccone V, Puri PL (2010) Epigenetic regulation of skeletal myogenesis. Organogenesis 6:48–53
Saito Y, Miyashita S, Yokoyama A, Komaki H, Seki A, Maegaki Y, Ohno K (2007) Facioscapulohumeral muscular dystrophy with severe mental retardation and epilepsy. Brain Dev 29:231–233
Sandri M, El Meslemani AH, Sandri C, Schjerling P, Vissing K, Andersen JL, Rossini K, Carraro U, Angelini C (2001) Caspase 3 expression correlates with skeletal muscle apoptosis in Duchenne and facioscapulo-humeral muscular dystrophy. A potential target for pharmacological treatment? J Neuropathol Exp Neurol 60:302–312
Schwarzkopf M, Coletti D, Marazzi G, Sassoon D (2008) Chronic p53 activity leads to skeletal muscle atrophy and muscle stem cell perturbation. Basic Appl Myol 18:131–138
Snider L, Asawachaicharn A, Tyler AE, Geng LN, Petek LM, Maves L, Miller DG, Lemmers RJ, Winokur ST, Tawil R, van der Maarel SM, Filippova GN, Tapscott SJ (2009) RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: new candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum Mol Genet 18:2414–2430
Snider L, Geng LN, Lemmers RJ, Kyba M, Ware CB, Nelson AM, Tawil R, Filippova GN, van der Maarel SM, Tapscott SJ, Miller DG (2010) Facioscapulohumeral dystrophy: incomplete suppression of a retrotransposed gene. PLoS Genet 6:e1001181
Spurlock G, Jim HP, Upadhyaya M (2010) Confirmation that the specific SSLP microsatellite allele 4qA161 segregates with fascioscapulohumeral muscular dystrophy (FSHD) in a cohort of multiplex and simplex FSHD families. Muscle Nerve 42:820–821
Stadler G, Chen JC, Wagner K, Robin JD, Shay JW, Emerson CP Jr, Wright WE (2011) Establishment of clonal myogenic cell lines from severely affected dystrophic muscles—CDK4 maintains the myogenic population. Skelet Muscle 1:12
Sun CY, van Koningsbruggen S, Long SW, Straasheijm K, Klooster R, Jones TI, Bellini M, Levesque L, Brieher WM, van der Maarel SM, Jones PL (2011) Facioscapulohumeral muscular dystrophy region gene 1 is a dynamic RNA-associated and actin-bundling protein. J Mol Biol 12:397–416
Tawil R (2008) Facioscapulohumeral muscular dystrophy. Neurotherapeutics 5:601–606
Tawil R, Forrester J, Griggs RC, Mendell J, Kissel J, McDermott M, King W, Weiffenbach B, Figelwicz D, Group FSH-DY (1996) Evidence for anticipation and association of deletion size with severity in facioscapulohumeral muscular dystrophy. Ann Neurol 39:744–748
Thomas NST, Wiseman K, Spurlock G, MacDonald M, Üstek D, Upadhyaya M (2007) A large patient study confirming that facioscapulohumeral muscular dystrophy (FSHD) disease expression is almost exclusively associated with an FSHD locus located on a 4qA-defined 4qter subtelomere. J Med Genet 44:215–218
Tupler R, Berardinelli A, Barbierato L, Frants R, Hewitt JE, Lanzi G, Maraschio P, Tiepolo L (1996) Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J Med Genet 33:366–370
Tupler R, Perini G, Pellegrino MA, Green MR (1999) Profound misregulation of muscle-specific gene expression in facioscapulohumeral muscular dystrophy. Proc Natl Acad Sci USA 96:12650–12654
Upadhyaya M, Cooper DN (2004) Facioscapulohumeral muscular dystrophy: clinical medicine and molecular cell biology. Garland Science/BIOS Scientific, Abingdon
Upadhyaya M, Lunt PW, Sarfarazi M, Broadhead W, Daniels J, Owen M, Harper PS (1990) DNA marker applicable to presymptomatic and prenatal diagnosis of facioscapulohumeral disease. Lancet 336:1320–1321
van Deutekom JC, Bakker E, Lemmers RJ, van der Wielen MJ, Bik E, Hofker MH, Padberg GW, Frants RR (1996) Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q26: implications for genetic counselling and the etiology of FSHD1. Hum Mol Genet 5:1997–2003
van Geel M, Dickson MC, Beck AF, Bolland DJ, Frants RR, van der Maarel SM, de Jong PJ, Hewitt JE (2002) Genomic analysis of human chromosome 10q and 4q telomeres suggests a common origin. Genomics 79:210–217
van Koningsbruggen S, Straasheijm KR, Sterrenburg E, de Graaf N, Dauwerse HG, Frants RR, van der Maarel SM (2007) FRG1P-mediated aggregation of proteins involved in pre-mRNA processing. Chromosoma 116:53–64
van Overveld PG, Lemmers RJ, Sandkuijl LA, Enthoven L, Winokur ST, Bakels F, Padberg GW, van Ommen GJ, Frants RR, van der Maarel SM (2003) Hypomethylation of D4Z4 in 4q-linked and non-4q-linked facioscapulohumeral muscular dystrophy. Nat Genet 35:315–317
van Overveld PG, Enthoven L, Ricci E, Rossi M, Felicetti L, Jeanpierre M, Winokur ST, Frants RR, Padberg GW, van der Maarel SM (2005) Variable hypomethylation of D4Z4 in facioscapulohumeral muscular dystrophy. Ann Neurol 58:569–576
Wallace LM, Garwick SE, Mei W, Belayew A, Coppee F, Ladner KJ, Guttridge D, Yang J, Harper SQ (2010) DUX4, a candidate gene for facioscapulohumeral muscular dystrophy, causes p53-dependent myopathy in vivo. Ann Neurol 69:540–552
Wallace LM, Garwick SE, Tupler R, Harper SQ (2011) RNA interference improves myopathic phenotypes in mice over-expressing FSHD region Gene 1 (FRG1). Mol Ther [Epub ahead of print]
Wijmenga C, Frants RR, Brouwer OF, Moerer P, Weber JL, Padberg GW (1990) Location of the facioscapulohumeral muscular dystrophy gene on chromosome 4. Lancet 336:651–653
Wijmenga C, Hewitt JE, Sandkuijl LA, Clark LN, Wright TJ, Dauwerse HG, Gruter A, Hofker MH, Moerer P, Williamson R, van Ommen GB, Padberg GW, Frants RR (1992) Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat Genet 2:26–30
Winokur ST, Bengtsson U, Feddersen J, Mathews KD, Weiffenbach B, Bailey H, Markovich RP, Murray JC, Wasmuth JJ, Altherr MR (1994) The DNA rearrangement associated with facioscapulohumeral muscular dystrophy involves a heterochromatin-associated repetitive element: implications for a role of chromatin structure in the pathogenesis of the disease. Chromosome Res 2:225–234
Winokur ST, Bengtsson U, Vargas JC, Wasmuth JJ, Altherr MR (1996) The evolutionary distribution and structural organization of the homeobox-containing repeat D4Z4 indicates a functional role for the ancestral copy in the FSHD region. Hum Mol Genet 5:1567–1575
Winokur ST, Chen YW, Masny PS, Martin JH, Ehmsen JT, Tapscott SJ, van der Maarel SM, Hayashi Y, Flanigan KM (2003a) Expression profiling of FSHD muscle supports a defect in specific stages of myogenic differentiation. Hum Mol Genet 12:2895–2907
Winokur ST, Barrett K, Martin JH, Forrester JR, Simon M, Tawil R, Chung SA, Masny PS, Figlewicz DA (2003b) Facioscapulohumeral muscular dystrophy (FSHD) myoblasts demonstrate increased susceptibility to oxidative stress. Neuromuscul Disord 13:322–333
Wohlgemuth M, van der Kooi EL, van Kesteren RG, van der Maarel SM, Padberg GW (2004) Ventilatory support in facioscapulohumeral muscular dystrophy. Neurology 63:176–178
Wuebbles RD, Hanel ML, Jones PL (2009) FSHD region gene 1 (FRG1) is crucial for angiogenesis linking FRG1 to facioscapulohumeral muscular dystrophy-associated vasculopathy. Dis Model Mech 2:267–274
Wuebbles RD, Long SW, Hanel ML, Jones PL (2010) Testing the effects of FSHD candidate gene expression in vertebrate muscle development. Int J Clin Exp Pathol 3:386–400
Yang F, Shao C, Vedanarayanan V, Ehrlich M (2004) Cytogenetic and immuno-FISH analysis of the 4q subtelomeric region that is associated with facioscapulohumeral muscular dystrophy. Chromosoma 112:350–359
Yip DJ, Picketts DJ (2003) Increasing D4Z4 repeat copy number compromises C2C12 myoblast differentiation. FEBS Lett 537:133–138
Zeng W, de Greef JC, Chen YY, Chien R, Kong X, Gregson HC, Winokur ST, Pyle A, Robertson KD, Schmiesing JA, Kimonis VE, Balog J, Frants RR, Ball AR Jr, Lock LF, Donovan PJ, van der Maarel SM, Yokomori K (2009) Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet 5:e1000559
Zhang XY, Wang R, Ehrlich M (1985) Human DNA sequene exhibiting gamete-speeille hypomethylatlon. Nucl Acids Res 13:4837–4851
Zhang Y, Forner J, Fournet S, Jeanpierre M (2001) Improved characterization of FSHD mutations. Ann Genet 44:105–110
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Richards, M., Coppée, F., Thomas, N. et al. Facioscapulohumeral muscular dystrophy (FSHD): an enigma unravelled?. Hum Genet 131, 325–340 (2012). https://doi.org/10.1007/s00439-011-1100-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00439-011-1100-z