
Abstract
Background
In 2010, INF2 mutations were associated with autosomal-dominant focal segmental glomerulosclerosis (FSGS), clinically presenting with moderate proteinuria in adolescence. However, in the meantime, cases with more severe clinical courses have been described, including progression to end-stage renal disease (ESRD) during childhood. INF2 mutations in patients with isolated FSGS are clustered in exons 2 to 4, encoding the diaphanous inhibitory domain, involved in the regulation of the podocyte actin cytoskeleton.
Methods
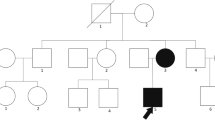
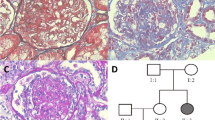
We report a family with 14 affected individuals (autosomal-dominant mode of inheritance), most of whom presented with nephrotic-range proteinuria, hypertension, and progressive renal failure. Four members received a kidney transplant without disease recurrence. Two patients underwent renal biopsy with the result of minimal-change glomerulopathy and IgA nephropathy respectively. We performed mutational analysis of ACTN4, CD2AP, COQ6, INF2, LAMB2, NPHS1, NPHS2, PLCE1, TRPC6, and WT1 in the index patient by next-generation sequencing. Additionally, in 6 affected and 2 unaffected family members target diagnostics were performed.
Results
We identified a novel heterozygous mutation c.490G>C (p.(Ala164Pro) in exon 3 of the INF2 gene in the index patient and 6 additionally examined affected family members. In silico analysis predicted it as “probably damaging”. Additionally, three patients and 2 unaffected relatives harbored a novel heterozygous variant in ACTN4 (c.1149C>G, p.(Ile383Met)) with uncertain pathogenicity.
Conclusion
Mutations in INF2 are associated with familial proteinuric diseases - irrespective of the presence of FSGS and in the case of rapid disease progression. Therefore, mutational analysis should be considered in patients with renal histology other than FSGS and severe renal phenotype.



Similar content being viewed by others
References
Büscher AK, Beck BB, Melk A, Hoefele J, Kranz B, Bamborschke D, Baig S, Lange-Sperandio B, Jungraithmayr T, Weber LT, Kemper MJ, Tonshoff B, Hoyer PF, Konrad M, Weber S, German Pediatric Nephrology Association (GPN) (2016) Rapid response to cyclosporin A and favorable renal outcome in nongenetic versus genetic steroid-resistant nephrotic syndrome. Clin J Am Soc Nephrol 11:245–253
Büscher AK, Konrad M, Nagel M, Witzke O, Kribben A, Hoyer PF, Weber S (2012) Mutations in podocyte genes are a rare cause of primary FSGS associated with ESRD in adult patients. Clin Nephrol 78:47–53
Sadowski CE, Lovric S, Ashraf S, Pabst WL, Gee HY, Kohl S, Engelmann S, Vega-Warner V, Fang H, Halbritter J, Somers MJ, Tan W, Shril S, Fessi I, Lifton RP, Bockenhauer D, El-Desoky S, Kari JA, Zenker M, Kemper MJ, Mueller D, Fathy HM, Soliman NA, SRNS Study Group, Hildebrandt F (2015) A single-gene cause in 29.5% of cases of steroid-resistant nephrotic syndrome. J Am Soc Nephrol 26:1279–1289
Brown EJ, Schlondorff JS, Becker DJ, Tsukaguchi H, Tonna SJ, Uscinski AL, Higgs HN, Henderson JM, Pollak MR (2010) Mutations in the formin gene INF2 cause focal segmental glomerulosclerosis. Nat Genet 42:72–76
Boyer O, Benoit G, Gribouval O, Nevo F, Tete MJ, Dantal J, Gilbert-Dussardier B, Touchard G, Karras A, Presne C, Grunfeld JP, Legendre C, Joly D, Rieu P, Mohsin N, Hannedouche T, Moal V, Gubler MC, Broutin I, Mollet G, Antignac C (2011) Mutations in INF2 are a major cause of autosomal dominant focal segmental glomerulosclerosis. J Am Soc Nephrol 22:239–245
Gbadegesin RA, Lavin PJ, Hall G, Bartkowiak B, Homstad A, Jiang R, Wu G, Byrd A, Lynn K, Wolfish N, Ottati C, Stevens P, Howell D, Conlon P, Winn MP (2012) Inverted formin 2 mutations with variable expression in patients with sporadic and hereditary focal and segmental glomerulosclerosis. Kidney Int 81:94–99
Munch J, Grohmann M, Lindner TH, Bergmann C, Halbritter J (2016) Diagnosing FSGS without kidney biopsy—a novel INF2-mutation in a family with ESRD of unknown origin. BMC Med Genet 17:73
Boyer O, Nevo F, Plaisier E, Funalot B, Gribouval O, Benoit G, Cong EH, Arrondel C, Tete MJ, Montjean R, Richard L, Karras A, Pouteil-Noble C, Balafrej L, Bonnardeaux A, Canaud G, Charasse C, Dantal J, Deschenes G, Deteix P, Dubourg O, Petiot P, Pouthier D, Leguern E, Guiochon-Mantel A, Broutin I, Gubler MC, Saunier S, Ronco P, Vallat JM, Alonso MA, Antignac C, Mollet G (2011) INF2 mutations in Charcot-Marie-Tooth disease with glomerulopathy. N Engl J Med 365:2377–2388
Rood IM, Bongers EM, Lugtenberg D, Klein IH, Steenbergen EJ, Wetzels JF, Deegens JK (2016) Familial focal segmental glomerulosclerosis: mutation in inverted formin 2 mimicking Alport syndrome. Neth J Med 74:82–85
Lee HK, Han KH, Jung YH, Kang HG, Moon KC, Ha IS, Choi Y, Cheong HI (2011) Variable renal phenotype in a family with an INF2 mutation. Pediatr Nephrol 26:73–76
Xie J, Hao X, Azeloglu EU, Ren H, Wang Z, Ma J, Liu J, Ma X, Wang W, Pan X, Zhang W, Zhong F, Li Y, Meng G, Kiryluk K, He JC, Gharavi AG, Chen N (2015) Novel mutations in the inverted formin 2 gene of Chinese families contribute to focal segmental glomerulosclerosis. Kidney Int 88:593–604
Liu Z, Blattner SM, Tu Y, Tisherman R, Wang JH, Rastaldi MP, Kretzler M, Wu C (2011) Alpha-actinin-4 and CLP36 protein deficiencies contribute to podocyte defects in multiple human glomerulopathies. J Biol Chem 286:30795–30805
Kos CH, Le TC, Sinha S, Henderson JM, Kim SH, Sugimoto H, Kalluri R, Gerszten RE, Pollak MR (2003) Mice deficient in alpha-actinin-4 have severe glomerular disease. J Clin Invest 111:1683–1690
Kaplan JM, Kim SH, North KN, Rennke H, Correia LA, Tong HQ, Mathis BJ, Rodriguez-Perez JC, Allen PG, Beggs AH, Pollak MR (2000) Mutations in ACTN4, encoding alpha-actinin-4, cause familial focal segmental glomerulosclerosis. Nat Genet 24:251–256
Kesselheim A, Ashton E, Bockenhauer D (2017) Potential and pitfalls in the genetic diagnosis of kidney diseases. Clin Kidney J. https://doi.org/10.1093/ckj/sfx075
Maas RJ, Deegens JK, Smeets B, Moeller MJ, Wetzels JF (2016) Minimal change disease and idiopathic FSGS: manifestations of the same disease. Nat Rev Nephrol 12:768–776
Habib R, Girardin E, Gagnadoux MF, Hinglais N, Levy M, Broyer M (1988) Immunopathological findings in idiopathic nephrosis: clinical significance of glomerular "immune deposits". Pediatr Nephrol 2:402–408
Hill GS, Karoui KE, Karras A, Mandet C, Duong Van Huyen JP, Nochy D, Bruneval P (2011) Focal segmental glomerulosclerosis plays a major role in the progression of IgA nephropathy. I. Immunohistochemical studies. Kidney Int 79:635–642
Yao J, Le TC, Kos CH, Henderson JM, Allen PG, Denker BM, Pollak MR (2004) Alpha-actinin-4-mediated FSGS: an inherited kidney disease caused by an aggregated and rapidly degraded cytoskeletal protein. PLoS Biol 2:e167
Bartram MP, Habbig S, Pahmeyer C, Hohne M, Weber LT, Thiele H, Altmuller J, Kottoor N, Wenzel A, Krueger M, Schermer B, Benzing T, Rinschen MM, Beck BB (2016) Three-layered proteomic characterization of a novel ACTN4 mutation unravels its pathogenic potential in FSGS. Hum Mol Genet 25:1152–1164
Acknowledgements
We would like to thank the family for their participation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Blood samples were obtained after informed consent was given by the following family members: III-9, III-10, III-11, III-18, IV-1, IV-2, IV-3, IV-4, and IV-5 (Fig. 1). Approval for this study was obtained from the Ethics Committee of the University Duisburg-Essen, Germany (09–3954-24.03.2009).
Conflicts of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Büscher, A.K., Celebi, N., Hoyer, P.F. et al. Mutations in INF2 may be associated with renal histology other than focal segmental glomerulosclerosis. Pediatr Nephrol 33, 433–437 (2018). https://doi.org/10.1007/s00467-017-3811-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00467-017-3811-4