
Abstract
Temozolomide (TMZ) is the standard chemotherapeutic agent for human malignant glioma, but intrinsic or acquired chemoresistance represents a major obstacle to successful treatment of this highly lethal group of tumours. Obtaining better understanding of the molecular mechanisms underlying TMZ resistance in malignant glioma is important for the development of better treatment strategies. We have successfully established a passage control line (D54-C10) and resistant variants (D54-P5 and D54-P10) from the parental TMZ-sensitive malignant glioma cell line D54-C0. The resistant sub-cell lines showed alterations in cell morphology, enhanced cell adhesion, increased migration capacities, and cell cycle arrests. Proteomic analysis identified a set of proteins that showed gradual changes in expression according to their 50% inhibitory concentration (IC50). Successful validation was provided by transcript profiling in another malignant glioma cell line U87-MG and its resistant counterparts. Moreover, three of the identified proteins (vimentin, cathepsin D and prolyl 4-hydroxylase, beta polypeptide) were confirmed to be upregulated in high-grade glioma. Our data suggest that acquired TMZ resistance in human malignant glioma is associated with promotion of malignant phenotypes, and our reported molecular candidates may serve not only as markers of chemoresistance but also as potential therapeutic targets in the treatment of TMZ-resistant human malignant glioma, providing a platform for future investigations.
Similar content being viewed by others


Introduction
Malignant gliomas are the most common type of primary brain tumours, and represent one of the most aggressive and lethal malignancies in adults [1]. The standard treatment of malignant glioma is multimodal, involving maximal surgical resection followed by concurrent radiation and chemotherapy using temozolomide (TMZ). Although this treatment combination has been shown to prolong survival, the prognosis of patients remains dismal with median survival time of just over 12 months [2]. Chemotherapeutics play an important role in the treatment of malignant glioma [3]. However, many patients continue to suffer from recurrent diseases due to de novo or acquired resistance against TMZ [4, 5].
The alkylating agent TMZ is currently the standard chemotherapeutic for newly diagnosed malignant gliomas [6]. It is well tolerated and has been shown to provide significant clinical benefits [7, 8]. Its major action is mediated through the induction of O6-G methylation, causing DNA damage which activates the mismatch repair mechanism. The latter then recognises a recurring error and triggers apoptosis [9, 10]. However, resistance to TMZ may occur due to the involvement of both intrinsic or acquired pathways [11, 12], and the presence of the blood–brain barrier, which impedes drug delivery [13]. Epigenetic modifications are frequently observed in innate chemoresistance in gliomas, such as the presence of high level of a DNA repair enzyme, O6-methylguanine methyltransferase (MGMT), that removes the methyl adducts and antagonizes the effects of TMZ [10]. Other mechanisms, including deficiency of the mismatch repair capacity [11] and robust base excision repair [14], have also been shown to confer innate resistance to TMZ. Furthermore, some tumours may display TMZ resistance that is independent of their MGMT levels or mismatch repair efficacy, suggesting that other unknown mechanisms could be involved.
Clinically, acquired TMZ resistance is a significant problem, with more than 90% of recurrent gliomas showing no response to repeated challenges with TMZ [15]. Some of the mechanisms that confer TMZ resistance include selection of pre-existing TMZ-resistant cells in the parental tumour [4], presence of a mitochondrial adaptive response to TMZ genotoxic stress [15] and dysregulation of glucose transporter and drug metabolising enzymes [16]. To improve the clinical efficacy of TMZ, we chose to identify molecular mechanisms that confer resistance, and to target these molecules or pathways in order to re-sensitize the tumours to TMZ therapy.
Proteomics may reveal such mechanisms by directly addressing the functional effectors of cellular, disease and treatment processes [17]. Over the last few years, there have been a number of published studies on glioma proteomics, including analysis of patient biopsy samples [18, 19], body fluids [20, 21], glioma cell lines [22, 23] and animal glioma models [24]. However, to our knowledge, none of these previous studies addressed the issue of TMZ resistance in glioma cells. Therefore, the aim of this study is to identify, by means of proteomics analysis, dysregulated protein candidates that may potentially be involved in the onset of TMZ resistance in human malignant gliomas. We firstly developed TMZ-resistant variants of human glioma cell lines. Protein expression changes associated with acquisition of TMZ resistance were evaluated by two-dimensional gel electrophoresis coupled with mass spectrometry (MS). We surmised that this approach would identify molecules that are associated with TMZ resistance in malignant gliomas. Our findings may have important implications for the development of novel treatment strategies aimed at improving the clinical efficacy of TMZ.
Materials and methods
Tissue samples
Patients included in this study are ethnic Chinese with either low- or high-grade glioma. A total of 14 resected tissues were included in this study. Prior to analysis, the histological classification for all tissues was confirmed according to the World Health Organization (WHO) brain tumour classification system [25].
Cell lines and culture conditions
Human malignant glioma cell lines, D54-MG (GBM, a gift from Dr. Darell D. Bigner, Duke University Medical Center, USA) and U87-MG (GBM, obtained from American Type Tissue Collection, Manassas, VA, USA), were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/F12 (1:1) and minimum essential medium (MEM)-α, respectively (GIBCO®; Life Technologies, Inc., Carlsbad, CA, USA). These media were supplemented with 10% heat-inactivated foetal bovine serum (FBS) (GIBCO®), 100 IU/ml penicillin and 100 μg/ml streptomycin (GIBCO®). Temozolomide (TEMODAL®) was obtained from Schering-Plough Co. Ltd. (Kenilworth, NJ, USA). Stock solutions were prepared in dimethyl sulphoxide (DMSO) at concentration of 10 mM and diluted in the cell culture medium to the final concentration.
The parental cell lines D54-MG and U87-MG, designated respectively as D54-C0 and U87-C0, were initially exposed to 100 μM TMZ for 2 weeks, and their resistant clones were isolated. These resistant clones were then repeatedly exposed to IC50 of TMZ for a period of 12 months. This procedure mimicked the clinical regimen of pulse-treatment method and ensured that the most resistant clones were selected. Acquired TMZ resistance was confirmed by sulforhodamine B (SRB) assay. Aliquot of cells was isolated monthly and maintained in low dose (100 μM) of TMZ. TMZ-resistant variants so generated were denoted according to the number of months of drug treatment (e.g. D54-P10 for cells isolated after 10 months of treatment). Both the parental (D54-C0 and U87-C0) and the passage controls (D54-C10 and U87-C6), which were not exposed to TMZ, were included for parallel analysis.
In vitro chemo-sensitivity assay
For the chemo-sensitivity assay, 5,000 cells/well were seeded in 96-well plates at 37°C with 5% CO2. After 24 h, the culture media were replaced with fresh media in the absence or presence of serial concentrations of TMZ. After 96 h, the sulforhodamine B (SRB) assay (Sigma-Aldrich, Saint Louis, MO, USA) was used to determine the surviving cell numbers as measured by absorbance (optical density, OD) at 490 nm. Results are expressed as the percentage of viable cells relative to the controls (cells without prior TMZ treatment). A dose-response curve was plotted, and IC50 values were calculated by derivation of the best-fit line, using three independent experiments performed in triplicate.
TUNEL assay
Apoptotic cells were determined using the in situ cell death detection kit POD (Roche Diagnostics, Indianapolis, IN, USA), according to the manufacturer’s protocol. This assay quantitatively determines DNA fragmentation at the free 3′-OH termini catalysed by the terminal deoxyribonucleotidyl transferase (TdT). Apoptotic indices were obtained by averaging the number of apoptotic cells within each of 10 randomly selected fields of view under light microscopy.
In vitro cell proliferation assay
5,000 cells/well were seeded in 96-well plates. Cells were serum-starved for 1 day and retreated with serum in medium before the experiment started. The relative density of cells was determined by the SRB assay on a daily basis for 5 days. Absorbance was measured at 490 nm in three independent experiments performed in triplicate.
Wound healing and adhesion assays
Wound healing assay was performed to assess cell migration. Assayed cells were grown to 100% confluency before a wound was created on the cell monolayer using a 10-μl pipette tip. Cell migration was then determined based on the movement of cells towards the created lesion. The closure of wound was observed after 12 and 18 h under light microscopy. For the adhesion assay, 2 × 105/ml cells in 100 μl were assayed by plating in triplicate in 96-well plates which were pre-coated with 5 μg/ml collagen I (Invitrogen, Carlsbad, CA, USA). Cell attachment was determined 1 h after seeding. Attached cells were fixed in 4% paraformaldehyde at room temperature for 15 min and stained with 5 mg/ml crystal violet (Accu-Chem Laboratories, Richardson, TX, USA) for 15 min. The purple crystals formed were dissolved in 20% sodium dodecyl sulphate (SDS; USB Corporation, Cleveland, OH, USA) for 30 min, and absorbance was measured at 570/655 nm.
Cell cycle analysis
Assayed cells (1 × 106) were harvested during the exponential growth phase. These cells were washed with phosphate-buffered saline (PBS), fixed in 80% ethanol and stored at −20°C overnight. After fixation, cells were washed twice with 1% FBS in PBS and resuspended in PBS solution containing and resuspended in FACS solution containing 200 μg RNase A (Sigma-Aldrich) and 1 μg propidium iodide (Invitrogen). The reaction was incubated in dark at 37°C for 30 min before analysis on Cytomics™ FC 500 flow cytometer (Beckman Coulter, Inc., Brea, CA, USA).
Two-dimensional gel electrophoresis (2-DE) analysis
Cell pellets of TMZ-treated and untreated samples were solubilised in 0.1 ml 40 mM Tris supplemented with 20 μg/ml RNase A (bovine pancreas source; USB Corporation), 50 U/ml DNase I (bovine pancreas source; USB Corporation), complete ethylenediamine tetraacetic acid (EDTA)-free protease inhibitors (Roche Diagnostics) and 0.5 mM phenylmethylsulphonylfluoride (PMSF) (USB Corporation), and lysed by vortexing. After centrifugation at 16,000g, the pellets were resuspended by vortexing in 0.1 ml extraction solution containing 8 M urea, 4% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulphonate (CHAPS), 40 mM Tris, 0.2% carrier ampholytes and 2 mM tributyl phosphine (these reagents were included in ReadyPrep sequential extraction kit; Bio-Rad, Hercules, CA, USA). After centrifugation at 16,000g for 60 min at 4°C, the supernatant was harvested as total protein lysates. The concentrations of these lysates were quantified using PlusOne 2-DE Quant kit (GE Biosciences, Buckinghamshire, England). Proteins from each sample were subjected to 2-DE as previously described [26, 27]. Briefly, 60 μg protein lysate was diluted in 200 μl sample buffer and applied on an 11-cm (pH 4–7) linear immobilized pH gradient (IPG) Immobiline dry strip (GE Biosciences) for first-dimension separation. In-sample active rehydration was performed, and samples were subjected to isoelectric focussing on IPGphor system (GE Biosciences). The same conditions were used as previously described [26, 28]. After equilibration, protein was electrophoresed on a 12.5% precast Ettan™ DALT gel (GE Biosciences) for second-dimension separation. The resulting gels were silver stained, dried and captured using GS-800 calibrated densitometer (Bio-Rad). Gel spots were detected, matched and normalised using PDQuest™ 2-DE analysis software (Bio-Rad), according to the manufacturer’s protocol. To enhance the high reproducibility of our data and improve consensus across triplicates gels, a group consensus tool was used. The volume of each gel spot was employed as an indicator of protein expression. The intensity of each spot was normalised by total valid spot volume, and is reported in parts per million (ppm). Student’s t-test was used to analyse variance of spot intensities in controls (parental cells and passage controls) and different TMZ-resistant sublines. For selecting protein spots for identification, only spots with significant expression difference (P < 0.05) in controls when compared with the TMZ-resistant variants as well as those which have shown correlation to their IC50 of TMZ were chosen for MS analysis.
Protein identification by MALDI-TOF/TOF
Protein spots of interest were excised manually with syringe needles, and the gel pieces were destained, washed, equilibrated and dried before trypsin digestion (MS grade; Promega, Madison, WT, USA) as previously described [26, 28]. Digestion was performed at 37°C overnight, and supernatants were pooled and purified using ZipTip® pipette tips (Millipore, Bedford, MA, USA). ABI 4800 proteomics analyser matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF)/TOF (Applied Biosystems Ltd., Foster City, CA, USA) operating in result-dependent acquisition mode was used to generate the MS/MS spectrum data. A maximum of the five most intense tandem mass spectra were collected for MS/MS, and the fragment ion masses were compared with the NCBI/Swiss-Prot database for protein identification using MASCOT version 2.1 (Matrix Science Inc., Boston, MA, USA).
Real-time quantitative polymerase chain reaction (qPCR)
Total RNA extracted from the parental controls and resistant counterparts were subjected to validation by qPCR. Purified RNA (1 μg) was reverse-transcribed to complementary DNA (cDNA) using TaqMan reverse-transcription reagents (Applied Biosystems Ltd.), following the manufacturer’s protocol. cDNA was then mixed with gene-specific primers (Supplementary Table 1) and Platinum® SYBR® Green qPCR SuperMix-UDG with ROX (Invitrogen), and gene amplification was carried out in ABI Prism 7900HT sequence detection system (Applied Biosystems Ltd.). Messenger RNA (mRNA) expression levels of each gene after normalisation to β-actin as internal control are expressed in relative fold ratios.
Results
Successful establishment of TMZ-resistant D54-MG
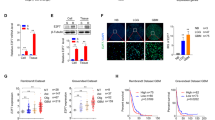
We successfully established a TMZ-resistant glioma cell model for proteomics analysis. Two resistant sublines, namely D54-P5 and D54-P10, were generated after treatment with TMZ for 5 and 10 months, respectively. The two resistant variants showed strong resistance to further TMZ treatment. When compared with their control counterparts, namely the parental line (D54-C0; IC50, 544.6 μM) and the passage control line (D54-C10; IC50, 520.7 μM), D54-P5 and D54-P10 showed ~2-fold (IC50, 1,251.2 μM; P = 0.046) and ~3-fold (IC50, 1,681.7 μM; P = 0.004) increase in their IC50 of TMZ, respectively (Figure 1a).

Development of TMZ-resistant D54-MG malignant glioma cells. a Survival curves of D54-C0 (parental D54-MG), D54-C10 (passage control, 10 months in culture), D54-P5 (TMZ-resistant variant, 5 months in culture) and D54-P10 (TMZ-resistant variant, 10 months in culture) cells, determined by drug sensitivity assay. IC50 of TMZ of each cell lines is tabulated underneath the graph. b Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay revealed more D54-C0 cells undergoing apoptosis when compared with its resistant D54-P10 counterpart after treatment with TMZ at different concentrations for 96 h. The above representative images show treatment of both D54-C0 and D54-P10 cells with 500 μM TMZ. Data were analysed by Student’s t-test and are reported as mean ± standard deviation (SD). *P < 0.05; **P < 0.01
To investigate whether the increase in TMZ resistance in glioma cells was associated with apoptosis, the numbers of apoptotic cells were compared between TMZ-sensitive (D54-C0) and TMZ-resistant (D54-P10) cells under different concentrations of TMZ (Figure 1b). Compared with D54-C0 cells, D54-P10 cells showed a significant decrease in the number of apoptotic cells (P < 0.05) when treated with 250 μM TMZ. At 500 μM and 1,000 μM TMZ (P < 0.01), the difference was even more pronounced, with a 5-fold difference in cell apoptosis (Fig. 1b). These results suggested a link between the onset of TMZ resistance and reduction in apoptosis.
Cell properties after acquisition of TMZ resistance
Long-term in vitro treatment of D54-C0 cells with TMZ was associated with a change in cell morphology. D54-P10 cells became more elongated, asymmetric and spike-like, with more varied size and shrinkage of the cytoplasm. Moreover, most of the D54-P10 cells exhibited pseudopodia and cytosolic granules (Fig. 2a). Besides, TMZ-resistant D54-P10 demonstrated a lower proliferation rate than TMZ-sensitive D54-C0 and D54-C10 (Fig. 2b). Chemotherapeutic agents typically target rapidly dividing cells, and are likely to be ineffective against slow-proliferative and/or quiescent cancer cells. Thus, this population (D54-P10) may indeed represent a subtype of cells that are resistant to TMZ. In fact, acquired resistance to chemotherapeutic drugs is often associated with an increase in malignant behaviour in cancer cells, because strong resistance can facilitate progression and invasion of cancer cells. The D54-P10 resistant subline also showed enhanced cell adhesion and migration capacities when compared with controls (Fig. 2c, d). Figure 2c demonstrates that D54-P10 cells had an almost 2-fold increase in cell adhesion (**P < 0.01). Faster wound closure time was also observed in D54-P10 cells when compared with both D54-C0 and D54-C10 controls at 12 h (Fig. 2d).

Alteration in morphology of D54-MG cells after acquisition of TMZ resistance. a Images show the morphological appearance of D54-C0, D54-C10 and D54-P10 cells under inverted microscope (upper panel) and after haematoxylin and eosin staining (lower panel). Original magnification, ×200. b Cell proliferation assay. D54-C0, D54-C10 and D54-P10 cells were serum-starved before being assayed for cell viability and growth using SRB assay. Cells were retreated with serum in medium before the measurement was started. Data were analysed by Student’s t-test and are reported as mean ± SD. *P < 0.05; **P < 0.01. c Cell adhesion assay was performed in D54-C0, D54-C10 and D54-P10 cells. Cell adhesive capabilities of each type of cell are expressed as the fold ratio of the number of adhered cells relative to the number of adhered D54-C0 cells. d Wound healing assay of D54-C0, D54-C10 and D54-P10 cells was performed to assess the extent of cell migration towards the artificially created wound. Images were taken at 12 and 18 h after wound creation on the cell monolayer. At 12 h, D54-P10 cells migrated faster towards the wound than D54-C0 and D54-C10 cells. e Cell cycle analysis of D54-C0, D54-C10 and D54-P10 cells was performed by flow cytometry. Long-term exposure to TMZ led to cell cycle arrest of D54-P10 cells, such that more cells remained in the G0/G1 phase. f Western blotting shows the levels of cell cycle regulators, cyclin B1, CDK4 and cyclin D1, in D54-C0, D54-C10 and D54-P10 cells. The images show representative results from triplicate experiments
Temozolomide is known to induce cell cycle arrests [29]. Hence, we examined whether alterations of cell cycle arrest were involved in the development of TMZ resistance. By means of flow cytometry and western blotting, the acquisition of TMZ resistance in D54-P10 cells was found to be associated with accumulation of cells at G0/G1 from 71.93% (D54-C0) or 68.67% (D54-C10) to 87.88% (D54-P10), and a decrease of cells in G2/M from 21.06% (D54-C0) or 19.23% (D54-C10) to 7.29% (D54-P10) (Fig. 2e). For the differential expression of cell cycle regulators, a higher level of cyclin B1, which is the regulatory subunit of the M phase promoting factor, was found in D54-P10 cells when compared with the controls. Inversely, lower levels of cyclin D1 and cyclin-dependent kinase 4 (CDK4), which are known to play important roles in the G1/S cell cycle checkpoint, were found in D54-P10 cells, suggestive of G1 phase arrest through inhibition of these proteins (Fig. 2f). These findings indicate that onset of TMZ resistance is mediated through loss of cell cycle check points and changes in programmed cell death.
Proteins involved in TMZ resistance
We examined the protein profiles in four cell lines—the two TMZ-resistant sublines (D54-P5 and D54-P10) and the parental (D54-C0) and passage (D54-C10) controls and looked for proteins that were differentially expressed. Comparisons of protein spots on gels revealed 98 protein spots that varied with significant differences (P < 0.05). Amongst these spots, 10 were verified to be valid protein spots with ppm greater than 1,000, and relative fold ratios that significantly correlated (P < 0.05) with the IC50 of TMZ of the treated cells. Importantly too, the spots were successfully verified by manual inspection. Of the 10 protein spots chosen for MS/MS, 9 were successfully identified. Figure 3 is a representative 2-DE gel showing the respective locations of these protein spots whose expressions varied within the 99th confidence level based on the variance of mean changes. The results of MS/MS and the detailed characterisation of the MS/MS data are presented in Table 1. The protein score, total ion score, confidence interval (CI, %), matched peptides and sequence coverage are reported for each assigned protein. In addition, the exact molecular weights and isoelectric points for these proteins mostly matched with those observed on the gel for each spot in question, providing additional verification of their identities. Individual mass spectrum of each protein is presented in Supplementary Fig. 1. In some instances, more than one protein spot was identified to be the same protein, suggesting the existence of post-translation modifications, degradation, or alternative splicing isoforms in these proteins. As a result, these nine spots actually referred to seven distinct proteins with higher expressions in resistant glioma cells (D54-P5 and D54-P10) than in the control lines (D54-C0 and D54-C10). The functions of these proteins and their respective expression ratios in each cell line are presented in Table 2. Besides, in order to enhance the reliability of our data, we performed Spearman’s rho correlation analysis between the raw intensities of each spot on the gels and the sample’s IC50 values. Results showed that, among the nine protein spots, three proteins (SSP 2601, SSP3213 and SSP3506) exhibited expressions that were strongly correlated with the IC50 values (P < 0.01, correlation coefficient >0.9). Others (SSP0308, SSP1608, SSP3403, SSP4514, SSP5428 and SSP5536) also demonstrated an association, showing correlation coefficient values >0.5.

Proteins associated with TMZ resistance in D54-P10 cells. A representative 2-DE gel image shows the profile of total protein extracted from D54-P10 cells. Nine proteins were recognised as spots of interest and exhibited significant differential expression between controls (D54-C0 and D54-C10) and resistant sublines (D54-P5 and D54-P10) using Student’s t-test
For further validation, the transcript expression of six out of these seven targets, including proliferating cell nuclear antigen (PCNA), vimentin (VIM), cathepsin D (CTSD), prolyl 4-hydroxylase, beta polypeptide (P4HB), mannose 6-phosphate receptor binding protein 1 (M6PRBP1) and ezrin (EZR), were subjected to qPCR analysis. In accordance with the proteomic data, upregulated transcript expression of these proteins was found to be associated with D54-P10 cells when compared with D54-C0 cells (Fig. 4). These proteins had previously been reported to be involved in tumoural processes, such as cell cycle alteration, cell proliferation, epithelial–mesenchymal transition (EMT), apoptosis and cell transformation. To demonstrate that the higher expression of these proteins was not limited to one particular cell line, their expression in another pair of TMZ-sensitive (U87-C0) and TMZ-resistant (U87-P6) malignant glioma cell lines was also studied using qPCR. Higher expression of these proteins was also found in U87-P6 resistant cells versus U87-C0 sensitive cells (Fig. 4), and thus they resembled the D54 cells. In particular, a 2-fold difference was found for PCNA, CSTD and P4HB (**Fig. 4). These results indicate that the proteins identified by our proteomics approach also have corresponding upregulation of their gene expression levels, and are associated with establishment of TMZ resistance in glioma cells of different origins.

Gene expression of PCNA, VIM, CTSD, P4HB, M6PRBP1 and EZR in TMZ-sensitive and TMZ-resistant D54-MG and U87-MG cells. qPCR was performed to investigate the gene expressions of PCNA, VIM, CTSD, P4HB, M6PRBP1 and EZR in TMZ-sensitive D54-C0 and U87-C0 cells and their respective TMZ-resistant D54-P10 and U87-P6 counterparts. Data for each cell line are presented as fold ratio relative to its respective control. Experiments conducted in triplicate and repeated at least twice. Student’s t-test was used to determine the level of significance. *P < 0.05; **P < 0.01
Upregulation of VIM, CTSD and P4HB in high-grade glioma
To elucidate the clinical significance of these identified proteins in human gliomas, three proteins (VIM, CTSD and P4HB) were chosen for assessment in five low-grade and nine high-grade glioma tissues. These proteins were chosen for analysis mainly after the overall evaluation of their relative fold ratios, MS scores and the degree of correlation to the IC50 values. Overall, by Western blotting (Fig. 5a, b) and immunohistochemical (IHC) staining (Fig. 5c), high expression level of these proteins was found in high-grade gliomas compared with low-grade tumours, and GBM, which is the most malignant form of gliomas, revealed the highest levels of expression, suggesting that these proteins may be related to the malignant properties of glioma.

Clinical significance of VIM, CTSD and P4HB in human malignant glioma. a Representative blot images demonstrate high expression levels of VIM, CTSD and P4HB in high-grade (HGG) compared with low-grade gliomas (LGG). Glioblastoma reveals the highest level of expression of the three proteins and is illustrated by asterisk. b Whiskers (min to max) diagram shows the densitometry data from Western blot analysis. Differences are expressed as ratio of the target protein in different grade of glioma to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) density. c VIM, CTSD and P4HB detection by IHC. Representative immunohistochemical staining of VIM, CTSD and P4HB in paraffin-embedded human glioma tissues. Low-grade glioma tissues seen in b, d, f are weakly to moderately stained, while in high-grade tissues, VIM, CTSD and P4HB stainings are strongly intense (A, C, E). Original magnification: ×100; ×400 (insets)
Discussion
Temozolomide has attracted considerable attention as an effective chemotherapeutic agent against malignant gliomas. Although significant clinical benefits have been achieved with the use of TMZ, patient outcomes remain unsatisfactory, as acquired resistance commonly develops and accounts for 90% of recurrent diseases [15]. Changes in MGMT level [30], mismatch repair status [31, 32], loss of p53 function [12] or activation of the Akt pathway [33] have been identified as responsible for the development of TMZ resistance. Despite extensive research, however, the mechanisms underlying acquired TMZ resistance are incompletely characterised, and many biological processes might exist independently of the aforementioned pathways.
Recently, Oliva et al. [15] reported that acquisition of TMZ resistance may be associated with remodelling of mitochondrial electron transport, whilst in Auger’s report, resistance to TMZ was related to selection of pre-existent resistant cells in the paternal tumour [4]. Although these studies have improved our understanding of the development of TMZ resistance in malignant gliomas, they were mainly conducted at genomic and transcriptional levels. In the present study, we took a step further to adopt a proteomics approach, based on the principle that proteins are the key structural and functional units in the body and are the final products capable of translating information between living cells. With our in vitro model, we demonstrated that glioma cells tended to grow slowly and develop significant changes in cell morphology after long-term exposure to TMZ, suggestive of changes in cell cycle and cytoskeleton regulation. Considering that proliferation rates depend on cell cycle progression, slow-proliferating cells may have lower activity of CDKs, which are critical regulators of cell cycle progression. Our results showed G1 phase arrest together with reduction of CDK4 regulator in the D54-P10 cell line, thus further reinforcing the reliability of our findings. Moreover, TMZ resistance was associated with increased adhesion and migration capacities, indicating that onset of chemoresistance may also promote cancer progression and other malignant phenotypes. In fact, cell adhesion-mediated drug resistance is thought to be a principal mechanism by which extracellular matrix proteins provide a survival advantage against cytotoxic drug-induced apoptosis [34]. We also observed cell cycle alterations after the development of TMZ resistance. It is of interest to note that, while previous studies have suggested the association of cell cycle alterations with p53 status [12], chk1- and chk2-mediated cell death prevention [35] and Akt pathway activation bypassing G2 arrest [33], none of the proteins identified in our proteomics analysis are known to be involved in these pathways.
Our study is the first to investigate and compare proteomics profiling with regards to TMZ resistance in human malignant glioma cells. We identified seven proteins associated with TMZ resistance. By comparing the resistant variants with not only the parental line but also the passage controls, we confirmed that these changes in protein expressions were the results of long-term exposure to TMZ rather than phenotypic changes following repeated passaging. Furthermore, the observed alterations in protein levels correlated well with changes in gene expressions.
There are several limitations to the present study. Our in vitro model may not be fully representative of the highly complex and heterogeneous in vivo microenvironment to which malignant gliomas are exposed. Although our model was designed to mimic pulse treatment with TMZ, it may differ from that found in a clinical setting due to the influences of in vivo pharmacokinetics and pharmacodynamics. Moreover, part of the clinical regimen currently employed involves the use of concomitant irradiation, which may alter cellular responses to TMZ. Although we have verified our findings in two cell lines, it is uncertain whether the same changes in protein expressions are universally present in all gliomas, which are now known to belong to a heterogeneous group of neoplasms with differing cellular lineages, genetic changes, biological behaviours and responses to chemotherapeutics.
Nevertheless, consistent with our observations of augmented malignant behaviours after chronic exposure to TMZ, several of the proteins (PCNA, VIM, CTSD, P4HB, M6PRBP1 and EZR) identified in this study are known to be associated with tumoural processes, such as cell cycle alteration, cell proliferation, EMT, apoptosis and cell transformation. For instance, PCNA is crucially involved in several aspects of DNA metabolism, as well as cell cycle regulation. It interacts with p21, which plays an essential role in DNA damage responses by inducing cell cycle arrest [36]. Moreover, B-cell lymphoma 2 (Bcl-2)-mediated chemoresistance in prostate cancers has been shown to be associated with an overexpression of PCNA that counteracts the altered expression of cell cycle regulators and controls cell cycle progression [37]. VIM is a type III intermediate filament protein that is expressed frequently in epithelial carcinomas and is correlated with tumour invasiveness and poor prognosis [38]. Upregulation of this protein enhances carcinoma cell migration and adhesion, which is in agreement with our findings. In malignant glioma, a significant increase in VIM expression levels was detected in cell cultures after treatment with a constant concentration of TMZ [39]. CTSD affects multiple tumour progression steps including proliferation, angiogenesis and apoptosis [40]. In glioma, CTSD is a potential serum marker of poor prognosis and is predictive of aggressive tumour behaviour [41]. Expression of this protein in relation to chemo-response has been reported also in neuroblastoma, with the effect of protecting cells against doxorubicin-induced cell death [42]. P4HB expression has previously been reported to be related to the invasive properties of malignant glioma. It is a chaperone protein that mediates integrin-dependent cell adhesion and plays important roles in cell invasion and migration [43]. ERp 57, a protein family member of P4HB, has previously been reported to be involved in chemoresistance in ovarian cancers [44]. These proteins have a range of functions within the endoplasmic reticulum, one of which is protection against cell apoptosis in response to chemotherapy [45]. M6PRBP1, also designated as cargo selection protein (TIP47), was characterised as a cargo selection device for mannose 6-phosphate receptors (MPRs) that directed their transport from endosomes to the trans-Golgi network [46]. Recently, Hocsak’s group showed that TIP47 was able to bind to mitochondria in oxidative stress, and inhibit mitochondrion-mediated cell death by protecting mitochondrial membrane integrity [47]. Over-expression of TIP47 was found to protect cells from taxol-induced cell death by stabilising the mitochondrial membrane system and by favourably affecting the expression of Bcl-2 homologues, suggesting that they may play important roles in the development of resistance against cytostatic agents [48]. EZR is a linker between plasma membrane and actin cytoskeleton, and can promote dissemination of tumour cells [49]. Over-expression of EZR is associated with the development of metastasis and poor outcome in osteosarcoma [50]. In comparative proteomic study of paclitaxel-resistant adenocarcinoma, overexpression of EZR was hypothesised to inhibit induction of apoptosis through activation of the Akt pathway [51].
In conclusion, our data show that long-term exposure to TMZ may lead to the development of TMZ resistance and the promotion of malignant phenotypes in human malignant glioma cells. Proteomics is a feasible approach for the identification of molecular candidates that may serve not only as markers of chemoresistance but also as potential therapeutic targets in the treatment of TMZ-resistant human malignant gliomas. Whether our protein candidates play roles in the development of TMZ resistance or simply represent by-stander changes remains to be determined by future work, including further in vivo studies and clinical correlations. The present study provides a foundation for future investigations on distinct protein candidates that may contribute to the development of chemoresistance in human malignant gliomas.
References
Tate MC, Aghi MK (2009) Biology of angiogenesis and invasion in glioma. Neurotherapeutics 6:447–457
Norden AD, Wen PY (2006) Glioma therapy in adults. Neurologist 12:279–292
Mason WP (2008) Emerging drugs for malignant glioma. Expert Opin Emerg Drugs 13:81–94
Auger N, Thillet J, Wanherdrick K, Idbaih A, Legrier ME, Dutrillaux B, Sanson M, Poupon MF (2006) Genetic alterations associated with acquired temozolomide resistance in SNB-19, a human glioma cell line. Mol Cancer Ther 5:2182–2192
a Fischer J, Costa Carvalho P, da Fonseca CO, Liao L, Degrave WM, a Carvalho M, Yates JR, Domont GB (2011) Chemo-resistant protein expression pattern of glioblastoma cells (A172) to perillyl alcohol. J Proteome Res 10:153–160
Hart MG, Grant R, Garside R, Rogers G, Somerville M, Stein K (2008) Temozolomide for high grade glioma. Cochrane Database Syst Rev (4):CD007415
Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, Belanger K, Brandes AA, Marosi C, Bogdahn U, Curschmann J, Janzer RC, Ludwin SK, Gorlia T, Allgeier A, Lacombe D, Cairncross JG, Eisenhauer E, Mirimanoff RO (2005) Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med 352:987–996
Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, Ludwin SK, Allgeier A, Fisher B, Belanger K, Hau P, Brandes AA, Gijtenbeek J, Marosi C, Vecht CJ, Mokhtari K, Wesseling P, Villa S, Eisenhauer E, Gorlia T, Weller M, Lacombe D, Cairncross JG, Mirimanoff RO (2009) Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol 10:459–466
Dehdashti AR, Hegi ME, Regli L, Pica A, Stupp R (2006) New trends in the medical management of glioblastoma multiforme: the role of temozolomide chemotherapy. Neurosurg Focus 20:E6
Nagasubramanian R, Dolan ME (2003) Temozolomide: realizing the promise and potential. Curr Opin Oncol 15:412–418
Friedman HS, Johnson SP, Dong Q, Schold SC, Rasheed BK, Bigner SH, Ali-Osman F, Dolan E, Colvin OM, Houghton P, Germain G, Drummond JT, Keir S, Marcelli S, Bigner DD, Modrich P (1997) Methylator resistance mediated by mismatch repair deficiency in a glioblastoma multiforme xenograft. Cancer Res 57:2933–2936
Bocangel DB, Finkelstein S, Schold SC, Bhakat KK, Mitra S, Kokkinakis DM (2002) Multifaceted resistance of gliomas to temozolomide. Clin Cancer Res 8:2725–2734
Fortin D (2004) The blood-brain barrier should not be underestimated in neuro-oncology. Rev Neurol (Paris) 160:523–532
Trivedi RN, Almeida KH, Fornsaglio JL, Schamus S, Sobol RW (2005) The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res 65:6394–6400
Oliva CR, Nozell SE, Diers A, McClugage SG 3rd, Sarkaria JN, Markert JM, Darley-Usmar VM, Bailey SM, Gillespie GY, Landar A, Griguer CE (2010) Acquisition of temozolomide chemoresistance in gliomas leads to remodeling of mitochondrial electron transport chain. J Biol Chem 285:39759–39767
Le Calve B, Rynkowski M, Le Mercier M, Bruyere C, Lonez C, Gras T, Haibe-Kains B, Bontempi G, Decaestecker C, Ruysschaert JM, Kiss R, Lefranc F (2010) Long-term in vitro treatment of human glioblastoma cells with temozolomide increases resistance in vivo through up-regulation of GLUT transporter and aldo-keto reductase enzyme AKR1C expression. Neoplasia 12:727–739
Niclou SP, Fack F, Rajcevic U (2010) Glioma proteomics: status and perspectives. J Proteomics 73:1823–1838
Jiang R, Mircean C, Shmulevich I, Cogdell D, Jia Y, Tabus I, Aldape K, Sawaya R, Bruner JM, Fuller GN, Zhang W (2006) Pathway alterations during glioma progression revealed by reverse phase protein lysate arrays. Proteomics 6:2964–2971
Li J, Zhuang Z, Okamoto H, Vortmeyer AO, Park DM, Furuta M, Lee YS, Oldfield EH, Zeng W, Weil RJ (2006) Proteomic profiling distinguishes astrocytomas and identifies differential tumor markers. Neurology 66:733–736
Petrik V, Saadoun S, Loosemore A, Hobbs J, Opstad KS, Sheldon J, Tarelli E, Howe FA, Bell BA, Papadopoulos MC (2008) Serum alpha 2-HS glycoprotein predicts survival in patients with glioblastoma. Clin Chem 54:713–722
Schuhmann MU, Zucht HD, Nassimi R, Heine G, Schneekloth CG, Stuerenburg HJ, Selle H (2010) Peptide screening of cerebrospinal fluid in patients with glioblastoma multiforme. Eur J Surg Oncol 36:201–207
Seyfried NT, Huysentruyt LC, Atwood JA 3rd, Xia Q, Seyfried TN, Orlando R (2008) Up-regulation of NG2 proteoglycan and interferon-induced transmembrane proteins 1 and 3 in mouse astrocytoma: a membrane proteomics approach. Cancer Lett 263:243–252
Bian XW, Xu JP, Ping YF, Wang Y, Chen JH, Xu CP, Wu YZ, Wu J, Zhou XD, Chen YS, Shi JQ, Wang JM (2008) Unique proteomic features induced by a potential antiglioma agent, Nordy (dl-nordihydroguaiaretic acid), in glioma cells. Proteomics 8:484–494
Rajcevic U, Petersen K, Knol JC, Loos M, Bougnaud S, Klychnikov O, Li KW, Pham TV, Wang J, Miletic H, Peng Z, Bjerkvig R, Jimenez CR, Niclou SP (2009) iTRAQ-based proteomics profiling reveals increased metabolic activity and cellular cross-talk in angiogenic compared with invasive glioblastoma phenotype. Mol Cell Proteomics 8:2595–2612
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Luk JM, Lam CT, Siu AF, Lam BY, Ng IO, Hu MY, Che CM, Fan ST (2006) Proteomic profiling of hepatocellular carcinoma in Chinese cohort reveals heat-shock proteins (Hsp27, Hsp70, GRP78) up-regulation and their associated prognostic values. Proteomics 6:1049–1057
Sun S, Poon RT, Lee NP, Yeung C, Chan KL, Ng IO, Day PJ, Luk JM (2010) Proteomics of hepatocellular carcinoma: serum vimentin as a surrogate marker for small tumors (< or = 2 cm). J Proteome Res 9:1923–1930
Sun S, Xu MZ, Poon RT, Day PJ, Luk JM (2010) Circulating Lamin B1 (LMNB1) biomarker detects early stages of liver cancer in patients. J Proteome Res 9:70–78
Hirose Y, Berger MS, Pieper RO (2001) p53 effects both the duration of G2/M arrest and the fate of temozolomide-treated human glioblastoma cells. Cancer Res 61:1957–1963
Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG (1999) Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer Res 59:793–797
Maxwell JA, Johnson SP, McLendon RE, Lister DW, Horne KS, Rasheed A, Quinn JA, Ali-Osman F, Friedman AH, Modrich PL, Bigner DD, Friedman HS (2008) Mismatch repair deficiency does not mediate clinical resistance to temozolomide in malignant glioma. Clin Cancer Res 14:4859–4868
Augustine CK, Yoo JS, Potti A, Yoshimoto Y, Zipfel PA, Friedman HS, Nevins JR, Ali-Osman F, Tyler DS (2009) Genomic and molecular profiling predicts response to temozolomide in melanoma. Clin Cancer Res 15:502–510
Hirose Y, Katayama M, Mirzoeva OK, Berger MS, Pieper RO (2005) Akt activation suppresses Chk2-mediated, methylating agent-induced G2 arrest and protects from temozolomide-induced mitotic catastrophe and cellular senescence. Cancer Res 65:4861–4869
Kohmo S, Kijima T, Otani Y, Mori M, Minami T, Takahashi R, Nagatomo I, Takeda Y, Kida H, Goya S, Yoshida M, Kumagai T, Tachibana I, Yokota S, Kawase I (2010) Cell surface tetraspanin CD9 mediates chemoresistance in small cell lung cancer. Cancer Res 70:8025–8035
Hirose Y, Berger MS, Pieper RO (2001) Abrogation of the Chk1-mediated G(2) checkpoint pathway potentiates temozolomide-induced toxicity in a p53-independent manner in human glioblastoma cells. Cancer Res 61:5843–5849
Cazzalini O, Scovassi AI, Savio M, Stivala LA, Prosperi E (2010) Multiple roles of the cell cycle inhibitor p21 (CDKN1A) in the DNA damage response. Mutat Res 704:12–20
Chaudhary KS, Abel PD, Stamp GW, Lalani E (2001) Differential expression of cell death regulators in response to thapsigargin and adriamycin in Bcl-2 transfected DU145 prostatic cancer cells. J Pathol 193:522–529
McInroy L, Maatta A (2007) Down-regulation of vimentin expression inhibits carcinoma cell migration and adhesion. Biochem Biophys Res Commun 360:109–114
Trog D, Yeghiazaryan K, Schild HH, Golubnitschaja O (2008) Up-regulation of vimentin expression in low-density malignant glioma cells as immediate and late effects under irradiation and temozolomide treatment. Amino Acids 34:539–545
Berchem G, Glondu M, Gleizes M, Brouillet JP, Vignon F, Garcia M, Liaudet-Coopman E (2002) Cathepsin-D affects multiple tumor progression steps in vivo: proliferation, angiogenesis and apoptosis. Oncogene 21:5951–5955
Fukuda ME, Iwadate Y, Machida T, Hiwasa T, Nimura Y, Nagai Y, Takiguchi M, Tanzawa H, Yamaura A, Seki N (2005) Cathepsin D is a potential serum marker for poor prognosis in glioma patients. Cancer Res 65:5190–5194
Sagulenko V, Muth D, Sagulenko E, Paffhausen T, Schwab M, Westermann F (2008) Cathepsin D protects human neuroblastoma cells from doxorubicin-induced cell death. Carcinogenesis 29:1869–1877
Goplen D, Wang J, Enger PO, Tysnes BB, Terzis AJ, Laerum OD, Bjerkvig R (2006) Protein disulfide isomerase expression is related to the invasive properties of malignant glioma. Cancer Res 66:9895–9902
Cicchillitti L, Della Corte A, Di Michele M, Donati MB, Rotilio D, Scambia G (2010) Characterisation of a multimeric protein complex associated with ERp57 within the nucleus in paclitaxel-sensitive and -resistant epithelial ovarian cancer cells: the involvement of specific conformational states of beta-actin. Int J Oncol 37:445–454
Fu Y, Li J, Lee AS (2007) GRP78/BiP inhibits endoplasmic reticulum BIK and protects human breast cancer cells against estrogen starvation-induced apoptosis. Cancer Res 67:3734–3740
Bulankina AV, Deggerich A, Wenzel D, Mutenda K, Wittmann JG, Rudolph MG, Burger KN, Honing S (2009) TIP47 functions in the biogenesis of lipid droplets. J Cell Biol 185:641–655
Hocsak E, Racz B, Szabo A, Mester L, Rapolti E, Pozsgai E, Javor S, Bellyei S, Gallyas F Jr, Sumegi B, Szigeti A (2010) TIP47 protects mitochondrial membrane integrity and inhibits oxidative-stress-induced cell death. FEBS Lett 584:2953–2960
Hocsak E, Racz B, Szabo A, Pozsgai E, Szigeti A, Szigeti E, Gallyas F Jr, Sumegi B, Javor S, Bellyei S (2010) TIP47 confers resistance to taxol-induced cell death by preventing the nuclear translocation of AIF and endonuclease G. Eur J Cell Biol 89:853–861
Heiska L, Carpen O (2005) Src phosphorylates ezrin at tyrosine 477 and induces a phosphospecific association between ezrin and a kelch-repeat protein family member. J Biol Chem 280:10244–10252
Di Cristofano C, Leopizzi M, Miraglia A, Sardella B, Moretti V, Ferrara A, Petrozza V, Della Rocca C (2010) Phosphorylated ezrin is located in the nucleus of the osteosarcoma cell. Mod Pathol 23:1012–1020
Sun QL, Sha HF, Yang XH, Bao GL, Lu J, Xie YY (2011) Comparative proteomic analysis of paclitaxel sensitive A549 lung adenocarcinoma cell line and its resistant counterpart A549-Taxol. J Cancer Res Clin Oncol 137:521–532
Acknowledgments
We would like to express our sincere gratitude for the insightful advice and support of Dr. Ching Fai Fung. The work was supported by a small project grant from the University of Hong Kong (project code 201007176020).
Conflict of interest
None to declare.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Table S1
qPCR primer sequences of the six potential targets (DOC 35 kb)
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Sun, S., Wong, T.S., Zhang, X.Q. et al. Protein alterations associated with temozolomide resistance in subclones of human glioblastoma cell lines. J Neurooncol 107, 89–100 (2012). https://doi.org/10.1007/s11060-011-0729-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-011-0729-8