
Abstract
Both the Levantine Corridor and the Horn of Africa route have figured prominently in early hominid migrations from Africa to Eurasia. To gauge the importance of these two African–Asian thoroughfares in the demic movements of modern man, we surveyed the mtDNA control region variation and coding polymorphisms of 739 individuals representing ten African and Middle Eastern populations. Two of these collections, Egypt and Yemen, are geographically close to the Levant and Horn of Africa, respectively. In this analysis, we uncover genetic evidence for the preferential use of the Levantine Corridor in the Upper Paleolithic to Neolithic dispersals of haplogroups H, J*, N1b, and T1, in contrast to an overwhelming preference in favor of the Horn of Africa for the intercontinental expansion of M1 during the Middle to Upper Paleolithic. Furthermore, we also observed a higher frequency of sub-Saharan mtDNA compared to NRY lineages in the Middle Eastern collections, a pattern also seen in previous studies. In short, the results of this study suggest that several migratory episodes of maternal lineages occurred across the African–Asian corridors since the first African exodus of modern Homo sapiens sapiens.
Similar content being viewed by others

Introduction
Modern human’s first Eurasian point of entry from Africa (54±8 kyr as estimated by Forster 2004) is believed by many to be at the far southwestern point of the Arabian Peninsula, located in present-day Yemen (Lahr and Foley 1994; Oppenheimer 2003; Forster 2004). This initial Red Sea crossing through the Bab el Mandab Strait or Cape of Sorrow (referred to as the Horn of Africa from this point forward) was most likely facilitated by several factors, including the shorter distance between the African and Arabian land masses (17 km vs. the 18 km observed today) as well as the much lower sea level during this time interval (80–60 m lower than that of present day) (Forster 2004; Siddall et al. 2003).
However, the question remains concerning the relative roles played by the Horn of Africa passage and the Levantine Corridor in subsequent forays out of Africa, or for that matter, the multiple pilgrimages back to the continental birth place of Homosapiens sapiens. A previous analysis (Luis et al. 2004) examining point and length polymorphisms in the non-recombining portion of the Y chromosome (NRY) revealed a marked asymmetry regarding the use of these two inter-continental corridors. The results of the study indicated that since the last glacial maximum (LGM) [25–15 kyr according to Fagan (1990)], the Levant was a major thoroughfare used in the spread of NRY markers E3b1-M78 and E3b3-M123 into Eurasia as well as the North African dissemination of K-M70 and R1*-M173 in the Upper Paleolithic (40–10 kyr) and G-M201, J-p12f2, R1a1-M17 and R1b3-269 during the Neolithic. In contrast, there is very little evidence of a Eurasian dispersal of NRY haplogroups via the Horn of Africa during these times. Furthermore, it was observed that the more recent arrival of the derivative M173 haplogroups into North Africa also occurred by way of the Levantine Corridor whereas the Bantu marker, M2, dispersed into Arabia and Asia via the East African slave trade routes during the last millennium.
The main objective of the current study is to assess the relative importance of the Levantine Corridor and the Horn of Africa as genetic conduits by using phylogeographic patterns of mitochondrial DNA (mtDNA) variation. To investigate which (if any) of these two pathways was favored in the dispersal of maternal lineages via African–Asian migrations during the Middle Paleolithic to Neolithic, we examine mtDNA HVI and HVII sequence motifs, coding-sequence polymorphisms, and the haplogroup frequency distribution of six Middle East populations. These collections comprise those representing the Levantine Corridor (Egypt and Jordan), and the Arabian Peninsula (Yemen, Oman, Qatar and United Arab Emirates or UAE for short).
A second aim of this study is to track more recent gene flow of sub-Saharan individuals along the East African trade routes as well as across the Central African Corridor. To accomplish this objective, we examined four groups from sub-Saharan Africa (Benin, Cameroon, Rwanda, and Kenya). In addition, the mtDNA sampling along the Levantine Corridor and the Horn of Africa as well as the sub-Saharan corridors is enriched by the assessment of Middle Eastern and African HVI motifs compiled from the available literature. This supplementary data also provides a wider geographical context in which to interpret our results.
Materials and methods
Populations analyzed
Details concerning the ten African and Middle Eastern populations examined in this study are listed in Table 1. In future discussion, Egypt and Jordan will be referred to as the Levant collections and the Oman, Qatar, UAE and Yemen as the Arabian Peninsula study groups. Family history of the participants was ascertained to select for individuals whose ancestors have lived in the targeted geographical area for at least two generations. The collection process was performed in strict compliance with NIH guidelines as well as with any other additional regulations imposed by the institutions involved. In addition, mtDNA haplogroup and control region sequence information from 15 published Eurasian and African populations was used in further analysis (see Table 1).
DNA extraction, PCR and sequencing
Genomic DNA was isolated from the peripheral leukocyte fraction of whole blood as previously described (Antunez de Mayolo et al. 1999). Both the HVI and HVII regions were PCR-amplified using primers described by Stoneking et al. (1991). Fragments were sequenced with Big Dye v 1.1® from Applied Biosystems in accordance with the recommended protocol. Sequenced samples were cleaned with spin filters (Centri Sep96® from Princeton Separations) and run on a 3100 Genetic Analyzer from ABI. The resulting sample sequences were aligned and compared to the revised Cambridge Reference Sequence (rCRS) (Anderson et al. 1981; Andrews et al. 1999).
Haplogroup assignment
Haplogroup assignment followed the basic classification scheme of Macaulay et al. (1999). Additional reports (Salas et al. 2002; Kivisild et al. 2004; Quintana-Murci et al. 2004; Salas et al. 2004) were consulted as needed to further delineate the haplotypes and assign them geographical labels (Western Eurasian, Eastern Eurasian, South Asian, and African). Restriction fragment length polymorphism (RFLP) analysis was performed in cases of haplogroup ambiguity or to confirm designations based solely upon rapidly evolving HVI mutations (Richards et al. 1998). PCR amplification protocols and primer sets followed the procedure described by Torroni et al. (1996). The haplogroup nomenclature used in the current study follows that of several previous analyses (Richards et al. 1998; Macaulay et al. 1999; Quintana-Murci et al. 1999; Salas et al. 2002).
Statistical analyses
A correspondence analysis (CA) using the Numerical Taxonomy System [NTSYSpc-2.02i package by Rohlf (2002) was performed on the haplogroup frequencies of the ten study populations together with those of the additional 15 published studies (Table 1).
Mean pairwise differences in sequence motifs (PD) and gene diversity (GD) (Nei 1987), a measure which takes into account the number of different sequence types and their corresponding frequencies, were calculated for each population with respect to HVI alone [nucleotide position (np) 16069 and nps 16090–16365] and both regions together (HVI + nps 00073–00398).
To assess whether intra-population sequence diversity conforms to the mathematical expectations of neutral theory (Kimura 1968), we conducted the Ewens-Watterson homozygosity test (Ewens 1972; Watterson 1978). It should be noted that although the Tajima’s D (Tajima 1989) and Fu’s Fs (Fu 1997) procedures are believed to be more powerful in detecting deviations from neutral theory compared to the Ewens-Watterson homozygosity test, these methods are based on the infinite sites model of mutation and therefore may not be applicable to mtDNA control region sequences in light of the high level of homoplasy and as well as the many nucleotide positions in which multiple substitution types are known to occur (30 HVI and HVII nucleotide positions in our data) (Kimura 1969, 1971; Watterson 1975; Fu 1997). The PD, GD, and neutrality tests were performed with the Arlequin Version 2.0 package (Schneider et al. 2000).
Population-specific and global (multi population) network analyses based on the available haplogroups and/or haplotypes were generated with the reduced median method (Bandelt et al. 1995) using the NETWORK 4.1 software program developed by Fluxus Technology Limited (http://www.fluxus-engineering.com). The weighting scheme for the nucleotide positions used in this analysis (nps 16090–16365) followed that of Richards et al. (1998). The time to the most recent common ancestor (TMRCA) was estimated by the product of rho (ρ), the mean transitional distance between lineages (Forster et al. 1996) and the human mtDNA mutation rate in years. The rate of one transition in 20,180 years for these nucleotide positions has been used in recent published studies (Richards et al. 2000; Saillard et al. 2000; Salas et al. 2002; Kivisild et al. 2004).
Other statistical analyses include the Baysean 0.95 credible region (0.95 CR) of the resulting haplogroup frequencies calculated using the SAMPLING program kindly provided by Vincent Macaulay and the chi-square procedures to test for significant differences in the observed frequencies. The results of the chi-square (χ2) are presented in the relevant sections below.
Results and discussion
Phylogeography and haplogroup diversity
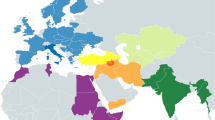
The resulting haplogroup/haplotype frequencies of the 739 individuals comprising the ten Middle Eastern and African populations is available online at the corresponding author’s website (online Table 1 in http://www.fiu.edu/~herrerar). The phylogeographic maps of Fig. 1, which are based on these frequencies, illustrate both the high level of mtDNA control region variation and the nonrandom geographical distribution of its variants. As is readily apparent from the geographical characterization of the haplogroups (Fig. 1a), the most dramatic difference lies along a north–south transect between the sub-Saharan and the Afro-Asiatic collections. The Benin, Cameroon, Rwanda, and Kenya haplogroup profiles are mostly composed of typically African L groups (L1, L2 and L3) whereas those of the Middle East are, as expected, characterized primarily by West Eurasian lineages (various members of HV, N, J–T and U). It is also interesting to note the longitudinal partitioning of the South Asian components, M*, U2a–c, R1 and/or R2 (Quintana-Murci et al. 2004), among the Afro-Asiatic groups [0.10 vs. 0.02 in the Arabian Peninsula and the Levant populations, respectively (χ2P<0.0005)].

Phylogeography of mtDNA haplogroups: geographic affiliations (a) and major haplogroup distribution (b). The geographical haplogroup assignments are as follows: West Eurasian HV, pre-HV, N, J-T, U-K, I, W, X; East Eurasian M-DE, A, B; South Asian: M*, U2a–c, U9, R1–R2; African L1, L2, L3, M1, U6. Although there is no scientific consensus regarding the origin of M1 (East African vs. South Asian), data from Kivisild et al. (2004) strongly suggest an Ethiopian origin, which is indicated in this figure
The gene diversity values, GD, of online Table 2 are not substantially different among the different regions under study (0.992, 0.989, 0.989, and 0.970 for the Arabian Peninsula, Levant, East and West Africa, respectively). The mean pairwise difference, PD, exhibits a slight east-to-west variation for both Afro-Asiatic and sub-Saharan factions (7.1 and 6.4 for the Arabian Peninsula vs. the Levant, respectively, and 8.8 and 7.2 for east vs. west sub-Saharan Africa, respectively). Also, the PD values display a minor latitudinal aspect (average scores of 8.0 and 6.8 for the sub-Saharan Africans vs. Middle East individuals, respectively). This observation can be explained by the high level of sequence variation inherent in the L haplogroups, which is expected since these are the oldest haplogroups (online Table 1 at http://www.fiu.edu/~herrerar). These patterns hold for indices based on both the HVI and HVII.
Correspondence analysis
A correspondence analysis including the ten study collections and the 15 additional African or Eurasian populations is shown in online Fig. 1 at http://www.fiu.edu/~herrerar. Axes 1 and 2 account for 34.8% of the total inertia (20.7 and 13.2%, respectively). The two-dimensional pattern displays four distinct clusters: the sub-Saharan Africans located in the most positive portion of axis 1 (a); the Indian–Pakistani populations partitioned in the upper-left quadrant (b) and the three Ethiopian populations (Amhara, Cushitic and Tigrai) sandwiched between the sub-Saharan cluster and the North African and Middle East populations (c), and a diffuse assembly of the North African and Middle East collections in the bottom-left quadrant (d). These fit well within the phylogeographical patterns established by previous genetic research (Cavalli-Sforza et al. 1994; Underhill et al. 2000; Salas et al. 2002; Kivisild et al. 2004). This corroboration suggests representative population sampling.
Networks phylogenies and time estimations
Global (encompassing haplogroup data from all relevant populations) and population-specific networks were constructed from the pertinent haplogroups and presented in the text (Figs. 2 and 3) or online (Figs. 2a–c and 3a–b at http://www.fiu.edu/~herrerar).
In addition, a more extensive compilation of population and haplogroup-specific TMRCAs (online Table 3) were generated from relationships that are generally star-like (Richards et al. 2000), a morphology characteristic of expansion from a single founder type. These networks are composed of at least six lineages (as in Richards et al. 2000) and contain mostly short branches. The TMRCAs of non star-like patterns may be artificially overestimated due to this apparent heterogeneity and thus are not included in online Table 3. Discussion of global and individual population-specific TMRCAs are presented below in the discussions of the relevant haplogroups.
Human migrations
One of the major aims of this study is to uncover genetic evidence for past migratory events across ancient African–Asian thoroughfares, the Levantine and Horn of Africa corridors, and to determine which of the two, if any, was the preferred passageway. In addition, demic movements of sub-Saharan individuals within Africa as well as into the Middle East via the East African trade routes are examined. This information is discussed in three parts: (a) Evidence for migrations through the Levantine Corridor; (b) Evidence for movements across the Horn of Africa; and (c) sub-Saharan lineages. The last section addresses both dispersal episodes within the African continent itself and recent inter-continental gene flow (within the last millennium or so) by way of the East African trade routes.
We discuss the Middle East/North African distribution (both frequency and individual HVI sequence types) of eight mtDNA haplogroups, H, J, J1, K, M1, N1b, (pre-HV)1 and T1. It has been established that seven of these haplogroups (All except for M1) originated in the Near East/Middle East (Richards et al. 1998, 2000; Achilli et al. 2004). Therefore, since none of these seven haplogroups evolved in Africa, it is logical to infer that the flow of the initial movement across African/Asian corridors transpired from Eurasia to Africa and not in the opposite direction.
Evidence for dispersals through the Levantine Corridor
There is a definite asymmetry (χ2P<0.0005) with respect to the H haplogroup distribution between the Levant populations (mean of 0.21 for Iraq, Israeli Druze, Jordan, Palestine, and Syria) and the Arabian Peninsula groups (mean of 0.10 for Qatar, UAE, Oman, and Yemen) (Macaulay et al. 1999; Richards et al. 2000; Al-Zahery et al. 2003; Achilli et al. 2004; Kivisild et al. 2004) as well as between Egypt and Ethiopia, two populations near the African entry/exit points of the two African/Asian routes [0.18 (0.95 CR 0.120–0.257) in Egypt versus 0.01 in Ethiopia, χ2P<0.0005] (Kivisild et al. 2004). The global H network (Fig. 2) exhibits a starburst arrangement of lineages around a large central node (HVI rCRS) whose sequence is common to 21 individuals from various Middle East populations including nine Egyptians (0.08), three Jordanians (0.08) but only one Yemeni (0.02). This topology indicates a widespread Eurasian H radiation from a Near East epicenter around 26.0±4.1 kyr (Fig. 2), which corroborates the expansion age of the Near Eastern H haplogroup estimated by Richards et al. (1998) (25.8±2.6 kyr). The TMRCA of the Spanish H samples (18.4±2.0 kyr in Achilli et al. 2004), which coincides substantially with that of Egypt (21.1±6.6 kyr in online Table 3 and online Fig. 2a), has been ascribed to an expansion event subsequent to a southward LGM movement to escape the harsh northern climate. It is conceivable that the presence of H in Egypt results from a parallel southern contraction in the Middle East. In support of this view, the Jordanian H TMRCA of 22.7±9.1 kyr (online Table 3) is consistent with a southbound migration during this time frame. Thus, the Near East origin of H, its substantial presence along the Levant, the LGM compatible time depths of the Egyptian H and Jordanian H expansions, and the ancestral lineage continuity between these two groups (0.08 for both) point to the transversal of this corridor from north to south during the LGM or shortly before. Furthermore, given the extremely low level of H (0.01) in the Ethiopian collection (Kivisild et al. 2004) and the lack of continuity (no shared HVI motifs or mutations) between the Yemini and Ethiopian H lineages, it is unlikely whether there was a late Upper Paleolithic dissemination of H lineages through the Horn of Africa. Although H is present in Yemen, it is not clear when, or if, Yemeni members of this haplogroup arrived in Ethiopia.

Global H network. TMRCA = 26±4.1 kyr. Add 16,000 to the numbers on the branches to obtain HVI nucleotide positions. Abbreviations are as follows: E Egypt, J Jordan, O Oman, Q Qatar, U United Arab Emirates, Y Yemen. Redundant mutations associated with alternate pathways are boxed
An alternative theory is that these lineages were introduced into Africa by way of the Strait of Gibraltar since there is a considerable proportion (0.37) of H individuals in Moroccan Berbers (Achilli et al. 2004). However, the Strait of Gibraltar was most likely a formidable barrier since paleoclimatological evidence suggests that the sea level between the Moroccan coast and southern tip of the Iberian Peninsula was too high even during the LGM (Fagan 1990). The presence of Moroccan Berber H individuals may stem from an initial westward migration of the Egyptian Berbers along the North African coast. This hypothesis is consistent with polymorphic H levels in three Tunisian Berber groups (a mean of 21.4 for the Matama, Sened, and Chenini-Douiret Berbers) (Fadhlaoui-Zid et al. 2004), which are positioned geographically between Egypt and Morocco.
The basal J* haplogroup (or the J2 of Kivisild et al. 2004) is present in the Levant and Arabian Peninsula collections of the current study at mean percentages of 0.06 and 0.04, respectively. This haplogroup was not detected in the current Yemeni sample and is found at very low levels (<0.01) in the Yemeni and Ethiopian groups of Kivisild et al. (2004). A widespread motif (nps 16069 and 16126) occupies the large ancestral node (seven individuals) in the basal J* cluster of the global collective J topology (Fig. 3). It is present in the two Levant and three of the four Arabian Peninsula collections (except for Yemen) of the current study. In addition to sharing the ancestral sequence, nearly all of the remaining Egyptian and Jordanian basal J* lineages deviate from this motif by only one or two mutations. The TMRCA of Middle East basal J* (which is comprised of polymorphic levels of Egyptians and Jordanians) is 32±8 kyr. This Middle East expansion time as well as the lineage similarity between Egypt and Jordan are consistent with a southward J diffusion along the Levant into Egypt encompassing the time interval from the Middle Paleolithic to the Neolithic.

Global J network. TMRCA for Global J=57.9±17.6 kyr; TMRCA for J*=32±8 kyr; TMRCAs for J1b=15.5±5.0 kyr. Add 16,000 to the numbers on the branches and to the ancestral motifs to obtain HVI nucleotide positions. Abbreviations are as follows: E Egypt, J Jordan, O Oman, Q Qatar, U United Arab Emirates. Redundant mutations associated with alternate pathways are boxed
On the other hand, no significant J* lineage continuity exists between Yemen and Ethiopia. There is a complete absence of the basal J* motif in the Yemeni collection (current study), a population with a considerable collective J frequency (0.22). There is also a lack of the J* ancestral motif in the Yemeni of Kivisild et al. (2004) and a very low level of a one-step derivative (<0.01). Furthermore, this ancestral J* lineage is found at a very low frequency (<0.01 for the ancestral lineage as well as any close derivative) in the Ethiopian population of Kivisild et al. (2004). It is unclear when J* was introduced into Ethiopia. Random genetic drift may have obliterated much of its path across the Horn of Africa during prehistoric times or it presence in Ethiopia could be the result of a recent arrival.
J1b is present at low frequencies in Egypt, Oman, UAE and Qatar (0.05–0.03) and represents a respectable contingent of Yemeni individuals [0.12 (0.95 CR 0.057–0.239)] of the current study. The resulting star-shaped Middle East pattern (see the J1b cluster in Fig. 3), which exhibits a large ancestral node (15 individuals), indicates an expansion event that emanated throughout this area at some point in time during the interval from the LGM to the Mesolithic or early Neolithic (TMRCA 15.5±5.0 kyr). The global J1b root sequence (nps 16029, 16126, 16145, 16222 and 16261) is present in Egypt as well as all Middle East populations except for Jordan. In addition, several J1b HVI motifs occur in Yemen, Oman, UAE, and Qatar. However, it is not clear where the J1b epicenter lies. There is also uncertainty regarding the route into Egypt, since no J1b lineages are detected in the Jordanians, the Druze of northern Israel (Macaulay et al. 1999) or in the Ethiopians (Kivisild et al. 2004). The very low frequency of J1b among the Matmata Berbers (0.02) and Tunisian Arabs (0.02) (Fadhlaoui-Zid et al. 2004) and its apparent absence in various northwest African groups including two populations geographically close to the Strait of Gibraltar (Moroccan Berbers and non-Berber Moroccans in Rando et al. 1998) adds complexity to this issue. It is possible that random genetic drift has erased J1b from many populations along its trail(s) (either through the Levant, from Ethiopia to Egypt and/or from North West Africa to Tunisia) after its introduction into Tunisia and Egypt. A more plausible explanation is that limited sampling (both with respect to number of individuals and geographically relevant locales) failed to detect the signature of this haplogroup through any or all of these pathways. Another possibility is that J1b evolved in situ after the Egyptian arrival of J. However, this latter premise would entail independent origins of the Egyptian and the Middle Eastern J1b. Since there are only three J1b sequence types detected in Egypt, there is no valid Egyptian J1b expansion date. As of now, there is no concrete evidence that a J1b dissemination occurred along any of the two African–Asian thoroughfares during the LGM to the Neolithic. Although the most parsimonious scenario would be an entry of J1b into Egypt by way of the Levantine Corridor, there is no way of gauging when this migration occurred.
The absence of N1b in our study populations except for low levels in Jordan [0.05 (0.95 CR 0.016–0.169)], Egypt [0.03 (0.95 CR 0.009–0.0172)] and UAE [0.02 (0.95 CR 0.008–0.065)] renders it difficult to achieve a detailed picture of its prehistoric Eurasian–African dispersal patterns. However, the strong lineage continuity between Jordan and Egypt illustrated in the global N1b of online Fig. 2c (no more than two mutations separating sequence types) suggests a Levant N1b conduit. Furthermore, the placement of a Jordanian sequence (nps 16145, 16176G and 16223) at the center of this topology suggests that the net direction of flow is from north to south. The estimated expansion time of the Egyptian/Jordanian cluster based on the global N1b network (20.2±10.7 kyr) spans the LGM to the early Neolithic. Although both time periods are associated with Asian to African back migrations (Camps-Fabrer 1974; McEvedy 1980; Camps 1982; Newman 1995; Forster 2004) the small number (n=4) of different lineage types available for analysis may impact the time estimation somewhat. The virtual absence of this haplogroup in Ethiopia and Yemen (both in the present study and that of Kivisild et al. 2004) suggests that the Horn of Africa passage was not involved in the N1b diffusion into Africa, although it is possible that a genetic bottleneck following a Yemen/Ethiopian transit may have reduced an already low N1b frequency to below the detection limits of the samples [n=50 for Yemen in present study and 270 and 115 for Ethiopia and Yemen in Kivisild et al. (2004), respectively].
In the present study, nearly all members of the T1 haplogroup are Egyptian [0.08 (0.95 CR 0.041–0.139)] except for one Yemeni [0.02 (0.95 CR 0.005–0.104)] and a single subject from UAE [<0.01 (0.95 CR 0.002–0.041). The HVI motif (nps 16126, 16163, 16186, 16189, and 16294), which forms the core of a star-like radiation of our global T1 network (online Fig. 3a), is found in two Egyptians and the single T1 Yemini of this study as well as one Yemeni (<0.01) and an Israeli Druze individual (0.02) from previous reports [Kivisild et al. (2004) and Macaulay et al. (1999), respectively]. This HVI sequence is the Near Eastern ancestral T1 motif described in Richards et al. (2000). A one-step derivative of this motif is observed in one Israeli Druze and distant versions (two and three mutations away from the ancestral) are found in the two Ethiopians (<0.01) of Kivisild et al. 2004.
The TMRCAs of the Egyptian (22.2±8.3 kyr for the Egyptian sub-cluster of online Fig. 3a) and global (26.3±8.3 kyr) networks as well as the high degree of T1 lineage similarity between the Israeli Druze (0.02) and the Egyptians (i.e., the presence of the Egyptian/global ancestral sequence in both populations and a one-step derivative in the Israeli Druze) are highly consistent with a southbound LGM migration along the Levantine Corridor. However, due to the lack of close continuity between the Ethiopian and Yemeni T1 lineages (≥2-step derivatives from Yemeni motifs in the Ethiopian collections), the unavailability of T1 expansion times and the near or total absence of T1 in Ethiopia, Yemen, UAE, Oman and Qatar, it is uncertain whether the Horn of Africa served as a major corridor for the dispersal of T1 into Africa during the LGM or at any other time period.
Evidence of migrations through the Horn of Africa
M1 frequencies are highest in Ethiopia (0.17), and are lower but still polymorphic in Yemen [0.06 (0.95 CR 0.022–0.162) in this study] and substantially reduced (≤0.03) in points west [Northwest Africa (Rando et al. 1998)], north [Middle East (Rando et al. 1998; Macaulay et al. 1999; Al-Zahery et al. 2003; Forster 2004; Quintana-Murci et al. 2004 and this study)] and south [East Africa (this study)]. Most of the M1 samples in this study belong to the M1a sub-clade whose HVI motif (nps 16129, 16189, 16223, 16249, 16311, and 16359) is shared by nine Ethiopian (0.03), one Kenyan [0.02 (0.95 CR 0.006–0.123) and one Israeli Druze (0.02). At the present time there is no consensus concerning the geographical genesis of this haplogroup. Nonetheless, the paucity of M1 in the Levant, its polymorphic presence in Yemen [0.06 (0.95 CR 0.022–0.162) in this study] and Ethiopia (0.17 in Kivisild et al. 2004) as well as a late Middle to early Upper Paleolithic expansion date in East Africa (25–47 kyr as reported by Maca-Meyer et al. 2001) and either an Indian or East African origin suggests a bias in favor of the Horn of Africa route over the Levantine Corridor for a pre-LGM M1 dispersal regardless of the direction of gene flow. However, the reduced presence of M1 in Egypt and Israeli Druze (≤0.03) may signal either a later, less extensive movement or an early migration through the Levant nearly erased by drift. Results of a recent study by Olivieri et al. (2006) suggest the later (more specifically, a back migration into Africa via the Levant sometime in the Middle to early Upper Paleolithic).
The geographical lineage distributions of K and (pre-HV)1, two mtDNA haplogroups of Eurasian origin (Richards et al. 1998, 2000; Achilli et al. 2004), are consistent with a dispersal into Africa by either route during the time interval involved. The overlapping TMRCAs of the star-shaped or mostly star-shaped K networks for Oman, UAE, and Egypt (17.9±14.2, 20.2±11.1 and 16.8±7.5 kyr, respectively, online Table 3) indicate time depths compatible with the LGM. The TMRCAs of the star-shaped (pre-HV)1 topologies of Egypt and Oman (15.7±6.7 and 20.2±6.5 kyr, respectively, in online Table 3) also encompass the LGM period. The substantially older TMRCA of the mostly star-shaped UAE (pre-HV)1 network (37.8±12.1 kyr in online Table 3) may be indicative of a southward movement.
Table 2 outlines the migration summaries for the haplogroups discussed above as well the NRY results from Luis et al. (2004). The NRY data suggested that the Levantine Corridor was the sole African–Asian route for pre-historical gene flow since the LGM. With respect to four of the six mtDNA haplogroups discussed above (H, J*, N1b, and T1), there also appears to have been a substantial preference for the use of the Levantine Corridor over the Horn of Africa from the Upper Paleolithic to the Neolithic. The close proximity of Egypt and the Levant collections in the CA analysis (online Fig. 1) versus the considerably greater distance exhibited between the Ethiopian populations and the Arab groups (especially Yemen) indicates a higher degree of mtDNA affinity observed among the former than that found among the latter groups. These results may reflect a greater amount of geneflow across the Levantine Corridor compared to the Horn of Africa. In contrast, given the possible East African or South Asian origin, the data of the current study suggests that the Horn of Africa may have been the major intercontinental thoroughfare for M1 in a much earlier episode (late Middle to early Upper Paleolithic).
A possible scenario is that an LGM back migration into Africa through the Levant involved a combination of several mtDNA haplogroups (H, J, K, N1b, (pre-HV)1 and T1) as well as some NRY haplogroups (K2-M70 and R1*173). Furthermore, post-LGM transversals through the Levantine Corridor may have also involved some of these mtDNA haplogroups (particularly K, N1b, (pre-HV)1 and T1) as well as other NRY groups, G-201 and Jp12f2 (see Table 2).
Notable is the predominance of evidence (both mtDNA and NRY) for spreads into Africa compared to dispersals toward Eurasia. Furthermore, these episodes of geneflow are relatively recent (spanning the latter stages of the Late Paleolithic to the Upper Neolithic). Earlier transit into Africa is not detected. These two observations may be related. It is possible that traces of older forays out of Africa may have been erased after repeated waves of subsequent migrations in either direction. It is also likely that those early demic movements towards Eurasia encompassed a limited number of individuals and/or episodes, which makes their northerly passage difficult to detect. In contrast, incursions back to Africa involving more technologically advanced humans may have allowed a much larger number of survivors, providing much stronger signatures that are easier to trace.
Sub-Saharan mtDNA lineages
The second part of the study focuses on the distribution and possible migration patterns of sub-Saharan Africans. For Benin, the exclusively sub-Saharan mtDNA composition (L1, L2, and L3) mirrors that of the NRY profile [YCC group E in Luis et al. (2004)]. Also, as in the NRY analysis, Cameroon is a mixture of predominantly sub-Saharan mtDNA haplogroups but harbors a minor Eurasian mtDNA component as well [0.07 (0.95 CR 0.017–0.319) of mtDNA haplogroup I]. The Rwanda Hutus contains negligible Eurasian elements with respect to both the mtDNA (Fig. 1a) and NRY lineages. However, in contrast to the completely African NRY profile in Kenya (A, B, and E), Eurasian mtDNA lineages (HV, N, and U) are present in 0.12 of this sample population (Fig. 1). The results of the present study also differ from the East African mtDNA distribution of Salas et al. (2002) in which Eurasian mtDNA markers are absent. A cross population comparison (Middle Eastern, Asian, and African) revealed that these Eurasian lineages are singletons and all are several mutations away from the next closest sequence within this expanded data set. They may be a consequence of historical contact with Europeans who arrived with these mtDNA sequence patterns in tow during the European colonization of Africa within the last few centuries (McEvedy 1980; Newman 1995).
Several mtDNA haplotypes detected in the present study are associated with the Bantu expansion (Vasina 1979; McEvedy 1980; Cavalli-Sforza et al. 1994; Newman 1995). These include L1a, L1e, L2a, L3b, and L3e (Bandelt et al. 1995; Chen et al. 1995; Watson et al. 1997; Alves-Silva et al. 2000; Bandelt et al. 2001; Salas et al. 2002). Two of these, L1a and L1e, are believed to have been incorporated in the Bantu gene pool at some point in East Africa (Salas et al. 2002). Today, these latter two haplotypes are found primarily in East Africa, although L1e barely attains polymorphic status even in this region [0.06 as reported]. The present data shows a distinct longitudinal partitioning of L1a (0.09 vs. 0.01, χ2P=0.01) but not for L1e (0.02 vs. 0) as seen in the online Table 1 and Fig. 1.
According to the literature (Salas et al. 2002), L2a is pervasive throughout the African continent, representing almost 0.25 of all sub-Saharan inhabitants. However, our data indicates a distinct prevalence in West Africans (a mean of 0.31, vs. that of 0.05 in East Africa, χ2P<0.0005).
L3b is found in 0.14, 0.13, and 0.24 of Benin, Rwandan, and Kenyan subjects, respectively. Two lineage types are particularly widespread. The first (nps 16124, 16223, 16278 and 16362) corresponds two the basal L3b sequence and is largely West African, while a second HVI motif, a subset of the first, incorporates a further transition at np 16093 and is predominantly East African. The global network in online Fig. 3b (TMRCA 19±8.2 kyr) depicts a radial morphology with the nps 16124, 16223, 16278 and 16362 motifs ancestral to several lineages including the predominantly East African motif described above. Nearly all members of each of these two HVI lineage types match at HVII as well. The geographical distribution of the ancestral versus the derivative lineage implies a west-to-east direction of dispersal (as that observed in the Bantu expansion) with respect to both haplotype evolution and demographic movement. However, there is no way to assign a date to the eastward expansion depicted in the phylogeny since there is not enough L3b sequence data to calculate population-specific TMRCAs for Kenya and Rwanda.
A longitudinal gradient is also observed with the Bantu mtDNA marker, L3e (0.21, 0.14, 0.11 and 0.07 for Benin, Cameroon, Rwanda and Kenya, respectively, as shown in online Table 1). A sequence from Benin (nps 16223, 16320) forms the center of a star-like radiation in the L3e phylogeny (not shown), which contains several one-step derivatives from Benin or Cameroon. Several downstream clusters are comprised of a mixture of East and West African motifs including those common to both regions. It is tempting to propose a west-to-east diffusion of this haplotype whose expansion in Benin began 30.2±7.2 kyr, however, there is no reliable time estimation for L2e in either Rwanda or Kenya from which to determine the net direction of gene flow.
The sub-Saharan (including East African) mtDNA component (L1, L2, L3 and M1, if an East African origin is considered) averages 0.17 in the Middle Eastern collections, (online Table 1 and Fig. 1). A previous study (Perreira et al. 2001) reported a similar sub-Saharan element in Arab Middle Eastern groups (a mean of approximately 0.18). Noteworthy is the considerable sub-Saharan faction in the Egyptian sample (0.24). There are no star-like network topologies to date the Middle East or Egyptian L1, L2, or L3 expansions, although the majority of this sub-Saharan genetic contribution is believed to have been made during the course of the Arab slave trade involving the transport of African slaves through the East African trade routes in the last 2,500 years (Richards et al. 2003). However, it is conceivable that in addition to this recent influx, the enriched Egyptian sub-Saharan mtDNA presence could also represent earlier contacts with sub-equatorial Africa. This geneflow transpired from centuries of commerce conducted through the extensive White Nile waterway, which, including all of its tributaries, extends north from its mouth on the Mediterranean Sea to its southern endpoint on the Zambezi coast. Also contributing to the Egyptian sub-Saharan component is the protracted sociopolitical interactions with the Nubian Empire to the south (McEvedy 1980; Newman 1995).
There is an asymmetrical presence of sub-Saharan mtDNA (including East African) versus sub-Saharan NRY markers (Luis et al. 2004) in Egypt (0.24 vs. 0.09, respectively, χ2P<0.0025). This is possibly the situation with Oman as well (0.17 vs. 0.10, respectively), although the corresponding χ2P>0.10. This bias, which was also reported in a previous study (Richards et al. 2003), has been attributed to the preferential assimilation of sub-Saharan females by Arab groups. Several historical observations support this contention. First, until the 19th century, a much higher number of females than males were transported from sub-Saharan Africa to Middle Eastern and South Asia, usually to be recruited into harems. These females often became part of the family and, due to the Arab practice of manumission, their offspring were considered free individuals (Lewis 1992; Segal 2001; Richards et al. 2003). Second, a considerable proportion of the African males were sent to the Arabian Peninsula populations as slaves that were subjected to castration and thus, unable to contribute to the Arab gene pool (Lewis 1992; Segal 2001; Richards et al. 2003).
Natural selection and haplogroup distribution
According to one group of researchers (Mishmar et al. 2003), regional distribution of mtDNA haplogroups throughout Eurasia and Africa may have been shaped by natural selection acting on the oxidative phosphorylation pathway in response to climatic conditions. In light of this possibility, we conducted the Ewens and Watterson’s homozygosity test of selective neutrality on the current HVI and HVII sequence data, which is equally applicable to both diploid and haploid data (Kimura 1968). The results, which are presented in online Table 2, indicate no significant departure from neutrality or population mutation–drift equilibrium for any of the ten collections with respect to the HVI alone and HVI and HVII combined at the α=0.05 level. However, the lack of statistical significance may also be due to the reduced power of this analysis (Fu 1997).
Conclusion
There is ample evidence of travel across the two Asian–African passageways. Furthermore, the distribution of four of the six haplogroups presented above (H, J*, N1b and T1) indicates the preferred usage of the Levantine Corridor over the Horn of Africa route. We propose that one or several Eurasia-to-Africa migrations incorporating these four mtDNA haplogroups as well as K and (pre-HV)1 and a few NRY haplogroups (K2-M70 and R1*173) most likely occurred during the LGM or earlier. In addition, the mtDNA haplogroups, N1b and T1, may have been associated with later migrations (Neolithic and/or Mesolithic) through the Levantine Corridor along with other NRY haplogroups (G-201 and Jp12f2). In contrast to the other haplogroups examined, the phylogeographic distribution of M1 in conjunction with the Ethiopian M1 TMRCA (Maca-Meyer et al. 2001) suggests a late middle to early Upper Paleolithic transversal across the Horn of Africa.
The mtDNA distribution of the sub-Saharan populations mirrors that of NRY results (Luis et al. 2004). The sub-Saharan L2a, L3b, and L3e frequency patterns suggest west-to-east population movements as occurred with the Bantu expansion (Vasina 1979; McEvedy 1980; Newman 1995). Also, two Bantu markers believed to be east sub-Saharan African in origin are more prevalent in Kenya and Rwanda versus the west African collections (Benin and Cameroon), an observation supporting the theorized population growth of the Bantu migrants in the eastern sub-equatorial Africa (Vasina 1979; McEvedy 1980; Newman 1995). There is a substantial sub-Saharan mtDNA faction in the six Middle Eastern populations similar to that found in Perreira et al. (2001). Also, there is an apparent asymmetry in favor of the mtDNA over the NRY sub-Saharan contribution to the Egyptian and possibly to that of the Omani gene pool as well, which suggests a greater biological and cultural assimilation of sub-Saharan females by Arab groups.
References
Achilli A, Rengo C, Magri C, Battaglia V, Olivieri A, Scozzari R, Cruciani F (2004) The molecular dissection of mtDNA haplogroup H confirms that the Franco-Cantabrian glacial refuge was a major source for the European gene pool. Am J Hum Genet 75:910–918
Alves-Silva J, Santos M, Guimarães P, Ferreira AC, Bandelt HJ, Pena SD, Prado VF (2000) The ancestry of Brazilian mtDNA lineages. Am J Hum Genet 67:444–461
Al-Zahery N, Semino O, Benuzzi G, Magri C, Passarino G, Torroni A, Santachiara-Benerecetti AS (2003) Y-chromosome and mtDNA polymorphisms in Iraq, a crossroad of the early human dispersal and of post-Neolithic migrations. Mol Phylogenet Evol 28:458–472
Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, Schreier PH, Smith AJ, Staden R, Young IG (1981) Sequence and organization of the human mitochondrial genome. Nature 290:457–465
Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N (1999) Reanalysis and revision of the Cambridge reference sequence for human mitochondrial. DNA Nat Genet 23:147
Antunez de Mayolo A, Antunez de Mayolo G, Thomas E, Reategui EP, Brown MD, Herrera RJ (1999) Worldwide distribution of a polymorphic Alu insertion in the progesterone receptor gene. In: Papiha SS, Deka R (eds) Genomic diversity: applications in human population genetics. Plenum Publishing, New York, pp 213–226
Bandelt HJ, Forster P, Sykes BC, Richards MB (1995) Mitochondrial portraits of human populations using median networks Genetics 141:743–753
Bandelt H-J, Alves-Silva J, Guimaraes P, Santos M, Brehm M, Pereira L, Coppa A, Larruga JM, Rengo C, Scozzari R, Torroni A, Prata MJ, Amorim A, Prado VF, Pena SDJ (2001) Phylogeography of the human mitochondrial L3e: a snapshot of African prehistory and the Atlantic slave trade. Ann Hum Genet 65:549–563
Camps G (1982) Beginnings of pastoralism and cultivation in north-west Africa and the Sahara: origins of the Berbers. In: Clark JD (ed) The Cambridge history of Africa, vol 1. Cambridge University Press, Cambridge, pp 548–623
Camps-Fabrer G (1974) Les civilisations préhistoriques de l´Afrique du Nord et du Sahara. Dion, París
Cavalli-Sforza LL, Piazza A, Menozzi P (1994) The history and geography of human genes. Princeton University Press, Princeton
Chen YS, Torroni A, Excoffier L, Santachiara-Benerecetti AS, Wallace DC (1995) Analysis of mtDNA variation in African populations reveals the most ancient of all human continent-specific haplogroups. Am J Hum Genet 57:133–149
Ewens WJ (1972) The sampling theory of selectively neutral alleles. Theor Popul Biol 3:87–112
Fadhlaoui-Zid K, Plaza S, Calafell F, Ben Amor M, Comas D, Bennamar El gaaied A (2004) Mitochondrial heterogeneity in Tunisian Berbers. Ann Hum Genet 68:222–233
Fagan B (1990) Journey from Eden: the peopling of our world. Thames and Hudson, New York
Forster P (2004) Ice ages and the mitochondrial DNA chronology of human dispersals: a review. Philos Trans R Soc Lond B Biol Sci 359:255–264
Forster P, Harding R, Torroni A, Bandelt H-J (1996) Origin and evolution of Native American mtDNA variation: a reappraisal. Am J Hum Genet 59:935–945
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147:915–925
Kimura M (1968) Evolutionary rate at the molecular level. Nature 217:624–626
Kimura M (1969) The number of heterozygous nucleotide sites maintained in a finite population due to steady flux of mutations. Genetics 61:893–903
Kimura M (1971) Theoretical foundation of population genetics at the molecular level. Theor Popul Biol 2:174–208
Kivisild T, Reidla M, Metspalu E, Rosa A, Brehm A, Pennarun E, Parik J, Gerberhiwot T, Usanga E, Villems R (2004) Ethiopian mitochondrial DNA heritage: tracking gene flow across and around the Gate of Tears. Am J Hum Genet 75:752–770
Lahr M, Foley R (1994) Multiple dispersals and modern human origins. Evol Anthropol 3:48–60
Lewis B (1992) Race and slavery in the Middle East: an historical enquiry. Oxford University Press, Oxford
Luis JR, Rowold DJ, Regueiro M, Caeiro B, Cinnioğlu C, Roseman C, Underhill PA, Cavalli-Sforza LL, Herrera RJ (2004) The Levant versus the Horn of Africa: evidence for bidirectional corridors of human migrations. Am J Hum Genet 74:532–544
Maca-Meyer N, Gonzalez A, Larruga J, Flores C, Cabrera V (2001) Major genomic mitochondrial lineages delineate early human expansions. BMC Genetics 2:13
Macaulay VA, Richards MB, Hickey E, Vega E, Cruciani F, Guida V, Scozzari R, Bonné-Tamir B, Sykes B, Torroni A (1999) The emerging tree of West Eurasian of West Eurasian mtDNAs: a synthesis of control region sequences and RFLPs. Am J Hum Genet 64:232–249
McEvedy C (1980) The penguin atlas of African history. Penguin Books Ltd, London
Mishmar D, Ruiz-Pesini E, Golik P, Macaulay V, Clark AG, Hosseini S, Brandon M, Easley K, Chen E, Brown MD, Sukernik RI, Olckers A, Wallace DC (2003) Natural selection shaped regional mtDNA variation in humans. Proc Natl Acad Sci USA 100:171–176
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Newman (1995) The peopling of Africa: a geographic interpretation. Yale University Press, New Haven
Olivieri A, Achilli A, Pala M, Battaglia V, Fornarino S, Al-Zahery N, Scozzari R, Cruciani F, Behar DM, Dugoujon JM, Coudray C, Santachiara-Benerecetti AS, Semino O, Bandelt HJ, Torroni A (2006) The mtDNA legacy of the Levantine early Upper Paleolithic in Africa. Science 314:1767–1770
Oppenheimer S (2003) Out of Eden: the peopling of the world. Constable, London
Perreira L, Macaulay V, Torroni A, Scozzari R, Prata MJ, Amorim A (2001) Prehistoric and historic traces of mtDNA of Mozambique: insights into the Bantu expansions and the slave trade. Ann Hum Genet 65:439–458
Quintana-Murci L, Semino O, Bandelt HJ, Passarino G, McElreavey K, Santachiara-Benerecetti AS (1999) Genetic evidence of an early exit of Homo sapiens sapiens from Africa through eastern Africa. Nat Genet 23:437–441
Quintana-Murci L, Chaix R, Wells RS, Behar DM, Sayar H, Scozzari R, Rengo C, Al-Zahery N, Semino O, Santachiara-Benerecetti AS, Coppa A, Ayub Q, Mohyuddin A, Tyler-Smith C, Mehdi SQ, Torroni A, McElreavey K (2004) Where West meets East: the complex mtDNA landscape of the southwest and central Asian corridor. Am J Hum Genet 74:827–845
Rando JC, Pinto F, Gonzalez AM, Hernadez M, Larruga JM, Cabrera VM, Bandelt HJ (1998) Mitochondrial DNA analysis of northwest African populations reveals genetic exchanges with European, near-eastern and sub-Saharan populations. Ann Hum Genet 62:531–550
Richards MB, Macaulay V, Bandelt HJ, Sykes BC (1998) Phylogeography of mitochondrial DNA in Western Europe. Ann Hum Genet 62:241–260
Richards M, Macaulay V, Hickey E, Vega E, Sykes B, Guida V, Rengo C et al (2000) Tracing European founder lineages in the Eastern mtDNA pool. Am J Hum Genet 67:1251–127
Richards M, Rengo C, Cruciani F, Gratix F, Wilson JF, Scozzari R, Macaulay V, Torroni A (2003) Extensive female lineage-mediated gene flow from sub-Saharan into Near Eastern Arab populations. Am J Hum Genet 72:0158–1064
Rohlf F (2002) NTSYSpc. Exeter Publishing Ltd, Setauket
Rosa A, Brehm A, Kivisild T, Metspalau M, Villems R (2004) MtDNA profile of West Africa Guineans: towards a better understanding of the Senegambia region. Ann Hum Genet 68:340–352
Saillard J, Forster P, Lynnerup N, Bandelt H-J, Nørby S (2000) mtDNA variation among Greenland Eskimos: the edge of the Beringian expansion. Am J Hum Genet 67:718–726
Salas A, Richards M, De la Fe T, Lareu M-V, Sobrino B, Sanchiz-Diz P, Macaulay V, Carracedo A (2002) The making of the African mtDNA landscape. Am J Hum Genet 71:1082–1111
Salas A, Richards M, Lareu M-V, Scozzari R, Coppa A, Torroni A, Macaulay V, Carracedo A (2004) The Africa diaspora: mitochondrial DNA and the Atlantic slave trade. Am J Hum Genet 74:454–465
Schneider S, Roessli D, Excoffier L (2000) Arlequin version 2.000: a software for population genetics data analysis. Genetics and Biometry Laboratory, University of Geneva, Switzerland
Segal R (2001) Islam’s black slaves. Atlantic, London
Siddall M, Rohling EJ, Almogi-Labin A, Hemleben C, Meischner D, Schmelzer I, Smeed DA (2003) Sea level fluctuations during the last glacial cycle. Nature 423:853–858
Stoneking M, Hedgecock D, Higuch RG, Vigilant L, Erlich HA (1991) population variation of human control region sequences detected by enzymatic amplification and sequence-specific oligonucleotide probes. Am J Hum Genet 48:370–382
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Torroni A, Huoponen K, Francalacci P, Petrozzi M, Morelli L, Scozzari R, Obinu D, Savontaus M-L, Wallace DC (1996) Classification of European mtDNAs from an analysis of three European populations. Genetics 144:1835–1850
Underhill PA, Shen P, Lin AA, Jin L, Passarino G, Yang WH, Kauffman E et al (2000) Y chromosome sequence variation and the history of human populations. Nat Genet 26:358–61
Vasina J (1979) Bantu in a crystal ball. Hist Afr 6:287–333
Watson E, Forster P, Richards M, Bandelt HJ (1997) Mitochondrial footprints of human expansions in Africa. Am J Hum Genet 61:601–704
Watterson G (1975) On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7:256–276
Watterson G (1978) The Homozygosity test of neutrality. Genetics 88:405–417
Acknowledgments
We are grateful to B. Caeiro for the Benin sample and to M. Tahir for the Jordan sample collections used in this study. We are also indebted to Rachel Chow and Erica Shepard for their work on the RFLP analysis and to Arif Kalantar for performing the HVI and HVII sequencing analysis on the UAE populations. Lastly, we are deeply grateful to all of the people who donated blood specimens for this and other similar projects.
Author information
Authors and Affiliations
Corresponding author
Additional information
D. J. Rowold and J. R. Luis have contributed equally to this manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Rowold, D.J., Luis, J.R., Terreros, M.C. et al. Mitochondrial DNA geneflow indicates preferred usage of the Levant Corridor over the Horn of Africa passageway. J Hum Genet 52, 436–447 (2007). https://doi.org/10.1007/s10038-007-0132-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-007-0132-7
Keywords
This article is cited by
-
Replication analysis of variants associated with multiple sclerosis risk
Scientific Reports (2020)
-
Population genetic diversity in an Iraqi population and gene flow across the Arabian Peninsula
Scientific Reports (2020)
-
Human maternal heritage in Andalusia (Spain): its composition reveals high internal complexity and distinctive influences of mtDNA haplogroups U6 and L in the western and eastern side of region
BMC Genetics (2014)
-
Multiplex genotyping system for efficient inference of matrilineal genetic ancestry with continental resolution
Investigative Genetics (2011)
-
Mitochondrial DNA and Y-chromosome microstructure in Tunisia
Journal of Human Genetics (2011)
