
Abstract
Rare syntenic conservation, sequence duplication, and the use of both DNA strands to encode genes are signature architectural features defining mitochondrial genomes of enoplean nematodes. These characteristics stand in contrast to the more conserved mitochondrial genome sizes and transcriptional organizations of mitochondrial DNAs (mtDNAs) derived from chromadorean nematodes. To address the frequency of gene rearrangement within nematode mitochondrial DNA (mtDNA), mitochondrial genome variation has been characterized within a more confined enoplean taxonomic unit, the family Mermithidae. The complete nucleotide sequences of the mosquito parasitic nematodes Romanomermis culicivorax, R. nielseni, and R. iyengari mtDNA have been determined. Duplicated expanses encompassing different regions of the mitochondrial genomes were found in each of these congeners. These mtDNA shared few rRNA and protein gene junctions, indicating extensive gene rearrangement within the Romanomermis lineage. Rapid structural changes are also observed at the conspecific level where no two individual nematodes carry the same haplotype. Rolling circle amplification was used to isolate complete mitochondrial genomes from individuals in local populations of Thaumamermis cosgrovei, a parasite of terrestrial isopods. Mitochondrial DNA length variants ranging from 19 to 34 kb are observed, but haplotypes are not shared between any two individuals. The complete nucleotide sequences of three haplotypes have been determined, revealing a constant region encoding most mitochondrial genes and a hypervariable segment that contains intact and pseudogene copies of several mitochondrial genes, duplicated to different copy numbers, resulting in mtDNA size variation. Constant rearrangement generates new T. cosgrovei mtDNA forms.
Similar content being viewed by others

Introduction
Nematodes are of ancient origin and constitute one of the most speciose animal phyla. Estimates of species numbers range from 100,000 to 100 million, although the upper limits may be exaggerated, perhaps by ten-fold; approximately 25,000 nematodes have now been described (Hugot et al. 2001; Blaxter 2003). Nematodes have engendered considerable interest in their population and evolutionary biology because they are remarkably adaptable to virtually every ecological habitat, resulting in a cosmopolitan distribution that features free-living and obligate plant and animal parasitic life histories. A paucity of conserved, easily scored morphological characters impeded early systematic analysis. With the advent of DNA sequencing, including mitochondrial DNA (mtDNA) sequence analysis, elucidating phylogenetic affinities among nematodes has dramatically accelerated. Nematode mitochondrial genomes typically encode an orthologous set of 12 (or 13) protein coding genes, two organelle-specific ribosomal RNA (rRNA) and 22 transfer RNA (tRNA) genes, a typical metazoan mtDNA gene content. Alignment of individual and catenated mitochondrial gene and protein sequences, and the precise order in which these same genes are positioned along this compact, circular genome, has contributed to the deduction of phylogenetic affinities among nematodes and has altered the way nematologists view the evolution of the phylum.
As of April 2010, complete mitochondrial genome sequences derived from 33 nematode genera were available in GenBank. The phylum is divided into two major taxonomic classes, the chromadorea and the enoplea. Sampling of nematode mtDNA sequences is biased; 26 GenBank entries are derived from the chromodorea whereas seven represent enoplean nematodes. Such a limited census represents a startling underrepresentation given the species-rich nature of the phylum. Nevertheless, this narrow sampling has unmasked the unexpected finding that the evolutionary dynamics of mitochondrial genomes dramatically differs between the two taxonomic classes.
Class distinctions in nematode mtDNA evolutionary dynamics
Chromadorean mtDNA molecules reveal, with a few exceptions, conservation of genome size, gene order, the absence of lengthy sequence duplications, and an exceptional asymmetry in transcriptional architecture as all protein coding genes are encoded on one of the two DNA strands. These mitochondrial genomes tend to be of a relatively constrained length, ranging from 13.6 to 20.5kb. Conservation in genome size is directly related to the absence of major sequence duplications. If repeated mtDNA segments are present, they tend to be short, variable number tandem repeats (VNTRs; Lunt et al. 1998) that do not encompass coding regions. Mitochondrial VNTRs have been successfully employed to address questions in the population dynamics of the phytopathogenic root-knot nematodes (Whipple et al. 1997).
Of the 26 available chromadorean genera mitochondrial protein coding and rRNA gene orders (without considering tRNA genes that transpose at an accelerated rate), 19 are fully syntenic; this common gene order has been termed G7; He et al. 2005) and is pictured below as a linear representation for illustrative purposes:
COI COII rrnL ND3 ND5 ND6 ND4L rrnS ND1 ATP6 ND2 CYTB COIII ND4
Beyond the common G7 transcriptional organization, several additional gene orders have been described within the chromadorea. One differs from G7 only by simple translocation of several gene clusters (GenBank accession NC_008534). Four others are unique and syntenic affinities with the common transcriptional organization(s) are not obvious; these “idiosyncratic” gene orders may be lineage specific (Kang et al. 2009). More extensive sampling will be required to further understand the molecular evolution of mitochondrial gene order within the chromadorea.
Standing in direct contrast to the relatively conserved nature of chromadorean mtDNA molecules, rare syntenic conservation, frequent kilobase-sized sequence duplication, and the use of both DNA strands to encode genes are signature architectural features defining mitochondrial genomes of enoplean nematodes. The lengthy duplications generate mtDNA molecules that range from 12.6 to approximately 34 kb; these include one of the smallest known animal mtDNAs (Xiphinema americanum encoding 17 of the expected 22 organelle-specific tRNAs; He et al. 2005) as well as some of the largest mtDNA molecules observed within the metazoa (Thaumamermis cosgrovei, featured below). Unlike short mtDNA VNTRs, lengthy repeated sequences within enoplean mitochondrial genomes often encompass gene coding regions, creating small mitochondrial multigene families, the generation of pseudogenes, and the opportunity for gene rearrangement inconsistent with a duplication-random loss mechanism (Boore and Brown 1998). The striking absence of syntenic relationships among these mitochondrial genomes suggests a rapid pace of gene order change within the enoplea.
Mitochondrial genome plasticity in the nematode family mermithidae
To address the occurrence and evolution of nematode mtDNA sequence rearrangement, we are studying mitochondrial genome variation within a more confined enoplean taxonomic unit, the family Mermithidae. Mermithid nematodes are obligate parasites of invertebrates, typically arthropods. The mitochondrial genome carried by mosquito parasitic nematode Romanomermis culicivorax was among the very first nematode mtDNA molecules to be characterized (Powers et al. 1986). Unexpectedly, this mtDNA molecule exhibited extensive size variation (26–32 kb) due to a 3.2 kb expanse duplicated to different copy numbers within individual mitochondrial genomes. This lengthy repeated sequence included genes for ND3 and several tRNAs; some repeating units were present in an inverted orientation relative to others, resulting in genes residing on both mtDNA strands. Romanomermis culicivorax mtDNA became the prototypical enoplean mitochondrial genome.
A comparative mitochondrial genomics study of the mermithid nematodes has now been undertaken for the purpose of understanding the molecular evolution of the unusual R. culicivorax mtDNA architecture. Moreover, it was anticipated that exploring the evolution of mermithid mtDNA might prove a useful paradigm for understanding the evolution of enoplean mitochondrial genomes. Mermithid nematode mtDNA variation at the family, intergeneric, and conspecific taxonomic levels are summarized here.
Rolling circle mtDNA amplification (Tang and Hyman 2005) coupled with conventional and high-throughput massively parallel sequencing has resulted in complete nucleotide sequences for 11 mermithid mitochondrial genomes including mtDNAs derived from three Romanomermis congeners (Wu 2007) and three T. cosgrovei individuals (Tang and Hyman 2007; Lewis and Hyman unpublished). Shared architectural features among enoplean mtDNA molecules are the presence of functional genes on both DNA strands and the presence of lengthy repeated mtDNA segments (though the repeats are not orthologous among any of the mitochondrial genomes). The difference in lengths of duplicated regions and copy numbers of these repeated sequences contribute to the wide size variation of enoplean mitochondrial genomes. Strikingly, there are no obvious syntenic relationships among any of the mermthid mtDNAs, nor are gene orders shared with two other enoplean mitochondrial genomes characterized by other laboratories (Tang 2006; Wu 2007).
Within the phylum Nematoda, the nematode class enoplea is considered to be ancestral, with the chromadorea a more contemporary monophyletic derivation. As such, it could be argued that the antiquity of the enoplea is reflected in the considerable divergence of mitochondrial gene order change relative to that observed among chromadorean nematodes. This possibility can be further tested by examining the conservation of mitochondrial gene orders between more recently diverged congeners.
Mitochondrial genome plasticity among mermithid congeners
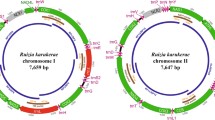
In general, congeners that have been sampled across the metazoa typically share identical or highly similar mtDNA protein coding and rRNA gene orders (Gissi et al. 2008). Complete synteny among congeners has been observed in all six genera sampled from the chromadorean nematodes. Information on intrageneric gene order variation is currently available in one Enoplid genus, Romanomermis. Surprisingly, each of the three Romanomermis congeners R. culicivorax, R. nielseni, and R. iyengari carry mitochondrial genomes with distinct gene orders (Fig. 1; Wu 2007). This observation suggests that mitochondrial gene order change is more rapid than can be easily explained by the presumed relative age of the chromadorean and enoplean lineages. What lineage specific forces are at work to engender such rampant mitochondrial gene order change among the Enoplids remains an open question.

Complete gene order of three Romanomermis sp. mitochondrial DNAs. Shared gene clusters are outlined with boxes and may represent an ancestral character state. Each gene cluster is shown by a different line styles. Genes in white boxes are transcribed left to right; –·–·–·boxed gene cluster is shared with a partial sequence from a fourth Romanomermis sp. mtDNA; and genes in shaded boxes are transcribed right to left. This figure was re-printed from (Wu 2007) with permission of the author
Hypervariation: an extreme example of conspecific mtDNA variation
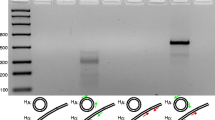
Thamamermis cosgrovei, an obligate mermithid parasitic nematode of the terrestrial isopod Armadillidium vulgare, carries an atypically large, size-variable mitochondrial genome of 19–34 kb. Remarkably, no two individuals share the same haplotype as revealed by rolling circle amplification of mtDNA and subsequent physical characterization by restriction enzyme analysis (Fig. 2; Tang and Hyman 2007); we have adopted the term “hypervariation” to describe this unexpected observation. For the purpose of illustrating the extent of hypervariation to the symposium audience, mtDNA analysis of six T. cosgrovei individuals randomly chosen from 43 recently characterized nematodes, each carrying a unique haplotype, are presented are presented in Fig. 2.

Haplotype hypervariation among T. cosgrovei individuals. Total cellular DNA was isolated from six T. cosgrovei individuals and used as template for rolling circle amplification of mtDNA. Pictured are restriction products of BglII-cleaved mtDNA size-fractionated by agarose gel electrophoresis. Arrows indicate BglII restriction products derived from the common region and shared among all haplotypes; these fragments represent the “common skeleton” of the T. cosgrovei mitochondrial genome containing all but one mitochondrial gene. The remaining bands that are not shared among haplotypes are derived from the hypervariable region as described in the text. Lane 1, 1 kb-ladder; Lanes 2–7, mtDNA
The T. cosgrovei mitochondrial genome encodes the same 36 genes (12 protein, 22 tRNA and 2 rRNA coding sequences) typically found in nematode mtDNAs (Tang and Hyman 2007). The ATPase subunit 8 gene is absent, as is the case for all other nematode mtDNAs characterized to date with the exception of Trichinella spiralis (Lavrov and Brown 2001). The T. cosgrovei mtDNA molecule can be divided into two expanses: a “constant” region, identified by a common ensemble of restriction enzyme products that is shared among all haplotypes and contains most of the coding regions, and a hypervariable segment that houses the small mitochondrial rRNA (rrnS) gene and four mitochondrial tRNA genes (trnF, trnH, trnV, trnW). This locus also contains partial, non-functional pseudogene copies of ND4, ATP6 and in some haplotypes a remnant rrnS copy. The frequent and large scale conspecific size variation (hypervariability) is a consequence of extensive DNA duplication within this region, coupled with frequent sequence rearrangement. Duplicated gene copies reveal a complex organization in that they are interspersed with six different DNA sequence motifs 91–293 bp in length of unknown function (elements A through E; Tang and Hyman 2007). Variation of both length and sequence organization of the hypervariable region can be ascribed to differences in the copy number and arrangement of different combinations of these genes, pseudogene copies, and non-coding sequence elements.
No shared haplotypes are found when individual T. cosgrovei nematodes are sampled from within local populations or from geographically isolated populations separated by 40–50 km. On occasion rare, multiply-infected isopod hosts can be recovered (Tang and Hyman, 2007). Individual nematodes harvested from the host as a “brood” appear to carry the same mtDNA haplotype. This observation suggests that (1) isopods may be passively infected by ingesting egg clutches from a single maternal source prior to egg hatching or (2) actively infected by recently hatched, multiple infectious J2 stage nematodes derived from a single maternal lineage, prior to dispersal in the soil.
Conclusions
Nematodes have been singled out as a taxon with mtDNA evolutionary dynamics that differ from most other metazoa (Blaxter 2002; Gissi et al. 2008). Among the criteria listed to support this premise is the experimental demonstration that nematode mitochondrial genomes undergo intra-molecular recombination (Lunt and Hyman 1997), a process that may be absent in many other metazoan lineages. Recombination processes seem the most plausible mechanism to explain the diversity of transcriptional organizations and hypervariation summarized here.
Current taxonomic status of the Nematoda bifurcates the phylum into two classes: the chromadorea and the enoplea. Gene orders for 26 chromadorean mitochondrial genomes can be inferred from complete nucleotide sequences available in public databases and reveal common features of a limited mtDNA size range, unidirectional transcription, and easily recognizable syntenic relationships. Of the 26 chromadorean genera with completely sequenced mitochondrial genomes, 19 share the same gene order (G7; He et. al. 2005). Conservation of gene order across wide distances within the chromadorea is consistent with the hypothesis that the chromadorea is a monophyletic group, as inferred from the nuclear 18S small subunit RNA gene sequence data (DeLey and Blaxter, 2002). The most common transcriptional organization (G7), or one of a very similar genetic architecture, may represent the mtDNA molecule transmitted at the ancestral speciation event leading generation of the chromadorea.
Why is it that the evolutionary dynamics of mitochondrial genomes appears to differ between the two major nematode taxonomic classes? While it is tempting to propose a bottleneck that generated a small population of progenitor mtDNA molecules and at present we are surveying an early window in chromadorean evolution, this hypothesis may be too simplistic. Mitochondrial genomes from but 26 chromadorean genera have been characterized, a sample size that may be too modest; the few “idiosyncratic” mitochondrial gene orders that have been described may ultimately prove not to be exceptional, indicating rates of change between the two nematode classes is not as different as currently perceived.
Evolutionary time alone appears not to be the exclusive factor in the observed class distinction between mitochondrial genome diversity levels. The rampant congener gene order variation associated with Romanomermis mtDNA suggests changes in mtDNA synteny can occur over more modest time intervals that define speciation events; hypervariation of T. cosgrovei mitochondrial genomes indicate that change can occur in real-time. Moreover, it cannot yet be ascertained if enoplean nematode mtDNA variation has been generated at a consistent trajectory through time or is episodic. Because congener comparisons appear to reflect mitochondrial genome plasticity at higher taxonomic levels (Gissi et al. 2008), it will be important to discover if Romanomermis is unique among the enoplean lineages with respect to the rapidity of mtDNA gene order change.
If the observed mtDNA plasticity is a consequence of genetic recombination (Lunt and Hyman 1997), requisite enzymatic machinery in the form of recombinases is likely involved. Mitochondrial gene order changes and related processes such as hypervariation within specific lineages may be due to a loss or gain of the ability to target these proteins to the organelle. Studies on the mechanistic enzymology of nematode mitochondrial genome rearrangement in species such as M. incognita and T. cosgrovei, where real-time mtDNA variation has been characterized (Lunt and Hyman, 1997; Tang and Hyman 2007) should provide another layer of understanding as to why the evolutionary dynamics of mitochondrial genomes varies throughout the phylum.
References
Blaxter ML (2003) Nematoda: Genes, genomes and the evolution of parasitism. Adv Parasit 54:101–195
Boore JL, Brown WM (1998) Big trees from little genomes: mitochondrial gene order as a phylogenetic tool. Curr Op Genet Devel 8:668–674
De Ley P, Blaxter ML (2002) Systematic position and phylogeny. In: Lee DL (ed) The Biology of Nematodes. London, Taylor and Francis, pp 1–30
Gissi C, Iannelli F, Pesole G (2008) Evolution of the mitochondrial genome of Metazoa as exemplified by comparison of congeneric species. Heredity 101:301–320
He Y, Jones J, Armstrong M, Lamberti F, Moens M (2005) The mitochondrial genome of Xiphinema americanum sensu stricto (Nematoda: enoplea): Considerable economization in the length and structural features of encoded genes. J Mol Evol 61:819–833
Hugot J-P, Baujard P, Morand S (2001) Biodiversity in helminthes and nematodes as a field of study: an overview. Nematology 3:199–208
Kang S, Sultana T, Eom KS, Park YC, Soonthornpong N, Nadler SA, Park J-K (2009) The mitochondrial genome sequence of Enterobius vermicularis (Nematoda:Oxyurida)–An idiosyncratic gene order and phylogenetic information for Chromadorean nematodes. Gene 429:87–97
Lavrov DV, Brown WM (2001) Trichinella spiralis mtDNA: A nematode mitochondrial genome that encodes a putative ATP8 and normally structured tRNAs and has a gene arrangement relatable to those of Coelomate metazoans. Genetics 157:621–637
Lunt DH, Hyman BC (1997) Animal mitochondrial DNA recombination. Nature 387:247
Lunt DH, Whipple LE, Hyman BC (1998) Mitochondrial DNA variable number tandem repeats (VNTRs): utility and problems in molecular ecology. Mol Ecol, pp. 1441–1455
Powers TO, Platzer EG, Hyman BC (1986) Large mitochondrial genome and mitochondrial DNA size polymorphism in the mosquito parasite Romanomermis culicivorax. Curr Genet 11:71–77
Tang S (2006) Comparative mitochondrial genomics of the Nematoda: The family Mermithidae represents rapid enoplean mitochondrial DNA evolution. Dissertation, University of California-Riverside
Tang S, Hyman BC (2005) Rolling-circle amplification of complete nematode mitochondrial genomes. J Nematol 37:236–241
Tang S, Hyman BC (2007) Mitochondrial DNA haplotype hypervariation within the isopod parasitic nematode Thaumamermis cosgrovei. Genetics 176:1139–1150
Whipple LE, Lunt DH, Hyman BC (1997) Mitochondrial DNA length variation in Meloidogyne incognita isolates of established genetic relationships: utility for nematode population studies. Fund Appl Nematol. 21:265–271
Wu Z (2007) Comparative mitochondrial genomics of Romanomermis congeners; implications for nematode phylogeny. Dissertation, University of California-Riverside
Acknowledgments
I wish to thank my former graduate students, Drs. Sha Tang and Zhen Wu for sharing their completed dissertation work on comparative mermithid mitochondrial genomics prior to publication. I wish to express my appreciation to Samantha C. Lewis, a current graduate student, for bringing high throughput DNA sequencing to my laboratory. This work was supported by the University of California-Riverside Institute for Integrative Genome Biology, the Committee on Research and the Agricultural Experiment Station.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Hyman, B.C., Lewis, S.C., Tang, S. et al. Rampant gene rearrangement and haplotype hypervariation among nematode mitochondrial genomes. Genetica 139, 611–615 (2011). https://doi.org/10.1007/s10709-010-9531-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-010-9531-3