
Abstract
While the central nervous system (CNS) was once thought to be excluded from surveillance by immune cells, a concept known as “immune privilege,” it is now clear that immune responses do occur in the CNS—giving rise to the field of neuroimmunology. These CNS immune responses can be driven by endogenous (glial) and/or exogenous (peripheral leukocyte) sources and can serve either productive or pathological roles. Recent evidence from mouse models supports the notion that infiltration of peripheral monocytes/macrophages limits progression of Alzheimer's disease pathology and militates against West Nile virus encephalitis. In addition, infiltrating T lymphocytes may help spare neuronal loss in models of amyotrophic lateral sclerosis. On the other hand, CNS leukocyte penetration drives experimental autoimmune encephalomyelitis (a mouse model for the human demyelinating disease multiple sclerosis) and may also be pathological in both Parkinson's disease and human immunodeficiency virus encephalitis. A critical understanding of the cellular and molecular mechanisms responsible for trafficking of immune cells from the periphery into the diseased CNS will be key to target these cells for therapeutic intervention in neurodegenerative diseases, thereby allowing neuroregenerative processes to ensue.
Similar content being viewed by others


Introduction
Certain sites in the body, in particular the central nervous system (CNS), eyes, gonads, and maternal–fetal interface, are known to exhibit limited immunological responses. This phenomenon represents a highly regulated active process that is referred to as immune privilege (Wenkel et al. 2000). As the CNS possesses limited regenerative capacity, it is invariably susceptible to damage mediated by inflammation. Accordingly, immune privilege serves to prevent this damage and to maintain neural health (Galea et al. 2007). While multiple mechanisms contribute to immune privilege, restriction of peripheral leukocyte traffic from the periphery to the CNS is a critically important one. Peripheral leukocyte migration and infiltration into the brain parenchyma is tightly regulated at the level of the blood–brain barrier (BBB) (Engelhardt 2008a). Despite this physical barrier and the brain's “privileged” immune status, neuroimmune surveillance by peripheral leukocytes does occur, both physiologically and in instances of disease. Our growing appreciation of the brain–immune interface has given rise to the field of neuroimmunology. In this article, we review the implications of CNS immune cell trafficking for the pathoetiology and potential treatment of neurodegenerative diseases.
Both endogenous and exogenous immune responses impact the healthy and diseased CNS. The endogenous brain immune response chiefly consists of brain-resident innate immune cells known as microglia (Streit et al. 2004; Town et al. 2005a). These myeloid cells are traditionally regarded as the resident macrophages of the CNS and are derived from monocyte precursors during embryogenesis (Alliot et al. 1999; Pessac et al. 2001). Microglia form the first line of defense against invading CNS pathogens and are often the first responders to CNS injury. Another endogenous CNS immune responder is the astrocyte, which is a neuroectoderm-derived cell that has limited innate immune properties, including secretion of certain acute-phase reactants and pro-inflammatory cytokines. The term “reactive gliosis” has been used to describe immune activation of glial cells, including microglia and astrocytes, which exist in a quiescent (resting) state in the healthy CNS. Following neural insult, these cells become activated and undergo hypertrophy accompanied by increased expression of cell surface immune antigens. Concomitantly, both activated microglia and astrocytes synthesize and release a myriad of pro-inflammatory cytokines, chemokines, complement proteins, proteinases, and reactive oxygen species. While this neuroinflammatory response may be beneficial for clearing infection and initiating tissue repair mechanisms, if left unresolved, it exposes sensitive neurons to elevated levels of potentially toxic molecules, leading to bystander injury. In fact, there is mounting evidence that chronic neuroinflammation plays a critical role in the pathoetiology of various neurodegenerative diseases including Alzheimer's disease (AD), multiple sclerosis (MS), and Parkinson's disease (PD; Town et al. 2005a, b; Akiyama et al. 2000; Kim and Joh 2006; Dheen et al. 2007; Town 2009).
In addition to CNS endogenous immunity, discrete populations of exogenous, peripherally derived immune cells can traffic to the CNS, particularly during disease states. These populations are derived from bone marrow precursors, whereas endogenous microglia are mostly replaced by local CNS progenitors (Eglitis and Mezey 1997; Brazelton et al. 2000; Mezey et al. 2000; Priller et al. 2001; Ajami et al. 2007; Mildner et al. 2007). Peripheral immune cell migration into the CNS resembles typical leukocyte extravasation into other organs, involving stochastic induction of mechanisms governing chemoattraction, cellular rolling, adhesion, and diapedesis across the vascular wall. Yet, infiltration of these cells into the CNS is more complex, owing to the existence of the BBB. Recent reports have furthered our understanding of this unique structure, establishing functionally independent roles as a cellular vs. metabolic barrier (Bechmann et al. 2007). Recruitment of peripheral immune cells at the level of the BBB is now known to occur primarily at post-capillary venules (Ransohoff et al. 2003). Infiltration at these sites is believed to involve two distinct events: (1) transmigration across the vascular wall into the perivascular space and (2) progression through the glia limitans (a network of astrocyte foot processes) into the brain parenchyma (Owens et al. 2008). Suppression of any one of the mechanisms governing these events may impede brain entry and consequently subvert immune surveillance.
CNS endogenous and exogenous immunity do not function in isolation from one another; rather, there is a dynamic interplay between these two arms. Following microglial and astrocytic activation, chemokines and cytokines released locally diffuse into the bloodstream thereby attracting leukocytes to the site of inflammation and upregulating the expression of cellular adhesion molecules, which are necessary for attachment and transmigration across post-capillary venules (Engelhardt 2008b). Once migrated across the endothelium, leukocytes enter an enlarged perivascular compartment, known as the Virchow–Robin space. It is in this region that leukocytes are “re-stimulated,” maximizing their invasive potential. However, the vast majority of infiltrating peripheral leukocytes are retained in this compartment and do not penetrate the glia limitans, unless they are recruited to the diseased CNS (Bechmann et al. 2007; Ransohoff et al. 2003; Tran et al. 1998).
Beneficial actions of central nervous system immune cell infiltrates
Infiltrating monocytes/macrophages as an anti-amyloid force in Alzheimer's disease
AD is the most common form of dementia, affecting an estimated 5.2 million Americans in 2008, and prevalence is projected to increase to more than 13.2 million by 2050 (Hebert et al. 2003; Plassman et al. 2007). This chronic, progressive neurodegenerative disorder is pathologically earmarked by: (1) deposition of amyloid-beta (Aβ) peptides as β-amyloid plaques, (2) neuronal injury, and (3) low-level, chronic neuroinflammation characterized by activated glial cells (Fig. 1). As first proposed by Hardy and Allsop (1991), the “amyloid cascade” hypothesis purports that mismetabolism and deposition of Aβ peptides as β-amyloid plaques is the principal etiopathological event in AD, which sets into motion downstream events culminating in neuronal demise. Although neuroinflammation was initially thought to be epiphenomenon, multiple lines of evidence now show that it is directly involved in AD pathoetiology. The most compelling data come from epidemiologic studies showing that exposure to nonsteroidal anti-inflammatory drugs is inversely associated with risk for AD (t' Veld et al. 2001; Szekely et al. 2004, 2007). While microglia are capable of phagocytosing Aβ in vitro, ultrastructural studies in AD patients suggest that these cells are not competent to take up and clear β-amyloid in vivo (Wisniewski et al. 1989, 1991; Frackowiak et al. 1992).

Confocal microscopy reveals activated microglia (green signal) clustered around Aβ deposits (magenta signal) in an 18-month-old Tg2576 mouse model of AD. Reproduced from Town et al. (2008)
Although AD lacks prominent infiltrates of peripheral leukocytes present in prototypical autoimmune neurodegenerative diseases such as MS (Town et al. 2005b; Aisen 2000), AD patients suffering rare comorbidity of stroke have been shown to exhibit brain-infiltrating macrophages. In early pioneering studies, Wisniewski and colleagues demonstrated that these macrophages contained β-amyloid fibrils (Wisniewski et al. 1991; Frackowiak et al. 1992; Jucker and Heppner 2008), suggesting a productive plaque clearance response mediated by these infiltrating innate immune cells. These findings laid the groundwork for recent studies examining the role of peripheral macrophages in β-amyloid clearance from the brain. Specifically, Jucker and Rivest generated bone marrow chimeras to follow peripheral immune cells in transgenic AD mouse models. Following irradiation of the APP23 AD model mouse line and subsequent transplantation of chimeric bone marrow into these animals, they demonstrated that a small percentage (about 1%) of brain-resident microglia were peripheral recruits, and about 20% of β-amyloid plaques were associated with blood-borne macrophages (Jucker and Heppner 2008; Stalder et al. 2005; Simard et al. 2006). While these intriguing reports begged the question of whether a relatively small percentage of brain-infiltrating monocytes/macrophages played a functional role in restricting cerebral amyloidosis, recent studies by Ajami and Mildner have suggested that the act of irradiation, by itself, results in BBB damage and leukocyte recruitment to the otherwise normal CNS (Ajami et al. 2007; Mildner et al. 2007). Further, those authors concluded that local microglial cells accounted for the majority of the innate immune response to CNS injury. Thus, (1) the specificity of monocyte/macrophage infiltration in response to CNS disease and (2) the origin–peripheral vs. CNS–resident of these potentially neurodegenerative disease-limiting macrophages/microglia is somewhat controversial.
Even so, Mildner and colleagues observed mainly chemokine receptor CCR2+ monocytes/macrophages populating CNS sites after irradiation (2007). This observation becomes important when considering a study from El Khoury and colleagues, who crossed an AD mouse model with animals deficient in CCR2, thereby interrupting brain recruitment of peripheral monocytes. These bigenic mice had markedly diminished infiltrates of microglia/macrophages near β-amyloid plaques and demonstrated heavier brain amyloid burden than parental AD model mice (El Khoury et al. 2007). Butovsky and colleagues (2007) took a different approach and selectively ablated dendritic-like innate immune cells by systemic injection of diphtheria toxin in mice genetically engineered to target toxicity to CD11c+ cells. They found that ablation of these CD11c+ cells resulted in greater cerebral amyloid burden, suggesting a normal amyloid clearance role for these immune cells. While these studies suggested that peripheral innate immune cells were an important anti-amyloid force, direct evidence for this was lacking.
Two recent reports provide positive evidence that peripheral monocytes/macropahges can act to restrict β-amyloid plaques. In the first paper (Town et al. 2008), we found that transforming growth factor beta (TGF-β)-Smad 2/3 signaling in innate immune cells serves the maladaptive role of suppressing brain infiltration of peripheral monocytes/macrophages. TGF-β, a pleiotropic polypeptide cytokine, plays a pivotal role in immune suppression and functions to maintain an “immune privileged” environment in the CNS. This is particularly apparent in conditions of neurodegenerative disease such as AD, where TGF-β1 mRNA has been shown to be increased by approximately 3-fold vs. nondemented control brains (Wyss-Coray et al. 1997, 2001). Importantly, genetic blockade of TGF-β-Smad 2/3 signaling in innate immune cells of AD model mice mitigates AD-like pathology, including cerebral amyloidosis and brain inflammation. In this model, TGF-β-Smad 2/3 blockade occurs at the level of peripheral macrophages (and not in brain-resident microglia), resulting in their brain entry (Fig. 2) and clearance of β-amyloid. Thus, it seems that removing an immunosuppressive TGF-β signal to peripheral macrophages “sensitizes” these cells to enter into the brain, where they take up a professional phagocyte role to clear β-amyloid deposits. Importantly, this beneficial anti-amyloid effect does not come at the cost of increased brain inflammation, as these peripheral macrophage immigrants (1) display a CD45+CD11b+Ly-6C- cell surface phenotype known to mark “anti-inflammatory” macrophages (Geissmann et al. 2003) and (2) occur in association with increased anti-inflammatory interleukin (IL)-10 levels in brains of Tg2576 AD model mice crossed with mice deficient in TGF-β-Smad 2/3 innate immune signaling (Town 2009; Town et al. 2008).

Confocal microscopy showing vascular Aβ deposits (green signal) and infiltrating blood-borne macrophages (red signal) in an 18-month-old Tg2576 AD mouse model deficient in TGF-β-Smad 2/3 innate immune signaling. Reproduced from Town et al. (2008)
In a second recent study, Hawkes and McLaurin (2009) performed two complementary in vivo experiments designed to elucidate the putative role of the perivascular macrophage subset in a pathology known as cerebral amyloid angiopathy (CAA; defined as presence of β-amyloid deposits in cerebral vessels), which occurs in 83% of AD cases (Ellis et al. 1996). In one model, they administered liposome-encapsulated clodronate, an intracellular toxin, to the TgCRND8 mouse model of AD in order to deplete perivascular macrophages. Using this approach, they showed that removal of perivascular macrophages was associated with significantly (~5-fold) increased CAA pathology in AD model mice. In a second set of experiments, the authors stimulated perivascular macrophage turnover by administering chitin to TgCRND8 mice. They noted an approximate 3-fold reduction in thioflavin-S positive β-amyloid deposits in cerebral vessels using this experimental approach. Thus, perivascular macrophages (likely of peripheral origin) represent a unique macrophage subset capable of restricting vascular β-amyloid deposits.
Brain recruitment of leukocytes in West Nile virus encephalitis limits infection
West Nile virus (WNV) is a mosquito-transmitted single-stranded RNA virus that is the most common cause of epidemic viral encephalitis in North America and has recently become a pandemic (Campbell et al. 2002; Debiasi and Tyler 2006; Gould and Fikrig 2004; Gubler 2007). WNV is both neurotrophic (capable of infecting neurons) and neurovirulent (damaging to the CNS); yet, most infections in humans are asymptomatic, while the elderly and immunocompromised are particularly at risk for life-threatening brain disease, including meningitis and encephalitis (Campbell et al. 2002; Davis et al. 2006). In the mouse model of lethal WNV encephalitis, animals first develop systemic infection, and WNV then crosses the BBB, culminating in encephalitis and ultimately death within weeks (Wang et al. 2001, 2004). During the initial systemic phase of WNV infection, antiviral immune responses including induction of type I interferons (Anderson and Rahal 2002; Brinton 2001; Gilfoy and Mason 2007), humoral immunity (Diamond et al. 2003a, b), and cellular immunity including γδ T cells (Wang et al. 2003a), CD4+ T cells (Kulkarni et al. 1991; Sitati and Diamond 2006), and CD8+ T cells (Shrestha and Diamond 2004, 2007; Shrestha et al. 2006; Sitati et al. 2007; Wang et al. 2003b) serve to limit viral infectivity.
After establishing a replicative pool in the periphery, WNV migrates into the CNS, usually 4–7 days after infection (Wang et al. 2004; Diamond et al. 2003a, b). Viral CNS penetration typically coincides with encephalitis, characterized by CNS entry of macrophages, CD4+ T cells, CD8+ T cells, natural killer T cells, and dendritic cells (Glass et al. 2005; Fig. 3a). However, it was previously unclear whether CNS infiltration of leukocytes played a deleterious or beneficial role in lethal WNV encephalitis. Glass and coworkers (2005) found that CNS expression of the chemokine receptor CCR5 and its ligand CCL5 were increased following WNV infection of wild-type mice, and this was associated with infiltration of peripheral leukocytes including CD4+ and CD8+ T cells, natural killer cells, and macrophages bearing the CCR5 receptor. These authors undertook a genetic approach to infect CCR5-deficient mice vs. wild-type animals with WNV, and they found that CCR5 deficient animals were more susceptible to lethal WNV encephalitis, had increased brain viral burden, and had markedly reduced brain leukocyte traffic compared to their wild-type counterparts. Interestingly, these authors were able to reverse this pathogenic CCR5-deficient phenotype by adoptively transferring CCR5 wild-type spleen cells into CCR5-deficient mice (Glass et al. 2005). In a related report, McCandless and colleagues (2008) examined brain tissues from mice or humans with WNV encephalitis for expression of the chemokine CXCL12 and its receptor, CXCR4. A prior study in mice showed that expression of CXCL12 at the BBB retained infiltrating monocytes within the perivascular spaces of the CNS microvasculature, thereby limiting monocyte entry into the CNS parenchyma (McCandless et al. 2006). They found a reduction in CXCL12 expression and an increase in CXCR4 levels in WNV-infected brains. To determine whether increased CXCR4 levels following WNV encephalitis restricted CNS leukocyte traffic and thereby promoted viral infectivity, these authors treated mice with a CXCR4 antagonist beginning at the time of WNV infection. They noted increased survival in treated mice following infection that was associated with enhanced migration of WNV-specific CD8+ T cells into the brain parenchyma, which likely neutralized the virus (McCandless et al. 2008).

a Confocal image of a wild-type C57BL/6 mouse brain 6 days after WNV infection. Green signal indicates infiltrating leukocytes, red signal shows WNV-infected brain cells, and blue signal shows DAPI cell nuclei. b Brain flow cytometry results 6 days after WNV infection of wild-type vs. Tlr7-deficient mice. Note the reduction in brain-infiltrating immune cell subsets in Tlr7-deficient mice. Reproduced from Town et al. (2009) with permission from Cell Press
It is interesting that most WNV infection in humans is asymptomatic (Campbell et al. 2002; Davis et al. 2006), underscoring that host immune mechanisms must be in place to suppress WNV infection. We hypothesized that Toll-like receptor 7 (Tlr7), a phylogenetically conserved, germline-encoded pattern recognition receptor that plays a key role in initiating innate immune responses to single-stranded RNA (Qureshi and Medzhitov 2003; Yamamoto et al. 2004; Lund et al. 2004), may serve to limit WNV infectivity. To test this hypothesis, we infected Tlr7-deficient mice with a dose of WNV at which 50% of wild-type mice succumb to mortality (LD50) and found that 91% of Tlr7-deficient mice died. Similarly, infection of MyD88-deficient mice (which lack Tlr7-mediated innate immune responses) with the same dose of WNV resulted in 85% mortality. Both Tlr7- and MyD88-deficient mice had increased viral burden in the blood, brain, and spleen, and while Tlr7-deficient animals manifested increased systemic levels of most pro-inflammatory innate immune cytokines, they had reduced abundance of IL-12/23 mRNA both systemically and in the brain. These effects occurred with reduced homing and infiltration of CD45+ leukocytes and CD11b+ monocytes/macrophages into brains of both Tlr7- (Fig. 3b) and MyD88-deficient mice, despite increased brain viral burden in these knockouts. These data led us to hypothesize that WNV-induced, Tlr7-dependent IL-12 or IL-23 production in the CNS might serve to recruit peripheral monocytes/macrophages to neutralize the virus. Several lines of evidence support this notion: (1) IL-23, but not IL-12, was able to directly induce macrophage chemotaxis in an in vitro migration assay, (2) Tlr7-deficient macrophages had reduced abundance of the shared IL-12/23 receptor subunit (IL-12Rβ1) and failed to home to IL-23 in vitro, and (3) mice-deficient in IL-23, but not IL-12, phenocopied Tlr7 and MyD88 knockouts on reduced survival and leukocyte infiltration and homing to the CNS after challenge with lethal WNV encephalitis (Town et al. 2009; Finberg and Wang 2009; van Ooij 2009). Thus, a model emerges where microglia first detect CNS-invading WNV by using Tlr7 to sense viral single-stranded RNA. By signaling through MyD88, microglial cells then secrete high levels of IL-23, which attracts infiltrating monocytes/macrophages and other leukocytes to the CNS, where they function to neutralize the virus.
West Nile virus “Aikido”: using host innate immunity against the host
While the above evidence clearly implicates CNS leukocyte infiltrates as beneficial in the host's fight against lethal WNV encephalitis, just as in the Japanese martial art “Aikido,” the virus has evolved an elegant form of defense against host innate immunity. We hypothesized that Tlr3, an innate immune pattern recognition receptor that recognizes viral double-stranded RNA (Alexopoulou et al. 2001; Town et al. 2006), may function to militate against WNV infectivity. As a positive single-stranded RNA virus, WNV produces double-stranded RNA during its life cycle, and we predicted that Tlr3-deficient mice would fail to mount an innate immune response to the virus and thus be more susceptible to lethal encephalitis. Strikingly, these knockout mice were resistant (40% survival) to a dose of WNV at which 0% of wild-type mice survived (LD100). Interestingly, Tlr3-deficient mice had increased viral load in the circulation but reduced production of innate immune cytokines including IL-6 and tumor necrosis factor-alpha (TNF-α) versus wild-type counterparts. In parallel, Tlr3 knockouts had reduced viral burden and inflammatory pathology in the brain after WNV infection, and they also showed markedly reduced neuronal pathology and a strikingly intact BBB compared with wild-type mice. It is well known that certain innate immune cytokines, including IL-6 and TNF-α, are able to “permeabilize” the BBB when produced systemically at high levels. To test whether IL-6 or TNF-α responses to WNV infection might act in this way, we infected IL-6- or TNF-α receptor-I-deficient mice with WNV and monitored animals for mortality and BBB compromise. While we did not detect a difference between IL-6-deficient and wild-type control mice, TNF-α receptor I knockouts demonstrated significantly reduced mortality after LD100 WNV challenge associated with reduced leakiness of the BBB (Wang et al. 2004; Diamond and Klein 2004). Thus, while CNS entry of activated leukocytes is a critical host defense mechanism against WNV, the virus has evolved to combat host immunity by using the host's own systemic pro-inflammatory cytokine response to open the BBB in order to gain access to and infect neurons.
Neuroprotective role of peripheral T cell infiltrates in amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is a rapidly progressive, ultimately fatal neuromuscular disease pathologically characterized by degeneration of neurons in the motor cortex, brain stem, and spinal cord (Rowland and Shneider 2001). This primarily late-onset neurodegenerative disease affects an estimated 20,000 Americans, and ~5–10% of these cases are purely heritable forms of the disease (Wijesekera and Leigh 2009). Although the mechanisms underlying motor neuron loss are not completely understood, genetic models of ALS have implicated free radical damage, gliosis, and excitotoxicity as disease-propagating factors (Pasinelli and Brown 2006). The role of the peripheral immune system—specifically infiltrating T cells—in the pathoetiologic neuroinflammatory component of ALS has only recently been examined. Initial reports suggested that the presence of these autoreactive T cells may be epiphenomenon or represent a secondary response with minor deleterious immune effects following neuronal injury (Holmoy 2008).
However, two recent independent reports suggest the converse: that a secondary T cell response may actually be beneficial in the context of ALS. In the first study by Beers and coworkers, ALS model mice expressing mutant copper–zinc superoxide dismutase (SOD1), the most common mutation linked to familial ALS, were crossed with recombination activating gene-2 deficient mice, which lack functional T cells and B cells. Similarly, Chiu and colleagues suppressed T-cell-mediated adaptive immune responses in ALS model mice by crossing SOD1 mutant animals with T-cell receptor β-chain deficient mice, which lack thymocyte development. Interestingly, both bigenic strains in these studies evidenced reduced microgliosis and accelerated rates of neurodegeneration (Beers et al. 2008; Chiu et al. 2008). Thus, these studies suggest that infiltrating CD4+ T cells in particular may confer neuroprotection by shifting the trophic/cytotoxic balance of glia in favor of the former. Interestingly, it was previously shown that (presumed infiltrating) DCs and the chemokine monocyte chemoattractant protein 1 were increased in spinal cord tissue from ALS patients and in the mutant SOD1 mouse model of ALS (Henkel et al. 2004, 2006). It is unclear whether these types of innate immune responses are protective or deleterious in ALS. Yet, combined evidence from the above studies can be interpreted as supporting the notion that promoting productive dialog between the adaptive and innate arms of the immune system via engaging infiltrating CD4+ T cells may militate against ALS pathology. However, it should be noted that this is a relatively new area of exploration, and we await further confirmation of this hypothesis.
Deleterious effects of central nervous system leukocyte traffic
Auto-aggressive T cells cooperate with antigen-presenting cells to initiate demyelination in multiple sclerosis
MS is the most common CNS autoimmune disorder, affecting over 250,000 Americans and over one million individuals worldwide (Steinman 1996). The clinical symptoms of MS are attributable to inflammatory lesions in CNS white matter regions that frequently lead to blindness, loss of motor control, and sensory dysfunction. The pathology of MS is due to an attack on the CNS by the immune system, resulting in demyelination of neuronal axons. Clinical symptoms manifest due to an inability of nerve cells in the brain and spinal cord to communicate via action potentials that propagate down axon fibers. It had long been presumed that antigens present on myelin-producing cells were protected from the immune system owing to residing in the “immune privileged” CNS. However, myelin-reactive T cells that likely recognize a structurally similar viral antigen (known as “molecular mimicry”) are present in the peripheral blood of normal individuals, suggesting that the pathology of the disease may be due to maladaptive CNS entry of autoimmune T cells (Fujinami and Oldstone 1985; Wucherpfennig and Strominger 1995).
The active lesions of MS are characterized by immune cell infiltrates composed of macrophages, T cells, and B cells (Traugott et al. 1982), although it is widely accepted that CNS invasion of encephalitogenic CD4+ T cells is the key pathoetiologic event in MS. Experimental autoimmune encephalomyeltits (EAE) is the most widely used mouse model of MS and is established by immunizing animals with myelin antigen(s) in the presence of adjuvant and pertussis toxin, resulting in (1) development of autoaggressive T cells, (2) CNS leukocyte infiltration, and (3) demyelination in the brain and particularly the spinal cord (Fujinami and Oldstone 1985). Activation of endogenous brain microglia occurs in both human disease and in the EAE mouse model (Greter et al. 2005). Once activated, it is believed that resident microglia chemoattract peripheral pro-inflammatory T helper type 1 (Th1) cells into the CNS. After infiltrating into the CNS, these cells interact with activated microglia, which serve as antigen-presenting cells (APCs; Greter et al. 2005). The importance of T cell/microglial cell interaction in the immunopathology of EAE is underscored by a report from Heppner and colleagues (2005). The authors generated CD11b-HSVTK transgenic mice, which express herpes simplex thymidine kinase in macrophages and microglia, thereby allowing deletion of these cells following ganciclovir treatment. They definitively showed that abrogation of microglial activation inhibited development and maintenance of inflammatory CNS lesions.
Once infiltrated into the CNS of MS patients or EAE model mice, a key molecular event underlying immunopathology is interaction between the CD40 receptor and its cognate ligand, CD40 ligand (CD40L). Activated Th1 cells express CD40L and interact with CD40-bearing reactive microglia in active MS or EAE lesions (Gerritse et al. 1996; Grewal et al. 1996; Tan et al. 1999; Howard et al. 1999; Becher et al. 2001). Early studies indicated that the interaction between CD40+ microglia and CD40L+ T cells initiated production of interferon (IFN)-γ by reactive T cells, which in turn acted as a stimulus for increased expression of CD40 on microglia. This feed-forward mechanism would lead to enhanced production of IFN-γ by activated T cells and TNF-α by reactive microglia, the result of which would be chronic activation of APCs via the CD40 pathway (Tan et al. 1999). This immune interaction is pathogenic, as treatment with anti-CD40L antibody or genetic deficiency in CD40L have both been shown to prevent disease development and reduce disease progression in EAE mice (Gerritse et al. 1996; Grewal et al. 1996; Howard et al. 1999). Further, recent preclinical assessment of therapeutic blocking antibodies against human CD40 in a nonhuman primate model of MS showed efficacy when administered early in disease development as well as after the onset of brain inflammation (‘t Hart et al. 2008). These encouraging results suggest that blockade of the CD40–CD40L immune interaction may be a viable therapeutic approach for MS by opposing pro-inflammatory T-cell activation.
Transfer of an expanded population of myelin-reactive encephalitogenic CD4+ Th1 lymphocytes into naïve recipient mice can also induce EAE. In order to recognize their target, these T cells depend on interaction with APCs expressing self myelin antigen in the context of major histocomptibility complex (MHC) class II. Utilizing a mouse model in which MHC class II expression was restricted to dendritic cells (DCs), Greter and coworkers (2005) demonstrated that DCs are capable of licensing CNS immune invasion. In this system, a population of DCs associated with the meninges and CNS blood vessels permitted encephalitogenic T cells to recognize self-myelin antigen, leading to CNS invasion and subsequent immunopathology. These DCs have been reported in normal human brain and potentially represent an interface between the CNS and the immune system in the context of EAE-type brain inflammation (Matyszak and Perry 1996). Interestingly, TGF-β signaling likely acts as an immunosuppressive signal to DCs, thereby mitigating DC-induced CNS leukocyte infiltration. Specifically, targeted functional inactivation of the TGF-β receptor in DCs results in a high frequency of T cells in the CNS along with increased levels of pro-inflammatory cytokines and acute-phase reactants in EAE mice (Laouar et al. 2008).
In addition to CNS infiltration of CD4+ T cell subsets, CD8+ T cell infiltrates are present during the terminal stage of EAE (Laouar et al. 2008). In early work, Sun and colleagues showed that a CD8+ T cell subset could suppress induction of EAE in Lewis rats, suggesting that these cells were counter-regulatory to encephalitogenic Th1 cells (Sun et al. 1988). However, mice deficient in CD8 show less mortality but a higher frequency of relapse than wild-type animals, suggesting that CD8+ T lymphocytes may contribute as both effectors and regulators in EAE (Koh et al. 1992). While it is generally accepted that MHC class II-bearing APCs and Th cells are the main players in the pathogenesis of MS, new evidence has implicated MHC class I, namely human leukocyte antigen-A3 (HLA-A3) and HLA-A3-restricted CD8+ T cells in the pathogenesis of MS (Friese et al. 2008). These results provide a molecular mechanism for the contribution of CD8+ T cells to the pathogenesis of EAE and MS, as interaction between CD8+ T cells and APCs bearing MHC class I molecules reveals a complex network of MHC interactions that may contribute to onset of EAE and MS. The description of myelin-derived peptides that are MHC class I restricted and recognized by CD8+ T cells represents a new avenue toward exploration of the role of CD8+ T cells in demyelinating diseases.
Interleukin-12 versus Interleukin-23 and Interleukin-17 in the pathogenesis of demyelinating disease
IL-12 plays a dominant role in orchestrating differentiation of Th1 effectors and mediating Th1-associated autoimmune diseases (Adorini 1999; Adorini et al. 1997). IL-12 is a heterodimeric protein (known as p70) secreted predominantly by activated APCs and consists of a pair of disulfide-linked subunits designated p40 and p35 (Becher et al. 2002). However, the “common chain” p40 subunit of IL-12 is shared with another heterodimeric cytokine, IL-23, which is comprised of p40 and p19 subunits (Oppmann et al. 2000). This sharing of the p40 subunit between full-length IL-12 and IL-23 heterodimers has made it difficult to determine the specific contribution of these cytokines to MS and EAE. However, using gene-targeting approaches, Becher and colleagues (2002) demonstrated that mice deficient in the p35 subunit of IL-12 were susceptible while p40 null mice were resistant to EAE. In a later complementary study, Cua and coworkers (2003) demonstrated that mice specifically lacking IL-23, and not IL-12, were resistant to EAE. These authors found that the interaction between IL-23-producing CNS DCs and T cells regulates encephalitogenicity during the effector phase of EAE (Cua et al. 2003). When taken together, these studies strongly implicate IL-23—and not IL-12—in EAE immunopathogenesis.
There is also mounting evidence supporting the role of IL-23 in the immunopathogenesis of MS. For example, IL-23 expression is elevated in DCs from MS patients compared to healthy donors (Vaknin-Dembinsky et al. 2006). Furthermore, IL-23 promotes expression of the cytokine IL-17, resulting in differentiation of CD4+ T helper 17 (Th17) effector cells from naïve T cells (Luger et al. 2008). Th17 lymphocytes are capable of transmigrating across BBB endothelial cells expressing IL-17 and are now regarded as a pathogenic T cell in MS due to their ability to functionally interact with and recruit multiple pathogenic leukocyte subsets to the CNS, including DCs and CD4+ lymphocytes (Ifergan et al. 2008; Kebir et al. 2007). Yet, it is now generally thought that Th1 and Th17 lymphocyte subsets are independently capable of inducing CNS autoimmune disease (Steinman 2008). However, there likely exists a complicated paradigm where APCs producing IL-23 and other T-cell polarizing cytokines promote Th1 and Th17 effector responses, resulting in CNS trafficking of multiple autoaggressive leukocyte subsets.
Role of peripheral immune cell infiltrates in Parkinson's disease
PD is the most common movement disorder, affecting over one million Americans and over four million individuals worldwide (Dorsey et al. 2007). After AD, it is the next most common progressive neurodegenerative disorder affecting elderly individuals over 65 years of age (Dorsey et al. 2007). Pathologically, PD is characterized by: (1) preferential loss of dopaminergic neurons, (2) the presence of Lewy bodies (cytoplasmic inclusions composed principally of abnormal α-synuclein), and (3) reactive gliosis. These three pathological hallmarks all occur within a small region of the midbrain known as the substantia nigra pars compacta (SNpc). While genetic and pharmacologic models of the disease have established oxidative stress, mitochondrial, and proteosomal dysfunction as disease-perpetrating events, the mechanism mediating dopaminergic deficit in sporadic PD remains unknown (Dauer and Przedborski 2003). The search for a PD pathoetiologic mechanism has uncovered the immune system—particularly innate neuroinflammatory responses—as a potential candidate (Czlonkowska et al. 2002; Whitton 2007). In addition, recent reports have also implicated the adaptive immune system in PD pathogenesis.
Although PD is not generally considered an autoimmune disease, a recent study by Benner and colleagues (2008) demonstrated that modified “self” epitopes such as nitrated α-synuclein (N-α-Syn) can overcome immunological tolerance and activate peripheral leukocytes in cervical lymph nodes following administration of the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) to wild-type mice. In addition, adoptive transfer of T cells from syngeneic donors immunized with N-α-Syn exacerbated dopaminergic neuronal loss following exposure to MPTP. Importantly, these purified CD3+ populations of T cells were capable of trafficking to and infiltrating the SNpc (Benner et al. 2008). In accordance with these findings and those of the seminal work of McGeer and coworkers (1988), Brochard and colleagues (2009) reported that T cells are largely recruited to the SNpc in the brains of patients with PD. These brain-invading lymphocytes consisted of a heterogeneous population of both CD8+ and CD4+ cells. A similar profile of peripheral leukocyte infiltration was also present in MPTP mouse models of PD, further validating the relevance of the model to the human syndrome (Brochard et al. 2009). Interestingly, both this experimental model and the clinical syndrome exhibit BBB dysfunction (Kortekaas et al. 2005). However, it should be noted that while the BBB appears to be “leaky” following exposure to MPTP, it is unlikely that this phenomenon is sufficient to grant peripheral leukocytes direct access to the diseased brain. Rather, a dysfunctional BBB is likely one of many steps involved in recruitment of peripheral immune cells to the PD brain.
To ascertain which T cell subsets might contribute to PD pathogenesis, Brochard and colleagues (2009) went on to examine dopaminergic neuronal loss in mice deficient in one or both subsets of T cells following MPTP intoxication. Interestingly, mice lacking complete cell-mediated immunity or CD4+, but not CD8+, T cells were protected against MPTP-induced injury, suggesting a pathogenic role of CD4+ T cells in the MPTP mouse model of PD. Upon further elucidation of this pathogenic role, CD4+ T cells were found to arbitrate dopaminergic neuronal loss via a Fas/FasL-dependent mechanism (Brochard et al. 2009). In addition, a recent report by Reynolds and colleagues further substantiated the detrimental activities of CD4+/CD25− cells by demonstrating the ability of these effector T cells to exacerbate microgliosis following exposure to N-α-Syn. This study also examined the potentially beneficial activities of infiltrating CD4+/CD25+ regulatory T cells (Tregs). These CNS infiltrating Tregs were capable of suppressing microgliosis by secreting cytokines that mitigate inflammatory responses or by directly inducing microglial apoptosis via a similar Fas/FasL-dependent mechanism as described above (Reynolds et al. 2009). While studies investigating the role of Tregs in PD have not definitively established protective vs. deleterious activities at various stages during disease, it is likely that the Treg lymphocyte subset has the potential to be mobilized to abrogate PD pathology (Reynolds et al. 2007, 2009). Taken together, these results suggest the existence of highly regulated, adaptive immunopathogenic mechanisms at work in PD that may ultimately be harnessed as therapeutic(s).
Detrimental role of peripheral immune cell infiltrates in HIV-associated neurocognitive dysfunction
Neurological disease arising from human immunodeficiency virus (HIV) remains a frequent complication of this viral infection. Specifically, HIV-associated neurocognitive dysfunction (HAND), which in itself is comprised of a spectrum of neurological impairments, continues to plague patients with advanced HIV infection despite the advent of highly active antiretroviral therapy (Ghafouri et al. 2006; Reger et al. 2002). Although the pathological mechanisms responsible for HAND are unclear, an abundance of clinical and laboratory investigations suggest that HIV encephalitis (HIVE), advancing age, and even comorbid neurodegenerative disease may interact in an additive or synergistic manner resulting in the clinical phenotype of this disorder (Ghafouri et al. 2006; Gonzalez-Scarano and Martin-Garcia 2005). It is well known that peripheral HIV-infected monocytes/macrophages infiltrate the CNS, resulting in reactive gliosis, formation of multinucleated giant cells, and neuronal loss characteristic of HIVE (Ghafouri et al. 2006; Gonzalez-Scarano and Martin-Garcia 2005; Budka 1991). However, the role of invading T-cell populations in HIVE-mediated neurodegeneration has only recently been investigated.
Two studies by Petito and coworkers examined infiltrating T lymphocytes in the brains of end-stage-acquired immunodeficiency syndrome (AIDS) patients with and without HIVE. In their earlier work, they found that hippocampal effector/memory CD45RO+ T lymphocytes were significantly increased in individuals with HIVE. Interestingly, these CD4+ and CD8+ T cells were observed in direct contact with neurons, underscoring the potential for lymphocyte-mediated neuronal injury or possibly trans-receptor-mediated neuronal infection (Petito et al. 2003). In their follow-up report, these authors observed cytotoxic CD8+ T cells in the parenchyma of AIDS patients with HIVE. The perineuronal localization of these T cells and their associated cytotoxic granules again suggested T lymphocyte-mediated neuronal killing (Petito et al. 2006). As indirect evidence of this pathogenic mechanism, Liu and colleagues demonstrated that adoptive transfer of Tregs attenuated reactive gliosis, viral replication, and neuronal loss in an HIVE mouse model. Importantly, adoptive transfer of effector T cells in this model had contrary effects (Liu et al. 2009). Although it is generally accepted that Tregs suppress antiviral immune responses in the periphery, it is likely that these same responses may directly or indirectly target neurons and even exacerbate HIV dissemination in the CNS. Therefore, specifically gating the subsets of T lymphocytes that are granted access to the HIV-infected brain may help resolve infection and consequently prevent or treat HAND.
Concluding remarks
In this review, we have explored both beneficial and deleterious impacts of peripheral immune cell traffic into the CNS depending on disease state. The AD brain responds to pathology by increasing “immune privilege” barriers that restrict CNS leukocyte traffic, including upregulation of TGF-β1 expression. While TGF-β1 is directly neurotrophic (Tesseur et al. 2006), this response may ultimately be maladaptive as it precludes brain entry of peripheral monocytes/macrophages. Overcoming this barrier by blocking innate immune TGF-β-Smad 2/3 signaling results in robust brain entry of peripheral macrophages and restriction of cerebral amyloidosis. In WNV encephalitis, infiltration of peripheral leukocytes, including CD4+ and CD8+ T cells and monocytes/macrophages, into the CNS also serves a beneficial role to seek out and neutralize the virus, thereby limiting CNS infectivity. However, the host's frontline innate immune defense is also capitalized upon by the virus to permeabilize the BBB, allowing viral entry into the CNS. Thus, aggressive pro-inflammatory innate immune responses must be kept in check to limit WNV infection of the CNS and to minimize neuronal bystander injury. In models of ALS, CD4+ T lymphocytes infiltrate the CNS where they may fine-tune glial responses to promote a neuroprotective microenvironment. In the context of MS and the EAE mouse model, infiltration of autoaggressive leukocytes including Th1 and Th17 CD4+ T cell subsets promotes brain inflammation and associated CNS injury. Thus, the pathology of MS and EAE underscores the importance of restricting “runaway autoimmunity” leading to CNS lesions. The role of peripheral leukocyte traffic to the brain is just beginning to be explored in PD, which was classically thought to be a neurodegenerative disease without an immune component. However, CD4+ and CD8+ T cells are detected in PD patient brains, and deletion of CD4+ T cells rescues mice from PD-like effects of MPTP intoxication. Finally, the impact of cytotoxic T-cell infiltration in HAND is viewed with increasing interest, as CD8+ T cells and their cytotoxic granules are present in the brains of late-stage AIDS patients with HIVE, suggesting T lymphocyte-mediated neuronal loss.
What emerges from studies reviewed in this article is that peripheral immune responses in the CNS are neither inherently beneficial nor damaging. However, controversy still surrounds whether peripheral immune responses directed toward self-antigen in the CNS are beneficial, a concept known as “autoimmune neuroprotection” or “protective autoimmunity” (Schwartz and Kipnis 2005). A number of recent reports touch on this debate, suggesting that early autoaggressive immune responses may accelerate the pathogenesis of various neurodegenerative diseases (Liu et al. 2009; Reynolds et al. 2009; Planas and Chamorro 2009). These and other studies also find that curbing this response via Treg-mediated immunosuppression affords protection against neurodegeneration, as appears to be the case with EAE, PD and HIVE (Liu et al. 2009; Reynolds et al. 2007; Kohm et al. 2002). Conversely, an alternative view suggests that dysregulation of autoaggressive immune responses may allow neurodegenerative mechanisms to persist. This is apparent in studies examining the adverse activities of Tregs in models of viral encephalitis and ALS (Bai et al. 2009; Goldstein et al. 2009). Failure to engage the appropriate immune responses may prevent resolution of neuroinsults, resulting in a common feature of these neurodegenerative diseases: chronicity. In the final analysis, CNS leukocyte traffic may serve either beneficial or deleterious roles, depending on the brain disease state (Fig. 4). A critical understanding of the cellular and molecular mechanisms responsible for this intriguing and important interface between the CNS and the immune system will likely lead to novel therapeutics in the future.

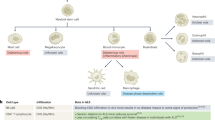
Model for detrimental versus beneficial roles of CNS immune cell traffic. See text for references
References
Adorini L (1999) Interleukin-12, a key cytokine in Th1-mediated autoimmune diseases. Cell Mol Life Sci 55:1610–1625
Adorini L, Aloisi F, Galbiati F et al (1997) Targeting IL-12, the key cytokine driving Th1-mediated autoimmune diseases. Chem Immunol 68:175–197
Aisen PS (2000) Anti-inflammatory therapy for Alzheimer's disease. Neurobiol Aging 21:447–448 discussion 451-443
Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–1543
Akiyama H, Barger S, Barnum S et al (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21:383–421
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413:732–738
Alliot F, Godin I, Pessac B (1999) Microglia derive from progenitors, originating from the yolk sac, and which proliferate in the brain. Brain Res Dev Brain Res 117:145–152
Anderson JF, Rahal JJ (2002) Efficacy of interferon alpha-2b and ribavirin against West Nile virus in vitro. Emerg Infect Dis 8:107–108
Bai F, Town T, Qian F, et al. (2009) IL-10 signaling blockade controls murine West Nile virus infection. PLoS Pathogens, in press
Becher B, Durell BG, Miga AV, Hickey WF, Noelle RJ (2001) The clinical course of experimental autoimmune encephalomyelitis and inflammation is controlled by the expression of CD40 within the central nervous system. J Exp Med 193:967–974
Becher B, Durell BG, Noelle RJ (2002) Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest 110:493–497
Bechmann I, Galea I, Perry VH (2007) What is the blood-brain barrier (not)? Trends Immunol 28:5–11
Beers DR, Henkel JS, Zhao W, Wang J, Appel SH (2008) CD4+ T cells support glial neuroprotection, slow disease progression, and modify glial morphology in an animal model of inherited ALS. Proc Natl Acad Sci U S A 105:15558–15563
Benner EJ, Banerjee R, Reynolds AD et al (2008) Nitrated alpha-synuclein immunity accelerates degeneration of nigral dopaminergic neurons. PLoS ONE 3:e1376
Brazelton TR, Rossi FM, Keshet GI, Blau HM (2000) From marrow to brain: expression of neuronal phenotypes in adult mice. Science 290:1775–1779
Brinton MA (2001) Host factors involved in West Nile virus replication. Ann N Y Acad Sci 951:207–219
Brochard V, Combadiere B, Prigent A et al (2009) Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J Clin Invest 119:182–192
Budka H (1991) Neuropathology of human immunodeficiency virus infection. Brain Pathol 1:163–175
Butovsky O, Kunis G, Koronyo-Hamaoui M, Schwartz M (2007) Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer's disease model. Eur J NeuroSci 26:413–416
Campbell GL, Marfin AA, Lanciotti RS, Gubler DJ (2002) West Nile virus. Lancet Infect Dis 2:519–529
Chiu IM, Chen A, Zheng Y et al (2008) T lymphocytes potentiate endogenous neuroprotective inflammation in a mouse model of ALS. Proc Natl Acad Sci USA 105:17913–17918
Cua DJ, Sherlock J, Chen Y et al (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421:744–748
Czlonkowska A, Kurkowska-Jastrzebska I, Czlonkowski A, Peter D, Stefano GB (2002) Immune processes in the pathogenesis of Parkinson's disease - a potential role for microglia and nitric oxide. Med Sci Monit 8:RA165–177
Dauer W, Przedborski S (2003) Parkinson's disease: mechanisms and models. Neuron 39:889–909
Davis LE, DeBiasi R, Goade DE et al (2006) West Nile virus neuroinvasive disease. Ann Neurol 60:286–300
Debiasi RL, Tyler KL (2006) West Nile virus meningoencephalitis. Nat Clin Pract Neurol 2:264–275
Dheen ST, Kaur C, Ling EA (2007) Microglial activation and its implications in the brain diseases. Curr Med Chem 14:1189–1197
Diamond MS, Klein RS (2004) West Nile virus: crossing the blood-brain barrier. Nat Med 10:1294–1295
Diamond MS, Shrestha B, Marri A, Mahan D, Engle M (2003a) B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J Virol 77:2578–2586
Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M (2003b) A critical role for induced IgM in the protection against West Nile virus infection. J Exp Med 198:1853–1862
Dorsey ER, Constantinescu R, Thompson JP et al (2007) Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 68:384–386
Eglitis MA, Mezey E (1997) Hematopoietic cells differentiate into both microglia and macroglia in the brains of adult mice. Proc Natl Acad Sci U S A 94:4080–4085
El Khoury J, Toft M, Hickman SE et al (2007) Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med 13:432–438
Ellis RJ, Olichney JM, Thal LJ et al (1996) Cerebral amyloid angiopathy in the brains of patients with Alzheimer's disease: the CERAD experience, Part XV. Neurology 46:1592–1596
Engelhardt B (2008a) The blood-central nervous system barriers actively control immune cell entry into the central nervous system. Curr Pharm Des 14:1555–1565
Engelhardt B (2008b) Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci 274:23–26
Finberg RW, Wang JP (2009) Antiviral responses: different roles for different tolls. Immunity 30:173–175
Frackowiak J, Wisniewski HM, Wegiel J, Merz GS, Iqbal K, Wang KC (1992) Ultrastructure of the microglia that phagocytose amyloid and the microglia that produce beta-amyloid fibrils. Acta Neuropathol 84:225–233
Friese MA, Jakobsen KB, Friis L et al (2008) Opposing effects of HLA class I molecules in tuning autoreactive CD8+ T cells in multiple sclerosis. Nat Med 14:1227–1235
Fujinami RS, Oldstone MB (1985) Amino acid homology between the encephalitogenic site of myelin basic protein and virus: mechanism for autoimmunity. Science 230:1043–1045
Galea I, Bechmann I, Perry VH (2007) What is immune privilege (not)? Trends Immunol 28:12–18
Geissmann F, Jung S, Littman DR (2003) Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity 19:71–82
Gerritse K, Laman JD, Noelle RJ et al (1996) CD40-CD40 ligand interactions in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A 93:2499–2504
Ghafouri M, Amini S, Khalili K, Sawaya BE (2006) HIV-1 associated dementia: symptoms and causes. Retrovirology 3:28
Gilfoy FD, Mason PW (2007) West Nile virus-induced interferon production is mediated by the double-stranded RNA-dependent protein kinase PKR. J Virol 81:11148–11158
Glass WG, Lim JK, Cholera R, Pletnev AG, Gao JL, Murphy PM (2005) Chemokine receptor CCR5 promotes leukocyte trafficking to the brain and survival in West Nile virus infection. J Exp Med 202:1087–1098
Goldstein MR, Mascitelli L, Pezzetta F (2009) The double-edged sword of statin immunomodulation. Int J Cardiol 135:128–130
Gonzalez-Scarano F, Martin-Garcia J (2005) The neuropathogenesis of AIDS. Nat Rev Immunol 5:69–81
Gould LH, Fikrig E (2004) West Nile virus: a growing concern? J Clin Invest 113:1102–1107
Greter M, Heppner FL, Lemos MP et al (2005) Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 11:328–334
Grewal IS, Foellmer HG, Grewal KD et al (1996) Requirement for CD40 ligand in costimulation induction, T cell activation, and experimental allergic encephalomyelitis. Science 273:1864–1867
Gubler DJ (2007) The continuing spread of West Nile virus in the western hemisphere. Clin Infect Dis 45:1039–1046
Hardy J, Allsop D (1991) Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci 12:383–388
Hawkes CA, McLaurin J (2009) Selective targeting of perivascular macrophages for clearance of beta-amyloid in cerebral amyloid angiopathy. Proc Natl Acad Sci U S A 106:1261–1266
Hebert LE, Scherr PA, Bienias JL, Bennett DA, Evans DA (2003) Alzheimer disease in the US population: prevalence estimates using the 2000 census. Arch Neurol 60:1119–1122
Henkel JS, Engelhardt JI, Siklos L et al (2004) Presence of dendritic cells, MCP-1, and activated microglia/macrophages in amyotrophic lateral sclerosis spinal cord tissue. Ann Neurol 55:221–235
Henkel JS, Beers DR, Siklos L, Appel SH (2006) The chemokine MCP-1 and the dendritic and myeloid cells it attracts are increased in the mSOD1 mouse model of ALS. Mol Cell Neurosci 31:427–437
Heppner FL, Greter M, Marino D et al (2005) Experimental autoimmune encephalomyelitis repressed by microglial paralysis. Nat Med 11:146–152
Holmoy T (2008) T cells in amyotrophic lateral sclerosis. Eur J Neurol 15:360–366
Howard LM, Miga AJ, Vanderlugt CL et al (1999) Mechanisms of immunotherapeutic intervention by anti-CD40L (CD154) antibody in an animal model of multiple sclerosis. J Clin Invest 103:281–290
Ifergan I, Kebir H, Bernard M et al (2008) The blood-brain barrier induces differentiation of migrating monocytes into Th17-polarizing dendritic cells. Brain 131:785–799
in t' Veld BA, Ruitenberg A, Hofman A et al (2001) Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med 345:1515–1521
Jucker M, Heppner FL (2008) Cerebral and peripheral amyloid phagocytes–an old liaison with a new twist. Neuron 59:8–10
Kebir H, Kreymborg K, Ifergan I et al (2007) Human TH17 lymphocytes promote blood-brain barrier disruption and central nervous system inflammation. Nat Med 13:1173–1175
Kim YS, Joh TH (2006) Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson's disease. Exp Mol Med 38:333–347
Koh DR, Fung-Leung WP, Ho A, Gray D, Acha-Orbea H, Mak TW (1992) Less mortality but more relapses in experimental allergic encephalomyelitis in CD8-/- mice. Science 256:1210–1213
Kohm AP, Carpentier PA, Anger HA, Miller SD (2002) Cutting edge: CD4+CD25+ regulatory T cells suppress antigen-specific autoreactive immune responses and central nervous system inflammation during active experimental autoimmune encephalomyelitis. J Immunol 169:4712–4716
Kortekaas R, Leenders KL, van Oostrom JC et al (2005) Blood-brain barrier dysfunction in parkinsonian midbrain in vivo. Ann Neurol 57:176–179
Kulkarni AB, Mullbacher A, Blanden RV (1991) Functional analysis of macrophages, B cells and splenic dendritic cells as antigen-presenting cells in West Nile virus-specific murine T lymphocyte proliferation. Immunol Cell Biol 69(Pt 2):71–80
Laouar Y, Town T, Jeng D et al (2008) TGF-beta signaling in dendritic cells is a prerequisite for the control of autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 105:10865–10870
Liu J, Gong N, Huang X, Reynolds AD, Mosley RL, Gendelman HE (2009) Neuromodulatory activities of CD4+CD25+ regulatory T cells in a murine model of HIV-1-associated neurodegeneration. J Immunol 182:3855–3865
Luger D, Silver PB, Tang J et al (2008) Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med 205:799–810
Lund JM, Alexopoulou L, Sato A et al (2004) Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A 101:5598–5603
Matyszak MK, Perry VH (1996) The potential role of dendritic cells in immune-mediated inflammatory diseases in the central nervous system. Neuroscience 74:599–608
McCandless EE, Wang Q, Woerner BM, Harper JM, Klein RS (2006) CXCL12 limits inflammation by localizing mononuclear infiltrates to the perivascular space during experimental autoimmune encephalomyelitis. J Immunol 177:8053–8064
McCandless EE, Zhang B, Diamond MS, Klein RS (2008) CXCR4 antagonism increases T cell trafficking in the central nervous system and improves survival from West Nile virus encephalitis. Proc Natl Acad Sci U S A 105:11270–11275
McGeer PL, Itagaki S, Akiyama H, McGeer EG (1988) Rate of cell death in parkinsonism indicates active neuropathological process. Ann Neurol 24:574–576
Mezey E, Chandross KJ, Harta G, Maki RA, McKercher SR (2000) Turning blood into brain: cells bearing neuronal antigens generated in vivo from bone marrow. Science 290:1779–1782
Mildner A, Schmidt H, Nitsche M et al (2007) Microglia in the adult brain arise from Ly-6ChiCCR2+ monocytes only under defined host conditions. Nat Neurosci 10:1544–1553
Oppmann B, Lesley R, Blom B et al (2000) Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13:715–725
Owens T, Bechmann I, Engelhardt B (2008) Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol 67:1113–1121
Pasinelli P, Brown RH (2006) Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci 7:710–723
Pessac B, Godin I, Alliot F (2001) Microglia: origin and development. Bull Acad Natl Med 185:337–346 discussion 346-337
Petito CK, Adkins B, McCarthy M, Roberts B, Khamis I (2003) CD4+ and CD8+ cells accumulate in the brains of acquired immunodeficiency syndrome patients with human immunodeficiency virus encephalitis. J Neurovirol 9:36–44
Petito CK, Torres-Munoz JE, Zielger F, McCarthy M (2006) Brain CD8+ and cytotoxic T lymphocytes are associated with, and may be specific for, human immunodeficiency virus type 1 encephalitis in patients with acquired immunodeficiency syndrome. J Neurovirol 12:272–283
Planas AM, Chamorro A (2009) Regulatory T cells protect the brain after stroke. Nat Med 15:138–139
Plassman BL, Langa KM, Fisher GG et al (2007) Prevalence of dementia in the United States: the aging, demographics, and memory study. Neuroepidemiology 29:125–132
Priller J, Flugel A, Wehner T et al (2001) Targeting gene-modified hematopoietic cells to the central nervous system: use of green fluorescent protein uncovers microglial engraftment. Nat Med 7:1356–1361
Qureshi ST, Medzhitov R (2003) Toll-like receptors and their role in experimental models of microbial infection. Genes Immun 4:87–94
Ransohoff RM, Kivisakk P, Kidd G (2003) Three or more routes for leukocyte migration into the central nervous system. Nat Rev Immunol 3:569–581
Reger M, Welsh R, Razani J, Martin DJ, Boone KB (2002) A meta-analysis of the neuropsychological sequelae of HIV infection. J Int Neuropsychol Soc 8:410–424
Reynolds AD, Banerjee R, Liu J, Gendelman HE, Mosley RL (2007) Neuroprotective activities of CD4+CD25+ regulatory T cells in an animal model of Parkinson's disease. J Leukoc Biol 82:1083–1094
Reynolds AD, Stone DK, Mosley RL, Gendelman HE (2009) Proteomic Studies of Nitrated Alpha-Synuclein Microglia Regulation by CD4+CD25+ T Cells. J Proteome Res 8:3497–3511
Rowland LP, Shneider NA (2001) Amyotrophic lateral sclerosis. N Engl J Med 344:1688–1700
Schwartz M, Kipnis J (2005) Protective autoimmunity and neuroprotection in inflammatory and noninflammatory neurodegenerative diseases. J Neurol Sci 233:163–166
Shrestha B, Diamond MS (2004) Role of CD8+ T cells in control of West Nile virus infection. J Virol 78:8312–8321
Shrestha B, Diamond MS (2007) Fas ligand interactions contribute to CD8+ T-cell-mediated control of West Nile virus infection in the central nervous system. J Virol 81:11749–11757
Shrestha B, Samuel MA, Diamond MS (2006) CD8+ T cells require perforin to clear West Nile virus from infected neurons. J Virol 80:119–129
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49:489–502
Sitati EM, Diamond MS (2006) CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J Virol 80:12060–12069
Sitati E, McCandless EE, Klein RS, Diamond MS (2007) CD40-CD40 ligand interactions promote trafficking of CD8+ T cells into the brain and protection against West Nile virus encephalitis. J Virol 81:9801–9811
Stalder AK, Ermini F, Bondolfi L et al (2005) Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci 25:11125–11132
Steinman L (1996) Multiple sclerosis: a coordinated immunological attack against myelin in the central nervous system. Cell 85:299–302
Steinman L (2008) A rush to judgment on Th17. J Exp Med 205:1517–1522
Streit WJ, Mrak RE, Griffin WS (2004) Microglia and neuroinflammation: a pathological perspective. J Neuroinflammation 1:14
Sun D, Ben-Nun A, Wekerle H (1988) Regulatory circuits in autoimmunity: recruitment of counter-regulatory CD8+ T cells by encephalitogenic CD4+ T line cells. Eur J Immunol 18:1993–1999
Szekely CA, Thorne JE, Zandi PP et al (2004) Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology 23:159–169
Szekely CA, Town T, Zandi PP (2007) NSAIDs for the chemoprevention of Alzheimer's disease. Subcell Biochem 42:229–248
‘t Hart BA, Hintzen RQ, Laman JD (2008) Preclinical assessment of therapeutic antibodies against human CD40 and human interleukin-12/23p40 in a nonhuman primate model of multiple sclerosis. Neurodegener Dis 5:38–52
Tan J, Town T, Paris D et al (1999) Activation of microglial cells by the CD40 pathway: relevance to multiple sclerosis. J Neuroimmunol 97:77–85
Tesseur I, Zou K, Esposito L et al (2006) Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology. J Clin Invest 116:3060–3069
Town T (2009) Alternative Abeta immunotherapy approaches for Alzheimer's disease. CNS Neurol Disord Drug Targets 8:114–127
Town T, Nikolic V, Tan J (2005a) The microglial "activation" continuum: from innate to adaptive responses. J Neuroinflammation 2:24
Town T, Tan J, Flavell RA, Mullan M (2005b) T-cells in Alzheimer's disease. Neuromolecular Med 7:255–264
Town T, Jeng D, Alexopoulou L, Tan J, Flavell RA (2006) Microglia Recognize Double-Stranded RNA via TLR3. J Immunol 176:3804–3812
Town T, Laouar Y, Pittenger C et al (2008) Blocking TGF-beta-Smad2/3 innate immune signaling mitigates Alzheimer-like pathology. Nat Med 14:681–687
Town T, Bai F, Wang T et al (2009) Toll-like receptor 7 mitigates lethal West Nile encephalitis via interleukin 23-dependent immune cell infiltration and homing. Immunity 30:242–253
Tran EH, Hoekstra K, van Rooijen N, Dijkstra CD, Owens T (1998) Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J Immunol 161:3767–3775
Traugott U, Scheinberg LC, Raine CS (1982) Multiple sclerosis: heterology among early T cells and Tg cells. Ann Neurol 11:182–186
Vaknin-Dembinsky A, Balashov K, Weiner HL (2006) IL-23 is increased in dendritic cells in multiple sclerosis and down-regulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol 176:7768–7774
van Ooij C (2009) Innate immunity: Not-so-lucky 7 for West Nile virus. Nat Rev Micro 7:1
Wang T, Anderson JF, Magnarelli LA, Wong SJ, Koski RA, Fikrig E (2001) Immunization of mice against West Nile virus with recombinant envelope protein. J Immunol 167:5273–5277
Wang T, Scully E, Yin Z et al (2003a) IFN-gamma-producing gamma delta T cells help control murine West Nile virus infection. J Immunol 171:2524–2531
Wang Y, Lobigs M, Lee E, Mullbacher A (2003b) CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J Virol 77:13323–13334
Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA (2004) Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med 10:1366–1373
Wenkel H, Streilein JW, Young MJ (2000) Systemic immune deviation in the brain that does not depend on the integrity of the blood-brain barrier. J Immunol 164:5125–5131
Whitton PS (2007) Inflammation as a causative factor in the aetiology of Parkinson's disease. Br J Pharmacol 150:963–976
Wijesekera LC, Leigh PN (2009) Amyotrophic lateral sclerosis. Orphanet J Rare Dis 4:3
Wisniewski HM, Wegiel J, Wang KC, Kujawa M, Lach B (1989) Ultrastructural studies of the cells forming amyloid fibers in classical plaques. Can J Neurol Sci 16:535–542
Wisniewski HM, Barcikowska M, Kida E (1991) Phagocytosis of beta/A4 amyloid fibrils of the neuritic neocortical plaques. Acta Neuropathol 81:588–590
Wucherpfennig KW, Strominger JL (1995) Molecular mimicry in T cell-mediated autoimmunity: viral peptides activate human T cell clones specific for myelin basic protein. Cell 80:695–705
Wyss-Coray T, Masliah E, Mallory M et al (1997) Amyloidogenic role of cytokine TGF-beta1 in transgenic mice and in Alzheimer's disease. Nature 389:603–606
Wyss-Coray T, Lin C, Yan F et al (2001) TGF-beta1 promotes microglial amyloid-beta clearance and reduces plaque burden in transgenic mice. Nat Med 7:612–618
Yamamoto M, Takeda K, Akira S (2004) TIR domain-containing adaptors define the specificity of TLR signaling. Mol Immunol 40:861–868
Acknowledgements
The authors would like to thank K. Pattie Figueroa, Cedars-Sinai Medical Center, and Jun Tan, University of South Florida, for helpful discussion. This work was supported by the National Institutes of Health/National Institute on Aging (4R00AG029726-02 and 5R00AG029726-03, to T. Town) and faculty start-up funds to T. Town from the Department of Neurosurgery and the Department of Biomedical Sciences at Cedars-Sinai Medical Center. T. Town is the inaugural holder of the Ben Winters Endowed Chair in Regenerative Medicine.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Rezai-Zadeh, K., Gate, D. & Town, T. CNS Infiltration of Peripheral Immune Cells: D-Day for Neurodegenerative Disease?. J Neuroimmune Pharmacol 4, 462–475 (2009). https://doi.org/10.1007/s11481-009-9166-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-009-9166-2
Keywords
- brain
- central nervous system
- neuroinflammation
- neuroimmunology
- leukocyte
- lymphocyte
- regulatory T cell
- monocyte
- macrophage
- cytokine
- chemokine
- transforming growth factor
- tumor necrosis factor
- interleukin-17
- interleukin-23
- Alzheimer's disease
- Parkinson's disease
- West Nile encephalitis
- multiple sclerosis
- experimental autoimmune encephalomyelitis
- human immunodeficiency virus
- amyotrophic lateral sclerosis