
Abstract
In cancer, Wnt/β-catenin signaling is ubiquitously referred to as an “oncogenic” pathway that promotes tumor progression. This review examines how the regulation and downstream effects of Wnt/β-catenin signaling in cancer varies depending on cellular context, with a focus on malignant melanoma. We emphasize that the cellular homeostasis of Wnt/β-catenin signaling may represent a more appropriate concept than the simplified view of the Wnt/β-catenin pathway as either oncogenic or tumor-suppressing. Ultimately, a more refined understanding of the contextual regulation of Wnt/β-catenin signaling will be essential for addressing if and how therapeutic targeting of this pathway could be leveraged for patient benefit.
Similar content being viewed by others

Introduction
At least two overlapping signaling pathways are regulated by the family of secreted glycoproteins known as Wnts [1]. The highly conserved “canonical” Wnt/β-catenin signaling pathway is activated by the binding of Wnt ligand to the receptors frizzled (FZD) and low-density lipoprotein receptor–related protein 5/6 (LRP5/6), triggering a series of downstream events that culminate in the cytosolic accumulation and nuclear translocation of the multifunctional protein β-catenin. Interaction of β-catenin with transcription factors of the TCF and LEF family results in the regulation of certain target genes that mediate the ultimate effects of this pathway on cellular processes including cell fate, proliferation, and migration. There are also one or more “non-canonical” or β-catenin–independent Wnt signaling pathways that are less well understand, and that act in a β-catenin–independent manner leading to changes to cytoskeletal dynamics, adhesion, and motility [1]. Interestingly, β-catenin–independent Wnt signaling can antagonize Wnt/β-catenin signaling in development [2], regeneration [3], and cancer [4, 5], highlighting the complex interplay between downstream effectors of Wnt signaling.
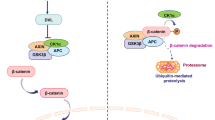
Wnt pathways have been intimately linked to cancer ever since the original realization that the mouse mammary oncogene int-1 is a homologue of the Drosophila wingless (Wg) gene [6], resulting in the portmanteau family designation of “Wnt.” Subsequent studies have implicated Wnt signaling in almost every major disease and cancer model, reflecting the importance of major developmental pathways in the pathogenesis of adult disease processes [7, 8]. Most dramatically, almost all colorectal carcinomas harbor inactivating mutations in the gene for adenomatous polyposis coli (APC), which forms a complex with AXIN and glycogen synthase kinase 3-β (GSK3B) that normally phosphorylates β-catenin to target the protein for proteasomal degradation. Mutations or loss of APC in colorectal carcinoma therefore prevent degradation of β-catenin and consequently lead to constitutive activation of the pathway. Further studies using both cell-based models and transgenic animal models have validated the essential role of Wnt dysregulation in the formation of colorectal cancer, establishing this disease paradigm as a primary model for studying the molecular mechanisms of Wnt/β-catenin signaling in oncogenesis [9, 10].
Since the initial demonstration that Wnt signaling regulates the stability and translocation of β-catenin [11], the immunohistochemical detection of nuclear β-catenin in both laboratory models and in patient tumors has been widely employed as a surrogate for demonstrating activation of the Wnt/β-catenin pathway [1]. In several cancer models including colorectal carcinoma, breast cancer, and esophageal carcinoma, the presence of nuclear β-catenin in cancer tissue compared to normal tissue has implicated this signaling pathway in cancer biology. Further studies have observed that the presence of nuclear β-catenin can predict decreased survival in these cancers, solidifying the importance of this pathway in oncogenesis and in cancer progression.
Not surprisingly, Wnt/β-catenin signaling has also been implicated in a broad variety of noncancerous medical conditions. Genetic polymorphisms in LRP5/6 that decrease Wnt/β-catenin signaling have been linked to altered bone density [12], metabolic syndrome [13], and to Alzheimer’s disease [14]. In normal tissues and organs, without genetic polymorphisms or mutations, Wnt/β-catenin signaling is activated in every animal that displays regeneration, and β-catenin signaling is also activated in traumatic brain injury, which does not display extensive regeneration [15]. Moreover, it is clear that attenuating β-catenin signaling delays regeneration while augmenting β-catenin signaling often enhances the rate of regeneration, as determined by analysis of tail fin regeneration in zebrafish, and liver regeneration in both mouse and zebrafish [16]. Given that regeneration employs progenitor cells it should come as no surprise that Wnts regulate embryonic stem cells, though there is not a consensus on the precise roles.
The increasing body of literature on Wnt/β-catenin signaling in disease has generated tremendous interest in the potential therapeutic targeting of this pathway. Until recently, the only modulator of Wnt/β-catenin signaling approved by the US Food and Drug Administration was lithium chloride, which prevents the degradation of β-catenin by inhibiting its phosphorylation by GSK3B. More recent studies have identified small molecule activators as well as inhibitors of Wnt/β-catenin signaling that may eventually have therapeutic utility in patients [17, 18, 19••, 20, 21, 22••].
In parallel with studies on Wnt/β-catenin signaling in cancer and other diseases, substantial progress has also been made in understanding how this pathway regulates developmental processes such as melanocyte differentiation. Wnt/β-catenin signaling is a major regulator of the pigmented cell lineage, playing a major role in determining the fate of neural crest cells and its derivative pigment cell lineages. Wnt/β-catenin signaling directly regulates the expression of microphthalmia transcription factor (MITF), a major determinant of both melanocyte development and melanoma progression [23–25]. Wnt3a ligand is one of only three factors required to differentiate a pluripotent human embryonic stem cell into a functional melanocyte, further highlighting the critical role of this pathway in pigment cell biology [26]. Not surprisingly, the Wnt/β-catenin pathway has been implicated in the pathogenesis of both benign melanocytic nevi as well as in malignant melanoma.
Not as Oncogenic as We Thought
In the last two decades since the initial identification of activated Wnt/β-catenin signaling in the murine breast cancer model, the role of this pathway in promoting proliferation has fostered the prevailing view that Wnt/β-catenin signaling is uniformly “oncogenic” [27]. Does the existing literature support that Wnt/β-catenin signaling is truly “oncogenic” in melanoma? The Wnt/β-catenin pathway would not fit the original definition of an oncogene as a gene or pathway that causes cancer when aberrantly activated, since the forced expression of a melanocyte-specific, nondegradable, constitutively active β-catenin mutant in either transgenic or Cre/lox systems is not enough to induce melanoma in mice [28]. As our understanding of cancer has advanced, the term “oncogenic” has seemingly broadened to include any gene or pathway implicated in cancer progression, a benchmark that is more difficult to define. The finding that constitutive activation of Wnt/β-catenin signaling increases the proliferation of murine melanoma cells in vitro, accompanied by MITF-dependent increases in clonogenic growth, implicates this pathway as a promoter of melanoma progression [29]. Likewise, the activation of this pathway acts in concert with activation of Ras to promote increased tumor formation in transgenic mice, further suggesting a role in tumor promotion [28]. In contrast to these studies in cell culture and mouse-based models, there have been several recent reports that activation of Wnt/β-catenin signaling in patient tumors, as monitored by increased levels of nuclear β-catenin, correlates with an improved rather than poorer prognosis [5, 30–32]. Consistent with the data seen in human patients, forced expression of Wnt3a in B16 melanoma cells leads to decreased proliferation in vitro and in vivo, along with the upregulation of genes associated with melanocyte differentiation that are frequently lost with melanoma progression [5]. Furthermore, almost all benign nevi are positive for nuclear β-catenin, and studies have observed a loss of nuclear β-catenin with melanoma progression to metastases [31]. Since the rate of transformation of nevi to melanomas is estimated to be very low [33], these collective observations from patients bring up the possibility that Wnt/β-catenin signaling may not be oncogenic in any sense, but rather is required to maintain a homeostatic balance that, when disrupted or lost, can lead to early melanoma transformation.
Another interesting aspect of Wnt/β-catenin signaling in melanoma involves β-catenin–independent Wnt signaling activated in many (but not all) contexts by WNT5A. WNT5A was first linked to disease as a gene that was more highly expressed in aggressive and late-stage melanomas [34], and the immunohistochemical detection of high WNT5A in tumors was subsequently correlated with decreased patient survival [35]. Studies on WNT5A in melanoma have focused largely on the effects of this ligand on cell motility, based on the precedent that WNT5A regulates the movement of cells during the convergent-extension phase of vertebrate gastrulation [2, 36]. Interestingly, at least two studies have shown that WNT5A inhibits the transcription of Wnt/β-catenin target genes in melanoma, recapitulating the ability of β-catenin–independent Wnt signaling to inhibit Wnt/β-catenin signaling that was first observed in developmental models [4, 5]. This finding suggests that the acquisition of increased WNT5A with later-stage tumors may be involved with the inhibition and/or loss of Wnt/β-catenin signaling seen during melanoma progression from benign nevi to metastases.
If one looks at the available data from patient-based studies of tumor tissue, the notion that Wnt/β-catenin signaling may not be universally oncogenic is supported by studies in a growing number of diseases. In the case of medulloblastoma, all patients with extensive nuclear β-catenin staining had a 5-year overall survival rate of 92.3% versus a rate of 65.3% in patients with nucleonegative tumors [37]. Furthermore, in a study of 72 children, all six children who had an activating mutation in the CTNNB1 gene were alive and free of disease 5 years following diagnosis, compared to a survival rate of 53.7% for the CTNNB1 wild-type group [38•]. In addition, these favorable activating mutations of β-catenin correlate with a gene expression profile that distinguishes it from other medulloblastoma subtypes [39]. More limited studies in prostate cancer [40], ovarian cancer [41], and even in late-stage colorectal carcinoma [42] also show that the presence of active Wnt/β-catenin signaling is correlated with improved patient outcomes.
Ultimately, debates on whether Wnt/β-catenin signaling is oncogenic are less relevant than understanding whether the therapeutic activation of this pathway could be beneficial for various diseases, including melanoma and other cancers. Interestingly, forced activation of Wnt/β-catenin signaling in the setting of Kras-driven murine pancreatic cancer models can antagonize the development of pancreatic intraepithelial neoplasia lesions, and drive the development of tumors that resemble more benign human solid pseudopapillary neoplasms rather than more malignant pancreatic ductal adenocarcinoma [43••, 44]. This type of intriguing observation invites the hypothesis that Wnt/β-catenin signaling can be leveraged clinically to prevent aggressive tumors at the expense of potentially acquiring tumors that are more benign and treatable. Similarly, the improved survival seen with activating β-catenin mutations in medulloblastoma, which is largely a Hedgehog-driven tumor, may also reflect the ability of Wnt/β-catenin signaling to promote cell fates that are less aggressive, a hypothesis supported by observations with genome-wide transcriptional profiling of patient tumors. Although in theory the idea of activating Wnt/β-catenin signaling in cancer patients could be achieved, further studies will be needed to address the timing, strength, and duration of Wnt/β-catenin signaling required for the desired outcome.
In melanoma, activation of Wnt/β-catenin signaling through the forced expression of Wnt3a or through treatment of cells with soluble Wnt3a ligand results in the decreased tumor cell proliferation in vitro and in vivo, as well as the transcriptional upregulation of genes associated with melanocyte differentiation [5]. Some of these genes, such as Trpm1/melastatin, are lost with melanoma progression. The ability of Wnt/β-catenin signaling to drive the expression of certain differentiation-related genes likely reflects its role as a major regulator of melanocyte development. Further studies will ultimately address whether the therapeutic activation of Wnt/β-catenin signaling in human melanoma patients is either practicable or beneficial.
Wnt and Cellular Homeostasis
Perhaps the time has come when we should not categorize signaling pathways as either oncogenic or tumor-suppressive, but instead view them in the context of cellular homeostasis. Most definitions of cellular homeostasis include the property of cells to maintain a viable and healthy state through the constant adjustment of various inputs such as biochemical pathways. In this context, it is not hard to envision a role for Wnt/β-catenin signaling in maintaining cell fate and regulating proliferation for both cancerous and noncancerous cells. The transcriptional response of Wnt/β-catenin signaling itself illustrates the importance of ongoing homeostasis, since many of the classical β-catenin–dependent target genes, including DKK1, AXIN2, TCF7, and LEF1 , can function as feedback inhibitors to modulate the pathway.
There are several well-studied examples where Wnt/β-catenin signaling maintains normal adult tissue, including in intestinal crypts and in hair follicle units. Benign melanocytic nevi are similar, since they are almost all positive by immunohistochemistry for nuclear β-catenin. Taking into consideration that nevi are thought (based on histology of patient tumors) to be precursors for a significant portion of melanomas, it is not so surprising that in some systems, the presence of active Wnt/β-catenin signaling may be needed for melanoma development. The dysregulation of Wnt/β-catenin homeostasis is reflected in the observation that nuclear β-catenin decreases with melanoma progression. Further studies will be needed to determine whether this loss of signaling represents a cause or effect of melanoma-genesis.
Unlike colon cancer, where mutation or loss of APC plays a primary role in constitutive activation of the pathway, studies have found that mutations leading to constitutive Wnt/β-catenin signaling in melanoma are rare. As a result, it is thought that the activation of Wnt/β-catenin signaling in benign nevi and melanomas is likely driven by Wnt ligand, either secreted by tumor cells themselves or from cells in the surrounding environment. Based on studies in cell-based systems, the signaling induced by secreted Wnt ligand is more susceptible to regulation than constitutive activation of the pathway by downstream mutations, suggesting that from a therapeutic point of view, the cause of activated Wnt/β-catenin signaling in different cancer contexts is an essential consideration. Additionally, in cases like melanoma where Wnt/β-catenin homeostasis is largely mediated by Wnt ligand, the role of this pathway in cancer progression may be more difficult to dissect since it likely involves complex interactions between Wnt/β-catenin signaling and other pathways activated by genetic amplification, deletion, or mutation.
Conclusions
Accumulating evidence suggests that Wnt/β-catenin signaling is subject to regulation by cellular, temporal, and spatial contexts that make it difficult to generalize the end results of pathway activation or inhibition. To this end, it is overly simplistic to think of the pathway as oncogenic or tumor-suppressive. While it is easy to draw conclusions from experiments in cell culture dishes or in mouse models, these findings should be reconciled particularly with data on Wnt/β-catenin activation in human tumors.
In studying the role of Wnt/β-catenin signaling in melanoma, we need to address two critical questions. First, how well do the mouse models of melanoma recapitulate what is seen in patient tumors with regard to the loss of Wnt/β-catenin signaling during tumor progression? Second, how reliable is our evaluation of Wnt/β-catenin activation in patient tumors using nuclear β-catenin as our sole measurement? The context-specific nature of Wnt/β-catenin signaling may preclude the development of universally applicable assays based on gene targets, but further studies could identify context-specific readouts that could be used in melanoma or other diseases to validate the presence or absence of nuclear β-catenin and provide a robust indicator of pathway activation in tumors. By developing reliable models and a faithful set of cell and tissue assays for human diseases, we can begin to fully grasp the cellular, spatial, and temporal contexts that determine the consequences of Wnt/β-catenin homeostasis and its dysregulation during disease.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Chien AJ, Conrad WH, Moon RT: A Wnt survival guide: from flies to human disease. J Invest Dermatol 2009, 129:1614–1627.
Torres MA, Yang-Snyder JA, Purcell SM, et al.: Activities of the Wnt-1 class of secreted signaling factors are antagonized by the Wnt-5A class and by a dominant negative cadherin in early Xenopus development. J Cell Biol 1996, 133:1123–1137.
Stoick-Cooper CL, Weidinger G, Riehle KJ, et al.: Distinct Wnt signaling pathways have opposing roles in appendage regeneration. Development 2007, 134:479–489.
Dissanayake SK, Olkhanud PB, O’Connell MP, et al.: Wnt5A regulates expression of tumor-associated antigens in melanoma via changes in signal transducers and activators of transcription 3 phosphorylation. Cancer Res 2008, 68:10205–10214.
Chien AJ, Moore EC, Lonsdorf AS, et al.: Activated Wnt/beta-catenin signaling in melanoma is associated with decreased proliferation in patient tumors and a murine melanoma model. Proc Natl Acad Sci U S A 2009, 106:1193–1198.
Rijsewijk F, Schuermann M, Wagenaar E, et al.: The Drosophila homolog of the mouse mammary oncogene int-1 is identical to the segment polarity gene wingless. Cell 1987, 50:649–657.
Moon RT, Kohn AD, De Ferrari GV, Kaykas A: WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet 2004, 5:691–701.
Chien AJ, Moon RT. WNTS and WNT receptors as therapeutic tools and targets in human disease processes. Front Biosci 2007, 12:448–457.
Moon RT, Bowerman B, Boutros M, Perrimon N: The promise and perils of Wnt signaling through beta-catenin. Science 2002, 296:1644–1646.
Maretto S, Cordenonsi M, Dupont S, et al.: Mapping Wnt/beta-catenin signaling during mouse development and in colorectal tumors. Proc Natl Acad Sci U S A 2003, 100:3299–3304.
Yost C, Torres M, Miller JR, et al.: The axis-inducing activity, stability, and subcellular distribution of beta-catenin is regulated in Xenopus embryos by glycogen synthase kinase 3. Genes Dev 1996, 10:1443–1454.
van Meurs JB, Trikalinos TA, Ralston SH, et al.: Large-scale analysis of association between LRP5 and LRP6 variants and osteoporosis. JAMA 2008, 299:1277–1290.
Mani A, Radhakrishnan J, Wang H, et al.: LRP6 mutation in a family with early coronary disease and metabolic risk factors. Science 2007, 315:1278–1282.
De Ferrari GV, Papassotiropoulos A, Biechele T, et al.: Common genetic variation within the low-density lipoprotein receptor-related protein 6 and late-onset Alzheimer’s disease. Proc Natl Acad Sci U S A 2007, 104:9434–9439.
White BD, Nathe RJ, Maris DO, et al.: Beta-catenin signaling increases in proliferating NG2+ progenitors and astrocytes during post-traumatic gliogenesis in the adult brain. Stem Cells 2010, 28:297–307.
Stoick-Cooper CL, Moon RT, Weidinger G: Advances in signaling in vertebrate regeneration as a prelude to regenerative medicine. Genes Dev 2007, 21:1292–1315.
Zhang Q, Major MB, Takanashi S, et al.: Small-molecule synergist of the Wnt/beta-catenin signaling pathway. Proc Natl Acad Sci U S A 2007, 104:7444–7448.
Teo JL, Ma H, Nguyen C, et al.: Specific inhibition of CBP/beta-catenin interaction rescues defects in neuronal differentiation caused by a presenilin-1 mutation. Proc Natl Acad Sci U S A 2005, 102:12171–12176.
••Huang SM, Mishina YM, Liu S, et al.: Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 2009, 461:614–620. Tankyrase provides a novel means for small molecule–based inhibition of Wnt/β-catenin signaling, and the novel compound XAV939 identified in this study will serve as an important tool for studying Wnt in different model systems.
Gandhirajan RK, Staib PA, Minke K, et al.: Small molecule inhibitors of Wnt/beta-catenin/lef-1 signaling induces apoptosis in chronic lymphocytic leukemia cells in vitro and in vivo. Neoplasia 2010, 12:326–335.
Fujii N, You L, Xu Z, et al.: An antagonist of dishevelled protein-protein interaction suppresses beta-catenin-dependent tumor cell growth. Cancer Res 2007, 67:573–579.
••Chen B, Dodge ME, Tang W, et al.: Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer. Nat Chem Biol 2009, 5:100–107. Using chemical genetic screening, these authors identified two classes of compounds that could inhibit Wnt signaling through either regulation of Wnt processing or regulation of AXIN stability.
Dorsky RI, Raible DW, Moon RT: Direct regulation of nacre, a zebrafish MITF homolog required for pigment cell formation, by the Wnt pathway. Genes Dev 2000, 14:158–162.
Dorsky RI, Moon RT, Raible DW: Environmental signals and cell fate specification in premigratory neural crest. Bioessays 2000, 22:708–716.
Dorsky RI, Moon RT, Raible DW: Control of neural crest cell fate by the Wnt signalling pathway. Nature 1998, 396:370–373.
Fang D, Leishear K, Nguyen TK, et al.: Defining the conditions for the generation of melanocytes from human embryonic stem cells. Stem Cells 2006, 24:1668–1677.
Thomas DM: Wnts, bone and cancer. J Pathol 2010, 220:1–4.
Delmas V, Beermann F, Martinozzi S, et al.: Beta-catenin induces immortalization of melanocytes by suppressing p16INK4a expression and cooperates with N-Ras in melanoma development. Genes Dev 2007, 21:2923–2935.
Widlund HR, Horstmann MA, Price ER, et al.: Beta-catenin-induced melanoma growth requires the downstream target Microphthalmia-associated transcription factor. J Cell Biol 2002, 158:1079–1087.
Maelandsmo GM, Holm R, Nesland JM, et al.: Reduced beta-catenin expression in the cytoplasm of advanced-stage superficial spreading malignant melanoma. Clin Cancer Res 2003, 9:3383–3388.
Kageshita T, Hamby CV, Ishihara T, et al.: Loss of beta-catenin expression associated with disease progression in malignant melanoma. Br J Dermatol 2001, 145:210–216.
Bachmann IM, Straume O, Puntervoll HE, et al.: Importance of P-cadherin, beta-catenin, and Wnt5a/frizzled for progression of melanocytic tumors and prognosis in cutaneous melanoma. Clin Cancer Res 2005, 11:8606–8614.
Tsao H, Bevona C, Goggins W, Quinn T: The transformation rate of moles (melanocytic nevi) into cutaneous melanoma: a population-based estimate. Arch Dermatol 2003, 139:282–288.
Bittner M, Meltzer P, Chen Y, et al.: Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000, 406:536–540.
Da Forno PD, Pringle JH, Hutchinson P, et al.: WNT5A expression increases during melanoma progression and correlates with outcome. Clin Cancer Res 2008, 14:5825–5832.
Moon RT, Campbell RM, Christian JL, et al.: Xwnt-5A: a maternal Wnt that affects morphogenetic movements after overexpression in embryos of Xenopus laevis. Development 1993, 119:97–111.
Ellison DW, Onilude OE, Lindsey JC, et al.: beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. J Clin Oncol 2005, 23:7951–7957.
•Fattet S, Haberler C, Legoix P, et al.: Beta-catenin status in paediatric medulloblastomas: correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J Pathol 2009, 218:86–94. This study confirmed previous work showing that activating mutations of β-catenin (CTNNB1) can impressively predict improved patient prognosis, providing strong evidence that activation of Wnt signaling in medulloblastoma is not “oncogenic.”
Kool M, Koster J, Bunt J, et al.: Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 2008, 3:e3088.
Horvath LG, Henshall SM, Lee CS, et al.: Lower levels of nuclear beta-catenin predict for a poorer prognosis in localized prostate cancer. Int J Cancer 2005, 113:415–422.
Gamallo C, Palacios J, Moreno G, et al.: beta-Catenin expression pattern in stage I and II ovarian carcinomas: relationship with beta-catenin gene mutations, clinicopathological features, and clinical outcome. Am J Pathol 1999, 155:527–536.
Elzagheid A, Buhmeida A, Korkeila E, et al.: Nuclear beta-catenin expression as a prognostic factor in advanced colorectal carcinoma. World J Gastroenterol 2008, 14:3866–3871.
••Morris JP, Cano DA, Sekine S, et al.: Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest 2010, 120:508–520. Using an established transgenic mouse model for pancreatic adenocarcinoma, these authors show that targeted activation of Wnt/β-catenin signaling can block the formation of pre-neoplastic lesions mediated by activation of Kras, which is a hallmark event in almost all pancreatic ductal adenocarcinoma.
Heiser PW, Cano DA, Landsman L, et al.: Stabilization of beta-catenin induces pancreas tumor formation. Gastroenterology 2008, 135:1288–1300.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Lucero, O.M., Dawson, D.W., Moon, R.T. et al. A Re-evaluation of the “Oncogenic” Nature of Wnt/β-catenin Signaling in Melanoma and Other Cancers. Curr Oncol Rep 12, 314–318 (2010). https://doi.org/10.1007/s11912-010-0114-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11912-010-0114-3