
Summary
Introduction Modeling and simulation of pharmacokinetics and pharmacodynamics has previously been shown to be potentially useful in designing Phase I programs of novel anti-cancer agents that show hematological toxicity. In this analysis, a two-stage model-based trial design was evaluated retrospectively using data from the Phase I program with the aurora kinase inhibitor barasertib. Methods Data from two Phase I trials and four regimens were used (n = 79). Using barasertib-hydroxy QPA plasma concentrations and neutrophil count data from only study 1A, a PKPD model was developed and subsequently used to predict the MTD and a safe starting dose for the other trials. Results The PKPD model based on data from the first study adequately described the time course of neutrophil count fluctuation. The two-stage model-based design provided safe starting doses for subsequent phase I trials for barasertib. Predicted safe starting dose levels were higher than those used in two subsequent trials, but lower than used in the other trial. Discussion The two-stage approach could have been applied safely to define starting doses for alternative dosing strategies with barasertib. The limited improvement in efficiency for the phase I program of barasertib may have been due to the fact that starting doses for the studied phase I trials were already nearly optimal. Conclusion Application of the two-stage model-based trial design in Phase I programs with novel anti-cancer drugs that cause haematological toxicity is feasible, safe, and may lead to a reduction in the number of patient treated at sub-therapeutic dose-levels.
Similar content being viewed by others


Introduction
During Phase I development of novel anticancer drugs, it is common to evaluate various administration regimens in parallel dose escalation studies. As a result, the information obtained from one trial may not be available to guide the design of the other Phase I trials, as they are often conducted simultaneously. Previously, our group has presented a two-stage model-based design for the Phase I program of novel anticancer agents with dose limiting haematological toxicity [1]. The proposed model-based design consists of a first stage which is the conduct of an initial Phase I dose escalation trial using a single dosing regimen. During a model-based interim analysis, data from the initial Phase I trial (stage 1) are then used to optimize the design of subsequent trials using alternative dosing regimens (stage 2). The interim analysis consists of the development of a pharmacokinetic (PK) and pharmacodynamic (PD) population model and subsequent trial simulations. These trial simulations predict the maximum tolerated dose (MTD), and safe doses to start dose escalation with alternative administration regimens. The PD model describing the time course of neutrophil counts was developed by Friberg et al. [2] This semi-mechanistic model has been applied to describe the time course of myelosuppression in patients following the administration of several anticancer drugs [3–11] and has shown consistency in its system-related parameters among a wide variety of anti-cancer agents and regimens [2, 10].
In a simulation study, we have previously explored the performance of the two-stage model-based trial designs [1]. Five compounds with diverse PKPD characteristics (different models and parameters) were selected to evaluate the approach. The simulation analysis showed that the approach could be applied safely, and improved the efficiency of the dose escalation process by reducing the number of patients required at dose levels below the MTD by 27%. Furthermore, data from the phase I program with indisulam was used to retrospectively evaluate the performance using real data, which showed that the approach provides more efficient safe starting doses for the trials in the second stage.
In the current study, the two-stage model-based design is evaluated retrospectively using data from another real compound, barasertib. This compound is an investigational anticancer agent (an aurora kinase inhibitor) that has shown activity against tumor cell-lines and human xenografts [12–14]. Barasertib is a soluble phosphate prodrug developed for parenteral administration that is rapidly activated by phosphatase cleavage in serum to release the more active drug, barasertib-hydroxy quinazoline pyrazol anilide (hQPA) [13, 15]. Barasertib has been investigated in a phase I program consisting of several dose escalation studies designed to test various dosing regimens. In these studies, hematological toxicity was observed as the main dose limiting toxicity [16].
Methods
Data
In total, three phase I studies of barasertib in solid tumours have been performed, evaluating four different dosing regimens of barasertib (Table 1). In these phase I studies, haematological toxicity was the dose limiting toxicity (DLT) and, as no other clinically significant DLTs were reported, this was the only factor influencing dose escalation [16]. Regimens that were investigated in these trials were (number of patients included): 1A: 2 h infusion, weekly (n = 19); 1B: 2 h infusion, 2-weekly (n = 23 in dose escalation, n = 17 additional at the MTD); 2A: 48-hour infusion, 2-weekly (n = 20); 2B: 2 h infusion on day 1 and day 2, 2-weekly (n = 15), see Tables 2 and 3. Administration of granulocyte colony-stimulating factor was not allowed during these trials.
Starting doses for the clinical trials were determined based on FDA guidance [17]. This document states that the maximal starting dose should be based one tenth of the lethal dose in rodents (LD10), using the appropriate conversion factors for species and, correcting for body weight on an allometric basis. Following this guidance, experiments in rodents and dogs were used to determine a safe starting dose in humans. Allowing some margin for low body surface areas in oncology patients and variations in body weight, the safe starting doses were determined to be 100 mg, 50 mg, and 50 mg for the 2-hour, 48-hour infusion schedule, respectively. The clinical trials for barasertib already incorporated some form of a two-stage design as the MTDs obtained in part 1A and 2A were used as starting doses in 1B and 2B, respectively.
Blood sampling for PK analysis was performed pre-infusion, and at 0.25 h, 0.5 h, 0.75 h, 1 h, 1.5 h, 2 h, 3 h, 4 h, 6 h, 8 h, 10 h, 12 h, 20 h and 22 h following the end of the first infusion, appropriate for this drug with a terminal half-life of about 5 h (metabolite). During the next three infusions, if undertaken, blood samples were taken pre-infusion, 5 min before the end of infusion and 1 h, 3 h and 6 h following the end of infusion. Preclinical experiments indicated rapid and total conversion of barasertib to the more active metabolite barasertib hydroxy-quinazoline pyrazole anilide (barasertib-hQPA) [18]. Therefore, bioanalysis included determination of concentration of both barasertib and barasertib-hQPA, which was performed using a validated high performance liquid chromatography with tandem mass spectrometry (HPLC-MS/MS). Haematological analysis (including absolute neutrophil count, ANC) was performed routinely at least every 7 days, and more often when neutropenia was observed, using standard laboratory procedures. For study 2, an extra day of PK sampling was added to the schedules on day 3.
Analysis
Figure 1 is a schematic representation of the design of the current analysis. Plasma concentration data and neutrophil count data from one of the administration regimens (study 1A) was used to construct a PKPD model. This information model was subsequently used for trial simulations of the other dosing regimens, for which the encountered MTDs were recorded, and from which a safe starting dose was calculated. The model-based design was evaluated for its ability to predict safe starting doses and reduce the number of patients treated at doses below the MTD.

Schematic representation of analysis approach. MTD maximum tolerable dose, NTD Non-tolerated dose, SSD Safe starting dose
PKPD analysis
Model estimation was performed with non-linear mixed effects modeling using NONMEM, version VI, level 2.0 (Icon Development Solutions, Ellicott City, MD, USA) with g77 as Fortran compiler, and Piraña as modeling environment [19]. The Laplacian estimation method with interaction was used for estimating the model parameters. Judgment of model fit was done using goodness-of-fit plots. Nested models were tested for significant improvement in fit at a significance level of p < 0.01, corresponding to a decrease in objective function value (OFV) of 6.67. Further model evaluation was based on model convergence, model stability (condition number) and estimation characteristics such as the successful completion of the covariance step. Three significant digits was considered to be the minimal acceptable precision. Visual predictive checks (VPC), created using PsN and Xpose were used to evaluate the performance of final models [20, 21]. Data in the VPC were binned, so that sufficient data in each bin was available to allow evaluation of differences between observed and simulated data [22].
Data from part A of study 1 were used to develop i) a population PK model describing the time profile of barasertib(−hQPA) plasma concentrations and ii) a semi-physiological population pharmacodynamic model describing the time course of drug-related neutropenia. It was attempted to construct models for both barasertib and barasertib-hQPA timecourse. The PKPD model was developed using a sequential approach, i.e. the PK sub-model was fitted to the PK data alone, and then the PD sub-model was fitted to the PD data alone conditional on the fitted PK model [23]. For PK concentrations below the lower limit of quantification (LLOQ), the likelihood of observing an LLOQ was calculated and used in fitting the PK model to the data (method M3. according to Beal et al.) [24].
Time courses of neutropenia were described by a semi-physiological model, introduced by Friberg et al. [2]. This model comprises a progenitor compartment for proliferating blood cells, linked to a series of three compartments representing the maturation chain in the bone marrow and leading to the central circulation compartment. The models is defined by the following parameters: mean transition time (MTT), a feedback parameter (γ), baseline absolute neutrophil count (ANC0), and drug effect parameters (Emax/EC50 or Slope). These parameters were all estimated. Aurora kinase inhibitors are key regulatory roles at critical points of the cell cycle, hence AZD1152-hQPA affects dividing cells. This justifies the model structure that was used, in which the effects of AZD1152-hQPA was implemented on the progenitor compartment. Both slope and Emax models were evaluated. The absolute neutrophil counts (ANC) were log transformed prior to analysis. All parameters were assumed to be log-normally distributed in the study population. Consequently, between subject variability (BSV) was estimated using an exponential function (P i = θi · exp(ηi)) to describe the individual deviation (ηi) from the population typical value (θi). An additive residual error model related model predicted ANC to the (log transformed) observed data. Standard errors and measures of correlation between parameter estimates were obtained from the covariance step in NONMEM
Simulation studies
The PK and PD models developed from study 1A data were used to simulate study parts 1B, 2A and 2B. The results of the simulation studies were used to predict the MTD and a safe starting dose for these regimens. All simulations were performed in R (http://cran.r-project.org, version 2.7.0), supplied with packages for solving the non-linear system of differential equations of the PK and PD models (odesolve) and sampling from multi-variate normal distributions (MASS) to allow for incorporation of uncertainty in the estimation of model parameters (for both fixed an random effects), and correlation between random effects [25]. It was confirmed that the odesolve package produced exactly the same results as the ODE-solver in NONMEM.
Dose escalation trials for studies 1B, 2A and 2B were simulated 200 times, thereby accounting for between-subject variation in model parameters. To account for uncertainty in the estimation of the model parameters, this step was repeated 200 times, each time with a new set of PKPD parameters drawn from the variance-covariance matrix of the final model. Courses of neutrophil counts were simulated with added residual variability, and virtual sampling was performed every 7 days. Neutrophil baseline was simulated based on the estimated baseline ANC0 and BSV in ANC0. Datasets were created on the fly based on the dose-escalation algorithm, and mimicking the clinical sampling schedule for neutrophil measurements, i.e. if the neutrophil count on an occasion was found to be lower than 1.5·109 cells (corresponding to a grade 2 toxicity [26]), the measurement was repeated 3 days later, according to clinical practice. A dose limiting toxicity (DLT) due to neutropenia was defined in the study protocol as one of two criteria:
-
any neutropenia of common toxicity (CTC, NCI version III) grade 4, or
-
neutropenia of CTC grade ≥3 with fever (38.5°C).
The former criterion was scored by evaluating the nadir of simulated neutrophil counts (including residual variability) for the first cycle. The latter criterion was not included in the simulation algorithm, as the occurrence of febrile neutropenia was only sparsely encountered in the actual trials (1A: 0%, 1B: 8%, 2A: 10%, 2B: 0% of included patients over the entire study period), and were in all cases already scored as a grade 4 neutropenia toxicity.
In silico dose escalation was performed according to the barasertib Phase I program using an accelerated titration design consisting of two phases [27]. In the first phase, 1-patient cohorts were studied, and 100% dose escalation were performed. If a toxicity of grade 2 or higher was encountered, a second dose escalation phase was initiated, with 3-patient cohorts. In this phase, doses were escalated by 50%, or by 25% if serious (grade 3 or 4) neutropenia was observed. If on a dose level ≥ 2 DLTs were encountered at a particular dose level, the previous (lower) dose level was expanded to a maximum number of 6 patients. Dose de-escalation was repeated until a cohort of six patients experienced less than two DLTs. This dose level was defined as the MTD, while the dose level just above was labelled the non-tolerated dose level (NTD).
One tenth of the non-tolerated dose in humans, predicted from preclinical experiments, is traditionally considered a safe starting dose for dose escalation studies. In the simulated escalation trials, the starting dose for the subsequent trial was determined by multiplying the NTD level obtained in the previous trial by 10%, and perturbed by a randomly generated factor, drawn from a normal distribution with geometric standard deviation (SD) of 1.178 to account for uncertainty [1]. The geometric SD was calculated from predicted and empirically determined non-tolerated doses of 21 anti-cancer drugs [28–31]. The predicted MTD and NTD for each regimen was defined as the median value of all simulated MTDs and NTDs for that regimen, respectively. The 5th percentile of the prediction interval of all simulated barasertib MTDs was considered a safe starting dose, because there is a 95% probability this starting dose will be lower than the MTD [1].
In addition, simulations of studies 1B, 2A and 2B were performed using the starting doses used in the actual trials, and with parameter estimates based on data from all studies (post hoc). This was done to evaluate whether the two-stage design was more efficient, in terms of numbers of patients included, than the original design.
Validation criteria
The predicted MTDs cannot be formally tested against the outcomes of the clinical studies because the study outcomes are often an imprecise estimates of the true MTDs. Therefore, data from Phase I studies 1A, 1B, 2A and 2B were combined and used to obtain post hoc estimates of the PK and PD parameters for barasertib-hQPA. These new parameter estimates were subsequently used to perform the same simulation experiments as described above, and to calculate MTDs and safe starting doses. If the post hoc prediction of the MTD were within the prospectively determined 90% confidence intervals, it could be concluded that the two-stage model-based design could have been successfully applied to the Phase I development of barasertib. In accordance with the criteria used in the earlier publication, the two-stage model-based design was considered successful if all selected starting doses were below the NTDs found in the post hoc simulated trials, and if the number of patients treated at a dose level below the recommended dose could have been reduced, i.e. if the predicted starting doses were higher than those used for the actual studies [1].
The exact reduction in number of patients cannot be calculated retrospectively, as it is not possible to repeat the actual trial with exactly the same patients. Therefore, another simulation study was initiated, in which it was evaluated if the two-stage approach could reduce the number of patients in the phase I studies. In these simulations, the safe starting doses predicted in the first stage were used as the starting dose, while it was assumed that the post-hoc model and parameter estimates described the true population. The number of patients needed in each simulated clinical trials was recorded, and the mean number was compared to the number of patients that were actually included in the trial.
Results
PKPD model development (based on data from 1A)
In Fig. 2, the sequence of patient inclusion is presented visually for all regimens.

Sequence of patient inclusion in each barasertib Phase I study. Trials 1B and 2B were conducted after completion of studies 1A and 1B, respectively
PK data was available from all patients, collected from the first four cycles. From the different trials, 569 (1A), 1179 (1B), 458 (2A), and 485 (2B) blood samples were available for PK analysis. The precentage of BLQ datapoints in the dataset used was only 4.0% (106 out of 2,639 samples). From the dataset, eight trough levels were discarded, as they were more likely to have been taken post-dose than pre-dose. For all patients where PD measurements were available, also PK data was available. For PD analysis, 210, 422, 117 and 200 neutrophil counts were available from the respective studies.
The dense PK sampling schedule that was used allowed the identification of a three compartment mamillary model for the metabolite, with a central and two peripheral compartments, and linear elimination from the central compartment fitted the data best. Due to the rapid conversion of the parent drug to its metabolite, combined modelling of both barasertib and barasertib-hQPA did not improve the model, and therefore only concentration data from barasertib-hQPA was used, assuming instantaneous and complete conversion from the parent compound. The final parameter estimates of the model are given in Table 3.
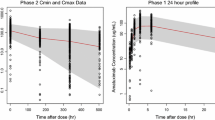
The PKPD model for neutropenia when implemented with an Emax model led to high uncertainty for the EC50 parameter (CV >100%), and did not improve model fit over a slope model. Therefore, the slope model was implemented and estimated. For the parameters Slope, MTT and ANC0, BSV could be estimated, as well as a negative correlation in BSV in MTT and ANC0 of 33% (CV 21%). Estimation of a full covariance matrix for BSV was not supported by the data. The parameter estimates for the PK and PD models are presented in Table 1. The VPC in Fig. 3 shows that the model adequately describes observed timecourse of neutrophils and the occurrence of myelosuppression observed in the trial, as both the observed median and boundaries of the 90% confidence interval were contained within the model prediction intervals for these statistics at almost all timepoints. Patients with grade 4 neutropenia are likely to drop-out of treatment. This may affect the VPC in two ways: the prediction interval (PI) may be downwardly biased, and the confidence interval around the edges of the PI may be widened due to availability of data. The latter was circumvented by binning on the number of available datapoints instead of on nominal timepoints. Furthermore, the VPC shows only the first treatment cycle, in which only 2, 4, 2, and 4 patients dropped out for studies 1A, 1B, 2A, and 2B respectively. It is therefore expected that the VPC was not affected much, and we did not account for patient drop-out in the VPC. A small negative correlation in individual parameter estimates was present for MTT and ANC0 but not between other parameters. The system specific parameters (MTT and γ) were very similar to those presented for other drugs that used the same PD model: MTT was estimated at 109 h (reported range 90.4–135 hs), and γ at 0.172 (reported range 0.121–0.239) [2].

Visual predictive check of (log transformed) neutrophil count data for PKPD model conditioned on data from study 1A. The points represent the observed ANCs. The black solid line connects the observed median values per bin, while the dotted lines represent the observed 5th and 95th percentile of the observations. Grey areas indicate the 95% confidence interval of the median, and 5th and 95th percentiles of the predicted neutrophil counts. Binning was done by count, using eight bins
Post-hoc PKPD model development (based on data from all trials)
The PK model developed using data from study 1A was used as a starting model for the post hoc PK analysis. Simpler models with no, or less peripheral compartments were significantly worse in fitting the barasertib-hQPA concentration data, while incorporating non-linearity in PK parameters did not improve fit. Again, combined modelling of a metabolite and parent did not improve model fit. Therefore, the same PK model was used in the simulation studies, although with updated parameter estimates (Table 1), and using an updated covariance matrix to account for uncertainty in parameter estimates (both fixed and random effects) in the simulation studies. The post hoc analysis contained sufficient data to support the estimation of an Emax model, and resulted in improved model fit. A sigmoidal Emax model did not described the data better than a non-sigmoidal one. Therefore, the Hill coefficient was fixed to 1. For the parameters EC50, MTT and ANC0, the incorporation of BSV was supported by the data, as well as the estimation of a full covariance block for the BSV random effects. The VPCs for the description of neutrophil time counts, shown in Fig. 4, show that the model adequately described observed time-course of myelosuppression observed in the trials as both median and 90% CI of the observed fall within their respective prediction intervals obtained by simulation. Again, at some timepoints in the VPC signs of model misfit were noted, however these were likely due to the small number of patients in the dataset, and hence the uncertain estimate of the 90% confidence interval of the observed data.

Visual predictive checks of (log transformed) neutrophil count data from posthoc analysis. The points represent the observed ANCs. The black solid lines connects the observed median values per bin, while the dotted lines represent the observed 5th and 95th percentile of the observations. Grey areas indicate the 95% confidence interval of the median, and 5th and 95th percentiles of the predicted neutrophil counts. Binning was done by count, using 8 bins
Performance of two-stage model-based design
As is presented in Fig. 5, for all studies, the 90% CI of all post hoc determined MTDs contained the observed MTD. In this figure, discrepancies between observed and post hoc values are indicative of the observed value not reflecting the true value, i.e. differing from the population mean by random chance. Here, the median of the post hoc determined MTDs was similar to those observed in the actual studies, except for study 1B, in which the post hoc MTD was determined to be 25% lower than was observed in the trial. On the other hand, discrepancies between post hoc estimates and predicted values are indicative of malperformance of the two-stage approach. The discrepancy between predicted and post hoc determined MTDs, was largest for study 2A, for which the predicted median MTD was about a factor two higher than its median post hoc estimate.

Observed, predicted and post hoc determined MTD, NTD, and starting doses for each study
The predicted safe starting doses (5th percentile of MTD predictions) were lower than the post hoc determined MTDs for all studies, except for 2A, which was similar. They were also lower than the observed MTDs for studies 1B and 2B, but again similar for study 2A. Thus, the first criterion for determining success of the proposed two-stage model-based design in this trial was met: the selected safe starting doses for the subsequent trials were below the median MTD levels, and thus well below the median NTD levels found in the post hoc simulated trials. This implies that, the two-stage model-based approach could have been safely implemented clinically for all trials.
The predicted safe starting doses were higher than the starting dose levels used in the actual studies, except for study 1B, for which the predicted safe dose level was about 25% lower than used in the actual study. Therefore, the second objective, predicting an efficient starting dose with the aim of reducing the number of patients treated at a dose level below the recommended dose, was met for trials 2A and 2B, but not for 1B. Trial simulation using the post hoc model parameter estimates revealed that reductions of 60%, 30%, and 0% (overall 40%) in the number of patients for the trials 1B, 2A, and 2B might have been possible using the two-stage approach.
Discussion
Novel approaches to phase I trial design have been presented to improve efficiency in the development of novel anti-cancer agents, such as the Bayesian continuous reassessment methods (CRM) [32], and escalation with overdose control (EWOC) [33]. It has been shown using simulation studies that these methods can reduce the overall number of patients in dose escalation trials, as well as the number of patients treated at sub-therapeutic doses [33, 34]. However, less than 2% of the phase I oncology trials between 1991 and 2006 were performed using other than conventional dose-escalation designs [35]. Important reasons for the limited adoption of more sophisticated designs may be the unfamiliarity of clinicians with advanced approaches such as CRM and EWOC, and the fact that these approaches require data analysis during the trial, which may pose operational or logistic difficulties, and requires close collaboration with the project statisticians. Also, if constraints are imposed on the dose escalation procedure, the benefit of using Bayesian approaches over conventional up-down designs is limited. Constraints may include e.g. limitation of dose escalation to an increase of 100% for the next dose level, escalation that is performed in three patient cohorts, and the use of conservative starting dose levels. Because of the constraints, the sophisticated method becomes quite similar to a conventional design. Hence, the differences between the conventional dose-escalation design and the sophisticated study designs in terms of reduction of numbers of patients and dose levels to be studied are expected to be limited. However, such designs do provide a more accurate estimate of the MTD [36].
In the current analysis we retrospectively evaluated a two-stage model-based phase I design, as we have described earlier [1]. This approach does not alter the execution of the phase I trials itself, i.e. the trials are performed using conventional modified Fibonacci designs, but instead divides the phase I program in two stages. After completion of an initial first phase I trial, a population PKPD model is constructed, and trial simulations are performed with the aim of reducing the number dose levels below the MTD, in order to efficiently identify the optimal dose to carry forward to phase II. In this article we tested the hypothesis that the model-based sequential phase I design is a safe and effective approach to increase this efficiency, based on a retrospective analysis of the phase I program of barasertib.
As stated, the major aim of the two-stage model-based trial design evaluated in this article is to reduce the number of patients treated at doses lower than the MTD while remaining safe for the subsequent trials. The approach of estimating safe starting doses on the basis of preclinical experiments is common practice in oncology, and was also used in the original design of the phase I program of barasertib. As the conventional dose escalation design remains unchanged in the proposed two-stage design, increases in efficiency can only be achieved by starting at higher dose levels than predicted from the preclinical experiments. However, the approach should also be safe: the predicted safe starting dose levels should be below the MTD.
Our retrospective analysis showed that these goals could have been achieved in the phase I program of barasertib. The two-stage approach could have been implemented safely since the predicted safe starting doses for the subsequent trials were all below the post-hoc determined MTD, which may be considered as more precise estimates of the ‘true MTD’. They were also below the observed MTDs. It was observed that the predicted safe starting dose for 2A was broadly similar to the observed MTD, which is clearly undesirable. It may be that the patient cohort around the MTD level in the actual 2A trial were more sensitive to the drug, or in worse condition than the average patient in trial 1A, and hence a low MTD was established. In fact, neutrophil counts at baseline were slightly lower for the 2A trial compared to the other trials. Of course, it may also be due to model misspecification, e.g. due to schedule dependency. The sampling schedules for determination of neutrophil countss were considered appropriate for demonstrating the maximum and minimum limits of the neutrophil profile i.e. the baseline, nadir and recovery points per cycle. However greater precision in defining the shape of the neutrophil profile would have been possible if additional sample points had been introduced.
The increase in efficiency was however not consistent for all three studies. For studies 2A and 2B, the predicted safe starting dose was higher than the one used in the clinical trials, implying that in these trials efficiency would have been increased. For trial 1B, the safe starting dose that was predicted was 25% lower than was actually used in the trial. An explanation for the two-stage approach proving of limited value for this specific trial, was the already optimal selection of the starting dose (one level below the MTD), and hence the low number of dose levels studied, and the low number of patients included (15). The optimal selection of the starting dose was likely due to the fact that this dose level was selected based on the MTD of trial 1A, i.e. the original design of the barasertib phase I program already incorporated a simple form of a two-stage design, which could not be improved by the model-based two-stage design.
For all actual clinical trials in the phase I program of barasertib, the starting doses were already chosen fairly efficiently, as the numbers of dose levels studied in these trials were low. A review of phase I trials oncology monotherapy trials with haematological toxicity as DLT, published in the last 12 months prior to preparation of this manuscript, revealed that a median of 7 dose levels (range 4–8) are generally investigated in phase I trials [37–47]. This is higher than the Phase I program of barasertib, which used 5, 3, 4, and 4 dose levels for the respective regimens. A median of 38% (range 13–72%) of patients in the cited studies were treated below the MTD, which is also high compared to those in the barasertib Phase I program (5.2%, 17.5%, and 33.3%, and 26.6% for the respective trials). These comparisons show that the starting doses in the barasertib Phase I trial program were already chosen closer to the MTD than is common, and hence only limited benefit from the proposed two-stage design could be realised. In addition, only limited benefits in terms of dose level/patient numbers reductions could be achieved as the starting doses for studies 1B and 2B were already based on the MTD found in studies 1A and 2A.
The actual performance of the two-stage method in terms of reducing the numbers of patients treated below the MTD can only be investigated in simulation studies, or in large prospective trials which compare the design against a conventionally designed trial. While the latter approach is not likely to be implemented due to practical and ethical considerations, the former approach has been performed previously, and has resulted in an overall 27% decrease of patients [1]. If however, it is assumed that for barasertib the post-hoc model and parameters estimates to describe the ‘true’ PKPD behavior of the patient population, it is possible to establish if the advanced design would be able to reduce patient numbers. These trial simulations were performed, mimicking the escalation designs used in the actual trials, and demonstrated a reduction 40% reduction in patients for the second stage of the phase I program. However, this may be a too optimistic estimate, as model misspecification is disregarded in such trial simulations. Interesting was the finding that for trial 1B, from the simulation analysis based on the data from the first stage, a reduction in the number of patients of 60% was estimated, although a lower starting dose was proposed than used in the actual trial. This may be linked to the fact that the predicted (and post-hoc estimated) MTD was lower than the MTD in the actual trial. Similarly, the failure to improve the efficiency in trial 2A may have been due to differences in predicted and observed MTD for this trial. In the simulations, the escalation algorithm from the study protocol was implemented, so no differences in escalation efficiency could have occurred, and differences in overall efficiency are due to a differences in selection of the starting dose, or differences in MTD.
It was noted that MTD in the four studies were different, and thus that design can influence the MTD. For example, the MTD in study 2A (48-hour infusion) was observed to be considerably lower than determined in the other trials (2-hour infusions). If the concentration-effect relationship is characterized sufficiently well by the model, typical neutrophil-time profiles can be simulated for each study design to investigate severity of neutropenia between the different designs. If the dosing regimens to be investigated are very different from the one used for model development, the unbiased and precise description of the relationship between drug exposure and effect on progenitor cell proliferation is of key importance. In the trial that was used for model development in this analysis, only few dose levels were studied (4), which might have precluded the conditioning of an Emax model for the drug effect. Moreover, it is likely that the small number of patients (19) led to a biased estimate of drug effect. In comparison with our previous published analysis, using data from the Phase I program of indisulam, where predictions were based on 26–42 patients per trial, and covered larger dose-ranges (ratio lowest/highest level: 12.5–33.3) compared to barasertib (ratio lowest/highest level: 3.25–9) [1]. Therefore, it is important to include uncertainty in model parameters in the simulation analysis, as otherwise biased parameter estimates may lead to the prediction of possibly toxic ‘safe’ starting dose levels.
The MTDs that were obtained in the post-hoc analysis for each trial can be considered as more precise and less biased estimates of the ‘true’ MTD. As the escalation cohorts in the conventional modified Fibonacci designs consist of only three patients, and six in expanded levels, imprecision may be expected in the determination of the MTD. Moreover, generally only discrete dose levels are studied, which decreases the precision of the MTD estimate. The proposed approach for obtaining a model-based MTD still suffers to some extent from these sources of bias since the post-hoc estimation step is based on data from the actual trials. However, the fact that determination of MTD will be based on parameter estimates obtained using all patient data simultaneously, instead of discrete toxicity grades from limited numbers of patients, bias will likely be reduced. Therefore, the MTDs predicted from the post-hoc analysis can be considered a more precise estimate of the ‘true’ MTDs for the trials. Additionally, the model-based approach provides a measure of the uncertainty of the predicted MTD. Overall, the median MTDs obtained in the post-hoc analysis were very close to those observed in the actual study, with only the one established for trial 1B being estimated about 25% lower in the post-hoc analysis than as determined in the actual study.
Conclusion
The work presented in this article shows that the application of the two-stage model-based approach to the design of Phase I programs with novel anti-cancer agents is feasible, and may be able to reduce the number of patients that are treated at less than efficacious dose levels. It was shown here that it could have been implemented safely in the barasertib phase I program. As the chosen starting doses for the different schedules were already nearly optimal in this Phase I program were already nearly optimal, and the number of dose escalations steps was low, the improvement in efficiency was limited here. It is our opinion however, based on these and previous results, that the two-stage approach can be applied safely, to reduce Phase I development timeframes for novel anti-cancer agents.
References
Zandvliet AS et al (2009) Two-stage model-based clinical trial design to optimize phase I development of novel anticancer agents. Invest New Drugs
Friberg LE et al (2002) Model of chemotherapy-induced myelosuppression with parameter consistency across drugs. J Clin Oncol 20:4713–4721
Sandström M et al (2005) Model describing the relationship between pharmacokinetics and hematologic toxicity of the epirubicin-docetaxel regimen in breast cancer patients. J Clin Oncol 23:413–421
Sandström M et al (2006) Population analysis of the pharmacokinetics and the haematological toxicity of the fluorouracil-epirubicin-cyclophosphamide regimen in breast cancer patients. Cancer Chemother Pharmacol 58:143–156
Latz JE et al (2006) Clinical application of a semimechanistic-physiologic population PK/PD model for neutropenia following pemetrexed therapy. Cancer Chemother Pharmacol 57:427–435
Latz J et al (2006) A semimechanistic-physiologic population pharmacokinetic/pharmacodynamic model for neutropenia following pemetrexed therapy. Cancer Chemother Pharmacol 57:412–426
Trocòniz I et al (2006) Phase I dose-finding study and a pharmacokinetic/pharmacodynamic analysis of the neutropenic response of intravenous diflomotecan in patients with advanced malignant tumours. Cancer Chemother Pharmacol 57:727–735
van Kesteren C et al (2005) Semi-physiological model describing the hematological toxicity of the anti-cancer agent indisulam. Invest New Drugs 23:225–234
Zandvliet AS et al (2007) PK/PD Model of indisulam and capecitabine: interaction causes excessive myelosuppression. Clin Pharmacol Ther
Kloft C et al (2006) Population pharmacokinetic-pharmacodynamic model for neutropenia with patient subgroup identification: comparison across anticancer drugs. Clin Cancer Res 12:5481–5490
Hansson E et al (2009) Limited inter-occasion variability in relation to inter-individual variability in chemotherapy-induced myelosuppression. Cancer Chemother Pharmacol
Yang J et al (2007) AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood 110:2034–2040
Wilkinson RW et al (2007) AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clin Cancer Res 13:3682–3688
Oke A et al (2009) AZD1152 rapidly and negatively affects the growth and survival of human acute myeloid leukemia cells in vitro and in vivo. Cancer Res 69:4150–4158
Keen N et al (2005) Biological characterisation of AZD1152, a highly potent and selective inhibitor of aurora kinase activity. Clin Cancer Res 11:B220
Schellens JH et al (2006) Phase I and pharmacological study of the novel aurora kinase inhibitor AZD1152. J Clin Oncol (Meeting Abstracts) 24:3008
(2005) Guidance for industry estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy volunteers
Mortlock AA et al (2007) Discovery, synthesis, and in vivo activity of a new class of pyrazoloquinazolines as selective inhibitors of aurora B kinase. J Med Chem 50:2213–2224
Keizer RJ, Zandvliet AS, Huitema ADR (2008) A simple infrastructure and graphical user interface (GUI) for distributed NONMEM analysis on standard network environments. Abstracts of the Annual Meeting of the Population Approach Group in Europe
Jonsson EN, Karlsson MO (1999) Xpose–an S-PLUS based population pharmacokinetic/pharmacodynamic model building aid for NONMEM. Comput Methods Programs Biomed 58:51–64
Lindbom L, Pihlgren P, Jonsson EN (2005) PsN-Toolkit–a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Programs Biomed 79:241–257
Karlsson M, Holford N (2008) A tutorial on visual predictive checks. Abstracts of the Annual Meeting of the Population Approach Group in Europe
Zhang L, Beal SL, Sheiner LB (2003) Simultaneous vs. sequential analysis for population PK/PD data I: best-case performance. J Pharmacokinet Pharmacodyn 30:387–404
Beal SL (2001) Ways to fit a PK model with some data below the quantification limit. J Pharmacokinet Pharmacodyn 28:481–504
Novartis The Comprehensive R Archive Network
NCI (2006) Common terminology criteria for adverse events
Simon R et al (1997) Accelerated titration designs for phase I clinical trials in oncology. J Natl Cancer Inst 89:1138–1147
Pessina A et al (2003) Application of the CFU-GM assay to predict acute drug-induced neutropenia: an international blind trial to validate a prediction model for the maximum tolerated dose (MTD) of myelosuppressive xenobiotics. Toxicol Sci 75:355–367
Gómez SG et al (2003) Use of CFU-GM assay for prediction of human maximum tolerated dose of a new antitumoral drug: Yondelis (ET-743). Toxicol In Vitro 17:671–674
Masubuchi N, May RD, Atsumi R (2004) A predictive model of human myelotoxicity using five camptothecin derivatives and the in vitro colony-forming unit granulocyte/macrophage assay. Clin Cancer Res 10:6722–6731
Moneta D et al (2003) Predicting the maximum-tolerated dose of PNU-159548 (4-demethoxy-3′-deamino-3′-aziridinyl-4′-methylsulphonyl-daunorubicin) in humans using CFU-GM clonogenic assays and prospective validation. Eur J Cancer 39:675–683
O’Quigley J, Pepe M, Fisher L (1990) Continual reassessment method: a practical design for phase 1 clinical trials in cancer. Biometrics 46:33–48
Babb J, Rogatko A, Zacks S (1998) Cancer phase I clinical trials: efficient dose escalation with overdose control. Stat Med 17:1103–1120
Piantadosi S, Fisher JD, Grossman S (1998) Practical implementation of a modified continual reassessment method for dose-finding trials. Cancer Chemother Pharmacol 41:429–436
Rogatko A et al (2007) Translation of innovative designs into phase I trials. J Clin Oncol 25:4982–4986
Ahn C (1998) An evaluation of phase I cancer clinical trial designs. Stat Med 17:1537–1549
Goel S et al (2009) A phase I study of eribulin mesylate (E7389), a mechanistically novel inhibitor of microtubule dynamics, in patients with advanced solid malignancies. Clin Cancer Res 15:4207–4212
Veltkamp SA et al (2008) Clinical and pharmacologic study of the novel prodrug delimotecan (MEN 4901/T-0128) in patients with solid tumors. Clin Cancer Res 14:7535–7544
Steeghs N et al (2009) Phase I Pharmacokinetic and pharmacodynamic study of the aurora kinase inhibitor danusertib in patients with advanced or metastatic solid tumors. J Clin Oncol
Dahut WL et al (2009) Phase I study of oral lenalidomide in patients with refractory metastatic cancer. J Clin Pharmacol 49:650–660
de Bono JS et al (2008) Phase I pharmacokinetic and pharmacodynamic study of LAQ824, a hydroxamate histone deacetylase inhibitor with a heat shock protein-90 inhibitory profile, in patients with advanced solid tumors. Clin Cancer Res 14:6663–6673
Mita AC et al (2009) Phase I and pharmacokinetic study of XRP6258 (RPR 116258A), a novel taxane, administered as a 1-hour infusion every 3 weeks in patients with advanced solid tumors. Clin Cancer Res 15:723–730
Tan AR et al (2009) Phase I study of eribulin mesylate administered once every 21 days in patients with advanced solid tumors. Clin Cancer Res 15:4213–4219
Widemann BC et al (2009) Phase I trial and pharmacokinetic study of ixabepilone administered daily for 5 days in children and adolescents with refractory solid tumors. J Clin Oncol 27:550–556
Zhu AX et al (2009) Phase I and pharmacokinetic study of gimatecan given orally once a week for 3 of 4 weeks in patients with advanced solid tumors. Clin Cancer Res 15:374–381
Mross K et al (2008) Phase I dose escalation and pharmacokinetic study of BI 2536, a novel Polo-like kinase 1 inhibitor, in patients with advanced solid tumors. J Clin Oncol 26:5511–5517
Yamamoto N, Boku N, Minami H (2009) Phase I study of larotaxel administered as a 1-h intravenous infusion every 3 weeks to Japanese patients with advanced solid tumours. Cancer Chemother Pharmacol 65:129–136
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Keizer, R.J., Zandvliet, A.S., Beijnen, J.H. et al. Two-stage model-based design of cancer phase I dose escalation trials: evaluation using the phase I program of barasertib (AZD1152). Invest New Drugs 30, 1519–1530 (2012). https://doi.org/10.1007/s10637-011-9694-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10637-011-9694-5