
Abstract
Individual differences in the rate of aging are determined by the efficiency with which an organism transforms resources into metabolic energy thus maintaining the homeostatic condition of its cells and tissues. This observation has been integrated with analytical studies of the metabolic process to derive the following principle: The metabolic stability of regulatory networks, that is the ability of cells to maintain stable concentrations of reactive oxygen species (ROS) and other critical metabolites is the prime determinant of life span. The metabolic stability of a regulatory network is determined by the diversity of the metabolic pathways or the degree of connectivity of genes in the network. These properties can be empirically evaluated in terms of transcriptional changes in gene expression. We use microarrays to investigate the age-dependence of transcriptional changes of genes in the insulin signaling, oxidative phosphorylation and glutathione metabolism pathways in mice. Our studies delineate age and tissue specific patterns of transcriptional changes which are consistent with the metabolic stability–longevity principle. This study, in addition, rejects the free radical hypothesis which postulates that the production rate of ROS, and not its stability, determines life span.
Similar content being viewed by others


Introduction
Aging in its broadest sense is a time-related process resulting in a progressive loss of function in an adult individual. The process is described by an increased probability of mortality with advancing age. This progressive impairment of physiological function with time has both a genetic and an environmental component (Finch 1990). The environmental component is indicated by the dramatic increases in mean life span observed in human populations during the last 150 years. The influence of genetic effects on survival is indicated by the large interspecific variation in maximal life span. Within the mammalian lineage, for example, maximal life span ranges from 4 years in the case of mice to 122 years in humans. There also exist significant variation in mortality rates: an exponential increase in mortality with age is typical; however, in many species, mortality rate is known to abate with age at advanced ages (Vaupel et al. 1998). Two main questions arise from these empirical observations:
-
1.
What are the evolutionary mechanisms that underlie the large interspecific variations in maximal life span?
-
2.
What are the developmental processes that underlie the observed individual variations in maximum age of death?
These two questions have been addressed in a mathematical model of the aging process (Demetrius 2004). This model invokes developmental and evolutionary arguments to relate the stability of metabolic networks in cells—a molecular level property—with individual and interspecific variations in life span—organismic and population level properties. This article is concerned with the empirical evaluation of the predictions of the mathematical model.
Studies of evolutionary logic of aging and its molecular basis have a long history. We briefly delineate this history in order to better situate the prediction of the metabolic stability theory and its empirical support. We refer to (Dietz 2005; Olshansky and Rattan 2005) for a critical account of some of the concepts that underlie the various theories.
Evolutionary models of aging are concerned with explaining the interspecific variation in age-specific fecundity and mortality that characterize natural populations. These models necessarily revolve around the concept of Darwinian fitness, that is the efficiency with which organisms acquire and transform resources into net-offspring production. The explanatory power of these evolutionary models is largely dependent on the measure of fitness invoked (Dietz 2005; Demetrius and Ziehe 2007).
Evolutionary arguments which appeal to the claim that fitness is determined by the population growth rate (the Malthusian parameter) include the following three models, mutation-accumulation (Medawar 1952), antagonistic pleiotropy (Williams 1957), disposable soma (Kirkwood 1977). These qualitative studies of aging have each provided insights into various observed relations between fecundity and mortality schedules in populations. However, as delineated by Hamilton (1966), this class of models, because of the Malthusian condition they implicitly assume, entail that the intensity of selection declines with age—a property which is inconsistent with the existence of mortality plateaus, that is, the deceleration of mortality rates at extreme ages.
The evolutionary argument based on the analytical and empirical fact that Darwinian fitness is characterized by demographic stability (a property measured by the statistical parameter evolutionary entropy) is a quantitative model which analytically predicts empirically observed relations between age-specific fecundity and mortality schedules. In the context of this model, the intensity of selection is a convex function of age—a condition which explains the incidence of mortality plateaus (Demetrius 2001; Demetrius et al. 2007). Accordingly, evolutionary models based on entropy as Darwinian fitness, have a wider explanatory range than competing models of aging which appeal to the Malthusian parameter as the fitness measure (Dietz 2005; Braeckman et al. 2006; Demetrius and Ziehe 2007).
Developmental models of aging are concerned with the molecular and biochemical mechanisms that regulate cell functions. The thrust of these models is the characterization of the efficiency with which cells acquire and transform resources into metabolic function. Their explanatory power derives in part from the measures of metabolic efficiency adduced. The free radical theory of aging postulated by Harman (1956) cited the deleterious effect of ROS on cellular components to propose that the production rate of ROS regulates metabolic efficiency, and hence determines the rate of aging.
The metabolic stability theory of aging proposed in Demetrius (2004) derives partly from the observation that ROS has two kinds of effects on metabolic activity. First, these small diffusible molecules can interact with DNA and RNA to impair cell function. Second, the molecules can also act as second messengers in signal transduction processes (Finkel and Holbrook 2000). The dual effect of ROS suggests that the ability of the cell to maintain stable concentrations of ROS may play a critical role in maintaining the functional integrity of the cell and consequently, the rate of aging of the organism.
The metabolic stability theory of aging formalizes these ideas and appeals to studies of the evolutionary logic of aging (Demetrius 2001) to relate stability properties of the metabolic networks with individual and interspecific variations in life span.
The central concept in this study of the evolutionary and developmental aspects of aging is metabolic stability—the cellular analogue of the population parameter, demographic stability. Metabolic stability describes the capacity of a cell to maintain steady state concentrations of ROS and other critical metabolites in the face of random perturbations in the rate and activity of the enzymes in the metabolic networks.
In Demetrius (2004), analytical models of the dynamics of the regulatory process of the cellular level were integrated with the dynamics of the evolutionary process at the population level to derive:
The metabolic stability–longevity principle: The metabolic stability of the cells in the regulatory network is positively correlated with life span and is the prime determinant of the rate of aging.
This article will exploit comparative studies of age-related transcriptional profiles in different organs in mice to evaluate the empirical support for this prediction. Our empirical evaluation of the metabolic stability–longevity principle will be based on distinguishing between two classes of regulatory systems.
-
1.
Dissipative networks: These metabolic pathways consist of enzymes that transduce physiologic stimuli into special metabolic programs. A canonical example of this class are the mitochondrial enzymes. This network provides most of the ATP for cellular reactions and is responsible for the majority of ROS production in cells.
-
2.
Stabilizing networks: These pathways act primarily as homeostatic controls of the production of ROS and other metabolites involved in the various metabolic programs which occur in the mitochondria. Typical example of this class of network are the DNA repair enzymes, stress response pathways and, inflammatory response proteins.
A large body of empirical evidence indicates that variations in ROS concentration does lead to transcriptional changes in gene expression (Johnson et al. 1999). Consequently changes in the activity of the dissipative and stabilizing pathways will be positively correlated with transcriptional changes in gene expression. Now, the metabolic stability of dissipative and stabilizing networks will be both species-specific and tissue-specific.
Species-specificity derives from the metabolic stability principle, namely, stability is positively correlated with maximal life span, a property of the species. Tissue-specificity follows from the fact that the stability of the regulatory network will be contingent on the physiological or homeostatic condition of the tissue—whether it has a high degree of metabolic stability, with few age-regulated genes, as in the case of the brain, or a low degree of metabolic stability with a large number of age-regulated genes, for example, the heart. See for example Zahn et al. (2007).
These observations, when integrated with the metabolic stability–longevity principle entails the following dynamical properties of gene expression in dissipative and stabilizing networks. (1) Transcriptional changes in gene expression will vary with age. (2) The nature and magnitude of the changes will be both (a) tissue-specific—the percentage of regulated genes will be small in tissues such as the brain, an organ with a strong level of homeostasis, and (b) species-specific—the percentage of regulated genes will be relatively small in tissues of long-lived species, such as humans. (3) Transcriptional changes in gene expression will be down-regulated in dissipative networks and up-regulated in stabilizing networks.
The concept metabolic entropy, a measure of the diversity of the metabolic pathways of energy and information flow in cells, is positively correlated with metabolic stability (Demetrius and Manke 2005). Now the relative contribution of a gene to metabolic entropy increases with the connectivity of the gene, that is the number of interaction it has with other genes in the network (Manke et al. 2006). These observations can be allied with the metabolic stability–longevity principle to predict what we call:
The connectivity–lifespan principle: The sensitivity of life span to mutations in a gene in the network increases with the connectivity of the gene.
This article applies microarray-based analysis of gene expression and regulation in mice to assess the empirical validity for these predictions.
Experimentalists, motivated by an interest in understanding the dynamics of gene expression and its age-dependent effects have carried out studies complementary in some sense and overlapping in others, with the studies described in this paper. Earlier works (Lee et al. 1999; Cao et al. 2001; Jiang et al. 2001; Amador-Noguez et al. 2004; Fu et al. 2006; Edwards et al. 2007; Misra et al. 2007; Zahn et al. 2007) based on microarray studies using different platforms have demonstrated changes in gene expression with age. In all of these studies, various tissues derived from mice of varying ages and strains were analyzed. A consequence of this is the noticeable differential expression in age-regulated target genes.
The study (Zahn et al. 2007) provides the most extensive data to date. It encompasses 16 different tissues and 4 distinct ages of the same mouse strain. Their data suggest that there are indeed major differences in aging mechanisms between different tissues. Additionally, they also provide evidence that the nature and magnitude of transcriptional changes is age and tissue specific. An important finding is the significant differences in age-regulation in mice and humans, an observation consistent with the prediction made by Demetrius in (2004) that humans and mice differ in terms of their life-history and metabolic stability, hence they will age at different rates.
The studies described in this paper were inspired by our explicit interest in assessing the empirical validity of a mathematical model of the molecular mechanisms underlying the aging process. Consequently, our methodology and some of our findings go beyond certain results described by Zahn et al. A cornerstone of our study is the analysis of transcriptional changes in the glutathione metabolism, insulin signaling and oxidative phosphorylation networks. In the context of our model, glutathione metabolism and insulin signaling are stabilizing networks, whereas oxidative phosphorylation is a dissipative network. Our analysis of gene expression in these networks will be the basis for our evaluation of the metabolic stability theory of aging.
Materials and methods
Mice
Healthy wild type female C57BL6 mice were housed in a room with controlled photoperiod and temperature. Animals were given free access to water and pelleted diet. Duplicate brain, heart and kidney were collected from young (10–12 weeks) and aged (~14 months) mice. Mice were sacrificed by cervical dislocation; tissues were collected, flash frozen in liquid nitrogen and stored at −80°C.
RNA isolation
Total RNA was isolated from brain, heart and kidney using the NucleoSpin® RNA/Protein Kit (Macherey–Nagel, Düren, Germany) and following the manufacturers protocol. Tissue homogenization was performed using the TissueLyser and 5 mm Stainless Steel Beads (Qiagen, Hilden, Germany) and homogenizing 2 times for 30 /s and with a frequency of 30/s. The quantity and quality of the RNA was determined using the NanoDrop (NanoDrop Technologies, Wilmington, DE, USA).
Illumina bead chip hybridisations and analysis of mouse expression data
Biotin-labelled cRNA was produced using a linear amplification kit (Ambion, Austin, TX, US) with 400 ng of quality-checked total RNA as input. Chip hybridisations, washing, Cy3-streptavidin staining, and scanning were performed on the Illumina BeadStation 500 (Illumina, San Diego, CA, US) platform using reagents and following protocols supplied by the manufacturer. cRNA samples were hybridised onto Illumina mouse-6 BeadChips. We hybridised the following samples as biological duplicates: young brain, aged brain, young heart, aged heart, young kidney and aged kidney. All basic expression data analysis was carried out using the manufacturer’s software BeadStudio 1.0. Raw data were background-subtracted and normalized using the “rank invariant” algorithm. Normalized data were then filtered for significant expression on the basis of negative control beads. Selection for differentially expressed genes was performed on the basis of arbitrary thresholds for fold changes plus statistical significance according to the Illumina t-test error model (Kuhn et al. 2004). Differentially expressed genes were further filtered according to Gene Ontology terms or mapped to Kegg pathways using DAVID 2006 (http://david.abcc.ncifcrf.gov) and FatiGO+ (http://babelomics.bioinfo.cipf.es/fatigoplus/cgi-bin/fatigoplus.cgi). For analysis, we used GenBank accession numbers represented by the corresponding chip oligonucleotides as input. In addition, we compared our target gene lists to published data (Zahn et al. 2007).
Real-Time polymerase chain reaction
For reverse transcription, 1.0 μl (1 μg/μl) of RNA was added to 1.0 μl (50 μM) Oligo-dT primer plus 8.0 μl of RNase-free water (dH2O). The mixture was spun briefly and heated to 70°C for 5 min and cooled on ice. 10.0 μl of master mix were added including the following components per reaction: 4.0 μl of 5× RT buffer, 2.0 μl of 0.1 M DTT, 2.0 μl of (10 mM) dNTP, 1.0 μl (200 U/μl) Superscript II (Invitrogen) and 1.0 μl of dH2O. After pulse spinning, incubation was carried out at 42°C for 1.5 h. Real-Time polymerase chain reaction (Real-Time PCR) was performed in 96-Well Optical Reaction Plates (Applied Biosystems, Foster City, CA, US). The PCR mix in each well included 10 μl of SYBR®Green PCR Master Mix (Applied Biosystems), 5 μl dH2O, 1.5 μl each of the forward and reverse primers (5 ng/μl) and 2 μl of single strand cDNA (2.5 ng/μl) in a final reaction volume of 20 μl. Triplicate amplifications were carried out per gene with three wells serving as negative controls without template. GAPDH and ACTB were amplified along with the target genes as endogenous controls for normalization. The PCR reaction was carried out on the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems) using the following program, stage 1, 50°C for 2 min, stage 2, 95°C for 10 min, stage 3, 95°C for 15 s and 60°C for 1 min, for 40 cycles and, stage 4, 95°C for 15 s, 60°C for 15 s and 95°C for 15 s. The last heating step in stage 4 was performed with a ramp rate of 2% in order to enable the generation of a dissociation curve of the product. The output data generated by the Sequence Detection System 2 software were transferred to Microsoft Excel for analysis. The differential mRNA expression of each gene was calculated with the comparative Ct method recommended by the manufacturer.
Western blotting
Protein extracts were isolated from brain, heart and kidney using Lysis buffer (150 mM NaCl, 10 mM Tris–HCl, 1 mM EDTA, 1% Triton-X100; pH 7.8) and the TissueLyser (Qiagen). Protein concentrations were measured using the Bradford method. Western blotting was performed according to standard procedures and using chemiluminescence detection (ECL, Amersham). Antibodies used were Abcam ab22604 (Gpx1), Ambion #4300 (Gapdh), Calbiochem #DC03L (HRP-linked), as well as Amersham NA9310 (HRP-linked).
Results
Global data analysis
We provide experimental evidence at the molecular level based on a genomics approach to support the metabolic stability hypothesis. To identify and further analyse common and tissue-specific metabolic pathways and metabolism-related biological processes altered with age in mice, RNA was isolated from the brain, heart and kidney of young (10–12 weeks) and aged (~14 months) C57/Bl6 female mice. Whole-genome gene expression analyses employing the Illumina Bead Chip technology was then used to profile the transcriptomes of these tissues. We used the rank invariant method for normalisation and the t-test error model.
Data reproducibility is demonstrated by sample correlation and clustering (Fig. 1a, b). As expected the clustering of the samples shows a good correlation within the same tissue type. Furthermore, the correlation coefficient of samples of the same tissue is high (>0.9). However, the transcriptional difference between young and aged samples of the same tissue is minor—the correlation coefficients for biological replicates range from 0.97 to 0.99 and for young versus aged samples from 0.94 to 0.99. Of the three tissue types analysed, the heart samples show the best separation between young and aged tissues. The differences between samples of the same tissue type could also reflect heterogeneity between biological replicates indicating minor differences in mRNA levels between our two age groups. This is also reflected by the results of our differential gene expression analysis between young and aged tissues.

Results of the global expression data analysis. a Clustering of all samples analysed and b the corresponding linear correlation factors. c Numbers of significant (detection > 0.99 for at least one group and P value < 0.05) changes in gene expression between young and aged tissues with ratios of 1.3 and above
Normalized data were analysed for significant (detection > 0.99 for at least one group and P value < 0.05) changes in gene expression between young and aged samples with ratios of 1.3 and above (Full data provided as Table S1 in Electronic supplementary material). To identify a significant number of age-related regulated genes for further analysis, a ratio of 1.3 was used. High fold changes in gene expression levels (fold change > 10) are hardly or even never observed with aging (Lee et al. 1999; Cao et al. 2001; Jiang et al. 2001; Amador-Noguez et al. 2004; Fu et al. 2006; Edwards et al. 2007; Misra et al. 2007). For the chosen criteria, we found more regulated genes in heart (740) and kidney (695) than in brain (448) including duplicate counts for genes represented more than once on the chip. The total number of age-related genes is shown in Fig. 1c.
The age-related gene lists (corresponding Gene accession as input) were further analysed using the Gene Annotation Tools—DAVID (http://niaid.abcc.ncifcrf.gov/) and FatiGO+ (http://babelomics.bioinfo.cipf.es/fatigoplus/cgi-bin/fatigoplus.cgi) to identify altered pathways and Gene Ontologies. Table 1 lists significant metabolic processes (GO: biological process) in brain, heart and kidney for which the respective genes within these processes are differentially regulated in young and aged mice. Several metabolic processes are regulated in an age-dependent manner. As expected from the number of regulated genes, we found fewer and more global processes for brain (e.g., protein and cellular metabolism) than in heart (e.g., phosphate and cofactor metabolism) and kidney (e.g., hexose and cellular lipid metabolism). Table 1 shows all processes that include the word metabolism. It is therefore a subgroup of the original DAVID output where we found 24 regulated processes for brain (4 metabolic processes), 88 for heart (21 metabolic processes) and 79 for kidney (22 metabolic processes). Beside the metabolic-related processes we found for instance apoptosis and transport-related processes. Table 2 lists results for Kegg and Biocarta annotated pathways in heart. In brain, only one Kegg annotated pathway (neurodegenerative disorders) was found and in kidney glutathione metabolism, natural killer cell mediated cytotoxicity, and antigen processing and presentation showed significant regulation (data not shown). For the purpose of this study, we would like to focus on the heart where we see indications of differential expression of relevant gene components of glutathione metabolism, insulin signaling and oxidative phosphorylation.
Glutathione metabolism
Glutathione (GSH) plays a critical role in many biological processes (Maher 2005). The main function/requirement of GSH and its metabolism is to regulate homeostasis by decreasing reactive oxygen species (ROS), which are produced either endogenously e.g., during respiration in the mitochondria (Murakami 2006) or exogenously e.g., by UV radiation. Three major components constitute glutathione metabolism; biosynthesis of glutathione in its reduced monomeric form (GSH), antioxidant function by forming glutathione disulfate (GSSG; oxidized glutathione), and the detoxification by conjugating toxic rests (e.g., peroxidised lipids and xenobiotics) to glutathione.
Our data on aged heart, revealed up-regulated expression of key enzymes encoded by the following genes, Gclm, Gpx1, Gpx3, Gsta2 and Gstm2 (Table 3, Fig. 2a), which were also confirmed by Real-Time-PCR (Fig. 3a). In addition, protein expression of Gpx1 was evaluated by western blotting in all three tissues. Gpx1 protein expression in heart increased with age, whereas the expression in brain and kidney was unaffected, which was also observed for gene expression (Fig. 3b). An overlapping regulation of the transcriptome and proteome will not necessarily be observed for all genes, however the overall trend of regulation in different biological processes on the gene level may also be observed at the protein level.

Age-dependent changes in glutathione metabolism (a) insulin signaling (b) and oxidative phosphorylation (c). The FatiGO+ output shows regulated genes in glutathione metabolism, insulin signaling and oxidative phosphorylation using Kegg pathways. Two different target gene lists were processed simultaneously and are shown in different colours. Green and red boxes indicate increased and decreased expression levels respectively (Coloured version is provided in the online version). The corresponding genes are shown in Table 3

Independent confirmation of the array-derived data. a Real-Time PCR confirming the expression ratios for genes involved in glutathione metabolism in the heart. b Western blot based confirmation of Gpx1 protein expression in brain, heart and kidney. The upper part shows the protein expression for all three tissues with equally loaded amounts of protein (15 μg). In the second case, 30 μg protein was loaded for brain and heart and 15 μg for kidney. The expression of Gapdh was used to monitor equal loading of protein
The synthesis of glutathione involves a 2-step ligation reaction. First glutamate–cysteine ligase (Gcl) conjugates l-glutamate and cysteine to l-gamma-glutamylcysteine, which is then further ligated to glycine by glutathione synthetase (Gs). The modifier subunit of Gcl named Gclm is 1.6-fold up-regulated in aged heart. There are several isozymes of glutathione peroxidases (Gpx) encoded by the different genes which reduce lipid hydroperoxides to their corresponding alcohols and free hydrogen peroxide to H2O. These enzymes have varied cellular location and substrate specificity. For example, glutathione peroxidase 1 (Gpx1; 2.2-fold up-regulated in aged heart) is the most abundant and found in the cytoplasm of nearly all mammalian tissues, its preferred substrate is hydrogen peroxide H2O2. The expression pattern of glutathione peroxidase 3 (Gpx3), which is 1.4-fold up-regulated in aged heart, is not as ubiquitously expressed as Gpx1 (Maher 2005). Finally, the detoxification component of glutathione metabolism is established by different glutathione S-transferases (GST). The GST family of enzymes comprises a long list of cytosolic, mitochondrial, and microsomal proteins which are capable of multiple reactions with a multitude of substrates, both endogenous and xenobiotic. Members of the alpha (Gsta2) and mu (Gstm2) class are 1.7-fold and 1.6-fold up-regulated in aged heart, respectively (Table 3).
In aged brain and kidney we could not find this significant up-regulation of Gclm, Gpx1, Gpx3, Gsta2 and Gstm2. In brain, Gclm shows constant expression levels between young and aged samples, Gpx1 and Gpx3 seem to be slightly up-regulated with age and Gsta2 and Gstm2 are not detected. In kidney, all five genes are expressed but just Gsta2 is significantly down-regulated with age (data not shown).
Insulin signaling
Insulin is a key molecule implicated in the aging process and it is known to be involved in life extension in many mutant mouse models (Kenyon 2005). Its main function is the regulation of glucose homeostasis, but it also indirectly regulates lipid homeostasis (Fig. 2b).
We found regulation of key enzymes of the insulin signaling pathway in aged heart (Foxo1, Pdpk1 and Pfkl). 3-Phosphoinositide dependent protein kinase-1 (Pdpk1; 1.4-fold up-regulated in aged heart) has a central role in mediating a variety of cascades within the insulin signaling pathway by phosphorylating many diverse kinases (King and Newton 2004). Downstream of Pdpk1, forkhead box O1 (Foxo1; 2.1-fold up-regulated in aged heart), a member of the FOXO family of forkhead transcription factors, acts as a key regulator that induces the expression of genes involved in gluconeogenesis and glycolysis in response to insulin (Aoyama et al. 2006). Additionally, the phosphofructokinase Pfkl (1.4-fold up-regulated in aged heart) catalyses a key step in glycolysis, namely the conversion of d-fructose 6-phosphate to d-fructose 1,6-bisphosphate. Furthermore, mitogen activated protein kinase kinase 1 (Map2k1) and Rap guanine nucleotide exchange factor (GEF) 1 (Rapgef1) are 0.6-fold down- and 1.4-fold up-regulated in aged heart, respectively (Table 3).
Oxidative phosphorylation
Oxidative phosphorylation is a metabolic pathway that uses energy released by the oxidation of nutrients to produce adenosine triphosphate (ATP). We found down-regulation of enzymes in four of the five complexes in the electron transport chain in aged heart. As part of complex I, the NADH dehydrogenases Ndufa5, Ndufb2, Ndufb4 and Np15 were down-regulated 0.70-, 0.51-, 0.46- and 0.77-fold, respectively. Ubiquinol–cytochrome c reductase binding protein (Uqcrb; complex III) is 0.76-fold and the cytochrome c oxidases Cox7a2 and Cox7b (complex IV) are 0.71- and 0.51-fold down-regulated. Finally, the ATP synthases Atp5a1 and Atp5f1 of complex V are 0.72- and 0.76-fold down-regulated (Table 3, Fig. 2c).
Novel and conserved age-related target genes
To identify conserved age-related genes we compared our data to the target genes identified by the most comprehensive study on age-related gene expression (Zahn et al. 2007). The result of this comparison is presented in Table S2. In addition, we provide a list of putative novel age-related target genes expressed in brain, heart and kidney (Table S3).
Discussion
The metabolic stability–longevity principle asserts that metabolic stability, that is the ability of cells to maintain stable concentrations of ROS and other key metabolites in the regulatory network, is the prime determinant of life span.
This principle was given analytical support by formulating a mathematical model of the aging process based on a formal representation of the metabolic stability concept (Demetrius 2004). The theory focuses on two classes of regulatory pathways: networks which transduce physiological stimuli by stimulating mitochondrial activity—dissipative systems; and networks which are involved in the regulation of ROS and other metabolites generated by the mitochondrial enzymes—stabilizing networks. By appealing to the metabolic stability–longevity principle, and invoking a correlation between metabolic entropy—a measure of diversity of energy flow in the network, and metabolic stability—the resilience of ROS concentrations to random perturbation in enzymatic reaction rates, we predicted a series of correlations between the following pairs of properties. (1) Transcriptional changes in gene expression and the tissue type and the taxon status of the organism. (2) The direction of regulation (up and down) and the metabolic function (dissipative or stabilizing) of the regulatory networks. (3) The patterns of interactions or connectivity of the network and the sensitivity of life span to mutations that affect the activity of genes in the network.
The empirical design described in this article is aimed at assessing the validity of these predictions by a study of gene profiling in the glutathione metabolism, insulin signaling (stabilizing networks) and oxidative phosphorylation pathways (dissipative network).
The data set in our empirical design is qualitative and comprehensive. The quality of the data set was rigorously tested in several ways including numerous statistical tests. A comparison of the expression profiles derived from biological duplicates revealed some variation which may be accounted for by, (1) technical variability attributed to the loss of transcripts during cRNA generation and hybridisations, (2) biological variability which reflects the expected heterogeneity amongst mice of even the same strain.
Gene expression in brain, heart and kidney
One of the main predictions of the theory is that transcriptional changes in gene expression will be contingent on the demographic stability of the species and the intrinsic metabolic stability of the tissue.
Accordingly, patterns of gene expression will be species-specific: they will differ between mice (opportunistic species; characterized by weak demographic stability), and humans (equilibrium species defined by strong demographic stability). In addition, the percentage of regulated genes in the network will decrease with the metabolic stability of the tissue or organ type.
The comparative studies of transcriptional changes in mice and humans reported in Zahn et al. (2007) are consistent with the prediction that patterns of gene expression are species-specific. The study reported in this paper has focused uniquely on mice. We compared gene expression in the brain, heart and kidney in young and aged mice. Our data indicates that aging of mouse brain, heart and kidney is accompanied by changes in their metabolic state. Global gene expression analysis revealed regulation of a variety of metabolic processes at the transcriptional level in all three tissues. However, the target gene lists of age-regulated genes for the different tissues do not strongly overlap on the gene level which confirms the proposed variation of organ type and age predicted by the metabolic stability theory. Additionally, the percentage of regulated genes in the group of all expressed genes in the three tissues varies with 3.26% in brain, 7.74% in heart and 6.10% in kidney. Brain, as expected to be a metabolically stable organ seems to be less regulated with age than heart and kidney. This observation is consistent with the prediction of the theory. Other studies (Fu et al. 2006; Zahn et al. 2007) investigating different tissues in parallel also found differences in gene regulation between distinct tissues. In particular, the study by Zahn et al. (2007) supports our empirical work. Although their results show controversial numbers of regulated genes, this could be due to different threshold settings and the divergent goals of these studies.
These empirical observations should be contrasted with certain implications of the free radical theory of aging proposed by Harman (1956). This theory essentially ignores the fact that ROS is implicated as specific signaling molecules and is critical in maintaining normal cell functions. According to the free radical theory, ROS are toxic byproducts of metabolism in an aerobic milieu. Hence the production rate of ROS will be negatively correlated with life span and hence constitute the prime determinant of aging. This proposition entails that changes in gene expression will be largely independent of the function of the regulatory networks and the homeostatic condition of the various tissues. The comparative studies we have discussed in this article point to specificity of transcriptional changes in gene expression at the network, tissue and species level. This observation is inconsistent with the free radical theory.
Metabolic pathways and nature of regulation
An important prediction of the theory is the relation between the direction of regulation and the function of the network: Transcriptional changes in gene expression will be down-regulated in dissipative networks and up-regulated in stabilizing networks.
Our empirical evaluation of this prediction was based on heart where we found significant regulation of glutathione metabolism, insulin signaling and oxidative phosphorylation. The brain and kidney, in contrast to the heart, have a relatively high degree of metabolic stability and contain few age-regulated genes (Zahn et al. 2007). Accordingly, these tissues were not considered in our analysis.
Glutathione acts as an antioxidant and is thus involved in ROS regulation, whereas the insulin signaling pathway is involved in glucose homeostasis. These two pathways are stabilizing networks. Oxidative phosphorylation is a metabolic energy pathway involved in ROS production and thus constitutes a dissipative network. The metabolic stability theory predicts that transcriptional changes in gene expression will be up-regulated in the glutathione and insulin signaling pathways, and down-regulated in the oxidative phosphorylation network.
Our empirical studies are consistent with these predictions. Table 4 describes the three regulatory networks, their metabolic and regulatory activity and the direction of gene regulation. The subsequent sections “Glutathione metabolism and ROS regulation”, “Insulin signaling and regulation” and “Oxidative phosphorylation” discuss in detail the activity of various genes in the networks and the mechanism whereby the regulatory activity is achieved. A recent study (Yanker et al. 2008) documents transcriptional changes in age-related pathways in the brain in mice and humans. The directional changes in the dissipative and stabilizing networks are consistent with the predictions we have stated.
Glutathione metabolism and ROS regulation
In the heart, 5 relevant genes (Gclm, Gpx1, Gpx3, Gsta2 and Gstm2) of the glutathione metabolism network exhibit up-regulated expression with age. These genes are involved in all three components of the pathway—biosynthesis of glutathione in its reduced monomeric form (GSH), antioxidant function by forming glutathione disulfate (GSSG; oxidized glutathione), and the detoxification by conjugating toxic rests (e.g., peroxidised lipids and xenobiotics) to glutathione.
The synthesis of glutathione involves a two-step ligation reaction. First glutamate–cysteine ligase (Gcl) conjugates l-glutamate and cysteine to l-gamma-glutamylcysteine, which is then further ligated to glycine by glutathione synthetase (Gs). Gcl is the first rate-limiting enzyme of glutathione synthesis (Maher 2005). The enzyme consists of two subunits, a heavy catalytic subunit (glutamate–cysteine ligase catalytic subunit; Gclc) and a light regulatory subunit (glutamate–cysteine ligase modifier subunit; Gclm). In most tissues, Gclm is limiting, suggesting an increase in Gclm alone would increase l-gamma-glutamylcysteine synthesis (Chen et al. 2005). This therefore would imply that an increase of Gclm alone leads to increased levels of glutathione within the cell.
The antioxidant function of glutathione metabolism is accomplished by glutathione peroxidases-Gpx. The function of these enzymes is to reduce lipid hydroperoxides to their corresponding alcohols and to reduce free hydrogen peroxide to H2O. Therefore, elevated activities of Gpx might counteract increased levels of ROS and thus maintain homeostasis.
GSTs catalyse the conjugation of reduced glutathione to a wide variety of substrates. This activity is useful in the detoxification of endogenous compounds such as oxidised lipids as well as the metabolism of xenobiotics (Mannervik and Danielson 1988; Coles and Kadlubar 2005; Hayes et al. 2005).
Insulin signaling and regulation
Foxo1 is one of the regulated genes within the insulin signaling pathway (Table3). In C. elegans, the ortholog DAF-16 has been implicated in the aging process (Curran and Ruvkun 2007). The FOXO family of forkhead transcription factors are key regulators that induce the expression of genes involved in gluconeogenesis and glycolysis (Fig. 2b).
In addition, insulin signaling is known to mediate other signaling pathways such as TOR signaling. TOR is a serine/threonine protein kinase that regulates cell growth, cell proliferation, cell motility, cell survival, protein synthesis, and transcription (Hay and Sonenberg 2004; Beevers et al. 2006). Current research indicates that TOR integrates the input from multiple upstream pathways, including insulin growth factors (such as IGF-1 and IGF-2), and mitogens (Hay and Sonenberg 2004). TOR is related to increased respiration and extended life span by reducing TOR signaling in Saccharomyces cerevisiae and inhibition of this pathway leads to extended life span in several model organisms (Bonawitz et al. 2007). In this regard, it is therefore not surprising that mice deficient in S6K1 (an effector of TOR) are hypersensitive to insulin and are protected against age- and diet-induced obesity (Blagosklonny 2007).
Oxidative phosphorylation
Oxidative phosphorylation is a metabolic pathway that uses energy released by the oxidation of nutrients to produce ATP. It has been shown that administering rapamycin (an antagonist of TOR) to yeast increases expression of genes involved in the TCA cycle, mitochondrial ribosome biogenesis and the assembly of oxidative phosphorylation complexes (Shamji et al. 2000). Referring to Table 2, we observed decreases in oxidative phosphorylation, TCA cycle and ribosome where five of ten genes linked to the ribosome are of the large subunit of the mitochondrial ribosome. Although these studies investigated yeast, a review on TOR signaling implicates the involvement of this pathway in heart disease in mammals and also the involvement of TOR in the biogenesis of ribosomes (Wang and Proud 2006).
Network connectivity and the rate of aging
Analytical studies of the relation between metabolic entropy and metabolic stability can be integrated with the metabolic-stability life span principle to predict:
The network connectivity–lifespan principle: The sensitivity of life span to mutations in the gene of the regulatory network increases with the number of interactions or connectivity among genes in the network.
The insulin signaling, glutathione metabolism and oxidative phosphorylation pathways have varying degrees of connectivity. The networks are described in Fig. 4 and indicate that connectivity is highest in the insulin signaling pathway. Accordingly, mutations of genes in this pathway will have the largest effect on life span.

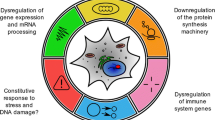
Proposed model of an age-related regulatory network operative in mouse heart. Based on our results, glutathione metabolism and insulin signaling are positively regulated whereas oxidative phosphorylation, ribosome and TCA cycle are negatively regulated in the aged mouse heart (Table 2). From the literature we found indications that increased insulin signaling leads to increased TOR signaling which then induces a reduction in the levels of expression of oxidative phosphorylation, ribosome and TCA cycle related genes (Shamji et al. 2000). The negative regulation of mitochondrial ribosomes additionally leads to decreased respiration functionality. Additionally, insulin signaling regulates (r.) glucose levels. Glucose is also linked to the TCA cycle via glycolysis/pyruvate and, the TCA cycle is then further linked to complex II (fumarate reductase) and to complex I (NADH dehydrogenase) of the respiration chain via fumarate/succinate and NADH, respectively. It has been proposed by Bonawitz et al. (2007) that decreases in oxidative phosphorylation lead to increases in ROS which eventually leads to an increase in glutathione metabolism
Our evaluation of the prediction will be based on our experimental observations and related studies. Although we have not shown the regulation of TOR on the RNA level or its protein phosphorylation status, we found many indications for a TOR-dependent regulation. This regulation includes the observed changes in gene expression of related pathways (insulin signaling, oxidative phosphorylation, ribosome and TCA cycle) and in addition, two important metabolites (glucose and ROS). An increase in insulin signaling leads to an increase in TOR signaling which leads, as discussed above, to decreases in oxidative phosphorylation, ribosome and TCA cycle. A decrease in oxidative phosphorylation alters ROS levels (Bonawitz et al. 2007) which in turn induces the regulation of glutathione metabolism. This cascade indicates that changes in insulin signaling will have more impact on the network than for instance changes in glutathione metabolism, as predicted by the theory.
Analyses by Promislow (2004) and Manke et al. (2006) are computational studies which provide additional support for the principle relating gene connectivity and life span.
Promislow in an analysis of the yeast interaction network compared patterns of connectivity for subsets of proteins associated with aging and certain other traits such as replicative senescence, the cell cycle, radiation sensitivity and DNA silencing, and salt tolerance. It was found that a protein’s connectivity is positively correlated with the number of traits it influences.
Manke et al. analysed functional data provided by gene knockouts and RNAi experiments in yeast and C. elegans. Proteins were classified as ‘viable’ or ‘lethal’ according to whether the organism survives the removal of the protein or not. The computational studies showed that proteins with the highest degrees of connectivity tend to be lethal.
The brace of numerical investigations underscore the validity of the connectivity–lifespan principle and suggest that candidates for genes associated with senescence are more likely to be located in proteins defined by a high degree of connectivity.
Conclusion
The rate of aging is determined by the efficiency with which organisms acquire and transform energy into metabolic work. The metabolic stability theory of aging appeals to a mathematical model of aging to show that metabolic stability, that is the capacity of a regulatory network to maintain stable concentrations of ROS and other critical metabolites, characterizes this metabolic efficiency, and is the main determinant of senescence. The model predicts that it is not the rate of production of ROS that determines the rate of aging, but the dynamic stability of concentration levels of ROS and other metabolites that regulate normal cell functions.
Transcriptional changes in gene expression constitute a biomarker of metabolic stability. Comparative studies of transcriptional gene regulation in glutathione metabolism, insulin signaling and oxidative phosphorylation, networks commonly associated with aging, have provided support for the metabolic stability theory. These studies reject the free radical theory of aging, and furthermore underscore the systemic effects of the aging process.
References
Amador-Noguez D, Yagi K, Venable S, Darlington G (2004) Gene expression profile of long-lived Ames dwarf mice and Little mice. Aging Cell 3:423–441. doi:10.1111/j.1474-9728.2004.00125.x
Aoyama H, Daitoku H, Fukamizu A (2006) Nutrient control of phosphorylation and translocation of Foxo1 in C57BL/6 and db/db mice. Int J Mol Med 18:433–439
Beevers CS, Li F, Liu L, Huang S (2006) Curcumin inhibits the mammalian target of rapamycin-mediated signaling pathways in cancer cells. Int J Cancer 119:757–764. doi:10.1002/ijc.21932
Blagosklonny MV (2007) An anti-aging drug today: from senescence-promoting genes to anti-aging pill. Drug Discov Today 12:218–224. doi:10.1016/j.drudis.2007.01.004
Bonawitz ND, Chatenay-Lapointe M, Pan Y, Shadel GS (2007) Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab 5:265–277. doi:10.1016/j.cmet.2007.02.009
Braeckman BP, Demetrius L, Vanfleteren JR (2006) The dietary restriction effect in C. elegans and humans: is the worm a one-millimeter human? Biogerontology 7:127–133. doi:10.1007/s10522-006-9003-4
Cao SX, Dhahbi JM, Mote PL, Spindler SR (2001) Genomic profiling of short- and long-term caloric restriction effects in the liver of aging mice. Proc Natl Acad Sci USA 98:10630–10635. doi:10.1073/pnas.191313598
Chen Y, Shertzer HG, Schneider SN, Nebert DW, Dalton TP (2005) Glutamate cysteine ligase catalysis: dependence on ATP and modifier subunit for regulation of tissue glutathione levels. J Biol Chem 280:33766–33774. doi:10.1074/jbc.M504604200
Coles BF, Kadlubar FF (2005) Human alpha class glutathione S-transferases: genetic polymorphism, expression, and susceptibility to disease. Methods Enzymol 401:9–42. doi:10.1016/S0076-6879(05)01002-5
Curran SP, Ruvkun G (2007) Lifespan regulation by evolutionarily conserved genes essential for viability. PLoS Genet 3:e56. doi:10.1371/journal.pgen.0030056
Demetrius L (2001) Mortality plateaus and directionality theory. Proc R Soc Biol Sci 268:2029–2037. doi:10.1098/rspb.2001.1739
Demetrius L (2004) Caloric restriction, metabolic rate, and entropy. J Gerontol A Biol Sci Med Sci 59:B902–B915
Demetrius L, Manke T (2005) Robustness and network evolution—an entropic principle. Phys A 346:682–696. doi:10.1016/j.physa.2004.07.011
Demetrius L, Ziehe M (2007) Darwinian fitness. Theor Popul Biol 72:323–345. doi:10.1016/j.tpb.2007.05.004
Demetrius L, Matthias Gundlach V, Ziehe M (2007) Darwinian fitness and the intensity of natural selection: studies in sensitivity analysis. J Theor Biol 249:641–653. doi:10.1016/j.jtbi.2007.08.016
Dietz K (2005) Darwinian fitness, evolutionary entropy and directionality theory. Bioessays 27:1097–1101. doi:10.1002/bies.20317
Edwards MG, Anderson RM, Yuan M, Kendziorski CM, Weindruch R, Prolla TA (2007) Gene expression profiling of aging reveals activation of a p53-mediated transcriptional program. BMC Genomics 8:80. doi:10.1186/1471-2164-8-80
Finch C (1990) Longevity, senescence and the genome. Chicago University Press, Chicago
Finkel T, Holbrook NJ (2000) Oxidants, oxidative stress and the biology of ageing. Nature 408:239–247. doi:10.1038/35041687
Fu C, Hickey M, Morrison M, McCarter R, Han ES (2006) Tissue specific and non-specific changes in gene expression by aging and by early stage CR. Mech Ageing Dev 127:905–916. doi:10.1016/j.mad.2006.09.006
Hamilton WD (1966) The moulding of senescence by natural selection. J Theor Biol 12:12–45. doi:10.1016/0022-5193(66)90184-6
Harman D (1956) Aging: a theory based on free radical and radiation chemistry. J Gerontol 11:298–300
Hay N, Sonenberg N (2004) Upstream and downstream of mTOR. Genes Dev 18:1926–1945. doi:10.1101/gad.1212704
Hayes JD, Flanagan JU, Jowsey IR (2005) Glutathione transferases. Annu Rev Pharmacol Toxicol 45:51–88. doi:10.1146/annurev.pharmtox.45.120403.095857
Jiang CH, Tsien JZ, Schultz PG, Hu Y (2001) The effects of aging on gene expression in the hypothalamus and cortex of mice. Proc Natl Acad Sci USA 98:1930–1934. doi:10.1073/pnas.98.4.1930
Johnson FB, Sinclair DA, Guarente L (1999) Molecular biology of aging. Cell 96:291–302. doi:10.1016/S0092-8674(00)80567-X
Kenyon C (2005) The plasticity of aging: insights from long-lived mutants. Cell 120:449–460. doi:10.1016/j.cell.2005.02.002
King CC, Newton AC (2004) The adaptor protein Grb14 regulates the localization of 3-phosphoinositide-dependent kinase-1. J Biol Chem 279:37518–37527. doi:10.1074/jbc.M405340200
Kirkwood TB (1977) Evolution of ageing. Nature 270:301–304. doi:10.1038/270301a0
Kuhn K, Baker SC, Chudin E, Lieu MH, Oeser S, Bennett H, Rigault P, Barker D, McDaniel TK, Chee MS (2004) A novel, high-performance random array platform for quantitative gene expression profiling. Genome Res 14:2347–2356. doi:10.1101/gr.2739104
Lee CK, Klopp RG, Weindruch R, Prolla TA (1999) Gene expression profile of aging and its retardation by caloric restriction. Science 285:1390–1393. doi:10.1126/science.285.5432.1390
Maher P (2005) The effects of stress and aging on glutathione metabolism. Ageing Res Rev 4:288–314. doi:10.1016/j.arr.2005.02.005
Manke T, Demetrius L, Vingron M (2006) An entropic characterization of protein interaction networks and cellular robustness. J R Soc Interface 3:843–850. doi:10.1098/rsif.2006.0140
Mannervik B, Danielson UH (1988) Glutathione transferases—structure and catalytic activity. CRC Crit Rev Biochem 23:283–337. doi:10.3109/10409238809088226
Medawar LB (1952) An unsolved problem of biology. Lewis, London
Misra V, Lee H, Singh A, Huang K, Thimmulappa RK, Mitzner W, Biswal S, Tankersley CG (2007) Global expression profiles from C57BL/6J and DBA/2J mouse lungs to determine aging-related genes. Physiol Genomics 31(3):429–440
Murakami S (2006) Stress resistance in long-lived mouse models. Exp Gerontol 41:1014–1019. doi:10.1016/j.exger.2006.06.061
Olshansky SJ, Rattan SI (2005) At the heart of aging: is it metabolic rate or stability? Biogerontology 6:291–295. doi:10.1007/s10522-005-2627-y
Promislow DE (2004) Protein networks, pleiotropy and the evolution of senescence. Proc Biol Sci 271: 1225–1234
Shamji AF, Kuruvilla FG, Schreiber SL (2000) Partitioning the transcriptional program induced by rapamycin among the effectors of the Tor proteins. Curr Biol 10:1574–1581. doi:10.1016/S0960-9822(00)00866-6
Vaupel JW, Carey JR, Christensen K, Johnson TE, Yashin AI, Holm NV, Iachine IA, Kannisto V, Khazaeli AA, Liedo P, Longo VD, Zeng Y, Manton KG, Curtsinger JW (1998) Biodemographic trajectories of longevity. Science 280:855–860. doi:10.1126/science.280.5365.855
Wang X, Proud CG (2006) The mTOR pathway in the control of protein synthesis. Physiology (Bethesda) 21:362–369. doi:10.1152/physiol.00024.2006
Williams GC (1957) Pleiotropy, natural selection, and the evolution of senescence. Evol Int J Org Evol 11:398–411. doi:10.2307/2406060
Yanker BA, Lu T, Loerch P (2008) The aging brain. Annu Rev Pathol Mech Dis 3:41–66. doi:10.1146/annurev.pathmechdis.2.010506.092044
Zahn JM, Poosala S, Owen AB, Ingram DK, Lustig A, Carter A, Weeraratna AT, Taub DD, Gorospe M, Mazan-Mamczarz K, Lakatta EG, Boheler KR, Xu X, Mattson MP, Falco G, Ko MS, Schlessinger D, Firman J, Kummerfeld SK, Wood WH, Zonderman AB, Kim SK, Becker KG (2007) AGEMAP: a gene expression database for aging in mice. PLoS Genet 3:e201. doi:10.1371/journal.pgen.0030201
Acknowledgments
We are indebted to Christian Regenbrecht for the assistance in performing the Western blot experiments and to Karsten Gülow, Andrew Hufton and Joel Cohen for their helpful comments on previous versions of the manuscript. Funding for this study was provided by the Max Planck Society.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Brink, T.C., Demetrius, L., Lehrach, H. et al. Age-related transcriptional changes in gene expression in different organs of mice support the metabolic stability theory of aging. Biogerontology 10, 549–564 (2009). https://doi.org/10.1007/s10522-008-9197-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10522-008-9197-8