
Abstract
We investigated the in vivo effects of a novel aquaporin 4 (AQP4) inhibitor 2-(nicotinamide)-1,3,4-thiadiazole, TGN-020, in a mouse model of focal cerebral ischemia using 7.0-T magnetic resonance imaging (MRI). Pretreatment with TGN-020 significantly reduced brain edema associated with brain ischemia, as reflected by percentage of brain swelling volume (%BSV), 12.1 ± 6.3% in the treated group, compared to (20.8 ± 5.9%) in the control group (p < 0.05), and in the size of cortical infarction as reflected by the percentage of hemispheric lesion volume (%HLV), 20.0 ± 7.6% in the treated group, compared to 30.0 ± 9.1% in the control group (p < 0.05). The study indicated the potential pharmacological use of AQP4 inhibition in reducing brain edema associated with focal ischemia.
Similar content being viewed by others
Introduction
Cerebral edema is one of the well recognized potentially serious complications of cerebral ischemia [1]. While edema is believed to be an important factor of the high morbidity and mortality of large territory ischemic strokes, clinical options for treating cerebral edema remain limited [2]. Accumulated evidence now indicates that aquaporin 4 (AQP4), the most abundant water channel in brain, plays an essential role in the pathogenesis of cerebral edema [3]. AQP4 belongs to the aquaporin family of transmembrane water and water/glycerol transporters [4] and is found in high concentrations in mammalian astrocytes, particularly in the periventricular region and subpial endfeet [5]. Genetic deletion of AQP4 ameliorates brain swelling due to ischemia [6]. Transgenic mice lacking the dystrophin-associated protein complex, by which end-foot pooled AQP4 is anchored [7], show less severe ischemic edema compared to wild-type mice [8]. Genetic deletion of α-syntrophin, which is a member of the dystrophin-associated protein complex, also reduces ischemic edema [9, 10].
Recently, we identified several chemical structures with AQP4 inhibitory effect [11, 12]. Using an in vitro bioassay, 2-(nicotinamide)-1,3,4-thiadiazole, TGN-020, was found to be the most potent AQP4 inhibitor among the agents studied [13]. In this study, we investigated the in vivo effects of TGN-020 on cerebral edema in a mouse model of focal cerebral ischemia using 7.0-T magnetic resonance imaging (MRI) in an attempt to find new pharmacological agents effective for treating ischemic cerebral edema.
Materials and methods
Animal preparation
All animal experiments were conducted in accordance with the National Institutes of Health guidelines for the care and use of animals in research [14], and used protocols approved by the University of Niigata Animal Care and Use Committee. Twenty-four adult male mice, C57/BL6 (weight 23–28 g) were maintained under standard laboratory conditions with a 12 h/12 h light/dark cycle. Food and water were available ad libitum, except for 10 h prior to MCA occlusion, during which food was withheld to prevent hyperglycemia.
Mice were anesthetized with 1–1.2% halothane in 30% oxygen and 70% nitrous oxide administered through a face mask, with the animals breathing spontaneously. Rectal temperature was maintained at 37 ± 0.5°C using a temperature control unit and heating pad during the anesthesia period. Oxygen saturation (SpO2) was monitored throughout the operation procedure using a pulse oxymeter Mouse Ox (STARR Life Sciences Co, Oakmont, PA, USA) with probe placement on the left thigh. Regional cerebral blood flow (rCBF) was measured continuously starting immediately prior to and throughout the 120-min interval of induced focal ischemia using laser-Doppler flowmetry (ALF21, Advance Co., Tokyo, Japan). The Doppler probe was affixed to the skull 1 mm posterior and 6 mm laterally to the Bregma. Study animals received a single injection, intra-peritoneally, of TGN-020 sodium salt, 200 mg/kg, dissolved in 0.1 ml normal saline, 15 min before induction of ischemia. Control animals received an identical volume of normal saline, intra-peritoneally. An additional seven mice underwent sham operation where only isolation of the carotid artery was performed.
Transient focal cerebral ischemia was induced using a modified version of the procedure described by Yang et al. [15]. The right middle cerebral artery (MCA) was occluded by introducing a 6-0 silicone-coated monofilament into the internal carotid artery to a point 6 mm distal to the internal carotid artery and pterygopalatine arterial bifurcation. Success of the MCA occlusion was confirmed in mice fulfilling the following three conditions: a greater than 93% of SpO2 throughout the operation procedure; a greater than 80% decrease in rCBF (CBF%) at 15–20 min following the ischemic insult relative to pre-ischemia level; and a neurological deficit score of 2 or greater at 30 min post-ischemia after allowing the mouse to regain consciousness. Neurological deficit scoring was done using the criteria established by Amiry-Moghaddam et al. [9]: normal motor function, 0; flexion of the torso and contralateral forelimb upon lifting the animal by the tail, 1; circling to the contralateral side but normal posture at rest, 2; leaning to the contralateral side at rest, 3; no spontaneous motor activity, 4. The filament was removed under anesthesia 2 h after induction of ischemia.
MRI
MRI studies were carried out 24 h after induction of ischemia on a 15-cm bore, 7-T horizontal magnet (Magnex Scientific, Abingdon, UK) equipped with a Varian Unity-INOVA-300 system (Varian Inc, Palo Alto, CA, USA) that had an actively shielded gradient. Anesthetized mice (1–1.2% halothane in 30% oxygen and 70% nitrous oxide administered through a face mask), breathing spontaneously, were positioned on their back in a tailor-made Plexiglas stereotaxic holder. The head was immobilized using ear and tooth bars. Rectal temperature was maintained at 37 ± 0.5°C using a specially designed temperature controlling system.
T2-weighted images were acquired using a fast-spin echo (FSE) sequence with ETE/TR = 80/2,000 ms; echo train, 4; field of view, 20 × 20 mm; matrix size, 256 × 256; and slice thickness, 1 mm without gap. 12 contagious slices were obtained to cover the entire brain volume.
Image and statistical analysis
Imaging data were transferred to a workstation (Blade1000, Sun Microsystems, Milpitas, CA, USA) and analyzed using MR Vision (MR Vision Co., Menlo Park, CA, USA). Quantitative indices of edema and infarction were expressed as percentage of brain swelling volume (%BSV) and hemispheric lesion volume (%HLV), respectively, as described by Gerriets et al. [16]. To correct the overestimation of infarcted area by cerebral edema, the relative size of the infarct area (% hemispheric lesion volume = %HLV) was derived as follows:
Corresponding volumes were then calculated for the total set of slices. Quantification of the effect of cerebral swelling was then expressed as the volume increase of the ipsilateral hemisphere (% brain swelling volume = %BSV) based on the following equation:
The size of the corresponding area on each imaging slice was quantified on the FSE image semi-automatically. Numerical data were subsequently subjected to two-way ANOVA with Fisher’s post hoc test; p less than 0.05 was regarded as being statistically significant.
Results
Physiological parameters and rCBF following ischemia induction monitored by laser-Doppler flowmetry were virtually identical between control and TGN-020 treated groups (Table 1). Three out of 17 mice did not fit the inclusion criteria (one mouse exhibited respiratory distress with an SpO2 under 94%, while the other two did not meet the minimum neurological deficit score). Thus, seven animals each of the vehicle and TGN-020 administered groups were analyzed by MRI. All mice meeting the inclusion criteria survived to the end of the MRI measurements.
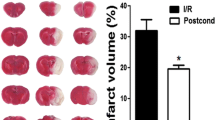
Representative FSE images are shown in Fig. 1. Results are summarized in Fig. 2. The %BSV of the TGN-020 group (12.1 ± 6.3%) was significantly smaller than that of the control group (20.8 ± 5.9%). Likewise, the %HLV of cortex in the TGN-020 group (20.0 ± 7.6%) was also significantly smaller than that in the control group (30.0 ± 9.1%). The basal ganglia infarction volume in treated and control animals showed essentially no difference (Table 1). All sham operated mice showed no apparent infarction area.

Representative FSE (TR = 2,000 ms, ETE = 80 ms) images of control (No. 3) and TGN-020 treated mouse (No. 5). Note that midline shift due to ischemic brain swelling is much less conspicuous in the TGN-020 treated mouse

a Effect of TGN-020 on brain swelling at 24 h after induction of transient (2 h) focal ischemia. *p < 0.05. b Effect of TGN-020 on infarct volume at 24 h after induction of transient (2 h) focal ischemia. T total, B basal ganglia, C cortex. *p < 0.05
Discussion
The current study convincingly demonstrated that pretreatment with the AQP4 inhibitor TGN-020 significantly reduced the volume of brain edema associated with ischemic injury. Ischemic edema is believed to be initiated by influx of Na+ associated with energy failure. Higher osmolarity conditions create the driving force for water influx into cells, resulting in ionic edema [17]. This early edema phase, so-called cytotoxic edema, is believed to last several hours before bulk leakage of water into the brain ensues, producing so-called vasogenic edema [18]. AQP4 is believed to play a significant role in the actual water flux in both processes.
Flux through AQP4 is bidirectional and purely dependent on osmolarity differences between the two spaces connected by AQP4. Theoretically, pretreatment with an AQP4 inhibitor can block water movement through these water channels, thereby reducing the main water influx associated with influx of Na+, and delaying the development of full-fledged ionic edema. Although speculative, halting the early process mediating brain edema, may interrupt the subsequent cascade of events that lead to further edema such as astrocytic process swelling, up-regulation of astrocytic volume-regulated anion channel (VRAC) [19, 20], or further influx of osmotically active ions [18]. An interesting observation in this study was the differential effect on infarct size by AQP4 inhibition. Whereas TGN-020 significantly reduced the cortical infarct volume (p < 0.05), it essentially had no effect on basal ganglia infarct volume. While entirely conjectural, the pathophysiologic mechanism underlying the observations may reflect the differential distribution of AQP4 channels in perivascular regions and cortical surfaces [3]. It is plausible that the difference in infarction volume between control and AQP4 treated animals denoted the effect of edema on infarction, and, therefore, represented potentially salvageable tissue by AQP4 inhibition. On the other hand, the similar %HLV, reflecting infarct volume for basal ganglia in both control and treated animals as well as for cortex in treated animals only, likely represented the direct result of ischemia.
In conclusion, we assessed the in vivo effect of a novel AQP4 inhibitor, TGN-020. Pretreatment with TGN-020 significantly reduced the severity of brain edema associated with brain ischemia. Further studies that better simulate clinical treatment conditions are essential. Nevertheless, AQP4 inhibition is a novel approach in reducing cerebral edema, and appears promising for the development of a unique class of pharmacologic agents effective in the clinical treatment of brain ischemia.
References
Hacke W, Schwab S, Horn M et al (1996) ‘Malignant’ middle cerebral artery territory infarction: clinical course and prognostic signs. Arch Neurol 53:309–315
Kimelberg HK (1995) Current concepts of brain edema. Review of laboratory investigations. J Neurosurg 83:1051–1059
Papadopoulos MC, Verkman AS (2007) Aquaporin-4 and brain edema. Pediatr Nephrol 22:778–784
Agre P (2007) The aquaporin water channels. Proc Am Thorac Soc 3:5–13
Nielsen S, Nagelhus EA, Amiry-Moghaddam M et al (1997) Specialized membrane domains for water transport in glial cells: high-resolution immunogold cytochemistry of aquaporin-4 in rat brain. J.Neurosci 17:171–180
Manley GT, Fujimura M, Ma T et al (2006) Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 6:159–163
Neely JD, Amiry-Moghaddam M, Ottersen OP et al (2001) Syntrophin-dependent expression and localization of aquaporin-4 water channel protein. Proc Natl Acad Sci USA 98:14108–14113
Vajda Z, Pedersen M, Fuchtbauer EM et al (2002) Delayed onset of brain edema and mislocalization of aquaporin-4 in dystrophin-null transgenic mice. Proc Natl Acad Sci USA 99:13131–13136
Amiry-Moghaddam M, Otsuka T et al (2003) An alpha-syntrophin-dependent pool of AQP4 in astroglial end-feet confers bidirectional water flow between blood and brain. Proc Natl Acad Sci USA 100:2106–2111
Liu X, Zhang W, Alkayed NJ, Froehner SC et al (2008) Lack of sex-linked differences in cerebral edema and aquaporin-4 expression after experimental stroke. J Cereb Blood Flow Metab 28:1898–1906
Huber VJ, Tsujita M, Yamazaki M et al (2007) Identification of arylsulfonamides as aquaporin 4 inhibitors. Bioorg Med Chem Lett 17:1270–1273
Huber VJ, Tsujita M, Kwee IL, Nakada T (2009) Inhibition of aquaporin 4 by antiepileptic drugs. Bioorg Med Chem 17:418–424
Huber VJ, Tsujita M, Nakada T (2009) Identification of aquaporin 4 inhibitors using in vitro and in silico methods. Bioorg Med Chem 17:411–417
National Research Council (1996) Guide for the care and use of laboratory animals. National Academies Press, Washington, DC
Yang GY, Chan PH, Chen J et al (1994) Human copper–zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke 25:165–170
Gerriets T, Stolz E, Walberer M et al (2004) Noninvasive quantification of brain edema and the space-occupying effect in rat stroke models using magnetic resonance imaging. Stroke 35:566–571
Simard JM, Kent TA, Chen M et al (2007) Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol 6:258–268
Mori K, Miyazaki M, Iwase H, Maeda M (2002) Temporal profile of changes in brain tissue extracellular space and extracellular ion (Na+, K+) concentrations after cerebral ischemia and the effects of mild cerebral hypothermia. J Neurotrauma 19:1261–1270
Kimelberg HK, Goderie SK, Higman S et al (1990) Swelling-induced release of glutamate, aspartate, and taurine from astrocyte cultures. J Neurosci 10:1583–1591
Rutledge EM, Aschner M, Kimelberg HK (1998) Pharmacological characterization of swelling-induced d-[3H]aspartate release from primary astrocyte cultures. Am J Physiol 274(6 Pt 1):C1511–C1520
Acknowledgments
The study was supported by grants from the Ministry of Education, Culture, Sports, Science, and Technology, and University of Niigata.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Igarashi, H., Huber, V.J., Tsujita, M. et al. Pretreatment with a novel aquaporin 4 inhibitor, TGN-020, significantly reduces ischemic cerebral edema. Neurol Sci 32, 113–116 (2011). https://doi.org/10.1007/s10072-010-0431-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10072-010-0431-1